

Abstract
Microbial communities in engineered terrestrial haloalkaline environments have been poorly characterized relative to their natural counterparts and are geologically recent in formation, offering opportunities to explore microbial diversity and assembly in dynamic, geochemically comparable contexts. In this study, the microbial community structure and geochemical characteristics of three geographically dispersed bauxite residue environments along a remediation gradient were assessed and subsequently compared with other engineered and natural haloalkaline systems. In bauxite residues, bacterial communities were similar at the phylum level (dominated by Proteobacteria and Firmicutes) to those found in soda lakes, oil sands tailings, and nuclear wastes; however, they differed at lower taxonomic levels, with only 23% of operational taxonomic units (OTUs) shared with other haloalkaline environments. Although being less diverse than natural analogues, bauxite residue harbored substantial novel bacterial taxa, with 90% of OTUs nonmatchable to cultured representative sequences. Fungal communities were dominated by Ascomycota and Basidiomycota, consistent with previous studies of hypersaline environments, and also harbored substantial novel (73% of OTUs) taxa. In bauxite residues, community structure was clearly linked to geochemical and physical environmental parameters, with 84% of variation in bacterial and 73% of variation in fungal community structures explained by environmental parameters. The major driver of bacterial community structure (salinity) was consistent across natural and engineered environments; however, drivers differed for fungal community structure between natural (pH) and engineered (total alkalinity) environments. This study demonstrates that both engineered and natural terrestrial haloalkaline environments host substantial repositories of microbial diversity, which are strongly shaped by geochemical drivers.
INTRODUCTION
Highly alkaline, saline environments pose numerous challenges for microbes, including maintaining a neutral cytoplasmic pH, regulating intracellular osmotic potential, and obtaining sufficient quantities of nutrients. The combination of stresses imposed by high-pH, high-salt environments require unique adaptations for survival and growth (1, 2). Extreme terrestrial, naturally formed alkaline and saline (haloalkaline) environments such as soda lakes and hot springs are now recognized as hot spots of microbial diversity (3–5), the investigation of which has yielded novel species and functional capacities (6–9) and reshaped our current understanding of microbial taxonomy, phylogeny, and evolutionary relationships (3, 4, 10–14) as well as enabling new, biotechnological applications (11, 15, 16). In comparison, anthropogenic, engineered haloalkaline environments, such as mine wastes and tailings facilities, have been poorly characterized to date and present substantial potential for the discovery of novel extremophiles and evolutionary lineages (16–21) as well as contributing to an expanded understanding of global microbial biogeography. Few studies have examined the composition of microbial communities in engineered terrestrial haloalkaline environments, and none have compared microbial communities among similar engineered haloalkaline environments. Furthermore, no study to date has compared microbial communities in engineered haloalkaline environments to those of their naturally formed counterparts. It is therefore unknown at present to what extent communities in engineered haloalkaline environments resemble those in natural haloalkaline environments and whether they share common drivers of microbial community structure. The low initial microbial diversity typically observed in mine wastes and tailings (22–25) provides an opportunity to study successional processes in extremophilic microbial communities and may lead to greater understanding of the origins and environmental drivers of high microbial diversity in geologically older, naturally developed extreme environments. Given the growing global production of tailings (7,125 × 109 kg [7,125 million tons {Mt}]/year worldwide [26]), characterization of microbial communities inhabiting engineered environments is also of importance in remediation and management.
Defining global microbial biogeographical patterns has been facilitated by the advent of culture-independent DNA-based methods for profiling microbial communities (27, 28). Although environmental microbial communities are well known to be heterogeneous, they are not necessarily random, as geochemical drivers of community structure are commonly observed (29–31). Salinity has been identified as the major driver of bacterial community composition and diversity across a wide variety of natural terrestrial environments, including those posing multiple stressors (hot springs, soda lakes, Arctic glaciers) (32). High-salinity environments such as soda lakes are associated with moderate to high species level bacterial diversity (5, 13, 32, 33), especially in microbial mats at lake edges, where a diversity of geochemical niches exist over small spatial scales and support highly diverse communities containing many species yet to be described and classified (3, 4, 34). Although no global-scale analysis of environmental drivers of fungal community composition and diversity exists, regional-scale studies identify pH as the major driver of fungal community structure in haloalkaline environments, whereas salinity dominates in circumneutral systems (35, 36). If geochemical parameters are important drivers of community structure in natural haloalkaline systems, then salinity and pH, respectively, should also be key drivers of bacterial and fungal community composition and diversity in engineered haloalkaline environments. Further, halo(alkali-)philic and tolerant (extremophile) species can be expected to dominate microbial communities inhabiting engineered haloalkaline environments.
This study uses bauxite residue (alumina refining tailings) as a model engineered haloalkaline environment in which to compare microbial community structures between natural and engineered systems and identify common environmental drivers of microbial community structure across these systems. Bauxite residue is produced globally in large volumes (estimated to be 70 to 120 Mt/year, of a total of 7,125 Mt/year of all tailings produced worldwide [26, 37]) across every continent except Antarctica. Bauxite residue is an alkaline (average pH 11.3), saline (average electrical conductivity [EC], 7.4 mS cm−1) tailings material produced during alumina refining, mostly composed of iron oxides (hematite, goethite), quartz, and titanium dioxides (rutile, anatase) (38). Major ions in pore water include Na+, Al(OH)4−, SO42−, CO32, and OH−, with minimal K+, Ca2+, and Mg2+ (39), giving it a geochemical profile similar to that of natural (sulfated) athalassohaline soda lakes. The objective of this study was to establish whether relationships exist between environmental characteristics and microbial community structure in extremely alkaline, saline, terrestrial engineered environments, consistent with those previously observed in natural alkaline, saline environments, through cultivation-independent approaches. To achieve this objective, samples from eight bauxite residue sites spanning a remediation gradient (see Materials and Methods for details) were directly assessed for both geochemical characteristics and microbial community structure (cultivation-independent techniques). Results from bauxite residue were then included into a larger data set incorporating published results from natural sulfated athalassohaline salt lakes, steel slag uranium mill tailings, and chromite ore processing residue as well as including a further direct assessment of an oil sands tailings residue sample (Fort McMurray, Alberta, Canada). We expected that microbial communities in bauxite residue would show a community structure similar to those of their geologically older, natural counterparts and other engineered haloalkaline environments, including a high proportion of novel operational taxonomic units (OTUs; matching reference database sequences at <97% similarity) within communities, and that salinity and pH would be identified as major drivers of bacterial and fungal community structure in bauxite residue, as observed in natural haloalkaline systems.
MATERIALS AND METHODS
Site history and sampling sites.
Bauxite residue samples were collected from three tailings storage facilities in Germany, Ireland, and Australia. Sample names were allocated sequentially as BR1 to BR8 along a remediation gradient (with “BR” standing for “bauxite residue”) according to their remediation status: unremediated (fresh, unamended tailings), poorly remediated (some weathering or amendments applied, but still no or patchy vegetation, lack of soil structure), and well remediated (weathering/amendments applied, with good vegetation cover or development of soil structure, as well as lower pH, salinity, and sodicity) (Table 1). An unremediated site (BR1, BR2) and a poorly remediated site (BR4, BR5) were sampled in Germany; a poorly remediated site under a sand cap was sampled in Australia (BR3, BR6); and a well-remediated site that had received compost, gypsum, sand, and tillage was sampled in Ireland (BR7, BR8) (Table 1). Sampling across a remediation gradient provided a range of environmental conditions as well as enabling investigation of microbial community succession during remediation. At all field storage facilities, samples for microbial community analyses were collected aseptically and immediately transferred to sterile, sealed containers on ice (for bulk community 16S rRNA sequencing). Samples were also collected for chemical and physical analyses (Table 1; see also Table S1 in the supplemental material); these were packed into plastic containers and shipped at ambient temperature.
TABLE 1.
Selected geochemical and physical properties of bauxite residue samplesa
| Sample name | Remediation status | Depth (cm) | Country | Age (yr) | pH | EC (μS cm−1) | TA (mol H+ kg−1) | W Na (% wt) | ESP (%) | MC (% wt) | Gravel (% wt) | Sand (% wt) | Silt (% wt) | Clay (% wt) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BR1 | Unremediated | 0–2 | Germany | 0 | 10.62 | 6,000 | 2.02 | 1.98 | 87 | 29 | 14 | 34 | 37 | 15 |
| BR2 | Unremediated | 90–100 | Germany | 0 | 10.95 | 3,720 | 1.06 | 1.32 | 85 | 35 | 17 | 24 | 40 | 19 |
| BR3 | Poorly remediated | 140–150 | Australia | 40 | 10.18 | 1,334 | 14.04 | 0.56 | 72 | 36 | 10 | 52 | 30 | 8 |
| BR4 | Poorly remediated | 90–100 | Germany | 1 | 4.12 | 1,964 | — | 0.57 | 10 | 66 | 11 | 65 | 24 | 1 |
| BR5 | Poorly remediated | 0–2 | Germany | 1 | 7.93 | 634 | — | 0.35 | 99 | 85 | 13 | 69 | 17 | 1 |
| BR6 | Poorly remediated | 0–2 | Australia | 40 | 9.72 | 342 | 0.14 | 0.08 | 3 | 4 | 11 | 82 | 7 | — |
| BR7 | Well remediated | 140–150 | Ireland | 15 | 10.32 | 1,100 | 1.14 | 0.17 | 16 | 30 | 20 | 47 | 25 | 8 |
| BR8 | Well remediated | 0–2 | Ireland | 15 | 7.67 | 512 | 0.66 | 0.04 | — | 34 | 14 | 73 | 12 | 1 |
Symbols and abbreviations: —, below detection limit; EC, electrical conductivity; TA, total alkalinity; W Na, water-extractable sodium; ESP, exchangeable sodium percentage; MC, moisture content. Additional properties are listed in Table S1 in the supplemental material.
DNA extraction, amplification, and sequencing.
Microbial DNA was extracted (MoBio PowerSoil DNA isolation kit, Carlsbad, CA) from frozen samples, and PCR was performed using modified versions of the universal bacterial 16S primers 27F (5′ AGR GTT TGA TCM TGG CTC AG 3′ [40]) and 519R (5′ GTN TTA CNG CGG CKG CTG 3′ [41]), the universal fungal primers ITS1 and ITS4 (42), multiplex identifiers (MIDs), and the HotStarTaq Plus master mix kit (Qiagen, Valencia, CA). The PCR was performed under the following conditions: 94°C (180 s); 28 cycles of 94°C (30 s), 53°C (40 s), and 72°C (60 s); and 72°C (300 s). Amplicon products were purified using the Agencourt AMPure PCR Purification system (Beckman Coulter, Indianapolis, IN) and sequenced using 454 FLX Titanium instruments and reagents (Roche Diagnostics Corporation, Bradford, CT) according to the manufacturer's guidelines.
Chemical and physical analyses.
Moisture content was determined by drying bauxite residue samples at 40°C to constant weight. A combination of dry sieving (for the 100- to 2,000-μm fraction) and laser sizing (for the <100-μm fraction; Malvern Mastersizer 2000; Malvern, United Kingdom) was used to determine particle size distribution. The pH and electrical conductivity (EC) of samples were measured in 1:5 soil/solution extracts (43), and total alkalinity was determined by the pH change of a buffer (44). Exchangeable cations were determined by silver thiourea extraction (43), and water-extractable elements (in 1:50 soil/water extracts) were subtracted from exchanged cations to correct for soluble salts. Extracts were analyzed by inductively coupled plasma optical emission spectrometry (ICP-OES) after filtering to <0.45 μm. Total N was determined by dry combustion-thermal conductivity furnace (LECO CNS-2000; LECO Corporation, St. Joseph, MI). Total and organic C concentrations were determined by inductive combustion-infrared absorption furnace (ELTRA CS-2000; Haan, Germany); inorganic C concentration was calculated by subtracting organic C from total C.
Bauxite residue community sequencing data analysis and statistical procedures.
Using QIIME (45), MIDs and primers were removed from bauxite residue DNA sequences, sequences under 150 bp or with ambiguous base calls or homopolymer runs exceeding 6 bp were removed, chimeras were removed, and OTUs were defined by clustering at 97% similarity using an open-reference OTU picking strategy. Although clustering at 97% similarity is commonly used to define “species” level groups, relatively short sequence reads may not allow unequivocal identification at the species level, and sequences showing ≥97% similarity to each other are therefore referred to as OTUs rather than species throughout this paper. Bauxite residue sequences were aligned using the Aligner tool within the Ribosomal Database Project Pyrosequencing pipeline (46, 47). Bacterial and fungal phylogenetic trees were constructed using FastTree (48). Taxonomic classification of OTUs was performed with BLASTn against a curated GreenGenes database (49). Best matches to cultured representatives based on percent identity and E value for each OTU sequence were identified within the NCBI GenBank RefSeq sequence database using BLASTn. The UniFrac tool (50) was used to perform principal components analysis of the bacterial and fungal communities in each sample and to cluster similar communities. Bacterial and fungal phylum abundances as a proportion of total sequence reads per sample (relative abundances) were plotted using the heatmap.2 function in the gplots package (51) for R (52). Rarefied species richness (Srar), Shannon diversity at the phylum (H′phy) and OTU (H′OTU) levels, and Simpson diversity at the phylum level (LOTU) were calculated using the vegan package (53) for R. Correlations between UniFrac principal component loadings, bacterial and fungal community composition metrics, and environmental characteristics (see Tables S2 to S6 in the supplemental material) were calculated to identify variables contributing to overall variation in microbial communities (bacterial and fungal) between samples, at a significance level (α) of 0.05 (two-tailed; Genstat release 12.1; VSN International).
Comparison of bauxite residue microbial communities with other haloalkaline microbial communities.
To assess similarity in microbial community composition between natural and engineered environments, a literature search was performed to identify published microbial community sequencing studies in natural and engineered terrestrial haloalkaline environments. Studies of microbial community structure in natural haloalkaline environments are abundant; however, we restricted analysis here to studies of sulfated athalassohaline salt lakes sediments (54, 55) (n = 6; Table 2) because these bear the greatest geochemical and physical similarity to bauxite residue (high pH, high Na+, high CO32−, high SO42−). Only three previous studies of microbial community composition in engineered terrestrial haloalkaline environments resulting from human industrial activities were found, in steel slag (56), uranium mill tailings (57), and chromite ore processing residue (58) (Table 2). To increase the number of engineered environments represented in this study (total, n = 7), an oil sands tailings sample collected under sterile conditions from Fort McMurray, Alberta, Canada (December 2012), was also included for DNA sequencing as per the protocol above.
TABLE 2.
Details of engineered and natural haloalkaline environments that were compared in this study
| Environment type | Accession no. (database) | Sequencing method | Primer pair | pH | Salinity (ppt) | Source or reference |
|---|---|---|---|---|---|---|
| Engineered haloalkaline environments | ||||||
| Bauxite residue | SRP049788 (Sequence Read Archive) | 454 pyrosequencing | 27F/519R | 4–11 | 0.17–3.3b | This study |
| Oil sands tailings | SRP055527 (Sequence Read Archive) | 454 pyrosequencing | 27F/519R | 8.3 | 0.66b | This study |
| Steel slag | AY396008–AY396023 (GenBank)a | Sanger | 28F/1492R | 12 | 2.4–5.1 | 56 |
| Uranium mill tailings | HE650716–HE650774 (EMBL) | Ion Torrent pyrosequencing | 786F/926R | 10 | —c | 57 |
| Chromite ore processing residue | FR687640–FR687744 (EMBL) | Sanger | 8F/907R | 9–14 | 100 | 58 |
| Natural haloalkaline environments | ||||||
| Kenyan soda lakes | AJ517850–AJ517908 (EMBL) | Sanger | 27F/519R | 8.5–11.5 | 50–350 | 54 |
| Tirez Lagoon, Spain | EU734574, EU725589–EU725602, EU722643–EU722714, FJ172052–FJ172100, FJ236710–FJ236714 (GenBank) | Sanger | 27F/1492R | 7.3–9.08 | ≥69 | 55 |
Note that only sites 1, 2, and 4 returned DNA sequences.
Converted from dS m−1, assuming 1 dS m−1 = 0.550 ppt salinity.
—, data not available.
To compare communities at the phylum level across natural and engineered haloalkaline environments, open-reference OTU picking was first applied to the bauxite residue and oil sands sequences, as this preserves all novel, uncultured microbial taxa. Phylum level microbial community compositions in these two environments were compared with published data (also generated by open-reference OTU picking) from other studies of natural (n = 6) and engineered (n = 3) environments as listed in Table 2. To compare communities at lower levels of taxonomic classification across studies of haloalkaline environments and identify common OTUs, closed-reference OTU picking was necessary due to differential coverage of sequences across the 16S rRNA gene between studies. After pooling all sequences retrieved via GenBank/EMBL (Table 2) and determined in this study, closed-reference OTU picking and taxonomic classification were performed with uclust (59) against a curated Greengenes database (49) with 97% similarity cutoff using QIIME (45). The disadvantage of closed-reference OTU picking is that it resulted in the discarding of novel uncultured sequences that did not match at ≥97% similarity to existing sequences in the database, and therefore it likely underestimates the number of OTUs shared across haloalkaline environments. However, as different regions of the 16S rRNA gene were sequenced across the published studies used for comparison, open-reference OTU picking, which does preserve novel taxa, was precluded.
Nucleotide sequence accession numbers.
The sequence data for the bauxite residue samples obtained from Ireland, Germany, and Australia (July to December 2012) have been submitted to the NCBI Sequence Read Archive under study accession no. SRP049788; the sequence data for the oil sands tailings sample from Fort McMurray, Alberta, Canada (December 2012), have been submitted to the NCBI Sequence Read Archive database under study accession no. SRP055527.
RESULTS AND DISCUSSION
Microbial community structure and diversity across haloalkaline systems.
Bauxite residue is a highly saline, sodic, fine-grained, and frequently waterlogged material, presenting a challenging environment for microbial survival. A total of 3,611 bacterial OTUs identified from 65,541 sequence reads were recovered from the eight samples; rarefied OTU richness (Srar) ranged between 103 and 1,032 OTUs/sample (see Table S2 in the supplemental material). The Shannon diversity index at the OTU level (H′OTU) was relatively low (H′OTU, 3.15 to 6.70; Table 3) compared to previously published data from naturally developed alkaline, saline environments (for salt/soda lakes, H′OTU = 1.56 to 7.83 [55, 60]; for geothermal springs, H′OTU = 2.5 to 10 [61]) but higher than other alkaline, saline tailings (for chromite ore processing residue, H′OTU = 0.75 to 2.32 [58]; for uranium mill tailings, H′OTU = 3.51 to 3.96 [25]; for oil sands tailings, H′OTU = 4.9 [62]), suggesting that bauxite residue communities are in a relatively early stage of community succession. Fungal DNA sequences were recovered from six of the eight samples, with a total of 31,362 sequence reads mapped to 823 OTUs. The two samples from which no fungal sequences were recovered were from an unremediated site (BR2) and a poorly remediated site (BR5). For samples with fungal reads, rarefied fungal OTU richness was very low (Srar, 7 to 126) and diversity at the OTU level was also low (H′OTU, 1.75 to 4.65) (see Table S2 in the supplemental material).
TABLE 3.
Metrics of bacterial community composition and structure across haloalkaline environments
| Environment type | Total no. of sequence reads | Total no. of OTUs | No. of known OTUsa (% of total OTUs) | OTU level Shannon diversity index (H′OTU)b | Source or reference |
|---|---|---|---|---|---|
| Engineered haloalkaline environments | |||||
| Bauxite residue | 65,541 | 3,611 | 367 (10) | 3.15–6.70 | This study |
| Oil sands tailings | 11,900 | 2,039 | 140 (7) | 8.29 | This study |
| Chromite ore processing residue | 99 | 26 | 12 (46) | 0.75–2.32 | 58 |
| Uranium mill tailings | 199 | 59 | 37 (63) | —c | 57 |
| Steel slag | 16 | 15 | 2 (13) | — | 56 |
| Natural haloalkaline environments | |||||
| Tirez Lagoon, Spain | 137 | 42 | 23 (55) | 1.56–1.88 | 55 |
| Kenyan soda lakes | 29 | 26 | 0 (0) | — | 54 |
“known” is defined as matching at ≥97% to a reference sequence in the GreenGenes database.
H′OTU designates Shannon diversity in discrete samples.
—, parameter unable to be determined on the basis of published data.
The overall bacterial community composition in bauxite residue (using open-reference OTU picking) was similar to compositions observed in some soda lakes and other alkaline, saline wastes in that the dominant phyla were Proteobacteria (40 to 80% relative abundance in bauxite residue), Firmicutes (2 to 22%), Actinobacteria (3 to 26%), Bacteroidetes (2 to 10%), and Cyanobacteria (≤1.5%) (14, 56–58, 60) (Fig. 1a; see also Table S7 in the supplemental material). Comparisons of fungal community structures were constrained by data availability: this is the first study to characterize fungal communities in bauxite residue and appears to also be the first in alkaline, saline tailings, limiting comparisons to previous studies of naturally occurring alkaline and/or saline environments. However, the fungal community structure observed here for bauxite residue was consistent at the phylum level with that observed in other hypersaline environments (36, 63, 64), dominated by Ascomycota (52 to 100% relative abundance in bauxite residue), with minor contributions from Basidiomycota (≤18%) (Fig. 1b; see also Table S8 in the supplemental material).
FIG 1.

Relative abundance (Rel. Abund.) of bacterial phyla (a) and fungal phyla (b) as a percentage of total sequence reads per sample. The dendrogram indicates the results of UniFrac environment clustering. Phyla representing <1% of reads per sample are not displayed. Samples BR1 and BR4 returned zero fungal sequence reads.
Closed-reference OTU picking also confirmed similarities in bacterial community composition at the phylum level (see Fig. S1 in the supplemental material). After closed-reference OTU picking, 560 bacterial OTUs were identified across all samples from natural environments (Tirez lagoon [55]) and engineered environments (bauxite residue, oil sands tailings, uranium mill tailings, chromite ore processing residue, and steel slag [56–58]), 367 of which were present in bauxite residue (Fig. 2). The relatively low number of OTUs identified in bauxite residue by closed-reference OTU picking (∼10% of total OTUs identified by open-reference OTU picking) reflects the high proportion of OTUs present in bauxite residue communities that are >3% dissimilar to reference sequences currently present in the GreenGenes database. At the genus and OTU levels, bacterial community composition diverged from those of other haloalkaline environments, with bauxite residue communities overall sharing only 23% of OTUs with one or more other alkaline, saline environments (Fig. 2; see also Table S9 in the supplemental material). Ten bacterial OTUs were identified as being shared across multiple haloalkaline environments, at moderate to high relative abundances, and persisted in bauxite residue microbial communities during remediation (Table 4). Not all shared OTUs were present in all bauxite residue samples during remediation (across samples BR1 to BR8), although the overall percentage of shared OTUs within individual bauxite residue communities was relatively stable during remediation, at around 30% of OTUs matched at ≥97% similarity to existing reference sequences (“known” OTUs) (see Table S9 and Fig. S2 in the supplemental material). Dispersal modulated by selection based on changing geochemical conditions (decreased salinity, sodicity; increased aeration) would account for the maintenance of some overlap between communities (not necessarily comprising the same taxa persisting over time) during tailings remediation. The preliminary analysis of the potential for common taxa to be hosted across haloalkaline environments presented here was limited by data availability (very few published DNA sequences from haloalkaline environments are available in a well-annotated, useful format through public databases), data comparability (amplicons in the few suitable studies available were generated from disparate regions of the 16S rRNA gene [Table 2], necessitating the use of closed-reference OTU picking), and data richness (the few studies available used mostly Sanger sequencing technology and returned ≤200 sequences per sample [Tables 2 and 3]).
FIG 2.
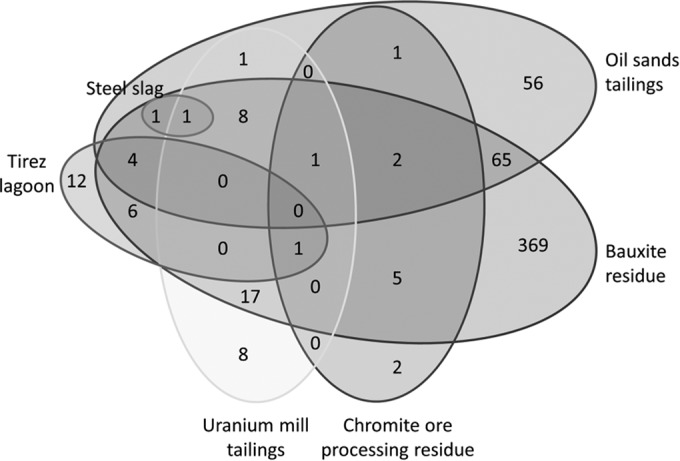
Shared OTUs between haloalkaline environmental samples from this study (bauxite residue, oil sands tailings) and previous studies (chromite ore processing residue, steel slag, uranium mill tailings, Tirez lagoon [55–58]).
TABLE 4.
Shared OTUs across natural and engineered haloalkaline environmentsa
| No. of environments and order; family; genus | Relative abundance in samples (% of total OTU counts) |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Bauxite residue |
Oil sands | COPR | Steel slag | Tirez lagoon | UMT | ||||||||
| BR1 | BR2 | BR3 | BR4 | BR5 | BR6 | BR7 | BR8 | ||||||
| Four environments | |||||||||||||
| Burkholderiales; Comamonadaceae | 14.8 | 25.2 | 2.62 | 0.66 | 1.35 | —b | 6.97 | 2.52 | 7.83 | — | 66.6 | — | 1.85 |
| Xanthomonadales; Flavobacteriaceae | 1.88 | 3.57 | 0.77 | — | — | 1.02 | 0.73 | 0.38 | 0.14 | 6.12 | — | — | 1.85 |
| Three environments | |||||||||||||
| Rhizobiales | 0.34 | 0.49 | 2.24 | 0.51 | 57.6 | 4.34 | 1.82 | 4.44 | 0.03 | — | 33.3 | — | — |
| Flavobacteriales; Flavobacteriaceae | 1.10 | 5.43 | 0.73 | — | — | — | 0.08 | — | 0.67 | — | — | 2.74 | — |
| Rhodobacterales; Rhodobacteraceae | 2.16 | 0.28 | 13.9 | — | 0.10 | 0.07 | 0.11 | 0.05 | 0.34 | — | — | 2.74 | — |
| Burkholderiales; Oxalobacteraceae | 1.61 | 0.75 | 0.43 | — | 0.79 | 0.20 | 0.11 | 0.27 | 0.58 | — | — | — | 9.26 |
| Pseudomonadales; Pseudomonadaceae; Pseudomonas | 0.68 | 0.36 | 0.04 | — | 0.07 | 1.36 | 0.40 | — | 2.88 | — | — | — | 5.56 |
| Two environments | |||||||||||||
| Pseudomonadales; Pseudomonadaceae | 1.88 | 4.17 | 1.38 | 0.95 | 0.88 | — | 0.38 | — | 1.44 | — | — | — | — |
| Actinomycetales | — | 0.36 | — | — | — | 0.07 | 0.28 | — | — | — | — | — | 3.70 |
| Bacillales; Staphylococcaceae; Staphylococcus | 3.35 | 1.24 | 0.02 | — | — | — | 0.05 | — | — | — | — | — | 3.70 |
These OTUs were present in more than one environment type, at a relative abundance of ≥1 % at least one environment, and present in at least three of the four bauxite residue remediation clusters. Phylogeny is given to the highest level of taxonomic classification returned from the GreenGenes database. Abbreviations: COPR, chromite ore processing residue; UMT, uranium mill tailings.
—, OTU was not present in this sample.
Consistent with the high proportion of novel taxa observed in the highly specialized communities from previously studied natural (100% of OTUs from Kenyan soda lakes; 45% of OTUs from Tirez Lagoon [Table 3]) and engineered (37% for uranium mill tailings to 93% for oil sands tailings communities [Table 3]) haloalkaline environments, here, 90% of bacterial OTUs and 73% of fungal OTUs from the bauxite residue samples could not be matched to existing 16S rRNA and ITS sequences from cultured representatives at >97% similarity (Tables 5 and 6), which reflects both the high diversity in haloalkaline environments and the relatively understudied nature of haloalkalitolerant microbial communities. In bauxite residue, phylogenetic hot spots for novel taxa were located within bacterial phyla Proteobacteria and Bacteroidetes, within which <12% of OTUs could be matched to sequences from cultured representatives, and fungal phyla Basidiomycota and Glomeromycota, within which <15% of OTUs could be matched to sequences from cultured representatives (Tables 5 and 6). Other hot spots were located in the minor phyla (accounting for <5% of all OTUs each), within which almost all OTUs were from uncultured taxa (Tables 5 and 6). Minor phyla hot spots included several candidate (GN02, GN04, TM7, OP11) and undescribed phyla.
TABLE 5.
Phylogenetic associations of bacterial OTUs within bauxite residue samples and their matches to cultured representative sequences in the NCBI GenBank RefSeq 16S rRNA databasea
| Phylum and class | Total no. of OTUs | % of total OTUs | Phylum/class OTUs matched at >97% to existing cultured representatives (% of OTUs within sample) |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| All samples | BR1 | BR2 | BR3 | BR4 | BR5 | BR6 | BR7 | BR8 | |||
| Proteobacteria | 1,820 | 50.40 | 11.76 | 50.00 | 58.82 | 47.06 | 63.64 | 11.49 | 5.68 | 14.27 | 9.60 |
| Alphaproteobacteria | 941 | 26.06 | 9.88 | 41.67 | 55.26 | 36.73 | 50.00 | 7.05 | 7.14 | 13.42 | 14.13 |
| Betaproteobacteria | 413 | 11.44 | 16.22 | 51.67 | 58.70 | 66.67 | 66.67 | 27.45 | 2.50 | 18.70 | 5.04 |
| Gammaproteobacteria | 290 | 8.03 | 16.90 | 75.00 | 84.00 | 53.33 | 100.00 | 36.00 | 4.17 | 18.13 | 4.84 |
| Deltaproteobacteria | 175 | 4.85 | 2.29 | 15.38 | 0.00 | 13.33 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| Epsilonproteobacteria | 1 | 0.03 | 100.00 | 100.00 | 100.00 | —b | — | — | — | — | — |
| Actinobacteria | 549 | 15.20 | 13.84 | 57.69 | 72.73 | 52.38 | 64.71 | 8.11 | 11.31 | 14.35 | 7.69 |
| Bacteroidetes | 285 | 7.89 | 7.37 | 46.67 | 42.86 | 37.04 | 0.00 | 2.86 | 9.52 | 4.91 | 5.56 |
| Cytophagia | 138 | 3.82 | 2.17 | — | 0.00 | 0.00 | — | 0.00 | 7.69 | 1.23 | 5.00 |
| Sphingobacteria | 99 | 2.74 | 7.07 | 0.00 | 0.00 | 30.00 | — | 14.29 | 14.29 | 3.51 | 3.70 |
| Flavobacteria | 38 | 1.05 | 26.32 | 75.00 | 85.71 | 58.33 | 0.00 | 0.00 | — | 23.81 | 20.00 |
| Bacteroidia | 10 | 0.28 | 10.00 | 33.33 | 0.00 | 0.00 | 0.00 | — | 0.00 | 0.00 | — |
| Firmicutes | 199 | 5.51 | 29.65 | 73.53 | 78.57 | 34.78 | 75.00 | 11.11 | 23.81 | 19.15 | 7.14 |
| Bacilli | 117 | 3.24 | 41.03 | 80.00 | 81.82 | 66.67 | 80.00 | 14.29 | 28.30 | 36.36 | 25.00 |
| Clostridia | 77 | 2.13 | 14.29 | 55.56 | 66.67 | 14.81 | 50.00 | 0.00 | 0.00 | 4.35 | 0.00 |
| Erysipelotrichi | 5 | 0.14 | 0.00 | — | — | 0.00 | — | 0.00 | — | 0.00 | 0.00 |
| Chloroflexi | 172 | 4.76 | 1.16 | — | — | 0.00 | 0.00 | 0.00 | 2.38 | 0.00 | 0.00 |
| Planctomycetes | 155 | 4.29 | 1.29 | 0.00 | — | 0.00 | — | 0.00 | 0.00 | 2.13 | 0.00 |
| Acidobacteria | 132 | 3.66 | 0.00 | — | — | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| Gemmatimonadetes | 78 | 2.16 | 0.00 | 0.00 | — | 0.00 | — | 0.00 | 0.00 | 0.00 | 0.00 |
| Verrucomicrobia | 47 | 1.30 | 0.00 | 0.00 | — | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| Totals | 3,437 | 95.18 | 10.50 | 52.27 | 60.32 | 38.78 | 60.19 | 8.07 | 7.85 | 10.78 | 7.52 |
Phyla representing <1% of total OTUs were excluded from this table.
—, no OTUs from this phylum were present in this sample.
TABLE 6.
Phylogenetic associations of fungal OTUs within bauxite residue samples and their matches to cultured representative sequences as recorded in the NCBI GenBank RefSeq 16S rRNA databasea
| Phylum and class | Total no. of OTUs | % of total OTUs | Phylum/class OTUs matched at >97% to existing cultured representatives (% of OTUs within sample) |
||||||
|---|---|---|---|---|---|---|---|---|---|
| All samples | BR2 | BR3 | BR5 | BR6 | BR7 | BR8 | |||
| Ascomycota | 662 | 81.53 | 30.51 | 33.70 | 71.43 | 23.53 | —b | 57.14 | 56.25 |
| Dothideomycetes | 136 | 16.75 | 40.44 | 58.33 | 66.67 | 57.14 | — | — | 0.00 |
| Eurotiomycetes | 32 | 3.94 | 21.88 | — | — | 0.00 | — | 66.67 | 50.00 |
| Lecanoromycetes | 1 | 0.12 | 0.00 | — | — | — | 0.00 | — | — |
| Leotiomycetes | 2 | 0.25 | 100.00 | — | — | — | — | — | 100.00 |
| Orbiliomycetes | 6 | 0.74 | 33.33 | — | — | 50.00 | 0.00 | — | 33.33 |
| Pezizomycetes | 9 | 1.11 | 0.00 | — | — | 0.00 | — | — | 0.00 |
| Saccharomycetes | 56 | 6.90 | 17.86 | 17.86 | 100.00 | — | — | — | — |
| Sordariomycetes | 361 | 44.46 | 29.09 | — | — | 22.94 | 28.29 | 55.81 | 60.00 |
| Unspecified | 59 | 7.27 | 54.40 | 0.00 | 0.00 | 0.00 | 25.00 | 66.67 | 75.00 |
| Basidiomycota | 66 | 8.13 | 15.15 | — | — | 6.67 | 27.78 | 0.00 | 16.67 |
| Agaricomycetes | 40 | 4.93 | 22.50 | — | — | 18.18 | 40.00 | 0.00 | 17.65 |
| Dacrymycetes | 13 | 1.60 | 0.00 | — | — | 0.00 | 0.00 | — | — |
| Tremellomycetes | 12 | 1.48 | 8.33 | — | — | 0.00 | 20.00 | — | 0.00 |
| Wallemiomycetes | 1 | 0.12 | 0.00 | — | — | — | 0.00 | — | — |
| Chytridiomycota | 29 | 3.57 | 0.00 | — | — | 0.00 | 0.00 | 0.00 | 0.00 |
| Glomeromycota | 41 | 5.05 | 9.76 | 0.00 | — | 7.69 | 0.00 | 0.00 | 14.29 |
| Unclassified | 10 | 1.23 | 20.00 | — | — | 14.29 | 0.00 | 0.00 | 66.67 |
| Totals | 808 | 100.00 | 26.85 | 32.63 | 71.43 | 18.46 | 28.12 | 41.18 | 42.06 |
Phyla representing <1% of total OTUs were excluded from this table. Note that no fungal sequences were recovered from samples BR1 and BR4; therefore, no data for these samples are shown here.
—, no OTUs from this phylum were present in this sample.
Salinity and total alkalinity are the major geochemical drivers of microbial community structure in engineered haloalkaline environments.
Environmental parameters were statistically significantly linked to 84% of variation in bacterial community composition in bauxite residues and 73% of variation in fungal community composition (see Tables S3, S5, S10, and S11 in the supplemental material). Hierarchical clustering grouped bauxite residue communities by remediation status (Fig. 1a and b), indicating that community structure responds to changes in geochemical and physical conditions. Salinity was significantly negatively correlated with bacterial OTU richness (Srar) and OTU level diversity (H′OTU), accounting for 31% of variation in bacterial community composition between samples in combination with sodicity and silt content through principal component 1 (PC1) (Fig. 3; see also Tables S3 and S10 and Fig. S7 in the supplemental material). This supports previous studies in natural environments (32) indicating that salinity is an important and consistent control on bacterial community composition and diversity across natural and engineered systems. Sodicity influenced bacterial and fungal communities differently: bacteria were sensitive to Na+ concentrations in pore water (as water-extractable Na), whereas fungi were sensitive to Na+ sorbed to mineral and organic matter surfaces (as exchangeable Na+) (see Tables S3 and S5 in the supplemental material). The close associations between fungi and soil particle surfaces and the limited motility of fungi may account for their sensitivity to exchangeable Na+.
FIG 3.

Bacterial community composition across sites as a function of their loading on principal components 1 (PC1) and 2 (PC2) as determined by UniFrac. The symbols legend refers to samples listed in Table 1. Axis labels refer to principal components (PCs) and the total proportion of variance explained by each component (%). Double-headed arrows and labels next to the top x and left y axes identify significant correlations between principal components and environmental variables. Double-headed arrows and labels next to the bottom x and right y axes identify significant correlations between principal components and microbial community composition and diversity variables. Bold text in boxes within graphs identifies inferred environmental characteristics associated with high and low loadings on each combination of principal components. Other principal components are displayed in Fig. S3 to S6 in the supplemental material.
Total alkalinity was the main environmental driver of fungal community composition in bauxite residue, significantly correlated with PC1, which accounted for 38% of the variation in fungal community composition among samples (Fig. 4; see also Tables S5 and S8 and Fig. S8 in the supplemental material). Decreases in total alkalinity (from as high as 14 mol H+ kg−1 to ≤1 mol H+ kg−1) across the remediation gradient significantly increased fungal community diversity and OTU richness and also decreased the dominance of putative alkaliphiles within the bacterial community (Fig. 4; see also Tables S5 and S6 in the supplemental material). Again, fungi have limited motility compared to bacteria, and the prolonged dissolution of minerals such as sodalite and calcite (38) over time in bauxite residue would select for alkalitolerant fungal species in the local pore environment.
FIG 4.

Fungal community composition across sites as a function of their loading on principal components 1 (PC1) and 2 (PC2) as determined by UniFrac. The symbols legend refers to samples identified in Table 1. Axis labels refer to principal components (PCs) and the total proportion of variance explained by each component (%). Double-headed arrows and labels next to the top x and left y axes identify significant correlations between principal components and environmental variables. Double-headed arrows and labels next to the bottom x and right y axes identify significant correlations between principal components and microbial community composition and diversity variables. Bold text in boxes within graphs identifies inferred environmental characteristics associated with high and low loadings on each combination of principal components.
In contrast to total alkalinity, pH was less influential on both bacterial and fungal community composition in this extremely saline and alkaline setting than expected from observations in natural soils (30, 31, 65). pH, which ranged from 4 to 11 across sites, did not correlate significantly with any principal component accounting for variation in the fungal community (see Table S5 in the supplemental material) and played only a minor role in bacterial community composition (statistically significantly correlated with PC5, accounting for 12% of variation in community structure in combination with exchangeable sodium percentage; see Table S3 in the supplemental material). The capacity of haloalkalitolerant species to acidify their local environment through the production of organic acids and exopolymeric substances may render them relatively insensitive to pore water pH but sensitive to total alkalinity once their buffering capacity is exhausted.
Implications for microbial biogeography: colonization and community assembly in natural and engineered haloalkaline environments.
Both bacterial and fungal community structures were strongly correlated with environmental conditions, identifying coherent geochemical-community structure linkages. Selection (based on environmental factors) appears to be more important in shaping bacterial than fungal communities in bauxite residue, because a greater proportion of variance in community structure could be explained by environmental factors and the major driver of bacterial community structure (salinity) was consistent across natural and engineered haloalkaline environments. This was not the case for fungal communities, which indicates that the relative importance of stochastic (neutral) versus deterministic (niche) processes during primary succession in tailings and mine wastes differs across kingdoms. As has been observed during primary succession in deglaciated soils, bacteria are less likely to be dispersal limited than fungi, and thus bacterial communities are more likely to be shaped by deterministic processes (including selection pressures from environmental factors consistent with those operating in natural haloalkaline environments) than fungal communities (66). The dominance of stochastic processes operating on fungal community assembly during the early stages of microbial community assembly may account for the smaller proportion of variation in fungal community structure, which could be statistically linked to environmental factors.
Extremophilic microbial communities in alkaline, saline bauxite residue shift from haloalkaliphile-dominated assemblages to assemblages comprising halotolerant, freshwater, and forest/grassland soil species as salinity, sodicity, and alkalinity decrease during remediation (Fig. 3 and 4; Table 1). Proportions of bacterial phyla typical of anoxic, saline environments (Firmicutes, Epsilonproteobacteria, Gammaproteobacteria) (14, 67, 68) significantly decreased and were replaced by phyla typical of freshwater and forest/grassland soil environments (Gemmatimonadetes, Planctomycetes) (30, 69–71) (Fig. 3). Additionally, the percentage of bacterial OTUs able to be matched to cultured representatives was lowest in well-remediated, diverse bauxite residue communities (Table 5). This held true across the community as a whole, as well as for individual phyla. The increase in novel OTUs (bearing <97% DNA similarity to cultured representatives) within communities during remediation (Table 5) indicates a role for dispersal and/or diversification (sensu Vellend [72) in bacterial community succession after tailings deposition. These results highlight that although alkaline, saline tailings are initially geochemically and biologically similar to naturally developed alkaline, saline environments, the weathering processes acting on tailings can rapidly shift these properties toward those of typical soils, with circumneutral pH, lower alkalinity and salinity, and a more diverse microbial community with decreased dominance of Gammaproteobacteria, Firmicutes, and Ascomycota.
The initial stages of microbial community colonization and assembly in engineered haloalkaline environments will therefore be of most value for understanding similar processes and the origins of high microbial diversity in natural analogues such as soda lakes. Geochemical shifts occurring during bauxite residue remediation are the reverse of shifts occurring during the development of thalassohaline (and some athalassohaline) soda lakes, which become progressively more saline and sodic during evapoconcentration of brines. Focusing on the initial stages of colonization in engineered haloalkaline environments will enable identification of how and when members of key functional guilds (nitrogen fixers, iron reducers, sulfate reducers, fermenters, etc.) are recruited and of their roles in the development of geochemically and spatially defined habitat niches based on the interaction of existing environmental characteristics with outputs of microbial metabolisms such as iron and sulfur reduction. This will improve current understanding of how the remarkable phylogenetic diversity over small spatial scales observed in natural haloalkaline environments (4, 34) has developed.
The investigation of microbial diversity and biogeography in extreme environments is also of importance in identifying more broadly the global environmental drivers of community composition and diversity. In natural soils, within the normal range of expected soil pH (4.5 to 8.5), Proteobacteria have been shown to increase in relative abundance as pH increases (31); however, in our study extending to highly alkaline pH (pH values ≤11), the relative abundance of Proteobacteria decreased as pH increased, with abundance of Alphaproteobacteria strongly negatively correlated with pH (see Table S4 in the supplemental material). This demonstrates that relationships between microbial community composition and environmental factors cannot be reliably extrapolated from agricultural, forest, or grassland soils to more-extreme environments and supports the inclusion of extreme environments in global biogeographical studies, as well as their separate characterization to elucidate successional processes that apply under conditions posing multiple challenges for microbial survival.
Conclusions.
This study demonstrated that although microbial communities are similar at the phylum level across natural and engineered terrestrial haloalkaline environments, they differed at the OTU level. Bauxite residue harbored substantial novel microbial taxa (90% of bacterial OTUs and >70% of fungal OTUs were unable to be matched to a reference sequence), consistent with other natural and engineered environments, while sharing a number of common OTUs. The presence of common OTUs across a range of natural and engineered alkaline, saline environments warrants further investigation for understanding community assembly and successional processes in salt and soda lakes. Of particular value in this approach is the opportunity to understand the origins of high microbial diversity in salt and soda lakes by investigating community assembly in engineered environments that provide a geochemically analogous, low-diversity initial setting.
Consistent geochemical drivers of bacterial, but not fungal, community structure across natural and engineered environments were identified in this study. Salinity was the major driver of bacterial community structure in bauxite residue, whereas total alkalinity was the major driver of fungal community structure, in contrast with natural haloalkaline environments, where pH is the major driver. Overall, 84% of variation in bacterial community composition in bauxite residues and 73% of variation in fungal community composition was linked to environmental factors. Microbial communities shifted from narrow, haloalkaliphile-dominated assemblages to more-diverse communities resembling those found in forest and grassland soils as salinity, sodicity, and alkalinity decreased in bauxite residue during remediation. Differences in microbial community compositions according to environmental conditions within bauxite residue tailings are likely driven by selection and imply links between the functional and physiological capacities of these microbes and their environment, which may be usefully exploited in the development of novel, microbially based bioremediation strategies for management of alkaline, saline wastes. The relative abundance of novel OTUs within bacterial communities increased during remediation in bauxite residue, implying that alleviation of the extreme geochemical conditions prevalent in unremediated tailings allows a more diverse community, distinct from that of natural analogues such as salt lakes or forest/grassland soils, to establish as an intermediate state during primary succession.
Supplementary Material
ACKNOWLEDGMENTS
We thank Rusal Aughinish Alumina Limited, Aluminum Oxid Stade, and Queensland Alumina Limited for assisting with sample collection and shipping.
This research was supported by funding from the International Aluminum Institute and the Natural Sciences and Engineering Research Council of Canada.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.01238-15.
REFERENCES
- 1.Grant WD, Sorokin DY. 2011. Distribution and diversity of soda lake alkaliphiles, p 27–54. In Horikoshi K. (ed), Extremophiles handbook. Springer, Tokyo, Japan. [Google Scholar]
- 2.Yumoto I, Hirota K, Yoshimune K. 2011. Environmental distribution and taxonomic diversity of alkaliphiles, p 55–79. In Horikoshi K. (ed), Extremophiles handbook. Springer, Tokyo, Japan. [Google Scholar]
- 3.Ley RE, Harris JK, Wilcox J, Spear JR, Miller SR, Bebout BM, Maresca JA, Bryant DA, Sogin ML, Pace NR. 2006. Unexpected diversity and complexity of the Guerrero Negro hypersaline microbial mat. Appl Environ Microbiol 72:3685–3695. doi: 10.1128/AEM.72.5.3685-3695.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harris JK, Caporaso JG, Walker JJ, Spear JR, Gold NJ, Robertson CE, Hugenholtz P, Goodrich J, McDonald D, Knights D, Marshall P, Tufo H, Knight R, Pace NR. 2013. Phylogenetic stratigraphy in the Guerrero Negro hypersaline microbial mat. ISME J 7:50–60. doi: 10.1038/ismej.2012.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lanzen A, Simachew A, Gessesse A, Chmolowska D, Jonassen I, Øvreås L. 2013. Surprising prokaryotic and eukaryotic diversity, community structure and biogeography of Ethiopian soda lakes. PLoS One 8:e72577. doi: 10.1371/journal.pone.0072577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vreeland RH, Litchfield CD, Martin EL, Eliot E. 1980. Halomonas elongata, a new genus and species of extremely salt-tolerant bacteria. Int J Syst Bacteriol 30:485–495. doi: 10.1099/00207713-30-2-485. [DOI] [Google Scholar]
- 7.Cayol JH, Ollivier B, Patel BKC, Prensier G, Guezennec J, Garcia JL. 1994. Isolation and characterization of Halothermothrix orenii gen. nov., sp. nov., a halophilic thermophilic, fermentative, strictly anaerobic bacterium. Int J Syst Bacteriol 44:534–540. doi: 10.1099/00207713-44-3-534. [DOI] [PubMed] [Google Scholar]
- 8.Switzer Blum J, Stolz JF, Oren A, Oremland RS. 2001. Selenihalanaerobacter shriftii gen. nov., sp. nov., a halophilic anaerobe from Dead Sea sediments that respires selenite. Arch Microbiol 175:208–219. doi: 10.1007/s002030100257. [DOI] [PubMed] [Google Scholar]
- 9.Sorokin DY, Tourova TP, Mussmann M, Muyzer G. 2008. Dethiobacter alkaliphilus gen. nov. sp. nov., and Desulfurivibrio alkaliphilus gen. nov. sp. nov.: two novel representatives of reductive sulfur cycle from soda lakes. Extremophiles 12:431–439. doi: 10.1007/s00792-008-0148-8. [DOI] [PubMed] [Google Scholar]
- 10.Franzmann PD, Wehmeyer U, Stackebrandt E. 1988. Halomonadaceae fam. nov., a new family of the class Proteobacteria to accommodate the genera Halomonas and Deleya. Syst Appl Microbiol 11:16–19. doi: 10.1016/S0723-2020(88)80043-2. [DOI] [Google Scholar]
- 11.Zavarzin GA. 1993. Epicontinental soda lakes as possible relict biotopes of terrestrial biota formation. Microbiology 62:473–479. [Google Scholar]
- 12.Jones BE, Grant WD, Duckworth AW, Owenson GG. 1998. Microbial diversity of soda lakes. Extremophiles 2:191–200. doi: 10.1007/s007920050060. [DOI] [PubMed] [Google Scholar]
- 13.Oren A. 2008. Microbial life at high salt concentrations: phylogenetic and metabolic diversity. Saline Syst 4:2. doi: 10.1186/1746-1448-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Antony CP, Kumaresan D, Hunger S, Drake HL, Murrell JC, Shouche YS. 2013. Microbiology of Lonar Lake and other soda lakes. ISME J 7:468–476. doi: 10.1038/ismej.2012.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rothschild LJ, Mancinelli RL. 2001. Life in extreme environments. Nature 409:1092–1101. doi: 10.1038/35059215. [DOI] [PubMed] [Google Scholar]
- 16.Agnew MD, Koval SF, Jarrell KF. 1995. Isolation and characterization of novel alkaliphiles from bauxite-processing waste and description of Bacillus vedderi sp. nov., a new obligate alkaliphile. Syst Appl Microbiol 18:221–230. doi: 10.1016/S0723-2020(11)80392-9. [DOI] [Google Scholar]
- 17.Phillips RW, Wiegel J, Berry CJ, Fliermans C, Peacock AD, White DC. 2002. Kineococcus radiotolerans sp. nov., a radiation-resistant, Gram-positive bacterium. Int J Syst Evol Microbiol 52:933–938. doi: 10.1099/ijs.0.02029-0. [DOI] [PubMed] [Google Scholar]
- 18.Fredrickson JK, Zachara JM, Balkwill DL, Kennedy D, Li SW, Kostandarithes HM, Daly MJ, Romine MF, Brockman FJ. 2004. Geomicrobiology of high-level nuclear waste-contaminated vadose sediments at the Hanford Site, Washington state. Appl Environ Microbiol 70:4230–4241. doi: 10.1128/AEM.70.7.4230-4241.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nedelkova M, Merroun ML, Rossberg A, Hennig C, Selenska-Pobell S. 2007. Microbacterium isolates from the vicinity of a radioactive waste depository and their interactions with uranium. FEMS Microbiol Ecol 59:694–705. doi: 10.1111/j.1574-6941.2006.00261.x. [DOI] [PubMed] [Google Scholar]
- 20.Ntougias S, Zervakis GI, Fasseas C. 2007. Halotalea alkalilenta gen. nov., sp. nov., a novel osmotolerant and alkalitolerant bacterium from alkaline olive mill wastes, and emended description of the family Halomonadaceae Franzmann et al. 1989, emend. Dobson and Franzmann 1996. Int J Syst Evol Microbiol 57:1975–1983. doi: 10.1099/ijs.0.65078-0. [DOI] [PubMed] [Google Scholar]
- 21.Piñón-Castillo HA, Brito EMS, Goñi-Urriza M, Guyoneaud R, Duran R, Nevarez-Moorillon GV, Gutiérrez-Corona JF, Caretta CA, Reyna-López GE. 2010. Hexavalent chromium reduction by bacterial consortia and pure strains from an alkaline industrial effluent. J Appl Microbiol 109:2173–2182. doi: 10.1111/j.1365-2672.2010.04849.x. [DOI] [PubMed] [Google Scholar]
- 22.Baker BJ, Banfield JF. 2003. Microbial communities in acid mine drainage. FEMS Microbiol Ecol 44:139–152. doi: 10.1016/S0168-6496(03)00028-X. [DOI] [PubMed] [Google Scholar]
- 23.Mendez MO, Neilson JW, Maier RM. 2008. Characterisation of a bacterial community in an abandoned semiarid lead-zinc mine tailing site. Appl Environ Microbiol 74:3899–3907. doi: 10.1128/AEM.02883-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yergeau E, Lawrence JR, Sanschagrin S, Waiser MJ, Korber DR, Greer CW. 2012. Next-generation sequencing of microbial communities in the Athabasca River and its tributaries in relation to oil sands mining activities. Appl Environ Microbiol 78:7626–7637. doi: 10.1128/AEM.02036-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dhal PK, Sar P. 2014. Microbial communities in uranium mine tailings and mine water sediment from Jaduguda U mine, India: a culture independent analysis. J Environ Sci Health A Tox Hazard Subst Environ Eng 49:694–709. doi: 10.1080/10934529.2014.865458. [DOI] [PubMed] [Google Scholar]
- 26.Mudd GM, Boger DV. 2013. The ever growing case for paste and thickened tailings—towards more sustainable mine waste management. J Aust Inst Mining Metall 2:56–59. [Google Scholar]
- 27.Oren A. 2002. Molecular ecology of extremely halophilic Archaea and Bacteria. FEMS Microbiol Ecol 39:1–7. doi: 10.1111/j.1574-6941.2002.tb00900.x. [DOI] [PubMed] [Google Scholar]
- 28.Wiegel J. 2011. Anaerobic alkaliphiles and alkaliphilic poly-extremophiles, p 81–97. In Horikoshi K. (ed), Extremophiles handbook. Springer, Tokyo, Japan. [Google Scholar]
- 29.Fierer N, Jackson RB. 2006. The diversity and biogeography of soil bacterial communities. Proc Natl Acad Sci U S A 103:626–631. doi: 10.1073/pnas.0507535103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lauber CL, Hamady M, Knight R, Fierer N. 2009. Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl Environ Microbiol 75:5111–5120. doi: 10.1128/AEM.00335-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rousk J, Bååth E, Brookes PC, Lauber CL, Lozupone C, Caporaso JG, Knight R, Fierer N. 2010. Soil bacterial and fungal communities across a pH gradient in an arable soil. ISME J 4:1340–1351. doi: 10.1038/ismej.2010.58. [DOI] [PubMed] [Google Scholar]
- 32.Lozupone CA, Knight R. 2007. Global patterns in bacterial diversity. Proc Natl Acad Sci U S A 104:11436–11440. doi: 10.1073/pnas.0611525104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mesbah NM, Abou-El-Ela SH, Wiegel J. 2007. Novel and unexpected prokaryotic diversity in water and sediments of the alkaline, hypersaline lakes of the Wadi An Natrun, Egypt. Microb Ecol 54:598–617. doi: 10.1007/s00248-006-9193-y. [DOI] [PubMed] [Google Scholar]
- 34.Schneider D, Arp G, Reimer A, Reitner J, Daniel R. 2013. Phylogenetic analysis of a microbialite-forming microbial mat from a hypersaline lake of the Kiritimati atoll, Central Pacific. PLoS One 8:e66662. doi: 10.1371/journal.pone.0066662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mohamed DJ, Martiny JBH. 2011. Patterns of fungal diversity and composition along a salinity gradient. ISME J 5:379–388. doi: 10.1038/ismej.2010.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu K, Ding X, Wang HF, Zhang X, Hozzein WN, Wadaan MAM, Lan A, Zhang B, Li W. 2014. Eukaryotic microbial communities in hypersaline soils and sediments form the alkaline hypersaline Huama Lake as revealed by 454 pyrosequencing. Antonie Van Leeuwenhoek 105:871–880. doi: 10.1007/s10482-014-0141-4. [DOI] [PubMed] [Google Scholar]
- 37.Power G, Gräfe M, Klauber C. 2011. Bauxite residue issues. I. Current management, disposal and storage practices. Hydrometallurgy 108:33–45. doi: 10.1016/j.hydromet.2011.02.006. [DOI] [Google Scholar]
- 38.Gräfe M, Power G, Klauber C. 2011. Bauxite residue issues. III. Alkalinity and associated chemistry. Hydrometallurgy 108:60–79. doi: 10.1016/j.hydromet.2011.02.004. [DOI] [Google Scholar]
- 39.Santini TC, Hinz C, Rate AW, Carter CM, Gilkes RJ. 2011. In situ neutralization of uncarbonated residue mud by cross layer leaching with carbonated bauxite residue mud. J Hazard Mater 194:119–127. doi: 10.1016/j.jhazmat.2011.07.090. [DOI] [PubMed] [Google Scholar]
- 40.Lane DJ. 1991. 16S/23S rRNA sequencing, p 115 In Stackebrandt E, Goodfellow M (ed), Nucleic acid techniques in bacterial systematics. John Wiley and Sons, New York, NY. [Google Scholar]
- 41.Turner S, Pryer KM, Miao VPW, Palmer JD. 1999. Investigating deep phylogenetic relationships among cyanobacteria and plastids by small subunit rRNA sequence analysis. J Eukaryot Microbiol 46:327–338. doi: 10.1111/j.1550-7408.1999.tb04612.x. [DOI] [PubMed] [Google Scholar]
- 42.White TJ, Bruns T, Lee S, Tailor S. 1990. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics, p 315–322. In Innins MA, Gelfand DH, Sninsky JJ, White TJ (ed), PCR protocols: a guide to methods and applications. Academic Press, San Diego, CA. [Google Scholar]
- 43.Rayment GE, Higginson FR. 1992. Australian laboratory handbook of soil and water chemical methods. Inkata Press, Melbourne, Australia. [Google Scholar]
- 44.Dobrowolski MP, Fey MV, Santini TC. 2011. Rapid determination of residual alkalinity in bauxite residue, p 38–44. In Travaux, vol 36 Proceedings of the International Seminar on Bauxite Residue. Mineral Information and Development Centre, Nagpur, India. [Google Scholar]
- 45.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Gonzalez Peña A, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM, Tiedje JM. 2009. The ribosomal database project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res 37:D141–D145. doi: 10.1093/nar/gkn879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nawrocki EP, Kolbe DL, Eddy SR. 2009. Infernal 1.0: inference of RNA alignments. Bioinformatics 25:1335–1337. doi: 10.1093/bioinformatics/btp157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Price MN, Dehal PS, Arkin AP. 2009. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol 26:1641–1650. doi: 10.1093/molbev/msp077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Hu P, Andersen GL. 2006. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72:5069–5072. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lozupone C, Hamady M, Knight R. 2006. UniFrac—an online tool for comparing microbial community diversity in a phylogenetic context. BMC Bioinformatics 7:371–385. doi: 10.1186/1471-2105-7-371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Warnes GR, Bolker B, Bonebakker L, Gentleman R, Liaw WHA, Lumley T, Maechler M, Magnusson A, Moeller S, Schwartz M, Venables B. 2013. gplots: various R programming tools for plotting data, v 2.11.3. http://CRAN.R-project.org/package=gplots. [Google Scholar]
- 52.R Core Team. 2013. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: http://www.R-project.org/. [Google Scholar]
- 53.Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O'Hara RB, Simpson GL, Solymos P, Stevens HH, Wagner H. 2013. vegan: community ecology package, v 2.0-7. http://CRAN.R-project.org/package=vegan. [Google Scholar]
- 54.Rees HC, Grant WD, Jones BE, Heaphy S. 2004. Diversity of Kenyan soda lake alkaliphiles assessed by molecular methods. Extremophiles 8:63–71. doi: 10.1007/s00792-003-0361-4. [DOI] [PubMed] [Google Scholar]
- 55.Montoya L, Vizioli C, Rodríguez N, Rastoll MJ, Amils R, Marin I. 2013. Microbial community composition of Tirez lagoon (Spain), a highly sulphated athalassohaline environment. Aquat Biosyst 9:19. doi: 10.1186/2046-9063-9-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Roadcap GS, Sanford RA, Jin Q, Pardinas JR, Bethke CM. 2006. Extremely alkaline (pH > 12) groundwater hosts diverse microbial community. Groundwater 44:511–517. doi: 10.1111/j.1745-6584.2006.00199.x. [DOI] [PubMed] [Google Scholar]
- 57.Bondici VF, Lawrence JR, Khan NH, Hill JE, Yergeau E, Wolfaardt GM, Warner J, Korber DR. 2013. Microbial communities in low permeability, high pH uranium mine tailings: characterisation and potential effects. J Appl Microbiol 114:1671–1686. doi: 10.1111/jam.12180. [DOI] [PubMed] [Google Scholar]
- 58.Brito EMS, Piñón-Castillo HA, Guyoneaud R, Caretta CA, Gutiérrez-Corona JF, Duran R, Reyna-López GE, Nevárez-Moorillón GV, Fahy A, Goñi-Urriza M. 2013. Bacterial biodiversity from anthropogenic extreme environments: a hyper-alkaline and hyper-saline industrial residue contaminated by chromium and iron. Appl Microbiol Biotechnol 97:369–378. doi: 10.1007/s00253-012-3923-5. [DOI] [PubMed] [Google Scholar]
- 59.Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 60.Hollister EB, Engledow AS, Hammett AJM, Provin TL, Wilkinson HH, Gentry TJ. 2010. Shifts in microbial community structure along an ecological gradient of hypersaline soils and sediments. ISME J 4:829–838. doi: 10.1038/ismej.2010.3. [DOI] [PubMed] [Google Scholar]
- 61.Sharp CE, Brady AL, Sharp GH, Grasby SE, Stott MB, Dunfield PF. 2014. Humboldt's spa: microbial diversity is controlled by temperature in geothermal environments. ISME J 8:1166–1174. doi: 10.1038/ismej.2013.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Golby S, Ceri H, Gieg LM, Chatterjee I, Marquez LLR, Turner RJ. 2012. Evaluation of microbial biofilm communities from an Alberta oil sands tailings pond. FEMS Microbiol Ecol 79:240–250. doi: 10.1111/j.1574-6941.2011.01212.x. [DOI] [PubMed] [Google Scholar]
- 63.Cantrell SA, Casillas-Martínez L, Molina M. 2006. Characterisation of fungi from hypersaline environments of solar salterns using morphological and molecular techniques. Mycol Res 110:962–970. doi: 10.1016/j.mycres.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 64.Gunde-Cimerman N, Ramos J, Plemenitaš A. 2009. Halotolerant and halophilic fungi. Mycol Res 113:1231–1241. doi: 10.1016/j.mycres.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 65.Taylor DL, Hollingsworth TN, McFarland J, Lennon NJ, Nusbaum C, Ruess RW. 2013. A first comprehensive census of fungi in soil reveals both hyperdiversity and fine-scale niche partitioning. Ecol Monogr 84:3–20. doi: 10.1890/12-1693.1. [DOI] [Google Scholar]
- 66.Schmidt SK, Nemergut DR, Darcy JL, Lynch R. 2014. Do bacterial and fungal communities assemble differently during primary succession? Mol Ecol 23:254–258. doi: 10.1111/mec.12589. [DOI] [PubMed] [Google Scholar]
- 67.Campbell BJ, Engel AS, Porter ML, Takai K. 2006. The versatile ε-proteobacteria: key players in sulphidic habitats. Nat Rev Microbiol 4:458–468. doi: 10.1038/nrmicro1414. [DOI] [PubMed] [Google Scholar]
- 68.Takai K, Moser DP, Onstott TC, Spoelstra N, Pfiffner SM, Dohnalkova A, Fredrickson JK. 2001. Alkaliphilus transvaalensis gen. nov., sp. nov., an extremely alkaliphilic bacterium isolated from a deep South African gold mine. Int J Syst Evol Microbiol 51:1245–1256. [DOI] [PubMed] [Google Scholar]
- 69.Janssen PH. 2006. Identifying the dominant soil bacteria taxa in libraries of 16S rRNA and 16S rRNA genes. Appl Environ Microbiol 72:1719–1728. doi: 10.1128/AEM.72.3.1719-1728.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wagner M, Horn M. 2006. The Planctomycetes, Verrucomicrobia, Chlamydiae and sister phyla comprise a superphylum with biotechnological and medical relevance. Curr Opin Biotechnol 17:241–249. doi: 10.1016/j.copbio.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 71.Fuerst JA, Sagulenko E. 2011. Beyond the bacterium: planctomycetes challenge our concepts of microbial structure and function. Nat Rev Microbiol 9:403–413. doi: 10.1038/nrmicro2578. [DOI] [PubMed] [Google Scholar]
- 72.Vellend M. 2010. Conceptual synthesis in community ecology. Q Rev Biol 85:183–206. doi: 10.1086/652373. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.