

Abstract
Riemerella anatipestifer is a well-described pathogen of waterfowl and other avian species that can cause septicemic and exudative diseases. In this study, we sequenced the complete genome of R. anatipestifer strain Yb2 and analyzed it against the published genomic sequences of R. anatipestifer strains DSM15868, RA-GD, RA-CH-1, and RA-CH-2. The Yb2 genome contains one circular chromosome of 2,184,066 bp with a 35.73% GC content and no plasmid. The genome has 2,021 open reading frames that occupy 90.88% of the genome. A comparative genomic analysis revealed that genome organization is highly conserved among R. anatipestifer strains, except for four inversions of a sequence segment in Yb2. A phylogenetic analysis found that the closest neighbor of Yb2 is RA-GD. Furthermore, we constructed a library of 3,175 mutants by random transposon mutagenesis, and 100 mutants exhibiting more than 100-fold-attenuated virulence were obtained by animal screening experiments. Southern blot analysis and genetic characterization of the mutants led to the identification of 49 virulence genes. Of these, 25 encode cytoplasmic proteins, 6 encode cytoplasmic membrane proteins, 4 encode outer membrane proteins, and the subcellular localization of the remaining 14 gene products is unknown. The functional classification of orthologous-group clusters revealed that 16 genes are associated with metabolism, 6 are associated with cellular processing and signaling, and 4 are associated with information storage and processing. The functions of the other 23 genes are poorly characterized or unknown. This genome-wide study identified genes important to the virulence of R. anatipestifer.
INTRODUCTION
Riemerella anatipestifer is a Gram-negative, non-spore-forming, nonmotile, capsule-like, rod-shaped bacterium. It is reported worldwide as the cause of epizootic infectious polyserositis in domestic ducks (1); it is also pathogenic for geese, turkeys, chickens, and other birds (2, 3). R. anatipestifer infection occurs in acute form in ducks less than about 8 weeks of age and in chronic form in older birds. It causes major economic losses in the duck industry by causing a high mortality rate, poor feed conversion, increased condemnations, and high treatment costs (4, 5).
Currently, 21 serotypes of R. anatipestifer have been identified by slide and tube agglutination tests using antisera (6). There is a large variation in virulence between different serotypes and strains, as assessed by mortality and morbidity rates (7). Infections with R. anatipestifer serotypes 1, 2, 3, 5, 6, 7, 8, 10, 11, 13, 14, and 15 have been reported in China, with serotypes 1, 2, and 10 being responsible for most of the major outbreaks (8). There is very little knowledge about the molecular bases of R. anatipestifer virulence, except for the virulence factors VapD, the Christie-Atkins-Munch-Peterson (CAMP) cohemolysin, and OmpA. VapD shows homology to virulence-associated proteins in other bacteria (9). The CAMP cohemolysin is a sialoglycoprotease produced during natural infection under certain intracellular conditions, and therefore, it is able to damage the host and facilitate the infection process (10). OmpA is a 42-kDa outer membrane protein that seems to be not only a predominant specific antigen (11) but also an adhesin that plays a critical role in colonization (12). Additionally, biofilm formation by R. anatipestifer may contribute to persistent infections in duck farms, as biofilm-producing isolates are more resistant to antibiotic and detergent treatments than planktonic isolates are (13).
Thus far, only limited genomic resources are available for R. anatipestifer, given that 21 serotypes have been identified. The complete genome sequences of strains DSM15868 (ATCC 11845), RA-GD, RA-CH-1, and RA-CH-2 have been released (14, 15). The established genomes are very similar in size and gene number. To comprehensively and systematically explore the genetic diversity and evolution of the virulence of R. anatipestifer strains, genome-wide profiling is needed. Here, we report the complete genomic sequence of R. anatipestifer serotype 2 strain Yb2, which was isolated in Jiangsu Province, China (8). Analysis of the complete genomic sequence revealed potential virulence factors and metabolic pathways in Yb2. Subsequently, a random transposon library containing 3,175 mutants was constructed and screened for Tn4351-induced virulence attenuation in animal experiments. Genes involved in bacterial virulence were identified and characterized. Further studies of virulence factors are invaluable for understanding R. anatipestifer pathogenesis, with the ultimate goal of disease prevention and control.
MATERIALS AND METHODS
Plasmids, bacterial strains, and growth conditions.
For the bacterial strains, plasmids, and primers used in this study, see Table S1 in the supplemental material. R. anatipestifer Yb2 is the wild-type strain used in this study (8). It was cultured in tryptic soy agar (TSA; Becton Dickinson, Franklin Lakes, NJ, USA) at 37°C for 24 h in 5% CO2 or tryptic soy broth (TSB; Becton Dickinson) at 37°C with shaking at 200 rpm for 8 to 12 h. Escherichia coli strain BW19851 containing plasmid pEP4351 was grown in Luria broth (LB; Becton Dickinson) or on LB agar containing 30 μg/ml chloramphenicol. To select for Tn4351-disrupted Yb2 mutants, erythromycin (0.5 μg/ml) and kanamycin (50 μg/ml) were added to TSA.
Animals.
One-day-old Cherry Valley ducklings were obtained from the ZhuangHang Duck Farm (Shanghai, China), housed in cages, and provided water and food ad libitum during the study. The animal experiments were conducted in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the Institutional Animal Care and Use Committee guidelines set by the Shanghai Veterinary Research Institute, Chinese Academy of Agricultural Sciences (CAAS). The protocol was approved by the Committee on the Ethics of Animal Experiments of the Shanghai Veterinary Research Institute, CAAS (permit no. 12-12). All surgeries were performed under sodium pentobarbital anesthesia, and all efforts were made to minimize suffering.
Genomic DNA extraction.
R. anatipestifer strain Yb2 was cultured in TSB at 37°C for 8 h with shaking until it reached the mid-logarithmic phase. Genomic DNA was extracted from bacterial cell pellets with the TIANamp Bacteria DNA kit (Tiangen, Beijing, China) in accordance with the manufacturer's instructions. DNA was resuspended in nuclease-free water and quantified with a NanoVue spectrophotometer (GE Healthcare, Little Chalfont, United Kingdom). DNA quality and RNA contamination were assessed by electrophoresis with 0.8% agarose gels.
Genome sequencing and assembly.
Genomic DNA of R. anatipestifer strain Yb2 was sequenced with the MiSeq Sequencing platform (Illumina, San Diego, CA, USA). A shotgun sequencing approach was adopted by employing two independent genomic libraries. A total of 804,600 reads, totaling 371,017,848 bases (average read length, 346 bp), or about 186-fold coverage of the genome, was obtained from a paired-end 400-bp (PE400) library. A sum of 142,150 reads, totaling 42,577,127 bases (average read length, 422 bp), was obtained from a mate-paired 8-kbp (MP8k) library, resulting in 22-fold genome coverage. Newbler software (version 2.8) (Roche, Indianapolis, IN, USA) was used for sequence assembly. The order of contigs was determined by multiplex PCR. Gaps were filled in first with GapCloser software and then by sequencing the PCR products (16). Low-quality regions of the genome sequence were resequenced to complete the sequencing.
Gene prediction and annotation.
Putative coding sequences (CDSs) were identified by Glimmer 3.0 (17). tRNA genes were predicted by tRNAscan-SE (18), while rRNA genes were predicted by RNAmmer1.2 (19). Other noncoding RNA (ncRNA) genes were obtained by comparing the genome sequences to the Rfam database (http://rfam.sanger.ac.uk) (20). The clustered regularly interspaced short palindromic repeats (CRISPR) recognition tool was used to predict direct repeat sequences and spacers (21). Functional annotation of CDSs was achieved by searching protein databases and a nonredundant protein database from the National Center for Biotechnology Information (NCBI). Clusters of orthologous groups (COGs) of proteins were used for functional classifications performed with the eggNOG (version 3) database (http://eggnog.embl.de/version_3.0/). Orthologs and paralogs were defined as proteins with >30% similarity (22). Metabolic pathways were constructed with the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (http://www.genome.jp/kegg/) (23, 24). Genomic islands (GIs) were detected by IslandViewer (25). The genome atlas was drawn with CGView (26).
Evolutionary analysis. (i) SNP calling.
To study the polymorphism and evolutionary rate of R. anatipestifer genes, we identified single nucleotide polymorphisms (SNPs) in the Yb2 genome against reference genomes of the DSM15868 (accession no. CP002346.1), RA-GD (CP002562.1), RA-CH-1 (CP003787.1), and RA-CH-2 (CP004020.1) strains. Genomic sequences with adjusted start positions were submitted to the NUCmer system (NUCleotide MUMmer version 3.07) for alignment (27, 28), and nonspecific records were filtered by delta-filter. After alignment of the sequenced reads with a reference genome, SNP calling was performed with show-snps software.
(ii) Colinearity analysis.
The reference genomes of R. anatipestifer strains DSM15868 (CP002346.1), RA-GD (CP002562.1), RA-CH-1 (CP003787.1), and RA-CH-2 (CP004020.1) were downloaded from the NCBI to identify conserved genomic regions in the presence of rearrangements and horizontal transfers. Prior to alignment, the genomic sequences were adjusted to a common start position. Mauve (version 2.3.1) was used to align the Yb2 genome with the four reference genomes and to draw a colinear atlas (29, 30).
(iii) Homology analysis and phylogenetic tree construction.
A local database was created and managed with formatdb software based on the four reference genomes (31). The query sequence was compared to the newly created database with the blastall (version 2.2.22) service (32). For phylogenetic tree construction, 16S rRNA gene sequences were aligned by using the default settings of Muscle (version 3.8.31) (33). The conserved alignment blocks were then extracted by Gblocks (version 0.91b) (34). A maximum-likelihood tree was built with Mega6 (version 6.05) by using the Kimura two-parameter module. The reliability of the phylogenetic tree branches was verified via bootstrap analysis (1,000 replications) (35).
Random transposon mutagenesis and isolation of mutants with attenuated virulence.
Tn4351 was introduced into R. anatipestifer wild-type strain Yb2 by conjugation from E. coli BW19851 as previously described (13). Briefly, donor and recipient cells were cultured to mid-logarithmic phase, mixed at a ratio of 4:5 (based on optical density at 600 nm), and filtered onto a 0.22-μm membrane (EMD Millipore, Billerica, MA, USA). The filter were placed on a TSA plate and incubated at 30°C for 8 h. After incubation, the cells was scraped off the filter, resuspended in 10 mM MgSO4, and plated onto TSA supplemented with erythromycin (0.5 μg/ml) and kanamycin (50 μg/ml) to select for potential transconjugants. Pairs of primers specific for R. anatipestifer (primers RA 16S rRNA-F/RA 16S rRNA-R) or transposon Tn4351 (primers Erm-F/Erm-R) were used to identify mutants by PCR amplification. PCRs were conducted with 2× Taq MasterMix (CWBIO, Beijing, China) with the following parameters: 94°C for 4 min; 30 cycles of 94°C for 40 s, 52°C for 40 s, and 72°C for 1 min; and 1 cycle of 72°C for 10 min. The mutants from which the R. anatipestifer 16S rRNA and Tn4351 were amplified were deemed to be Tn4351-disrupted mutants.
To obtain mutants with attenuated virulence, 18-day-old Cherry Valley ducklings were infected intramuscularly with 1 × 107 CFU of the mutants, which is equal to 100 median lethal doses (LD50s) of wild-type strain Yb2 (36). The infected ducklings were housed in separate cages with a 12-h light-dark cycle and free access to food and water during the study. The clinical symptoms and deaths of ducklings were recorded daily for 7 days, and mutants that failed to kill the susceptible ducklings were selected for further study.
Identification, sequencing, and bioinformatic analysis of the mutated genes.
Southern blotting was used to confirm a single Tn4351 insertion in the R. anatipestifer genome. The region of transposon Tn4351 was amplified as a 626-bp PCR product from pEP4351 with primers Tn4351-F/Tn4351-R. The probe was prepared with DIG-High Prime DNA labeling and detection starter kit I (Roche). Genomic DNA of the attenuated mutants was purified with the TIANamp Bacteria DNA kit (Tiangen), digested with XbaI, separated by gel electrophoresis, and transferred to a nylon membrane. Hybridization procedures were conducted by using standard molecular biology protocols.
The sites of transposon insertions in the attenuated mutants were determined by cloning the Tn4351-disrupted genes by genomic walking or inverse PCR. Genomic walking was performed with a genome walking kit (TaKaRa, Dalian, China) according to the manufacturer's instructions. Arbitrary primers (AP1, AP2, AP3, and AP4) provided in the kit, as well as specific primers (SP1, SP2, and SP3), were used to amplify the DNA adjacent to the site of insertion (13). For inverse PCR, genomic DNA was digested with HindIII and religated to form closed circles (37). The resulting circular molecules were used as templates to amplify the Tn4351 insertion region with the TN-1/IS4351-F or 340/341 primer pair (38).
After purifying the PCR products from agarose gels with the GeneJET gel extraction kit (Thermo Fisher Scientific, Waltham, MA, USA), the PCR products were sequenced by HuaGene Biotech Co., Ltd. (HuaGene, Shanghai, China). Sequences were compared to those in the existing NCBI database with BLAST (http://www.ncbi.nlm.nih.gov/BLAST/). Predictions regarding subcellular localization were made by using the PSORTb (version 3.0) server (http://www.psort.org/). Functional characterization of the proteins was predicted by searching against the eggNOG (version 3) database with BLASTP (http://eggnog.embl.de/version_3.0/).
Determination of LD50s.
The virulence of the attenuated mutants was evaluated by determining their LD50s. The attenuated mutants were grown for 8 to 10 h to mid-logarithmic phase and concentrated by centrifugation (1,500 × g, 10 min). The bacterial pellet was washed twice with phosphate-buffered saline (PBS, pH 7.4) and serially diluted to obtain different bacterial concentrations (CFU counts). Eighteen-day-old ducklings were injected intramuscularly with 107 to 1011 CFU of the attenuated mutants in 0.5 ml of PBS. The ducklings were monitored daily for clinical symptoms, and deaths were recorded until 7 days postinfection. The LD50 was calculated by an improved version of Karber's method (39).
Nucleotide sequence accession number.
The full genomic sequence of R. anatipestifer strain Yb2 has been submitted to the GenBank database (http://www.ncbi.nlm.nih.gov/GenBank/) under accession number CP007204.
RESULTS
Genome features of R. anatipestifer Yb2.
R. anatipestifer strain Yb2 has a single circular chromosome of 2,184,066 bp with a 35.73% GC content. There was no evidence of plasmids (Fig. 1; see Table S2 in the supplemental material). The genome encodes six rRNA operons, 38 tRNAs that represent all 20 amino acids, and 1 other ncRNA. CRISPR is a unique family of DNA direct repeat sequence that widely exist in prokaryotic genomes. There is one predicted CRISPR array in the Yb2 genome. In addition to seven pseudogenes, 2,021 open reading frames (ORFs) were identified in the Yb2 genome, and they had an average length of 976 bp and constituted 90.88% of the genome. Among these ORFs, 1,214 (60.1%) genes were classified into COG families comprising 20 functional categories (see Table S3 in the supplemental material). A total of 127 ORFs were classified into the “translation, ribosomal structure, and biogenesis” category, 119 ORFs were related to “cell wall/membrane/envelope biogenesis,” 478 ORFs belonged to “metabolism,” and most of the ORFs were related to “amino acid transport and metabolism,” “energy production and conversion,” and “coenzyme transport and metabolism.” With KEGG, 1,217 ORFs (60.2%) were assigned to 30 pathways (Fig. 2). Of these ORFs, 948 (77.9%) belonged to “metabolism,” and the majority of these were involved in the metabolism of amino acids, carbohydrates, cofactors, and vitamins. Other ORFs were assigned to pathways in “genetic information processing” (13.6%), “environmental information processing” (3.7%), and “human diseases” (2.1%).
FIG 1.

Summary of gene annotations and GC skew analysis of the Yb2 genome. From outer to inner, circles 1 and 2 show the CDSs (blue), rRNA operon (light purple), and tRNA information (reddish brown); circle 3 shows the GC contents; circle 4 shows the GC skew [(G − C)/(G + C); green, >0; purple, <0]; and circle 5 shows the coordinates of the Yb2 genome. The artificial start site is at 0 kbp.
FIG 2.

Summary of KEGG pathway classifications. The metabolic pathways were constructed by using the KEGG database. Values are the numbers of respective classifications.
GIs.
GIs are relatively large segments of DNA (usually 10 to 200 kb) that are present in the genomes of many bacterial species (40). A total of six GIs were predicted to be scattered throughout the single chromosome that makes up the Yb2 genome (see Table S4 in the supplemental material). Putative GI-1 contained mostly ORFs that encode hypothetical proteins, as well as two ORFs that encode a DNA-binding protein and a phage head morphogenesis protein. Additionally, there was one ORF that encodes each of the following: an N-acetylmuramoyl-l-alanine amidase, a phage head protein, and an aminopeptidase. Putative GI-2 included ORFs that encode hypothetical proteins, an integrase, an AAA (ATPases associated with diverse cellular activities) ATPase, restriction endonuclease EcoRI subunit R, and a transcriptional regulator. Putative GI-3 included only two ORFs for hypothetical proteins. Putative GI-4 included ORFs that encode peptidase M23, a transposase, a membrane protein, and kinase enzymes, such as a serine/threonine protein kinase, suggesting that this cluster may play a regulatory role in cellular processing. Putative GI-5 consisted of ORFs that encode a DNA-binding protein, restriction endonuclease subunit R, and hypothetical proteins. These two clusters may be involved in transcription and DNA recombination. ORFs in putative GI-6 encode multidrug resistance proteins such as LinF, chloramphenicol acetyltransferase, and a tetracycline resistance protein; metal transporters such as a mercury transporter, a multicopper oxidase, and a cation transporter; toxins such as RelE and YoeB; AraC and xenobiotic response element family transcriptional regulators; and two proteins involved in twitching motility.
Evolutionary analysis.
We performed SNP calling to determine polymorphic sites between Yb2 and the reference genomes of strains DSM15868, RA-GD, RA-CH-1, and RA-CH-2 (see Table S5 in the supplemental material). A comparison of Yb2 and RA-GD revealed 4,548 SNPs, 4,252 (93.5%) of which are located in coding regions. Similarly, of the 5,448 SNPs between Yb2 and RA-CH-2, 5,119 (94.0%) are located in coding regions. There are 78,163 SNPs between Yb2 and RA-CH-1 and 11,905 SNPs between Yb2 and DSM15868.
All five R. anatipestifer genomes were aligned by Mauve to investigate the degree of colinearity between them. The alignment revealed 50, 29, 203, and 42 colinearity blocks between Yb2 and DSM15868, RA-GD, RA-CH-1, and RA-CH-2, respectively. Genome structure rearrangements, which are shown in Fig. 3, illustrate a high level of synteny, with the exception of four inversions in the Yb2 genome.
FIG 3.

Schematic representations of R. anatipestifer genomic sequences. Each contiguously colored region is a colinear block, a region without rearrangement of the homologous backbone sequence. Lines between genomes trace each orthologous colinear block through every genome. The colinear blocks below a genome's center line represent segments that are inverted relative to the reference genome. The image shown was generated by the Mauve rearrangement viewer.
To investigate the phylogenetic relationships among the five R. anatipestifer strains, we analyzed their 16S rRNA gene sequences. The resulting phylogenetic tree revealed a strikingly short genetic distance between Yb2 and RA-GD (Fig. 4), indicative of very recent divergence, and a much greater distance between Yb2 and DSM15868.
FIG 4.
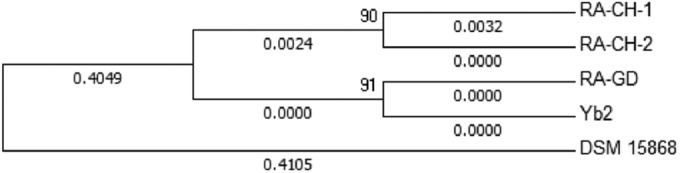
Phylogenetic relationships among R. anatipestifer strains DSM15868, RA-GD, RA-CH-1, RA-CH-2, and Yb2. The maximum-likelihood tree shown was generated with Mega6 (version 6.05) and is based on 16S rRNA gene sequences. Values are branch lengths.
Isolation of Tn4351-induced mutants with reduced virulence.
We introduced Tn4351 from E. coli BW19851 into R. anatipestifer via the delivery vector pEP4351 and obtained 3,175 erythromycin-resistant Yb2 colonies. Tn4351 insertion mutants were deemed defective in virulence on the basis of the loss of pathogenicity in our animal model. We identified 100 mutants with different virulence levels in the initial screening for transconjugants. Southern blot analysis revealed that 62% (62/100) of the attenuation mutants had a single Tn4351 insertion, while 30% (30/100) had two and 8% (8/100) had three insertions (data not shown).
Identification of genes disrupted by transposon insertion.
A set of 62 R. anatipestifer Yb2 mutants with virulence defects was obtained by Tn4351 mutagenesis. The DNA sequences flanking the transposon insertions were obtained by inverse PCR or genomic walking. The flanking sequences were then BLAST searched against the Yb2 genome (accession no. CP007204) and four other R. anatipestifer genome sequences in the NCBI database to determine which genes were interrupted by the transposon. We identified 49 mutated genes (Table 1), 4 of which had multiple mutations. The AS87_06670 gene, which encodes a hypothetical protein, was mutated in mutants RA712 and RA791; the AS87_01210 gene, which encodes a phosphoribosyl formylglycinamidine synthase, was mutated in mutants RA848, RA908, and RA1333; the AS87_09295 gene, which encodes a pyruvate dehydrogenase, was mutated in mutants RA1323 and RA2067; and the AS87_06745 gene, which encodes a hypothetical protein, was mutated in mutants RA1476 and RA2279. Tn4351 was located outside ORFs in mutants RA512, RA723, RA750, RA783, RA885, RA2330, RA2792, and RA3096 (data not shown).
TABLE 1.
Characterization of R. anatipestifer Yb2 mutants with attenuated virulence
| Mutant(s) | Locus taga | Description of gene | Gene product |
|
|---|---|---|---|---|
| Subcellular locationb | Function group (COG)c | |||
| RA38 | AS87_03285 | Phosphoribosylaminoimidazole carboxylase (NCAIR synthetase) | Cytoplasmic membrane | COG0026 F |
| RA55 | AS87_10600 | Phosphoadenosine phosphosulfate reductase | Cytoplasmic | COG0175EH |
| RA253 | AS87_06385 | Transcriptional regulator, LuxR family | Unknown | —d |
| RA256 | AS87_08785 | Gliding-motility protein Gldk | Unknown | COG1262 S |
| RA514 | AS87_04980 | Antitermination protein NusB | Cytoplasmic | COG0781 K |
| RA625 | AS87_01735 | PncA | Cytoplasmic | COG1335 Q |
| RA702 | AS87_04750 | Inosine-5-monophosphate dehydrogenase | Cytoplasmic | COG0517 R |
| RA712, RA791 | AS87_06670 | Hypothetical protein | Cytoplasmic | COG0457 R |
| RA756 | AS87_01090 | Hypothetical protein | Cytoplasmic membrane | — |
| RA772 | AS87_10710 | Hypothetical protein | Cytoplasmic | — |
| RA799 | AS87_00910 | Phosphoribosylaminoimidazole (AIR) synthetase | Cytoplasmic | COG0150 F |
| RA835 | AS87_00505 | Gliding-motility protein GldG | Unknown | COG3225 N |
| RA848, RA908, RA1333 | AS87_01210 | Phosphoribosylformylglycinamidine (FGAM) synthase | Cytoplasmic | COG0047 F |
| RA1072 | AS87_09335 | Membrane protein | Outer membrane | — |
| RA1323, RA2067 | AS87_09295 | Pyruvate dehydrogenase | Cytoplasmic | COG0508 C |
| RA1340 | AS87_04850 | Hypothetical protein | Unknown | — |
| RA1436 | AS87_10665 | Hypothetical protein | Unknown | — |
| RA1450 | AS87_04765 | Beta-carotene 15,15′-monooxygenase | Cytoplasmic membrane | — |
| RA1476, RA2279 | AS87_06745 | Hypothetical protein | Unknown | — |
| RA1633 | AS87_01220 | Amidophosphoribosyltransferase | Cytoplasmic | COG0034 F |
| RA1824 | AS87_05545 | Biotin synthase | Cytoplasmic | COG0502 H |
| RA1893 | AS87_09530 | 8-Amino-7-oxononanoate synthase | Cytoplasmic | COG0156 H |
| RA2041 | AS87_08840 | Holliday junction resolvase | Cytoplasmic | COG0816 L |
| RA2101 | AS87_02660 | DNA-binding protein | Cytoplasmic | COG0776 L |
| RA2145 | AS87_09440 | ATPase | Cytoplasmic | COG0714 R |
| RA2148 | AS87_09345 | Hypothetical protein | Unknown | — |
| RA2238 | AS87_08115 | Gliding-motility protein | Unknown | — |
| RA2241 | AS87_10760 | Amino acid transporter | Cytoplasmic membrane | COG0531 E |
| RA2257 | AS87_05585 | Beta-lactamase | Cytoplasmic | COG0607 P |
| RA2281 | AS87_08795 | Gliding-motility protein GldM | Unknown | — |
| RA2442 | AS87_04755 | Membrane protein | Cytoplasmic membrane | — |
| RA2607 | AS87_07960 | Dihydroorotase | Cytoplasmic | COG0044 F |
| RA2634 | AS87_01015 | Glycosyltransferase family 2 | Unknown | COG0463 M |
| RA2640 | AS87_04050 | Vi polysaccharide biosynthesis protein VipB/TviC | Cytoplasmic | COG0451 MG |
| RA2671 | AS87_00250 | PorT protein | Unknown | — |
| RA2672 | AS87_07135 | Hypothetical protein | Unknown | — |
| RA2673 | AS87_09985 | Hypothetical protein | Cytoplasmic | — |
| RA2742 | AS87_04845 | Hypothetical protein | Cytoplasmic | COG0807 H |
| RA2922 | AS87_10150 | Phosphoesterase | Cytoplasmic membrane | — |
| RA2928 | AS87_05075 | ATPase AAA | Cytoplasmic | COG0507L |
| RA3008 | AS87_09245 | Restriction endonuclease | Cytoplasmic | COG0732V |
| RA3034 | AS87_09000 | Ankyrin | Unknown | COG0666R |
| RA3052 | AS87_07625 | Fructose-bisphosphate aldolase | Cytoplasmic | COG0191 G |
| RA3059 | AS87_10020 | Hypothetical protein | Cytoplasmic | — |
| RA3094 | AS87_05990 | Hypothetical protein | Unknown | COG1002 V |
| RA3107 | AS87_08340 | TonB-dependent receptor | Outer membrane | COG4206H |
| RA3151 | AS87_05195 | Hypothetical protein | Outer membrane | — |
| RA3174 | AS87_00480 | Hypothetical protein | Outer membrane | COG1843N |
Based on the R. anatipestifer Yb2 genome (accession no. CP007204).
Subcellular locations were predicted by the PSORTb (version 3.0) server (http://www.psort.org/).
Functional characterization of the proteins was predicted by searching against the eggNOG (version 3) database with BLASTP.
—, no related COG. Functional categories: (i) information storage and processing (J, translation, ribosomal structure, and biogenesis; K, transcription; L, DNA replication, recombination, and repair); (ii) cellular processes and signaling (D, cell division and chromosome partitioning; V, defense mechanisms; O, posttranslational modification, protein turnover, chaperones; M, cell envelope biogenesis, outer membrane; N, cell motility; T, signal transduction mechanisms); (iii) metabolism (C, energy production and conversion; G, carbohydrate transport and metabolism; H, coenzyme transport and metabolism; P, inorganic ion transport and metabolism; E, amino acid transport and metabolism; F, nucleotide transport and metabolism; Q, secondary metabolite biosynthesis, transport, and catabolism); (iv) poorly characterized (R, general function prediction only; S, function unknown).
Bioinformatic analysis of the proteins encoded by the mutated genes.
We predicted the subcellular locations of the protein products of these 49 identified genes with PSORTb (version 3.0) software. Twenty-five proteins (51%) were predicted to be cytoplasmic, six (12%) were predicted to be cytoplasmic membrane proteins, and four (8%) were predicted to be outer membrane proteins. Fourteen proteins (29%) had unknown locations.
These proteins were further categorized on the basis of their putative functions (Table 1). Sixteen proteins (33%) were classified into the “metabolism” (C, G, E, F, H, P, and Q) category, of which 11 were related to “nucleotide transport and metabolism,” “coenzyme transport and metabolism,” and “energy production and conversion.” Cellular processes and signaling-related categories (D, V, O, M, N, and T) contained six proteins (12%). Information storage and processing categories (J, K, and L) included four proteins (8%), and poorly characterized COGs (R and S) contained five proteins (10%). Eighteen proteins (37%) could not be categorized.
LD50 determination.
To elucidate the effects of the mutations on Yb2 virulence, the LD50 of the attenuation mutants was assessed in Cherry Valley ducklings. Twenty-two mutants with different COG function classifications were selected for virulence evaluation. Deaths were observed from day 1 to day 7 postinfection. The LD50s are summarized in Table 2. Of these 22 mutants, 3 were attenuated 100-fold, 10 were attenuated 1,000-fold, 3 were attenuated 10,000-fold, and 5 were attenuated 100,000-fold compared with wild-type Yb2. The results indicate that the genes identified by Tn4351 mutagenesis screening are required for full virulence in Yb2.
TABLE 2.
LD50s of the virulence attenuation mutants in this study
| Disrupted genea | Description of gene | LD50 (CFU) | Virulence attenuation (fold)b |
|---|---|---|---|
| AS87_08795 | Gliding-motility protein GldM | 1.97 × 107 | 184 |
| AS87_01220 | Amidophosphoribosyltransferase | 2.88 × 107 | 269 |
| AS87_01210 | Phosphoribosylformylglycinamidine (FGAM) synthase | 5.75 × 107 | 537 |
| AS87_00250 | PorT protein | 1.24 × 108 | 1,160 |
| AS87_01015 | Glycosyltransferase family 2 | 1.27 × 108 | 1,190 |
| AS87_00910 | Phosphoribosylaminoimidazole (AIR) synthetase | 1.48 × 108 | 1,380 |
| AS87_04980 | Antitermination protein NusB | 2.43 × 108 | 2,270 |
| AS87_00505 | Gliding-motility protein GldG | 3.16 × 108 | 2,950 |
| AS87_05585 | Beta-lactamase | 3.16 × 108 | 2,950 |
| AS87_07625 | Fructose-bisphosphate aldolase | 4.16 × 108 | 3,890 |
| AS87_04765 | Beta-carotene 15,15′-monooxygenase | 4.27 × 108 | 3,990 |
| AS87_08785 | Gliding-motility protein Gldk | 7.5 × 108 | 7,000 |
| AS87_03285 | Phosphoribosylaminoimidazole carboxylase (NCAIR synthetase) | 1 × 109 | 9,300 |
| AS87_06385 | Transcriptional regulator, LuxR family | 3.83 × 109 | 35,800 |
| AS87_04750 | Inosine-5-monophosphate dehydrogenase | 8.13 × 109 | 76,000 |
| AS87_09295 | Pyruvate dehydrogenase | 1 × 1010 | 93,400 |
| AS87_10020 | Hypothetical protein | 1.10 × 1010 | 103,000 |
| AS87_09440 | ATPase | 1.76 × 1010 | 165,000 |
| AS87_04050 | Vi polysaccharide biosynthesis protein VipB/TviC | 1.91 × 1010 | 179,000 |
| AS87_01735 | PncA | 5.23 × 1010 | 488,000 |
| AS87_09530 | 8-Amino-7-oxononanoate synthase | 8.22 × 1010 | 768,000 |
| AS87_05545 | Biotin synthase | 2.18 × 1011 | 2,040,000 |
Based on the R. anatipestifer Yb2 genome (accession no. CP007204).
The measured LD50 of wild-type strain Yb2 was 1.07 × 105 CFU. Fold virulence attenuation was calculated by dividing the LD50 of the mutant strain by the LD50 of wild-type strain Yb2.
DISCUSSION
Only in recent years have complete genomes of R. anatipestifer strains been sequenced. The first published R. anatipestifer genome was that of DSM15868, and several R. anatipestifer genome sequences are available in GenBank. In this study, the complete genome sequence of R. anatipestifer serotype 2 virulent strain Yb2 was determined and compared to the reference genomes of R. anatipestifer strains DSM15868, RA-GD, RA-CH-1, and RA-CH-2. The Yb2 genome was found to be highly colinear with the four reference genomes, except for a predominant rearrangement involving four inversions (Fig. 3). Pairwise and reciprocal comparisons identified 4,548 SNPs between Yb2 and RA-GD, which is Yb2's closest phylogenetically related neighbor. The two genome sequences are 99.30% identical.
Strong variations in virulence, as assessed by mortality and morbidity rates in outbreaks, have been reported for different R. anatipestifer strains. Despite the devastating losses it has caused in the poultry industry, only a few virulence factors have been established for R. anatipestifer (10, 12). To better understand the potential pathogenicity of R. anatipestifer, genes involved in the full virulence of R. anatipestifer strain Yb2 were identified by genome-wide random transposon mutagenesis. A total of 49 genes were confirmed to be virulence associated, with protein products involved in metabolism, cellular processes and signaling, and information storage and processing. Transposon disruption of either gene attenuated Yb2 virulence by >100-fold. Of these, mutant strain RA2640, which showed significantly attenuated virulence due to inactivation of the AS87_04050 gene, has been previously investigated (41).
Four genes that encode outer membrane proteins were identified as virulence factors. In particular, genes AS87_09335 and AS87_08340, identified in mutants RA1072 and RA3107, respectively, are involved in transport processes. Both of them encode outer membrane receptor proteins, also named TonB-dependent receptors. The TonB-dependent receptors combine with an extracellular or intracellular messenger and transmit it into or out of the cytoplasm, leading to the transcriptional activation of target genes (42). The TonB protein of E. coli carries out high-affinity binding and energy-dependent uptake of specific substrates into the periplasmic space (43). The proteins that are currently known to act as TonB-dependent receptors include HpuAB, ShuA, lactoferrin-binding protein (Lbp), and transferrin-binding protein (Tbp) (44–47). Furthermore, TonB-dependent receptor TbdR1 has been identified as a cross-immunogenic antigen among R. anatipestifer serotypes 1, 2, and 10 (48). The TonB-dependent receptor TdrA of Pseudomonas fluorescens, which is a protective immunogen, is likely to be required for iron acquisition and optimal bacterial virulence (49). We speculate that the products of AS87_09335 and AS87_08340 may play a similar role in the pathogenicity of R. anatipestifer.
Cell invasion or disruption may be considered the first step of systemic disease development. It has been proposed that R. anatipestifer may gain entry into the systemic circulation through blood and tissue. Gene AS87_01015, identified in mutant RA2634, encodes a family 2 glycosyltransferase that is responsible for the transfer of nucleotide-diphosphate sugars to substrates such as lipopolysaccharide (LPS) and lipids. LPS, which functions as an endotoxin, an adhesin, and a modulator of the immune response, is an important virulence factor for numerous bacteria (50–52). Pseudomonas aeruginosa LPS was specifically bound by the cystic fibrosis transmembrane conductance regulator and endocytosed by epithelial cells, leading to rapid nuclear translocation of the transcription factor nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) (53). Therefore, it is attempting to speculate that AS87_01015 may affect invasion by inhibiting bacterial LPS assembly and capsule formation.
Five of our transposon-disrupted genes, namely, AS87_03285, AS87_04750, AS87_09910, AS87_01210, and AS87_01220, are involved in the de novo IMP biosynthetic process. The product of AS87_01210 performs the fourth step in IMP biosynthesis, while the protein encoded by AS87_09910 is the fifth enzyme in this pathway. The reaction catalyzed by the product of AS87_04750 is the first committed and rate-limiting step in the de novo synthesis of guanine nucleotides, and therefore, it plays an important role in cell growth regulation (54, 55). The protein encoded by AS87_01220 is the rate-limiting enzyme in the de novo pathway of purine ribonucleotide synthesis, and it is regulated by feedback inhibition by AMP and GMP (56). As a result of the disrupted genes, virulence was >200- to 80,000-fold lower in the mutants than in wild-type strain Yb2, suggesting that de novo IMP biosynthesis is important for the full virulence of R. anatipestifer. A previous study also showed that the purine biosynthetic enzyme PurH, which catalyzes the final steps in purine IMP biosynthesis, is required for the pathogenesis and virulence of Bacillus anthracis in guinea pigs (57).
Among the virulence genes we found, AS87_09530 and AS87_05545 were involved in the biotin synthetic process. The 8-amino-7-oxononanoate synthase encoded by AS87_09530 catalyzes the decarboxylative condensation of l-alanine with pimeloyl coenzyme A (pimeloyl-CoA) to form 8(S)-amino-7-oxononanoate, which is the first committed step in biotin biosynthesis (58). The predicted product of AS87_05545 is biotin synthase, which catalyzes the last step of the biotin biosynthetic pathway (59). Biotin is the essential cofactor of biotin-dependent carboxylases such as pyruvate carboxylase and acetyl-CoA carboxylase (60); therefore, it is necessary for cell growth, the production of fatty acids, and the metabolism of amino acids. Additionally, recent reports have revealed that biotin synthesis is essential for bacterial growth, infection, and survival during the latent phase of Mycobacterium marinum (61, 62). In this study, the mutants inactivated in biotin synthesis-related genes were attenuated >700,000- to 2,000,000-fold compared with wild-type strain Yb2, demonstrating that biotin synthesis plays an important role in the pathogenicity of R. anatipestifer.
Various antibiotics are currently used to prevent and control R. anatipestifer infection in ducks, but this accelerates the emergence of drug-resistant strains. We predicted a GI containing three antibiotic resistance genes (AS87_09605, AS87_09610, and AS87_09615) in this study. The products of these genes confer resistance to lincomycin, chloramphenicol, and tetracycline, respectively. In addition, three metal transporter genes (AS87_09680, AS87_09690, and AS87_09710) that are involved in transmembrane transport are also located on this GI. Furthermore, the product of AS87_09680 is a mercury transporter that is exposed to the cytoplasm and involved in the transport of Hg across the cytoplasmic membrane (63). This mercury transporter enables bacteria to respond to changes in the concentration of mercury in the environment (64). Our data may provide a genetic basis for drug resistance and persistence during R. anatipestifer infections. In summary, we completed a whole-genome sequence analysis of R. anatipestifer strain Yb2 and identified 49 virulence genes by genome-wide random transposon mutagenesis and in vivo screening. Our data provide the foundation for future studies of R. anatipestifer pathogenesis.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (31272591).
We thank the staff of Shanghai Personal Biotechnology Co., Ltd. (Shanghai, China), for sequencing the genome of R. anatipestifer strain Yb2.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.00828-15.
REFERENCES
- 1.Leavitt S, Ayroud M. 1997. Riemerella anatipestifer infection of domestic ducklings. Can Vet J 38:113. [PMC free article] [PubMed] [Google Scholar]
- 2.Glunder G, Hinz KH. 1989. Isolation of Moraxella anatipestifer from embryonated goose eggs. Avian Dis 18:351–355. [DOI] [PubMed] [Google Scholar]
- 3.Helfer DH, Helmboldt CF. 1977. Pasteurella anatipestifer infection in turkeys. Avian Dis 21:712–715. doi: 10.2307/1589432. [DOI] [PubMed] [Google Scholar]
- 4.Kardos G, Nagy J, Antal M, Bistyák A, Tenk M, Kiss I. 2007. Development of a novel PCR assay specific for Riemerella anatipestifer. Lett Appl Microbiol 44:145–148. doi: 10.1111/j.1472-765X.2006.02053.x. [DOI] [PubMed] [Google Scholar]
- 5.Sun N, Liu JH, Yang F, Lin DC, Li GH, Chen ZL, Zeng ZL. 2012. Molecular characterization of the antimicrobial resistance of Riemerella anatipestifer isolated from ducks. Vet Microbiol 158:376–383. doi: 10.1016/j.vetmic.2012.03.005. [DOI] [PubMed] [Google Scholar]
- 6.Pathanasophon P, Phuektes P, Tanticharoenyos T, Narongsak W, Sawada T. 2002. A potential new serotype of Riemerella anatipestifer isolated from ducks in Thailand. Avian Pathol 31:267–270. doi: 10.1080/03079450220136576. [DOI] [PubMed] [Google Scholar]
- 7.Brogden K. 1989. Pasteurella anatipestifer infection. Academic Press, London, United Kingdom. [Google Scholar]
- 8.Hu Q, Zhang Z, Miao J, Liu Y, Liu X, Ding C. 2001. The epidemiology study of Riemerella anatipestifer infection in Jiangsu and Anhui Provinces. Chin J Vet Sci Technol 31:12–13. doi: 10.3969/j.issn.1673. [DOI] [Google Scholar]
- 9.Chang C, Hung P, Chang Y. 1998. Molecular characterization of a plasmid isolated from Riemerella anatipestifer. Avian Pathol 27:339–345. doi: 10.1080/03079459808419349. [DOI] [PubMed] [Google Scholar]
- 10.Crasta KC, Chua K-L, Subramaniam S, Frey J, Loh H, Tan H-M. 2002. Identification and characterization of CAMP cohemolysin as a potential virulence factor of Riemerella anatipestifer. J Bacteriol 184:1932–1939. doi: 10.1128/JB.184.7.1932-1939.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Subramaniam S, Huang B, Loh H, Kwang J, Tan HM, Chua KL, Frey J. 2000. Characterization of a predominant immunogenic outer membrane protein of Riemerella anatipestifer. Clin Diagn Lab Immunol 7:168–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu Q, Han X, Zhou X, Ding C, Zhu Y, Yu S. 2011. OmpA is a virulence factor of Riemerella anatipestifer. Vet Microbiol 150:278–283. doi: 10.1016/j.vetmic.2011.01.022. [DOI] [PubMed] [Google Scholar]
- 13.Hu Q, Zhu Y, Tu J, Yin Y, Wang X, Han X, Ding C, Zhang B, Yu S. 2012. Identification of the genes involved in Riemerella anatipestifer biofilm formation by random transposon mutagenesis. PLoS One 7:e39805. doi: 10.1371/journal.pone.0039805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mavromatis K, Lu M, Misra M, Lapidus A, Nolan M, Lucas S, Hammon N, Deshpande S, Cheng J-F, Tapia R. 2011. Complete genome sequence of Riemerella anatipestifer type strain (ATCC 11845T). Stand Genomic Sci 4:144. doi: 10.4056/sigs.1553865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yuan J, Liu W, Sun M, Song S, Cai J, Hu S. 2011. Complete genome sequence of the pathogenic bacterium Riemerella anatipestifer strain RA-GD. J Bacteriol 193:2896–2897. doi: 10.1128/JB.00301-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, He G, Chen Y, Pan Q, Liu Y. 2012. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1:18. doi: 10.1186/2047-217X-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Delcher AL, Bratke KA, Powers EC, Salzberg SL. 2007. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 23:673–679. doi: 10.1093/bioinformatics/btm009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schattner P, Brooks AN, Lowe TM. 2005. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res 33:W686–W689. doi: 10.1093/nar/gki366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lagesen K, Hallin P, Rødland EA, Stærfeldt H-H, Rognes T, Ussery DW. 2007. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res 35:3100–3108. doi: 10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burge SW, Daub J, Eberhardt R, Tate J, Barquist L, Nawrocki EP, Eddy SR, Gardner PP, Bateman A. 2013. Rfam 11.0: 10 years of RNA families. Nucleic Acids Res 41(Database issue):D226–D232. doi: 10.1093/nar/gks1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grissa I, Vergnaud G, Pourcel C. 2007. The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinformatics 8:172. doi: 10.1186/1471-2105-8-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Powell S, Szklarczyk D, Trachana K, Roth A, Kuhn M, Muller J, Arnold R, Rattei T, Letunic I, Doerks T. 2012. eggNOG v3.0: orthologous groups covering 1133 organisms at 41 different taxonomic ranges. Nucleic Acids Res 40:D284–D289. doi: 10.1093/nar/gkr1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kanehisa M, Goto S. 2000. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. 2012. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res 40:D109–D114. doi: 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Juhas M, van der Meer JR, Gaillard M, Harding RM, Hood DW, Crook DW. 2009. Genomic islands: tools of bacterial horizontal gene transfer and evolution. FEMS Microbiol Rev 33:376–393. doi: 10.1111/j.1574-6976.2008.00136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stothard P, Wishart DS. 2005. Circular genome visualization and exploration using CGView. Bioinformatics 21:537–539. doi: 10.1093/bioinformatics/bti054. [DOI] [PubMed] [Google Scholar]
- 27.Delcher AL, Phillippy A, Carlton J, Salzberg SL. 2002. Fast algorithms for large-scale genome alignment and comparison. Nucleic Acids Res 30:2478–2483. doi: 10.1093/nar/30.11.2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL. 2004. Versatile and open software for comparing large genomes. Genome Biol 5:R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Darling AC, Mau B, Blattner FR, Perna NT. 2004. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res 14:1394–1403. doi: 10.1101/gr.2289704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Darling AE, Mau B, Perna NT. 2010. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 32.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. 2009. BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Talavera G, Castresana J. 2007. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol 56:564–577. doi: 10.1080/10635150701472164. [DOI] [PubMed] [Google Scholar]
- 35.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol 30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang X, Hu Q, Han X, Ding C, Yu S. 2012. Screening of serotype 2 Riemerella anatipestifer candidate strains for the production of inactivated oil-emulsion vaccine. Chin J Anim Infect Dis 20:5. doi: 10.3969/j.issn.1674-6422.2012.01.010. [DOI] [Google Scholar]
- 37.Ochman H, Gerber AS, Hartl DL. 1988. Genetic applications of an inverse polymerase chain reaction. Genetics 120:621–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alvarez B, Secades P, Prieto M, McBride M, Guijarro J. 2006. A mutation in Flavobacterium psychrophilum tlpB inhibits gliding motility and induces biofilm formation. Appl Environ Microbiol 72:4044–4053. doi: 10.1128/AEM.00128-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gu B, Li Y, Yu R, Wang X. 2009. Summary of median lethal dose and its calculation methods. China Occup Med 36:507–508, 511. [Google Scholar]
- 40.Hacker J, Kaper JB. 2000. Pathogenicity islands and the evolution of microbes. Annu Rev Microbiol 54:641–679. doi: 10.1146/annurev.micro.54.1.641. [DOI] [PubMed] [Google Scholar]
- 41.Wang X, Ding C, Wang S, Han X, Hou W, Yue J, Zou J, Yu S. 2014. The AS87_04050 gene is involved in bacterial lipopolysaccharide biosynthesis and pathogenicity of Riemerella anatipestifer. PLoS One 9:e109962. doi: 10.1371/journal.pone.0109962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koebnik R. 2005. TonB-dependent trans-envelope signalling: the exception or the rule? Trends Microbiol 13:343–347. doi: 10.1016/j.tim.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 43.Chimento DP, Kadner RJ, Wiener MC. 2003. The Escherichia coli outer membrane cobalamin transporter BtuB: structural analysis of calcium and substrate binding, and identification of orthologous transporters by sequence/structure conservation. J Mol Biol 332:999–1014. doi: 10.1016/j.jmb.2003.07.005. [DOI] [PubMed] [Google Scholar]
- 44.Rohde K, Gillaspy A, Hatfield M, Lewis L, Dyer D. 2002. Interactions of haemoglobin with the Neisseria meningitidis receptor HpuAB: the role of TonB and an intact proton motive force. Mol Microbiol 43:335–354. doi: 10.1046/j.1365-2958.2002.02745.x. [DOI] [PubMed] [Google Scholar]
- 45.Mills M, Payne SM. 1997. Identification of shuA, the gene encoding the heme receptor of Shigella dysenteriae, and analysis of invasion and intracellular multiplication of a shuA mutant. Infect Immun 65:5358–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ekins A, Khan AG, Shouldice SR, Schryvers AB. 2004. Lactoferrin receptors in Gram-negative bacteria: insights into the iron acquisition process. Biometals 17:235–243. doi: 10.1023/B:BIOM.0000027698.43322.60. [DOI] [PubMed] [Google Scholar]
- 47.Renauld-Mongénie G, Poncet D, Mignon M, Fraysse S, Chabanel C, Danve B, Krell T, Quentin-Millet M-J. 2004. Role of transferrin receptor from a Neisseria meningitidis tbpB isotype II strain in human transferrin binding and virulence. Infect Immun 72:3461–3470. doi: 10.1128/IAI.72.6.3461-3470.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hu Q, Ding C, Tu J, Wang X, Han X, Duan Y, Yu S. 2012. Immunoproteomics analysis of whole cell bacterial proteins of Riemerella anatipestifer. Vet Microbiol 157:428–438. doi: 10.1016/j.vetmic.2012.01.009. [DOI] [PubMed] [Google Scholar]
- 49.Hu Y-H, Dang W, Sun L. 2012. A TonB-dependent outer membrane receptor of Pseudomonas fluorescens: virulence and vaccine potential. Arch Microbiol 194:795–802. doi: 10.1007/s00203-012-0812-3. [DOI] [PubMed] [Google Scholar]
- 50.Lüneberg E, Mayer B, Daryab N, Kooistra O, Zähringer U, Rohde M, Swanson J, Frosch M. 2001. Chromosomal insertion and excision of a 30 kb unstable genetic element is responsible for phase variation of lipopolysaccharide and other virulence determinants in Legionella pneumophila. Mol Microbiol 39:1259–1271. doi: 10.1111/j.1365-2958.2001.02314.x. [DOI] [PubMed] [Google Scholar]
- 51.Morona R, Daniels C, Van Den Bosch L. 2003. Genetic modulation of Shigella flexneri 2a lipopolysaccharide O antigen modal chain length reveals that it has been optimized for virulence. Microbiology 149:925–939. doi: 10.1099/mic.0.26141-0. [DOI] [PubMed] [Google Scholar]
- 52.Schaeffer LM, McCormack FX, Wu H, Weiss AA. 2004. Bordetella pertussis lipopolysaccharide resists the bactericidal effects of pulmonary surfactant protein A. J Immunol 173:1959–1965. doi: 10.4049/jimmunol.173.3.1959. [DOI] [PubMed] [Google Scholar]
- 53.Schroeder TH, Lee MM, Yacono PW, Cannon CL, Gerçeker AA, Golan DE, Pier GB. 2002. CFTR is a pattern recognition molecule that extracts Pseudomonas aeruginosa LPS from the outer membrane into epithelial cells and activates NF-κB translocation. Proc Natl Acad Sci U S A 99:6907–6912. doi: 10.1073/pnas.092160899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.O'Gara MJ, Lee C-H, Weinberg GA, Nott JM, Queener SF. 1997. IMP dehydrogenase from Pneumocystis carinii as a potential drug target. Antimicrob Agents Chemother 41:40–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ye D, Lee C-H, Queener SF. 2001. Differential splicing of Pneumocystis carinii f. sp. carinii inosine 5′-monophosphate dehydrogenase pre-mRNA. Gene 263:151–158. doi: 10.1016/S0378-1119(00)00577-1. [DOI] [PubMed] [Google Scholar]
- 56.Yamaoka T, Yano M, Kondo M, Sasaki H, Hino S, Katashima R, Moritani M, Itakura M. 2001. Feedback inhibition of amidophosphoribosyltransferase regulates the rate of cell growth via purine nucleotide, DNA, and protein syntheses. J Biol Chem 276:21285–21291. doi: 10.1074/jbc.M011103200. [DOI] [PubMed] [Google Scholar]
- 57.Jenkins A, Cote C, Twenhafel N, Merkel T, Bozue J, Welkos S. 2011. Role of purine biosynthesis in Bacillus anthracis pathogenesis and virulence. Infect Immun 79:153–166. doi: 10.1128/IAI.00925-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Webster SP, Alexeev D, Campopiano DJ, Watt RM, Alexeeva M, Sawyer L, Baxter RL. 2000. Mechanism of 8-amino-7-oxononanoate synthase: spectroscopic, kinetic, and crystallographic studies. Biochemistry 39:516–528. doi: 10.1021/bi991620j. [DOI] [PubMed] [Google Scholar]
- 59.Fugate CJ, Jarrett JT. 2012. Biotin synthase: insights into radical-mediated carbon-sulfur bond formation. Biochim Biophys Acta 1824:1213–1222. doi: 10.1016/j.bbapap.2012.01.010. [DOI] [PubMed] [Google Scholar]
- 60.Serebriiskii IG, Vassin VM, Tsygankov YD. 1996. Two new members of the BioB superfamily: cloning, sequencing and expression of bioB genes of Methylobacillus flagellatum and Corynebacterium glutamicum. Gene 175:15–22. doi: 10.1016/0378-1119(96)00114-X. [DOI] [PubMed] [Google Scholar]
- 61.Yu J, Niu C, Wang D, Li M, Teo W, Sun G, Wang J, Liu J, Gao Q. 2011. MMAR_2770, a new enzyme involved in biotin biosynthesis, is essential for the growth of Mycobacterium marinum in macrophages and zebrafish. Microbes Infect 13:33–41. doi: 10.1016/j.micinf.2010.08.010. [DOI] [PubMed] [Google Scholar]
- 62.Salaemae W, Azhar A, Booker GW, Polyak SW. 2011. Biotin biosynthesis in Mycobacterium tuberculosis: physiology, biochemistry and molecular intervention. Protein Cell 2:691–695. doi: 10.1007/s13238-011-1100-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Howell SC, Mesleh MF, Opella SJ. 2005. NMR structure determination of a membrane protein with two transmembrane helices in micelles: MerF of the bacterial mercury detoxification system. Biochemistry 44:5196–5206. doi: 10.1021/bi048095v. [DOI] [PubMed] [Google Scholar]
- 64.Mindlin S, Kholodii G, Gorlenko Z, Minakhina S, Minakhin L, Kalyaeva E, Kopteva A, Petrova M, Yurieva O, Nikiforov V. 2001. Mercury resistance transposons of Gram-negative environmental bacteria and their classification. Res Microbiol 152:811–822. doi: 10.1016/S0923-2508(01)01265-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.