

Abstract
Persistent CB1 cannabinoid receptor activity limits neurotransmitter release at various synapses throughout the brain. However, it is not fully understood how constitutively active CB1 receptors, tonic endocannabinoid signaling, and its regulation by multiple serine hydrolases contribute to the synapse-specific calibration of neurotransmitter release probability. To address this question at perisomatic and dendritic GABAergic synapses in the mouse hippocampus, we used a combination of paired whole-cell patch-clamp recording, liquid chromatography/tandem mass spectrometry, stochastic optical reconstruction microscopy super-resolution imaging, and immunogold electron microscopy. Unexpectedly, application of the CB1 antagonist and inverse agonist AM251 [N-1-(2,4-dichlorophenyl)-5-(4-iodophenyl)-4-methyl-N-1-piperidinyl-1H-pyrazole-3-carboxamide], but not the neutral antagonist NESS0327 [8-chloro-1-(2,4-dichlorophenyl)-N-piperidin-1-yl-5,6-dihydro-4H-benzo[2,3]cyclohepta[2,4-b]pyrazole-3-carboxamine], significantly increased synaptic transmission between CB1-positive perisomatic interneurons and CA1 pyramidal neurons. JZL184 (4-nitrophenyl 4-[bis(1,3-benzodioxol-5-yl)(hydroxy)methyl]piperidine-1-carboxylate), a selective inhibitor of monoacylglycerol lipase (MGL), the presynaptic degrading enzyme of the endocannabinoid 2-arachidonoylglycerol (2-AG), elicited a robust increase in 2-AG levels and concomitantly decreased GABAergic transmission. In contrast, inhibition of fatty acid amide hydrolase (FAAH) by PF3845 (N-pyridin-3-yl-4-[[3-[5-(trifluoromethyl)pyridin-2-yl]oxyphenyl]methyl]piperidine-1-carboxamide) elevated endocannabinoid/endovanilloid anandamide levels but did not change GABAergic synaptic activity. However, FAAH inhibitors attenuated tonic 2-AG increase and also decreased its synaptic effects. This antagonistic interaction required the activation of the transient receptor potential vanilloid receptor TRPV1, which was concentrated on postsynaptic intracellular membrane cisternae at perisomatic GABAergic symmetrical synapses. Interestingly, neither AM251, JZL184, nor PF3845 affected CB1-positive dendritic interneuron synapses. Together, these findings are consistent with the possibility that constitutively active CB1 receptors substantially influence perisomatic GABA release probability and indicate that the synaptic effects of tonic 2-AG release are tightly controlled by presynaptic MGL activity and also by postsynaptic endovanilloid signaling and FAAH activity.
SIGNIFICANCE STATEMENT Tonic cannabinoid signaling plays a critical role in the regulation of synaptic transmission. However, the mechanistic details of how persistent CB1 cannabinoid receptor activity inhibits neurotransmitter release have remained elusive. Therefore, electrophysiological recordings, lipid measurements, and super-resolution imaging were combined to elucidate those signaling molecules and mechanisms that underlie tonic cannabinoid signaling. The findings indicate that constitutive CB1 activity has pivotal function in the tonic control of hippocampal GABA release. Moreover, the endocannabinoid 2-arachidonoylglycerol (2-AG) is continuously generated postsynaptically, but its synaptic effect is regulated strictly by presynaptic monoacylglycerol lipase activity. Finally, anandamide signaling antagonizes tonic 2-AG signaling via activation of postsynaptic transient receptor potential vanilloid TRPV1 receptors. This unexpected mechanistic diversity may be necessary to fine-tune GABA release probability under various physiological and pathophysiological conditions.
Keywords: 2-arachidonoylglycerol, endocannabinod, GABA, hippocampus, interneuron, TRPV1
Introduction
Neurons set their synaptic strength by using several signaling mechanisms that dynamically adjust parameters of synaptic transmission, including neurotransmitter release probability (Branco and Staras, 2009). Endocannabinoid signaling is one of the most widespread and efficient mechanisms to control release probability via presynaptic CB1 cannabinoid receptor activation (Lovinger, 2008). Accordingly, CB1 receptors are indispensable for various phasic forms of synaptic plasticity (Castillo et al., 2012) and have an essential function in the tonic control of neurotransmitter release (Losonczy et al., 2004; Hentges et al., 2005; Zhu and Lovinger, 2005; Neu et al., 2007; Oliet et al., 2007). However, although the molecular mechanisms accounting for phasic endocannabinoid signaling have been outlined in considerable detail previously (Ohno-Shosaku and Kano, 2014), our understanding of how tonic regulation of neurotransmitter release is organized and controlled by specific regulatory mechanisms is rather limited.
The lipid messengers anandamide and 2-arachidonoylglycerol (2-AG) are both released from postsynaptic neurons and act retrogradely on presynaptic CB1 receptors (Ohno-Shosaku and Kano, 2014). Their synaptic levels are determined by complex enzymatic interactions with numerous synthesis and degradation routes identified in the brain (Blankman and Cravatt, 2013). Because anandamide and 2-AG are known to participate differentially in specific behavioral processes (Long et al., 2009a), it is generally assumed that this molecular and functional complexity reflects division of labor in setting synaptic strength in the underlying brain circuits. However, it is not clear whether it is 2-AG or anandamide, which primarily contributes to the tonic control of neurotransmitter release at given circuit locations. For example, previous studies using culture preparations and pharmacological tools with limited target selectivity arrived at different conclusions concerning the identity of the endocannabinoid responsible for tonic action at hippocampal GABAergic synapses (Hashimotodani et al., 2007; Kim and Alger, 2010). Moreover, emerging in vitro and in vivo observations raised the possibility that constitutive receptor activity may also influence axonal CB1 distribution and tonically attenuate GABA release in the forebrain (Leterrier et al., 2006; Meye et al., 2013; Thibault et al., 2013).
Because the cannabinoid tone plays a major role in interneuron-type-specific and activity-dependent adjustment of GABAergic synaptic strength, a better understanding of the underlying signaling processes is important (Losonczy et al., 2004; Ali and Todorova, 2010; Lee et al., 2010). In addition, recent studies have reported selective, long-term pathological alterations in the tonic endocannabinoid control of GABA release in rodent models of major brain disorders, including autism, Huntington's disease, and seizures (Chen et al., 2003, 2007; Dvorzhak et al., 2013; Földy et al., 2013). Therefore, we used paired patch-clamp recordings in acute brain slices, in combination with mass spectrometry, stochastic optical reconstruction microscopy (STORM) super-resolution imaging, and immunogold electron microscopy, to identify the specific mechanisms that mediate the tonic control of GABA release in the mouse hippocampus. The findings indicate that constitutively active CB1 receptors play a considerable role in the regulation of GABA release probability and elucidate a functional interaction between intercellular 2-AG endocannabinoid and intracellular anandamide endovanilloid signaling in setting the strength of synaptic inhibition.
Materials and Methods
Animals.
All protocols related were approved by the Institutional Animal Care and Use Committee of the University of California, Irvine, and the Hungarian Committee of the Scientific Ethics of Animal Research (license number XIV-1-001/2332-4/2012), and all animal experiments were performed according to the Hungarian Act of Animal Care and Experimentation (1998, XXVIII, Section 243/1998), which are in accordance with the European Communities Council Directive of November 24, 1986 (86/609/EEC; Section 243/1998). All efforts were made to minimize pain and suffering and to reduce the number of animals used. Adult male C57BL/6 mice aged 4–7 weeks were used in the electrophysiological, anatomical, and analytical chemistry experiments. In the electrophysiological experiments testing the role of TRPV1, adult C57BL/6 wild-type and TRPV1−/− mice of either sex (The Jackson Laboratory) aged 4–7 weeks were used.
Electrophysiological recordings.
The procedures of electrophysiology experiments are similar to those described previously by Lee et al. (2010). Briefly, coronal dorsal hippocampal slices (300 μm; anteroposterior, −1.9 to −2.8 mm from bregma) were prepared from deeply anesthetized mice. After cutting, slices were incubated in sucrose-containing artificial CSF (ACSF) for 1 h (in mm: 85 NaCl, 75 sucrose, 2.5 KCl, 25 glucose, 1.25 NaH2PO4, 4 MgCl2, 0.5 CaCl2, and 24 NaHCO3). All electrophysiological recordings were made at 33°C in ACSF solution (in mm: 126 NaCl, 2.5 KCl, 26 NaHCO3, 2 CaCl2, 2 MgCl2, 1.25 NaH2PO4, and 10 glucose). Slices were visualized with an upright microscope (Olympus BX61WI or Nikon Eclipse FN1) with infrared differential interference contrast optics. Whole-cell recordings were obtained from the interneurons with patch pipettes (3–5 MΩ) filled with internal solution containing 126 mm K-gluconate, 4 mm KCl, 10 mm HEPES, 4 mm ATP-Mg, 0.3 mm GTP-Na, 10 mm phosphocreatine, and 0.2% biocytin, pH 7.2, 290 mOsm. The interneurons were located in the stratum radiatum of the CA1 subfield. Pyramidal cells (whole-cell voltage-clamp configuration, holding potential set to −70 mV) were recorded with internal solution containing the following (in mm): 40 CsCl, 90 K-gluconate, 1.8 NaCl, 1.7 MgCl2, 3.5 KCl, 0.05 EGTA, 10 HEPES, 2 MgATP, 0.4 Na2GTP, and 10 mm phosphocreatine, pH 7.2 (290 mOsm). In specific experiments, the calcium chelator BAPTA (10 mm) was also added to the internal solution. All drugs were obtained from Cayman Chemical, Tocris Bioscience, or Sigma-Aldrich. Recordings were made using MultiClamp700B amplifiers (Molecular Devices). Signals were filtered at 3 kHz using a Bessel filter and digitized at 10 or 20 kHz with Digidata 1440A or 1550 analog-to-digital interfaces (Molecular Devices). Series resistances were carefully monitored, and recordings were discarded if the series resistance changed >20% or reached 20 MΩ. The recorded traces were analyzed using the Clampfit 10 software (Molecular Devices). Action potentials in presynaptic interneurons were induced in current clamp by injecting 2 ms square pulses of 1.5–2 nA at 10 Hz frequency. Trains of 50 action potentials were performed once every minute. Values for the so-called “effective” unitary IPSC (euIPSC; Neu et al., 2007) amplitudes included both successful events and failures. To examine drug effects, unless stated otherwise, brain slices were treated with a drug or a mixture (JZL184, JZL184 + AM251, PF3845, PF3845 + JZL184, PF3845 + JZL184 + AM9810, or AM251 + AMG9810, dissolved in the recording ACSF solution) for 40 min before paired recordings were performed (note that the lipid level measurements were also performed after 40 min treatment of slices with one of the drug conditions or 0.1% DMSO control solution; for detailed information, see below, Endocannabinoid level measurements by liquid chromatography/tandem mass spectrometry). In specific experiments, acute drug effects (AM251, NESS0327, JZL184, JZL184 + NESS0327, WIN55,212-2, dissolved in the recording ACSF solution) were quantified by averaging three series of 50 trials each in the respective drug conditions. Depolarization-induced suppression of inhibition (DSI) of uIPSCs was produced by using a 0.5-s-long depolarizing pulse from −70 to 0 mV, and the IPSCs were compared between the pre-DSI period (−2 to 0 s; the latter being the start of the depolarizing pulse) and the DSI period (0–2 s after the end of the pulse).
Endocannabinoid level measurements by liquid chromatography/tandem mass spectrometry.
Acute hippocampal slices were prepared and treated exactly as hippocampal slices used for electrophysiological experiments. To reduce potential variability in endocannabinoid levels attributable to the different circuit architecture of dorsal and ventral hippocampal sections, five 300-μm-thick acute hippocampal slices were pooled in a single measured sample. To exclude potential hemispheric differences, these slices were collected in an alternate manner from the left and right hemisphere. Thus, endocannabinoid levels were always compared between two hippocampal tissue samples derived from an individual animal, with the tissue being treated by either an enzyme inhibitor or its corresponding vehicle control. Animals were housed, and brain slices were prepared in exactly the same manner and at the same time of the day from one liquid chromatography/tandem mass spectrometry (LC/MS/MS) experiment to the next, to further reduce biological variability in endocannabinoid levels. After pharmacological treatment, the hippocampal samples were frozen on dry ice and stored at −70°C until the LC/MS/MS measurement. Sample preparation for chromatography started with tissue incubation on ice for 30 min in 1 ml of methanol containing the deuterated internal standards 2-arachidonoyl-glycerol-d5 (100 ng/ml) and arachidonoyl-ethanolamide-d (1 ng/ml). After ultrasonic homogenization, samples were centrifuged on an Eppendorf miniSpin microtube centrifuge at 13,400 rpm for 15 min. The supernatant was transferred into a plastic tube and was diluted with 3 ml of ultrapure water. The samples were subjected to solid-phase extraction (SPE) sample clean-up according to the following protocol. First, the SPE cartridges (SUPELCO Discovery DSC-18 1 ml tubes, 100 mg) were conditioned with 2 ml of methanol and 2 ml of ultrapure water. Diluted samples were loaded at ∼0.5 ml/min flow rate to apply a gentle vacuum. In the consecutive washing step, 2 ml of ultrapure water and 2 ml of methanol/water (50:50 v/v%) were percolated through the sorbent material. Elution was performed with 0.7 ml of methanol. Eluates were diluted to initial HPLC eluent composition with 10 mm ammonium formate solution.
To measure endocannabinoid levels, we used a PerkinElmer Life and Analytical Sciences HPLC Series 200 system, which was coupled to an Applied Biosystems/Sciex 4000 QTRAP triple quadrupole/linear ion trap tandem mass spectrometer operated in positive electrospray ionization mode. The electronspray ionization ion source parameters were set as follows: curtain gas, 10; ion spray voltage, 5000 V; temperature, 500°C; collisionally activated dissociation gas, medium; gas 1, 50; gas 2, 40. Chromatographic separation was achieved with a Phenomenex Kinetex C18 column (50 mm × 3.00 mm, 2.6 μm, 100 Å) using methanol and 10 mm ammonium formate as elution solvents at a flow rate of 500 μl/min. The injection volume was 50 μl. The initial eluent condition was 80% methanol/20% buffer that was increased to 85% organic phase during 3 min and then further elevated to 95% during 2 min and was kept at this condition for 2 min. Afterward, the column was equilibrated to the initial condition. Analytes were detected in multiple reaction monitoring (MRM) mode at the following ion transitions and parameter settings: (1) 2-AG, MRM transition [mass/charge ratio (m/z), 379.4 → 287.2, 379.4 → 91.1], declustering potential (81 V), collision energy (23 V, 81 V), cell exit potential (10 V, 8 V); (2) 2-arachidonoylglycerol-d5, MRM transition (m/z, 384.4 → 287.2, 384.4 → 91.1), declustering potential (81 V), collision energy (23 V, 81 V), cell exit potential (10 V, 8 V); (3) arachidonoylethanolamide, MRM transition (m/z, 348.4 → 62.1, 348.4 → 90.9), declustering potential (51 V), collision energy (43 V, 63 V), cell exit potential (4 V, 8 V); and (4) arachidonoylethanolamide-d4, MRM transition (m/z, 352.4 → 66.0, 352.4 → 91.2), declustering potential (81 V), collision energy (41 V, 77 V), cell exit potential (6 V). The peak areas were determined with Analyst 1.4.2. software. The quantity of the analytes was calculated by comparing their peak areas with those of the deuterated internal standards, and it was normalized to the sample weight.
All endocannabinoid standards were purchased from Cayman Chemical. HPLC gradient-grade methanol was supplied by Merck. Ammonium formate was from Riedel-de Haën. Water was purified with a MilliQ Direct 8 system (Millipore).
Immunostaining for cell-type identification.
After in vitro recording with pipettes containing biocytin, slices were transferred into a fixative solution containing 4% paraformaldehyde and 0.2% picric acid in 0.1 mm phosphate buffer (PB). Slices were resectioned into 40–70 μm thin sections and tested for CB1 immunopositivity (Fukudome et al., 2004; CB1-GP-Af530-1; 1:5000, guinea pig; Frontiers Science). A secondary antibody conjugated to Alexa Fluor 488, raised in donkey against guinea pig (Invitrogen), were used to detect the location of the primary antibody, and streptavidin conjugated to DyLight 594 (1:500; Jackson ImmunoResearch) was used to visualize biocytin. All primary and secondary antibodies were diluted in Tris-buffered saline (TBS) containing 2% normal goat serum. The sections were then mounted in Vectashield (Vector Laboratories) and analyzed with a fluorescent microscope (Zeiss Axioscope 2 plus or Nikon Eclipse 80i) at low magnification and then with a confocal microscope (Zeiss LSM700 microscope or Nikon A1R confocal laser-scanning system built on a Ti-E inverted microscope and operated by NIS-Elements AR software) at high magnification. To evaluate interneuron morphology, Z-stacks were acquired throughout the whole axonal arborization of the recorded cells. In some cases, the biocytin-filled cells were visualized with 3,3′-diaminobenzidine tetrahydrochloride (0.015%) using a standard ABC kit (Vector Laboratories). Identification of CB1-expressing interneurons was done based on the distinct axonal morphology (perisomatic interneurons: axons branching almost exclusively the pyramidale layer; dendritic interneurons: axons ramifying predominantly in the stratum radiatum and to a lesser extent in the oriens) and the immunopositivity for CB1 (Vida et al., 1998; Cope et al., 2002; Pawelzik et al., 2002; Lee et al., 2010, 2014; Dudok et al., 2015).
Immunostaining for STORM imaging.
Immersion-fixed 300-μm-thick acute brain slices containing biocytin-filled cells were developed with Alexa Fluor 488–streptavidin, covered in Vectashield, and imaged on a Zeiss LSM780 confocal microscope using a Plan Apochromat 20× objective (0.8 numerical aperture), and the cell types were evaluated based on axonal morphology. After imaging, the slices containing interneurons selected for STORM imaging were washed and embedded in 2% agarose in distilled water, and 20-μm-thick coronal sections were cut with a Leica VT-1000S Vibratome in PB and transferred into 24-well tissue culture plates. Immunostaining was performed in a free-floating manner as described previously (Dudok et al., 2015). Briefly, sections were washed in TBS, blocked with 1% HSA (albumin from human serum; Sigma), and incubated overnight with an affinity-purified primary antibody (guinea pig anti-CB1, 1:1000; Fukudome et al., 2004). Sections were then washed in TBS and incubated with a secondary antibody (2 μg/ml) for 4 h, which was prepared for STORM imaging by using an unlabeled affinity-purified anti-guinea pig IgG (Jackson ImmunoResearch), and was tagged with Alexa Fluor 647 as reporter and Cy3 as activator. After washing in TBS and PB, sections were mounted and dried on coverslips. Finally, the samples were covered with freshly prepared imaging medium containing 5% glucose, 0.1 m mercaptoethylamine, 1 mg/ml glucose oxidase, and catalase (2.5 μl/ml aqueous solution from Sigma; ∼1500 U/ml final concentration) in Dulbecco's PBS (Sigma). Coverslips were sealed with nail polish and transferred onto the microscope after 10 min incubation in imaging medium. STORM imaging was performed for up to 3 h after covering the specimens.
Combined confocal/STORM image acquisition and processing.
Correlated STORM and confocal imaging, image processing, and analysis were performed as described previously (Dudok et al., 2015). Images were taken using a Nikon Ti-E inverted microscope equipped with CFI Apo TIRF 100× objective (1.49 numerical aperture), Nikon N-STORM system, a Nikon C2 confocal scan head, and an Andor iXon Ultra 897 EMCCD. First, the field of view containing biocytin-labeled axon terminals was selected, and a confocal stack was captured. Next, the astigmatic 3D STORM image of the anti-CB1 immunostaining was acquired, using 1000 cycles of one activator and three reporter frames, 30 ms each. The low-power activation and the similar imaging depth (∼5 μm) in tissue for all boutons enabled quantitative evaluation of the number of detected localization points (LPs), as demonstrated previously (Dudok et al., 2015).
The corresponding deconvolved confocal images and STORM molecule lists were overlaid using custom-written macros in NIH ImageJ. The outline of the analyzed bouton was determined using the confocal image of biocytin, and LPs from the STORM data belonging to the same bouton were identified. To quantify the internalization of CB1 receptors, the distance (d) of each CB1 LP was first measured from the 3D center of mass of all LPs within the given axon terminal. The radius of the terminal was calculated as the radius of a circle with the same area as the 2D area measured on the corresponding confocal image of the biocytin-containing axon terminal. The ratio of internalized receptors was estimated as the ratio of predicted plasma membrane-bound receptors over total receptors in untreated animals based on previous immunogold electron microscopy data (88%; Dudok et al., 2015). Subsequently, we determined the 88% percentile of the d/r values of all individual LPs from boutons of untreated cells and used this as a threshold to identify internalized LPs in boutons from control and treated samples. Visualization of STORM data was achieved by using the N-STORM module in NIS-Elements AR 4.2 software for 2D images and by using the Visual Molecular Dynamics 1.9.2 software for 3D renderings. The axon terminal surface was approximated by fitting a 3D convex hull on the LPs. LPs were visualized as atoms, and convex hull edges were visualized as bonds.
Immunogold staining for electron microscopy.
Two wild-type and two TRPV1−/− mice (5 weeks old) were anesthetized deeply with ketamine–xylazine (25 mg/ml ketamine, 5 mg/ml xylazine, and 0.1% w/w pipolphen in H2O; 1 ml/100 g, i.p.). Animals were perfused transcardially with 0.9% saline for 3 min, followed by 100 ml of 4% paraformaldehyde fixative in 0.1 m PB, pH 7.4, for 20 min. After perfusion, the brains were postfixed for 2 h and washed in PB. Hippocampal blocks were sliced into 50-μm-thick coronal sections with a Leica VTS-1000 vibratome. After slicing and extensive washing in 0.1 m PB, the sections were incubated in 10% sucrose for 15 min and 30% sucrose overnight, followed by freeze-thawing over liquid nitrogen four times. Subsequently, all washing steps and dilutions of the antibodies were performed in 0.05 m TBS, pH 7.4. After extensive washing in TBS, the sections were blocked in 1% human serum albumin for 45 min and then incubated in goat anti-TRPV1 (1:1000; sc-12498; Santa Cruz Biotechnology) for a minimum of 48 h at 4°C. After the primary antibody incubation, the sections were washed extensively in TBS before incubation in 0.8 nm gold-conjugated donkey anti-goat secondary antibody (1:50; Aurion) overnight at 4°C. Then the sections were silver intensified using the R-GENT SE-LM kit according to the protocol (Aurion). After development, the sections were treated with 1% osmium tetroxide in PB for 10 min on ice in the dark and dehydrated in an ascending series of ethanol and acetonitrile, before being embedded in Durcupan (ACM; Fluka). During dehydration, sections were treated with 1% uranyl acetate in 70% ethanol for 15 min on ice in the dark. Areas of interest from the CA1 stratum pyramidale or stratum radiatum were reembedded and resectioned for additional electron microscopic analysis. Ultrathin (60 nm) sections were collected on Formvar-coated single-slot grids and stained with lead citrate. Electron micrographs were taken at 40,000× magnifications with a Hitachi 7100 electron microscope. Finally, the images were collected and analyzed by an experimenter blind to the animal genotype.
To establish the precise subcellular distribution of TRPV1 at inhibitory symmetrical synapses in the hippocampal CA1 stratum pyramidale and stratum radiatum, high-resolution quantitative evaluation was performed in a population of >250 immunogold-labeled synapses in each animal. Superficial ultrathin sections were collected at a similar tissue depth (∼5 μm) to ensure equal probability of antibody penetration. Images were taken randomly of clearly identifiable symmetrical synapses projecting onto the soma or onto the distal thin dendrites of CA1 pyramidal cells. If a gold particle was present in the image, its location was determined within one of six compartments: (1) presynaptic (within the synaptic bouton); (2) postsynaptic (directly adjacent to the bouton on the postsynaptic cell soma); (3) somatic cytoplasmic (within the postsynaptic soma but not adjacent to the synaptic bouton); (4) mitochondrial (within mitochondria); (5) nuclear (within the cell nucleus); or (6) unknown. The χ2 tests for these subcellular compartments revealed similar numbers of gold particles in the two animals within each genotype in all six compartments. Therefore, data from the two animals belonging to the same genotype were pooled and compared with data obtained from the other genotype. The χ2 tests showed significantly more gold particles in the postsynaptic compartment of wild-type mice compared with TRPV1 knock-out animals in the case of perisomatic synapses, whereas dendritic symmetrical synapses did not contain specific TRPV1 immunolabeling. Gold particle numbers also did not differ between genotypes in the categories of presynaptic, somatic cytoplasmic, mitochondrial, and unknown. More gold particles were found over the nuclei of TRPV1 knock-out mice than wild-type mice, which likely reflects the fact that the antibody can bind to off-targets in a nonspecific manner when its real target protein is absent in the tissue.
To assess the subsynaptic location of these gold particles, we measured their distance from the postsynaptic membrane and allocated them to corresponding 100 nm “bins” to assess their proximity to the synapse. Distance measurements were performed using NIH ImageJ Software.
Statistics.
To analyze the data obtained from electrophysiological recordings, paired or unpaired (as appropriate) two-tailed Student's t tests were used. In cases in which the data did not show normal distribution based on the Shapiro–Wilk test or D'Agostino–Pearson test, then the Wilcoxon's signed-rank or Mann–Whitney U tests for paired and unpaired data, respectively, were used. Pearson's χ2 tests were performed for the validation of immunogold staining, and Kolmogorov–Smirnov test was applied for the subcellular distribution analysis of TRPV1-positive immunogold particles. ANOVAs were followed by Tukey–Kramer or Dunnet's test for mean comparisons. Data are presented as mean ± SEM. A p value <0.05 was considered as significant. Statistical analyses were performed using Origin Pro 8 and STATISTICA analysis software.
Results
Tonic cannabinoid control on GABA release at perisomatic synapses in the mouse hippocampus
Paired whole-cell patch-clamp recordings were performed in acute slice preparations from identified presynaptic CB1-positive interneurons and from postsynaptic pyramidal neurons located in the CA1 region of the mouse hippocampus (n = 164 pairs). The presynaptic interneurons were selected based on the position of their cell bodies in the stratum radiatum, their large soma size and multipolar shape, and their characteristic regular action potential firing pattern showing prominent accommodation in response to suprathreshold depolarizing current pulses (Fig. 1A). Importantly, each presynaptic interneuron was filled with biocytin during the electrophysiological recording and then visualized and identified after the recording (Fig. 1A). Post hoc immunostaining confirmed the presence of CB1 receptors on the axon terminals of all recorded interneurons used in this study (Fig. 1A).
Figure 1.

Inverse agonist application unmasks persistent CB1 receptor activity on the axon terminals of perisomatically projecting interneurons. A, Camera lucida reconstruction shows a perisomatically projecting GABAergic interneuron in the CA1 region of the mouse hippocampus. The neuron was filled with biocytin during electrophysiological recording. Although the dendritic tree (red) of this multipolar interneuron covers most hippocampal layers (Ori., stratum oriens; Pyr., stratum pyramidale; Rad., stratum radiatum), its axon arbor (blue) is restricted to the stratum pyramidale, in which somata of CA1 pyramidal neurons are concentrated. Scale bar, 50 μm. Example voltage traces in response to depolarizing and hyperpolarizing current steps (−200 and 150 pA, respectively, from −60 mV) recorded in whole-cell patch-clamp configuration depict spike frequency adaptation typical for CB1-positive interneurons. Calibration: 30 mV, 0.3 s. Indeed, double-immunofluorescence staining confirms that the biocytin-filled axon terminals (labeled by arrows) of this interneuron contain CB1 receptors. Scale bar, 10 μm. B–G, Averaged traces of presynaptic action potentials (top) evoked in a perisomatic interneuron and the respective postsynaptic responses (bottom) in CA1 pyramidal cells. Fifty consecutive uIPSCs (gray) and their averages (black) are presented before and during the application of the CB1 receptor antagonist/inverse agonist AM251 from the same cell pair (10 μm AM251 in B; 1 μm AM251 in E). Note the large increase in uIPSC amplitude and success rate of GABA synaptic transmission after AM251 treatment, indicating the presence of CB1-dependent tonic signaling at this connection. Summary data showing the proportion of successful transmissions and the euIPSC (including both failures and successes) amplitudes (10 μm AM251 in C, D; 1 μm AM251 in F, G; open circles represent individual pairs; green filled circles label averages). Ctrl, Control.*p < 0.05.
CB1-positive interneurons with the typical regular spiking pattern represent a morphologically heterogeneous population of several distinct GABAergic cell types in the hippocampus, which can be best characterized on the basis of the layer-specific distribution of their axonal arbors (Cope et al., 2002; Pawelzik et al., 2002; Klausberger and Somogyi, 2008; Nissen et al., 2010; Szabó et al., 2014b; Dudok et al., 2015; Lenkey et al., 2015). Recently, we reported that phasic and tonic forms of synaptic endocannabinoid signaling are markedly different at the output synapses of perisomatically versus dendritically targeting interneurons in the CA1 (Lee et al., 2010). Therefore, we characterized each interneuron according to the distribution of its axons, and only those were included in this study that could be unequivocally classified either as a perisomatic or a dendritic cell having an axon arbor predominantly distributed in either the stratum pyramidale or stratum radiatum, respectively.
Action potentials in presynaptic, perisomatically projecting, CB1-positive interneurons evoked uIPSCs in postsynaptic CA1 pyramidal neurons (Fig. 1B,E). In agreement with previous findings in rats (Neu et al., 2007; Lee et al., 2010), the proportion of presynaptic action potentials that resulted in successful postsynaptic events (“successes”) and the amplitudes of euIPSCs, including both successes and failures (Neu et al., 2007), were highly variable between individual connections, ranging from 17 to 91% and from 7 to 120.7 pA, respectively (Fig. 1C,D,F,G). Nevertheless, independent analysis of two randomly selected datasets obtained from perisomatic connections in our two different laboratories (Irvine and Budapest) revealed consistently similar mean values for both successes and euIPSC amplitudes under control conditions. Neither the proportion of successful postsynaptic events (Fig. 1C; Irvine: 63 ± 9%, n = 7 pairs; Fig. 1F; Budapest: 57 ± 11%, n = 6 pairs; t test, p = 0.68) nor the amplitude of euIPSCs (Fig. 1D; Irvine: 61.3 ± 15.4 pA, n = 7 pairs; Fig. 1G; Budapest: 49.9 ± 15 pA, n = 6 pairs; t test, p = 0.6) exhibited statistically significant differences between laboratories.
As expected from perisomatically projecting CB1-positive interneuron to CA1 pyramidal cell connections (Neu et al., 2007; Lee et al., 2010), application of the CB1 receptor antagonist/inverse agonist AM251 [N-1-(2,4-dichlorophenyl)-5-(4-iodophenyl)-4-methyl-N-1-piperidinyl-1H-pyrazole-3-carboxamide] (10 μm) unmasked the presence of a cannabinoid tone (Fig. 1B). Both the proportion of successful postsynaptic events (predrug control, 63 ± 9%; AM251, 78 ± 8%; Wilcoxon's signed-rank test, p < 0.05) and the amplitude of the euIPSCs (predrug control, 61.3 ± 15.4 pA; AM251, 103.1 ± 18.9 pA; paired t test, p < 0.05) were significantly increased after AM251 administration (Fig. 1C,D). Application of a lower dose of AM251 (1 μm; Fig. 1E–G) also augmented both the success rate (predrug control, 57 ± 11%; AM251, 69 ± 8%; Wilcoxon's signed-rank test, p < 0.05) and the euIPSC amplitude (predrug control, 49.9 ± 15 pA; AM251, 74.7 ± 22 pA; Wilcoxon's signed-rank test, p < 0.05), confirming that persistent CB1 receptor activity regulates release probability and synaptic strength at perisomatic GABAergic connections in the mouse hippocampus.
Constitutively active CB1 receptors control GABA release at perisomatic synapses
Persistent receptor activity can arise from continuous ligand availability or can be explained by a constitutively active receptor conformation, which spontaneously signals even in the absence of an endogenous ligand (Kenakin, 2004; Pertwee, 2005). With the help of high-resolution biosensor imaging, the former mechanism, i.e., tonic 2-AG endocannabinoid activation, has been shown recently to account for persistent CB1 receptor activity observed on the somatodendritic surface of cultured hippocampal neurons (Ladarre et al., 2014). This form of tonic CB1 activation was postulated to play an important indirect role in receptor targeting and in the regulation of axonal CB1 numbers (Leterrier et al., 2006). In addition, compelling evidence based on inverse CB1 agonists used in expression systems (Bouaboula et al., 1997; Pan et al., 1998) and in brain tissue (Mato et al., 2002) supports the notion that the latter mechanism, i.e., constitutive CB1 activity, may also have biological significance. However, the presence of constitutively active CB1 receptors on hippocampal GABAergic axon terminals has not been substantiated. Because AM251 is a mixed antagonist/inverse agonist of CB1 receptors (Pertwee, 2005), we next tested the effect of the CB1 neutral antagonist NESS0327 [8-chloro-1-(2,4-dichlorophenyl)-N-piperidin-1-yl-5,6-dihydro-4H-benzo2,3]cyclohepta[2,4-b]pyrazole-3-carboxamine] (Ruiu et al., 2003) on GABAergic synaptic transmission between perisomatic CB1-positive interneurons and postsynaptic CA1 pyramidal neurons (Fig. 2A). Unexpectedly, application of NESS0327 (0.5 μm) did not cause changes in the ratio of successful postsynaptic events (Fig. 2B; predrug control, 53 ± 6%; NESS0327, 53 ± 8%; n = 7 pairs; paired t test, p = 0.9) and in the amplitudes of euIPSCs (Fig. 2C; predrug control, 47.5 ± 12.5 pA; NESS0327, 48.8 ± 14.4 pA; n = 7 pairs; paired t test, p = 0.7). In contrast, NESS0327 at the same dose completely blocked the inhibitory effect of the synthetic CB1 agonist WIN55,212-2 [R-(+)-(2,3-dihydro-5-methyl-3-[(4-morpholinyl)methyl]pyrol[1,2,3-de]-1,4-benzoxazin-6-yl)(1-naphthalenyl) methanone monomethanesulfonate] (20 nm) on GABA release from CB1-positive perisomatic interneurons (Fig. 2D–F; euIPSC amplitude: predrug control, 63.6 ± 16.0 pA; NESS0327 + WIN55,212-2, 59.1 ± 11.1 pA; n = 3 pairs; paired t test, p = 0.54; successes: predrug control, 76.4 ± 1.9%; NESS0327 + WIN55,212-2, 78.4 ± 2.9%; n = 3 pairs; paired t test, p = 0.69). Importantly, the same dose of WIN55,212-2 alone strongly reduced GABA release from CB1-positive perisomatic interneurons (for details, see below). Moreover, NESS0327 at the same dose mostly diminished DSI (Fig. 2G), which is known to be dependent on the phasic activation of presynaptic CB1 receptors by postsynaptically released 2-AG (for review, see Ohno-Shosaku and Kano, 2014). Both the proportion of successful postsynaptic events (percentage of predepolarization successes: predrug control, 46.1 ± 12.0%; NESS0327, 76.0 ± 10.5%; paired t test, p = 0.02) and the amplitude of the euIPSCs (percentage of predepolarization uIPSC amplitude: predrug control, 36.3 ± 14.6%; NESS0327, 66.0 ± 15.7%; n = 5 pairs; paired t test, p = 0.03) were significantly increased after NESS0327 administration (Fig. 2H,I).
Figure 2.
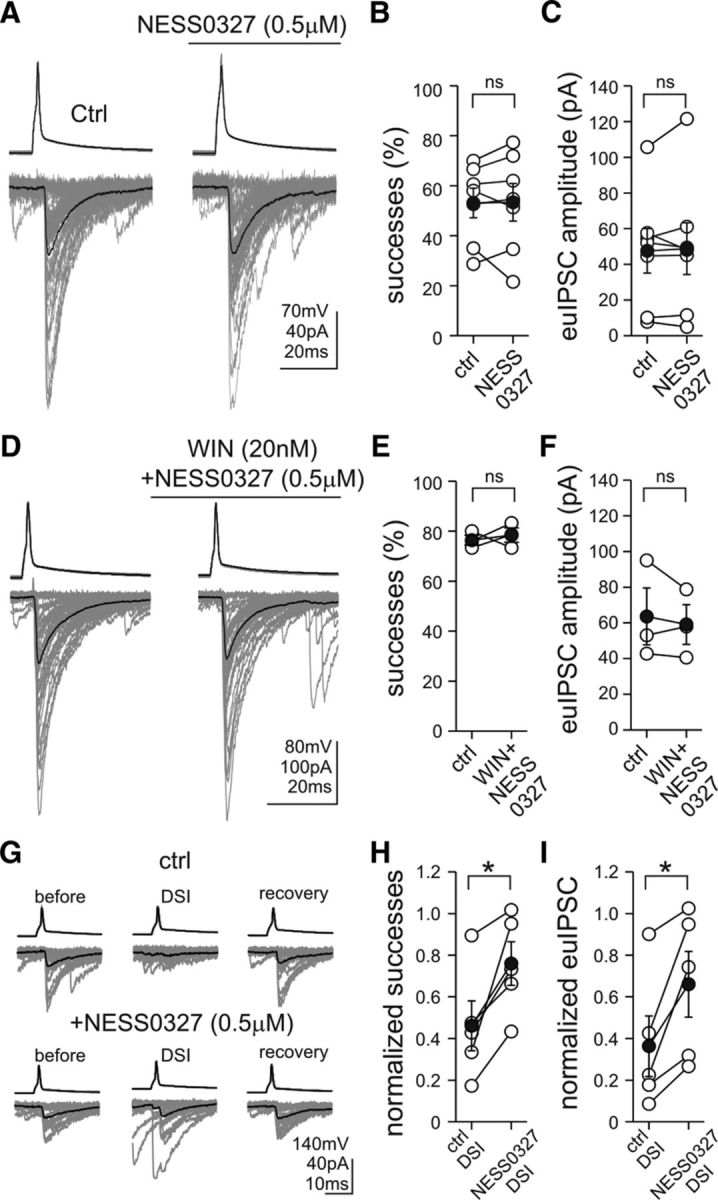
Neutral antagonist application inhibits phasic cannabinoid signaling, but it does not affect constitutive CB1 receptor activity at perisomatic synapses. A, Averaged traces of presynaptic action potentials (top) evoked in a perisomatic interneuron and the respective postsynaptic responses (bottom) in CA1 pyramidal cells illustrate that administration of the CB1 receptor neural antagonist NESS0327 (0.5 μm) did not cause changes in the ratio of successful postsynaptic events and the amplitudes of euIPSCs. Fifty consecutive uIPSCs (gray) and their averages (black) are presented before and during the application of NESS0327 (0.5 μm) from the same cell pair. B, C, Summary data showing the proportion of successful transmission and the euIPSC amplitudes (open circles represent individual pairs; black filled circles indicate averages). D, In the presence of NESS0327 (0.5 μm), administration of the CB1 receptor agonist WIN55,212-2 (WIN; 20 nm) failed to cause changes in the ratio of successful postsynaptic events and in the amplitudes of euIPSCs. E, F, Summary data showing the proportion of successful transmission and the euIPSC amplitudes (open circles represent individual pairs; black filled circles indicate averages). G, DSI was blocked by NESS0327 (0.5 μm). DSIs were evoked by 0.5-s-long depolarization pulses from −70 to 0 mV in the postsynaptic pyramidal cell. All euIPSCs were analyzed from 0 to 2 s after the end of the pulses. Twenty consecutive control uIPSCs and their averages (black) are shown for prepulse control (before), DSI, and full recovery (recovery). In the predrug control condition (ctrl), the depolarizing pulses almost completely reduced the proportion of successful transmission and the euIPSC amplitudes. In the same CB1-postive perisomatic interneuron–pyramidal cell pair, NESS0327 (0.5 μm) blocked DSIs. H, I, Summary data showing that NESS0327 significantly reduced DSIs (open circles represent individual pairs; black filled circles indicate averages). *p < 0.05; ns, not significant.
In summary, these findings demonstrate that the neutral antagonist NESS0327 at a dose of 0.5 μm effectively inhibits phasic CB1 activation evoked by exogenous and endogenous cannabinoids, but it does not affect tonic cannabinoid signaling. Thus, the tonic blockade of neurotransmitter release probability is likely to be caused primarily by the presence of constitutively active CB1 receptors on perisomatic GABAergic axon terminals.
Tonically active monoacylglycerol lipase controls 2-AG-mediated endocannabinoid signaling at perisomatic GABAergic synapses
Endocannabinoid levels and the activity of their metabolizing enzymes dynamically change according to specific physiological states, e.g., strictly follow diurnal phases (Vaughn et al., 2010; Liedhegner et al., 2014; Hanlon et al., 2015). Moreover, impaired inactivation of anandamide or 2-AG attributable to reduced fatty acid amide hydrolase (FAAH) or monoacylglycerol lipase (MGL) expression, respectively, leads to an elevated endocannabinoid tone, which is often associated with pathophysiological mechanisms (Sumislawski et al., 2011; Dincheva et al., 2015). Therefore, in the next set of experiments, we aimed to determine whether anandamide, 2-AG, or the two endocannabinoids together may also be involved in tonic endocannabinoid signaling at perisomatic GABAergic synapses by using pharmacological inhibitors of endocannabinoid-degrading enzymes. As above, we used the technically challenging and labor-intensive paired whole-cell patch-clamp recording approach, because interference with any aspects of endocannabinoid signaling using pharmacological tools may trigger complex perturbations at the network level. In addition, post hoc anatomical identification of every interneuron was performed to ensure that our measurements were focused on the specific GABAergic connections of interest, because CB1 receptors are only present on a subset of perisomatic GABAergic axon terminals (Katona et al., 1999). Therefore, the paired recording technique has distinct advantages compared with alternative approaches, such as the bulk electrical stimulation of mixed populations of fibers or the measurements of compound spontaneous inhibitory postsynaptic events (which are derived from more than one interneuron type). Electrophysiological measurements were complemented by LC/MS/MS to directly monitor changes in endocannabinoid levels in parallel with alterations in the magnitude of the synaptic endocannabinoid tone in similarly prepared and maintained control or drug-treated acute hippocampal slice preparations.
MGL is responsible for the inactivation of ∼85% of the total 2-AG content in the brain (Dinh et al., 2002; Blankman et al., 2007). Moreover, recent studies demonstrated that this serine hydrolase plays a regulatory role in endocannabinoid-mediated short-term DSI (Straiker and Mackie, 2009; Pan et al., 2011). Therefore, we first tested the effects of JZL184 (4-nitrophenyl 4-[bis(1,3-benzodioxol-5-yl)(hydroxy)methyl]piperidine-1-carboxylate), an irreversible inhibitor of MGL (Long et al., 2009b). Although JZL184 has a 300-fold selectivity for MGL over FAAH (Long et al., 2009b), the primary degrading enzyme of anandamide, we took precautions to reduce the potential off-target effects of this compound on anandamide levels. Besides using a low dose (100 nm), 2-AG and anandamide levels were also measured by LC/MS/MS. The data showed that a 40 min pretreatment of acute hippocampal slices with JZL184 evoked a robust 105% increase in hippocampal 2-AG levels compared with vehicle-treated slices (Fig. 3A; paired t test, p < 0.001; n = 9 animals in each group), whereas anandamide levels did not change significantly (Fig. 3A; paired t test, p > 0.05; n = 9 animals in each group).
Figure 3.

Persistent MGL activity limits the tonic endocannabinoid control of GABA release from the axon terminals of perisomatically projecting interneurons. A, Pretreatment of acute hippocampal slices with the irreversible MGL inhibitor JZL184 (100 nm) for 40 min evoked a robust increase in hippocampal 2-AG levels as measured by LC/MS. In contrast, the same treatment had no significant effect on anandamide levels. Data were normalized to either average control 2-AG or anandamide levels. B–D, Pretreatment of acute slices with JZL184, in a manner identical to that used for the LC/MS measurements, strongly decreased success rates and euIPSC amplitudes. The tonic inhibitory effect of MGL blockade on GABA release was absent when JZL184 was applied together with AM251. E, 2-AG levels were measured 5 min and 40 min after incubation of hippocampal slices in ACSF or in ACSF containing JZL184 (100 nm) by LC/MS. Unexpectedly, 2-AG levels increased tonically under basal conditions in the absence of any pharmacological perturbation (ACSF, 5 vs 40 min values are compared here). Similarly, 2-AG levels were elevated even more strongly in the presence of JZL184 (JZL184, 5 vs 40 min values are compared here). F–H, Acute administration of JZL184 (100 nm) dramatically reduced the strength of perisomatic GABAergic connections. The drop in success rates and euIPSC amplitudes reached significance and maximal effect after 12 and 16 min, respectively, of JZL184 application. The baseline levels of success rates and euIPSC amplitudes measured between 3 and 8 min after establishment of the paired recording did not exhibit significant changes during the following 20-min-long recording period (between 23 and 28 min) in the absence of any pharmacological perturbation (ctrl, G, H). Notably, the presence of BAPTA (10 mm) in the postsynaptic neuron or the coapplication of NESS0327 (0.5 μm) completely blocked the acute effect of MGL blockade (G, H). In G and H, the plots show summary data of control (n = 6), JZL184 (n = 4), JZL184 + BAPTA (n = 4), and JZL184 + NESS0327 (n = 4). Error bars were removed for clarity. The bar graphs with error bars display summary data at the time point 16 min after drug application. The numbers of individual experiments are shown in parentheses. Error bars represent SEM. *p < 0.05, ***p < 0.001; ns, not significant.
In accordance with the strong increase in 2-AG levels, preincubation of acute hippocampal slices with JZL184 (100 nm) caused a dramatic reduction in the strength of GABAergic transmission (Fig. 3B). Both the proportion of successful postsynaptic events (Fig. 3C; control, 58 ± 8%, n = 11 pairs; JZL184, 17 ± 3%, n = 10 pairs; Tukey–Kramer test, p < 0.001) and the amplitude of euIPSCs (Fig. 3D; control, 55.9 ± 12.2 pA, n = 11 pairs; JZL184, 8.9 ± 2.1 pA, n = 10 pairs; Tukey–Kramer test, p < 0.01) decreased significantly compared with vehicle-treated slices. The effects of JZL184 were CB1 receptor mediated, because application of AM251 (10 μm) completely reversed the synaptic effects of elevated 2-AG concentrations on the proportion of successful transmissions (Fig. 3C; 79 ± 10%; Tukey–Kramer test, p < 0.001; n = 4 pairs) and on the amplitude of euIPSCs (Fig. 3D; 87.3 ± 26.8 pA; Tukey–Kramer test, p < 0.01; n = 4 pairs). In accordance with the plausible scenario of 2-AG acting on CB1 receptors located presynaptically, the 1/CV2 value was reduced strongly during JZL184 pretreatment (from 1.27 ± 0.32 in control to 0.26 ± 0.07 in JZL184; Tukey–Kramer test, p < 0.05; n = 11 and 10 pairs), which was rescued by AM251 application (1.73 ± 0.6; Tukey–Kramer test, p < 0.05; n = 4 pairs).
These mass spectrometry and electrophysiology results indicated that 2-AG is mobilized constitutively in acute hippocampal slice preparations. To investigate more directly this possibility, we measured 2-AG levels in the absence of any pharmacological perturbation. Surprisingly, acute hippocampal slices produced 2-AG even under basal conditions, because comparison of two sets of samples kept in ACSF revealed 41% higher 2-AG levels in slices snap-frozen at a later time point (35 min; Fig. 3E). MGL can partially keep the tonic elevation of 2-AG levels under control, because the same measurement in the presence of JZL184 (100 nm) resulted in a much larger increase in 2-AG levels (89%; Fig. 3F). Next, we aimed to test directly the effect of the tonic release of 2-AG on GABAergic transmission. After establishing stable paired recording between a CB1-positive perisomatic interneuron and a CA1 pyramidal neuron, the baseline levels of successes and euIPSCs were measured, and then the effect of acute administration of JZL184 (100 nm) was followed through 20 min. Notably, blockade of MGL significantly reduced the strength of perisomatic GABAergic connections within 12 min of JZL184 application (Fig. 3F–H). The maximal decrease in the proportion of successful postsynaptic events was down to 23 ± 6% of the baseline levels (Fig. 3G), whereas the amplitude of euIPSCs was dropped to 29 ± 8% of the baseline values (Fig. 3H) at 16 min after drug application (one-way ANOVA with repeated measures; n = 4 pairs, p < 0.001 for both parameters).
Previous studies provided direct evidence that tonic endocannabinoid signaling is a homosynaptic phenomenon (Hentges et al., 2005; Zhu and Lovinger, 2005; Neu et al., 2007; Roberto et al., 2010). In these studies, the increased GABAergic synaptic transmission observed in the presence of 10 μm AM251 could be blocked by calcium chelation in the postsynaptic neuron. Therefore, we also recorded GABAergic events by using an intracellular pipette solution containing 10 mm BAPTA. Remarkably, the presence of BAPTA in the postsynaptic neuron vetoed the acute effect of MGL blockade (Fig. 3G,H), demonstrating that the postsynaptic neuron is tonically releasing 2-AG, which then affects GABA release probability in a homosynaptic manner. Specifically, both the success rate (95 ± 1% of baseline values; n = 4 pairs) and the euIPSC amplitude (86 ± 1% of baseline values; n = 4 pairs) remained unchanged (one-way ANOVA with repeated measures; n = 4 pairs, p > 0.05 for both parameters), when the MGL inhibitor was applied in the presence of BAPTA in the pipette that was used to record the uIPSCs from the postsynaptic cell. Finally, to fully exclude any potential confounding effect of inverse agonism by AM251, the neutral antagonist NESS0327 was used to prove unequivocally that tonic 2-AG activation of CB1 receptors was necessary to diminish GABAergic synaptic transmission (Fig. 3G,H). Indeed, when NESS0327 (0.5 μm) was coapplied with JZL184 (100 nm), neither the proportion of successful postsynaptic events (control, 92 ± 5% of baseline value; JZL184 + NESS0327, 94 ± 8%; n = 4 pairs) nor the amplitude of euIPSCs (control, 82.6 ± 9.2 pA of baseline value; JZL184 + NESS0327, 84.1 ± 15.2 pA; n = 4 pairs) were changed (one-way ANOVA with repeated-measures; p > 0.05 for both parameters).
Collectively, these findings demonstrate that 2-AG is generated constitutively in acute hippocampal slice preparations and that tissue levels of 2-AG are under the tonic control of MGL activity. As a result, persistent MGL activity plays a critical role in setting the strength of GABAergic synaptic transmission at the CB1-expressing GABAergic perisomatic inputs to pyramidal cells.
FAAH does not regulate tonic anandamide signaling at perisomatic GABAergic synapses
In accordance with the distinctive role of anandamide signaling in certain behavioral processes (Long et al., 2009a), emerging evidence indicates that specific forms of phasic and tonic endocannabinoid signaling at given brain circuit locations or under specific physiological conditions are mediated by anandamide (Azad et al., 2004; Kim and Alger, 2010; Huang and Woolley, 2012; Lerner and Kreitzer, 2012; Khlaifia et al., 2013; Mathur et al., 2013). Therefore, we next sought to determine whether anandamide is also involved in tonic endocannabinoid signaling at perisomatic GABAergic synapses in the mouse hippocampus. As mentioned above, the predominant degrading enzyme of anandamide is FAAH (Cravatt et al., 1996). Blockade of FAAH with the recently introduced selective inhibitor PF3845(N-pyridin-3-yl-4-[[3-[5-(trifluoromethyl)pyridin-2-yl]oxyphenyl]methyl]piperidine-1-carboxamide) (Ahn et al., 2009) at 1 μm evoked a striking and selective increase in anandamide levels by 107% (Fig. 4A; paired t test, p < 0.001; n = 4 animals) but was unable to cause significant changes in 2-AG levels (Fig. 4A; paired t test, p > 0.05; n = 4 animals). Throughout this study, PF3845 was used to perturb anandamide signaling because of its high selectivity for FAAH inhibition. For comparison purposes, the pretreatment effects of another widely used FAAH inhibitor, URB597 [3′-(aminocarbonyl)[1,1′-biphenyl]-3-yl)-cyclohexylcarbamate] (1 μm; Kathuria et al., 2003), on endocannabinoid levels were also measured. Similar to PF3845, URB597 also caused a large increase (103%) in anandamide levels (Fig. 4A; paired t test, p < 0.05; n = 5 animals) but not in 2-AG levels (Fig. 4A; paired t test, p > 0.05; n = 5 animals).
Figure 4.

FAAH does not directly mediate tonic endocannabinoid signaling at CB1-expressing perisomatic GABAergic synapses. A, Prevention of anandamide inactivation by using the selective FAAH inhibitor PF3845 (1 μm; 40 min) resulted in a large increase in hippocampal anandamide levels, whereas 2-AG levels remained unaffected in acute hippocampal slice preparations. Pretreatment with a second independent FAAH inhibitor, URB597 (1 μm; 40 min), caused a similar rise in anandamide levels, whereas 2-AG levels did not change. Data were normalized to either average control 2-AG or anandamide levels. B–D, Despite the elevated anandamide levels in hippocampal slices, inhibition of FAAH by PF3845 pretreatment (1 μm; 40 min) did not alter success rates or euIPSC amplitudes at perisomatic synapses. The numbers of individual experiments are shown in parentheses. Error bars represent SEM. *p < 0.05, ***p < 0.001; ns, not significant; ctrl, control.
Despite the highly elevated levels of anandamide, paired recordings from presynaptic perisomatically projecting CB1-positive interneurons and postsynaptic CA1 pyramidal neurons (Fig. 4B) revealed that the proportion of successful postsynaptic events (Fig. 4C; control, 60 ± 7%, n = 14 pairs; PF3845, 56 ± 8%, n = 12 pairs; unpaired t test, p > 0.05) and the amplitude of euIPSCs (Fig. 4D; control, 56.6 ± 9.5 pA, n = 14 pairs; PF3845, 54.9 ± 15.7 pA, n = 12 pairs; Mann–Whitney U test, p > 0.05) remained unchanged in the presence of PF3845 (1 μm).
These data together show that anandamide is also constitutively generated in hippocampal slice preparations and its tissue level is tonically controlled by FAAH activity. However, in contrast to the effects of the elevated 2-AG levels, highly increased anandamide levels have no direct effect on the strength of neurotransmission at perisomatic GABAergic synapses.
Neither MGL nor FAAH regulates GABA release at dendritic inhibitory synapses
Tonic endocannabinoid signaling is not present at the efferent synapses of dendritically projecting CA1 interneurons, despite the fact that the axon terminals of these interneurons do possess functional CB1 receptors (Lee et al., 2010). This was especially surprising because diacylglycerol lipase-α (DGL-α), the synthesizing enzyme of 2-AG (Bisogno et al., 2003), is concentrated mainly in the dendritic spine heads of pyramidal neurons (Katona et al., 2006; Yoshida et al., 2006). The finding that MGL tonically controls the synaptic effects of 2-AG at perisomatic GABAergic synapses raised the possibility that elevated MGL activity in dendritically projecting interneuron boutons could mask the presence of a weak tonic 2-AG effect at dendritic GABAergic synapses. To test this possibility, paired recordings were performed from presynaptic dendritically projecting CB1-positive interneurons (Fig. 5A) and postsynaptic CA1 pyramidal neurons in acute hippocampal slices.
Figure 5.

Blockade of endocannabinoid degradation does not affect synaptic functions at GABAergic synapses formed by CB1-expressing dendritically projecting interneurons. A, Camera lucida reconstruction illustrates a CB1-positive dendritically projecting GABAergic interneuron in the CA1 region of the mouse hippocampus. The dendrites (red) and most of the axons (blue) of this multipolar interneuron arborized in stratum radiatum (Rad.), but a few additional axon branches were also observed in the stratum pyramidale (Pyr.) and stratum oriens (Ori.). Scale bar, 50 μm. The example voltage traces in response to depolarizing and hyperpolarizing current steps (−100 and 150 pA, respectively, from −60 mV) show a characteristic regular spiking pattern with prominent spike frequency adaptation, an electrophysiological fingerprint of CB1-positive interneurons. Calibration: 30 mV, 0.3 s. Confocal images demonstrate that the biocytin-filled boutons (labeled by arrows) of this interneuron are indeed CB1 positive. Scale bar, 10 μm. B, Representative traces demonstrate that a low dose of the synthetic CB1 agonist WIN55,212-2 (20 nm) significantly reduced synaptic strength at both dendritic and perisomatic GABAergic connections. C, D, Application of WIN55,212-2 reduced the success rates and euIPSC amplitudes more strongly at perisomatic connections. E–J, Application of neither 1 μm AM251 nor 10 μm AM251 unmasked persistent CB1 activity on the axon terminals of CB1-expressing dendritic interneurons. K, Despite the presence of functional CB1 receptors on the axon terminals, GABAergic synaptic transmission originating from dendritic interneurons is not affected by inhibiting endocannabinoid hydrolysis by either the MGL inhibitor JZL184 (100 nm; 40 min) or the FAAH inhibitor PF3845 (1 μm; 40 min). L, M, Neither the proportion of successful postsynaptic events nor the amplitude of euIPSCs were altered by JZL184 or PF3845 pretreatment, indicating that the absence of dendritic tonic inhibition is not attributable to high MGL or FAAH activity at dendritic connections. The numbers of individual experiments are shown in parentheses. Error bars represent SEM. *p < 0.05, **p < 0.01; ns, not significant.
First, we tested the functionality of presynaptic CB1 receptors on dendritic connections (Fig. 5B) by applying a very low dose of the synthetic CB1 agonist WIN55,212-2 (20 nm), which could still readily reduce the success rate by 19 ± 7% (Fig. 5C; n = 6 pairs; Wilcoxon's signed-rank test, p < 0.05) and the euIPSC amplitudes by 35 ± 6% (Fig. 5D; n = 6 pairs; Wilcoxon's signed-rank test, p < 0.05). As expected (Lee et al., 2010), the same dose of WIN55,212-2 (20 nm) had significantly larger effects on perisomatic connections (for both parameters, Mann–Whitney U test, p < 0.05). The proportion of successful postsynaptic events originating from perisomatically projecting interneurons was inhibited by 42 ± 9% (Fig. 5C; n = 7 pairs; paired t test, p < 0.01), and the euIPSC amplitudes were decreased by 61 ± 7% (Fig. 5D; n = 7 pairs; Wilcoxon's signed-rank test, p < 0.05). Despite the latter data demonstrating the presence of functionally active CB1 receptors on the axon terminals of dendritically projecting interneurons, application of 1 or 10 μm AM251 could not uncover the presence of persistent CB1 activity on these axon terminals (Fig. 5E–J), in agreement with our previous findings in rats (Lee et al., 2010). Both the proportion of action potentials evoking successful postsynaptic events (predrug control, 80 ± 7%; 1 μm AM251, 76 ± 7%; n = 3 pairs, Wilcoxon's signed-rank test, p > 0.05; predrug control, 58 ± 6%; 10 μm AM251, 60 ± 8%; n = 4 pairs, Wilcoxon's signed-rank test, p > 0.05) and the amplitude of the euIPSCs (predrug control, 48 ± 6.8 pA; 1 μm AM251, 43.7 ± 4.9 pA; n = 3 pairs, Wilcoxon's signed-rank test, p > 0.05; predrug control, 20.5 ± 2.9 pA; 10 μm AM251, 20.1 ± 1.7 pA; n = 4 pairs, Wilcoxon's signed-rank test, p > 0.05) remained unchanged after AM251 administration.
In the next set of experiments, acute hippocampal slice preparations were pretreated with either vehicle or the MGL inhibitor JZL184 (100 nm; Fig. 5K). However, neither the proportion of successful postsynaptic events (Fig. 5L; control, 57 ± 12%, n = 5 pairs; JZL184, 57 ± 11%, n = 4 pairs; Tukey–Kramer test, p > 0.05) nor the amplitude of euIPSCs (Fig. 5M; control, 27.2 ± 13.0 pA, n = 5 pairs; JZL184, 33.8 ± 18.4 pA, n = 4 pairs; Tukey–Kramer test, p > 0.05) were changed significantly after blockade of MGL activity by JZL184 at dendritic connections. Given that the axon terminals of both perisomatically and dendritically projecting interneurons contain CB1 receptors (Figs. 1A, 5A), the presence of tonic 2-AG signaling at perisomatic, but not at dendritic, GABAergic synapses raised the possibility of an input-specific functional segregation between 2-AG and anandamide signaling. Thus, we also investigated the possibility whether tonic FAAH activity may regulate tonic anandamide signaling at dendritic GABAergic synapses. However, application of the FAAH inhibitor PF3845 (1 μm) did not affect the proportion of successful postsynaptic events (Fig. 5L; control, 57 ± 12%, n = 5 pairs; PF3845, 56 ± 12%, n = 4 pairs; Tukey–Kramer test, p > 0.05) or the amplitude of euIPSCs (Fig. 5M; control, 27.2 ± 13.0 pA, n = 5 pairs; PF3845, 21.9 ± 5.6 pA, n = 4 pairs; Tukey–Kramer test, p > 0.05) in dendritically projecting, CB1-expressing interneuron to pyramidal cell synapses.
These observations indicate the absence of constitutive CB1 activity, tonic endocannabinoid signaling, and the lack of functionally detectable tonic MGL and FAAH activities at dendritic GABAergic synapses in the mouse hippocampus. The interneuron-type specificity of tonic 2-AG signaling also indicates that the robust JZL184 effect on perisomatic GABAergic synaptic transmission was not the consequence of unidentified nonspecific network mechanisms because of a potential elevation of 2-AG throughout the hippocampal tissue.
Increased endocannabinoid tone affects synaptic functions without changing the number of presynaptic CB1 receptors
The above results were somewhat unexpected considering the general view that anandamide may be better suited to serve as a tonic endocannabinoid signal (Alger, 2012; Ohno-Shosaku and Kano, 2014; Piomelli, 2014). This notion is based in part on the slower time course of anandamide mobilization and the fact that anandamide displays partial agonism toward CB1, in contrast to 2-AG, which is a full agonist (Stella et al., 1997; Hillard, 2000). In accordance with this view, chronic elevations of 2-AG levels by long-term pharmacological or genetic blockade of MGL activity causes robust behavioral tolerance as a result of the massive downregulation of CB1 receptors, whereas chronically increased anandamide levels attributable to the inhibition of FAAH do not evoke similar adaptive responses (Schlosburg et al., 2010). These findings obtained at the systems level also suggest that any experimental perturbation that increases endocannabinoid levels may also indirectly affect CB1 receptor availability. Thus, any measured changes in synaptic strength could simply be the consequence of a change in receptor numbers.
We tested the possibility of alterations in receptor numbers after the increase in endocannabinoid tone using STORM super-resolution imaging (Rust et al., 2006; Dani et al., 2010; Dudok et al., 2015) at CB1 receptor-expressing perisomatic synapses onto CA1 pyramidal cells, i.e., at the synapse type that exhibits tonic endocannabinoid-mediated inhibition of GABA release. CB1-positive perisomatic interneurons were filled with biocytin during patch-clamp recordings in either vehicle-treated or enzyme (MGL or FAAH) inhibitor-treated acute hippocampal slices. The CB1 content of the biocytin-labeled axon terminals of interneurons was analyzed at the nanoscale level to distinguish surface and internalized receptors (Fig. 6A). Because the vast majority of CB1 receptors are found in the plasma membrane (Nyíri et al., 2005), fitting a convex hull onto the outermost LPs representing CB1 receptors could reliably identify the receptor population on the axon terminal surface (Dudok et al., 2015). Notably, an analysis of the nanoscale distribution of CB1 receptors in 303 individual axon terminals from 13 identified perisomatically projecting interneurons revealed no significant changes in the total receptor number (Fig. 6B) or in the ratio of internalized/surface membrane CB1 receptors (Fig. 6C) after JZL184 (100 nm) or PF3845 (1 μm) application for similar amounts of time as in the electrophysiological experiments described above (40 min; Tukey–Kramer test, p > 0.05 in all four measurements, n = 7 vehicle-treated cells compared to 3–3 JZL184- or PF3845-treated cells). These observations demonstrate that the pretreatment protocol used in the present study does not cause a change in CB1 receptor numbers on perisomatic GABAergic axon terminals, despite the strong increase in endocannabinoid levels attributable to the blockade of endocannabinoid degradation.
Figure 6.

Acute inhibition of endocannabinoid-degrading enzymes does not affect CB1 numbers on the plasma membrane of perisomatic interneuron boutons. A, CB1-positive perisomatic interneurons were filled with biocytin in acute hippocampal slice preparations that were pretreated with vehicle (0.1% DMSO; 40 min), JZL184 (100 nm; 40 min), or PF3845 (1 μm; 40 min) to determine whether increased 2-AG or anandamide levels affect the number of CB1 receptors on perisomatic axon terminals. Deconvolved confocal images of identified boutons of perisomatic interneurons that were visualized by biocytin labeling (orange) are shown together with the corresponding STORM images of CB1 immunostaining. LPs (green dots) in the raw STORM images represent the position of CB1 receptors in the axon terminals. To analyze the nanoscale distribution of CB1 receptors, individual LPs were first visualized in 3D and illustrated using Visual Molecular Dynamics. Based on the acquired 3D localization coordinates, a convex hull was then fitted onto the outermost CB1 LPs to approximate the plasma membrane. To distinguish membrane-attached LPs (green) and those that are likely intracellular receptors located away from the surface of the convex hull (blue), an internalization index was calculated (for details, see Materials and Methods). B, C, Summary data of the STORM super-resolution imaging experiment reveals that neither the total number of CB1 LPs nor the proportion of intracellular CB1 LPs are changed significantly by pretreatment of MGL or FAAH inhibitors, indicating that any observed changes in synaptic functions during pharmacological treatments are not attributable to an altered number of available CB1 receptors on the axon terminal surface. The number of individual experiments is shown in parentheses. Data were normalized to average control CB1 number of localization points (NLP) values. Error bars represent SEM. ns, Not significant; Ctrl, Control.
Tonic anandamide signaling interacts with tonic 2-AG signaling
The biological rationale of why the brain needs two endocannabinoid messengers is not fully understood (Di Marzo and De Petrocellis, 2012). A potential clue may be that the remarkable biochemical diversity of endocannabinoid synthesis routes (Blankman and Cravatt, 2013) provides an opportunity for specific signaling mechanisms to selectively recruit endocannabinoid synthesis under certain physiological or pathophysiological conditions. This also raises the possibility that coincident physiological signals may sometimes trigger the mobilization of both endocannabinoids at the same time, which then interact with each other.
Therefore, in the next set of experiments, we asked the question whether tonic 2-AG signaling at perisomatic GABAergic synapses could be fine-tuned by anandamide signaling. One potential explanation for the lack of effects of FAAH inhibition on GABAergic synaptic strength could stem from the fact that anandamide, as mentioned above, is a partial agonist of CB1, and hence it may not elicit a response by itself at these specific GABAergic synapses. As a consequence, it may either facilitate or occlude the effects on tonic 2-AG signaling depending on how 2-AG saturates the functionally relevant CB1 receptor pool on the axon terminals. Indeed, in paired recordings from CB1-positive perisomatic interneurons and CA1 pyramidal neurons (Fig. 7A), when MGL and FAAH were inhibited simultaneously by JZL184 (100 nm) and PF3845 (1 μm), both the proportion of successful postsynaptic events (Fig. 7B; JZL184, 17 ± 3%, n = 10 pairs; JZL184 + PF3845, 41 ± 8%, n = 6 pairs; unpaired t test, p < 0.05) and the amplitude of euIPSCs (Fig. 7C; JZL184, 8.9 ± 2.1 pA, n = 10 pairs; JZL184 + PF3845, 37.9 ± 11.0 pA, n = 6 pairs; unpaired t test, p < 0.05) were significantly larger compared with the selective blockade of MGL with JZL184 alone. Thus, this observation demonstrates that, although enhanced anandamide signaling is not effective in itself, it can readily interfere with tonic 2-AG signaling at perisomatic GABAergic synapses.
Figure 7.

Antagonistic interaction of endocannabinoid-degrading pathways fine-tunes tonic endocannabinoid signaling at CB1-positive perisomatic GABAergic synapses. A, Example traces of 50 consecutive postsynaptic events (bottom; individual uIPSCs are gray; averages are black) evoked by presynaptic action potentials (top) in CB1-positive perisomatically projecting interneurons demonstrate that the low synaptic efficiency of GABAergic transmission induced by pretreatment with the MGL inhibitor JZL184 (100 nm; 40 min) can be enhanced significantly by the simultaneous inhibition of FAAH activity (PF3845; 1 μm; 40 min). B, C, Both the proportion of successful synaptic events and the amplitude of euIPSCs were notably larger when JZL184 was coapplied with PF3845 compared with JZL184 applied alone. D, Prevention of the hydrolysis of both endocannabinoids resulted in significantly smaller levels of 2-AG compared with the effects of MGL inhibition alone. E, In contrast, coapplication of the two serine hydrolase inhibitors did not attenuate anandamide levels induced by PF3845 treatment alone. Data were normalized to either average 2-AG levels in the presence of JZL184 or average anandamide levels in the presence of PF3845. The numbers of individual experiments are shown in parentheses. Error bars represent SEM. *p < 0.05; ns, not significant.
These findings may result from an occlusion of 2-AG effects on presynaptic CB1 activation by anandamide. However, the facts that FAAH is postsynaptic, whereas MGL is presynaptic (Gulyás et al., 2004), and that brain 2-AG concentrations are 170 times greater than anandamide concentrations (Stella et al., 1997) do not support this scenario. Conversely, DGL-α, the synthesizing enzyme of 2-AG, also occurs postsynaptically in the somatodendritic domain of pyramidal neurons, in which FAAH is located (Gulyás et al., 2004; Katona et al., 2006; Yoshida et al., 2006). This molecular architecture indicates that postsynaptic elevation of anandamide levels may interfere with the postsynaptic mobilization of 2-AG. Indeed, coapplication of JZL184 (100 nm) with PF3845 (1 μm) significantly reduced the increase in 2-AG levels compared with MGL blockade alone (Fig. 7D; paired t test, p < 0.05; n = 5 slices). In striking contrast, increased anandamide levels induced by PF3845 pretreatment were not attenuated by the coapplication of PF3845 with JZL184 (Fig. 7E; paired t test, p > 0.05; n = 5 slices).
Together, these results uncovered that tonic 2-AG signaling at perisomatic GABAergic synapses in the hippocampus are controlled by the anandamide-degrading enzyme FAAH via potential postsynaptic interaction mechanisms affecting 2-AG synthesis.
TRPV1 channels are located on intracellular membrane cisternae at perisomatic symmetrical synapses
To explore how postsynaptic anandamide signaling may interfere with tonic retrograde 2-AG signaling, in the final set of experiments, we tested the hypothesis whether TRPV1 receptors represent a link between the two lipid signaling pathways at hippocampal GABAergic synapses. TRPV1 appeared to be a potential candidate, because anandamide is a full agonist of TRPV1 (Zygmunt et al., 1999), and anatomical evidence at the light microscopic level shows that TRPV1 is present in the somatodendritic domain of CA3 pyramidal neurons together with FAAH (Cristino et al., 2008). In addition, emerging evidence indicates that anandamide signaling elicits synaptic plasticity via postsynaptic TRPV1 activation in other brain areas (Chávez et al., 2010, 2014; Grueter et al., 2010; Puente et al., 2011), and, most importantly, anandamide inhibits metabotropic glutamate receptor-induced 2-AG synthesis and phasic endocannabinoid signaling in the striatum via TRPV1 activation (Maccarrone et al., 2008).
Although recent evidence showed that TRPV1 is found postsynaptically at excitatory synapses (Puente et al., 2011, 2015), there was no direct evidence as to whether this ion channel is also located at hippocampal GABAergic synapses, which would be a prerequisite for it to serve as the regulator of tonic endocannabinoid signaling at perisomatic inhibitory synapses. Therefore, we used immunogold electron microscopy to study the localization of TRPV1 at symmetrical synapses in the stratum pyramidale and stratum radiatum of the CA1 subregion of the hippocampus (Fig. 8A,B). In wild-type mice, immunogold particles representing the position of TRPV1 were found frequently in the cell body of CA1 pyramidal neurons (Fig. 8A). Moreover, the subcellular distribution analysis of TRPV1 immunolabeling revealed a significant population of this ion channel postsynaptically, adjacent to symmetrical, putative GABAergic synapses in the stratum pyramidale (Fig. 8A,C). In contrast, immunogold particles were found only occasionally in the latter postsynaptic compartment in TRPV1 knock-out mice (Fig. 8B,C), validating the specificity of the postsynaptic distribution pattern of TRPV1 at inhibitory synapses (Pearson's χ2 test, p < 0.001; n = 429 and 445 synapses in two wild-type and two knock-out mice, respectively). Specific TRPV1 immunolabeling was not found in the presynaptic GABAergic axon terminals or on mitochondria. Analysis of single 60 nm ultrathin sections, including normalization of the data to the nonspecific labeling within the same compartment in TRPV1 knock-out mice, revealed that 18% of perisomatic GABAergic synapses were positive for TRPV1. Given the size of CB1-positive perisomatic symmetrical synapses, which span several consecutive ultrathin sections, and considering their proportion among all symmetrical synapses in the stratum pyramidale of the CA1 subfield (35–40%; Takács et al., 2015), our data are consistent with the possibility that most, if not all, CB1-positive perisomatic synapses contain postsynaptic TRPV1 channels. Conversely, no significant difference was detected in the ratio of immunogold-containing dendritic GABAergic synapses in the stratum radiatum between WT and TRPV1 knock-out mice (6.08 and 3.7%; n = 115 and 108 dendritic synapses, respectively; p = 0.41, χ2 test).
Figure 8.
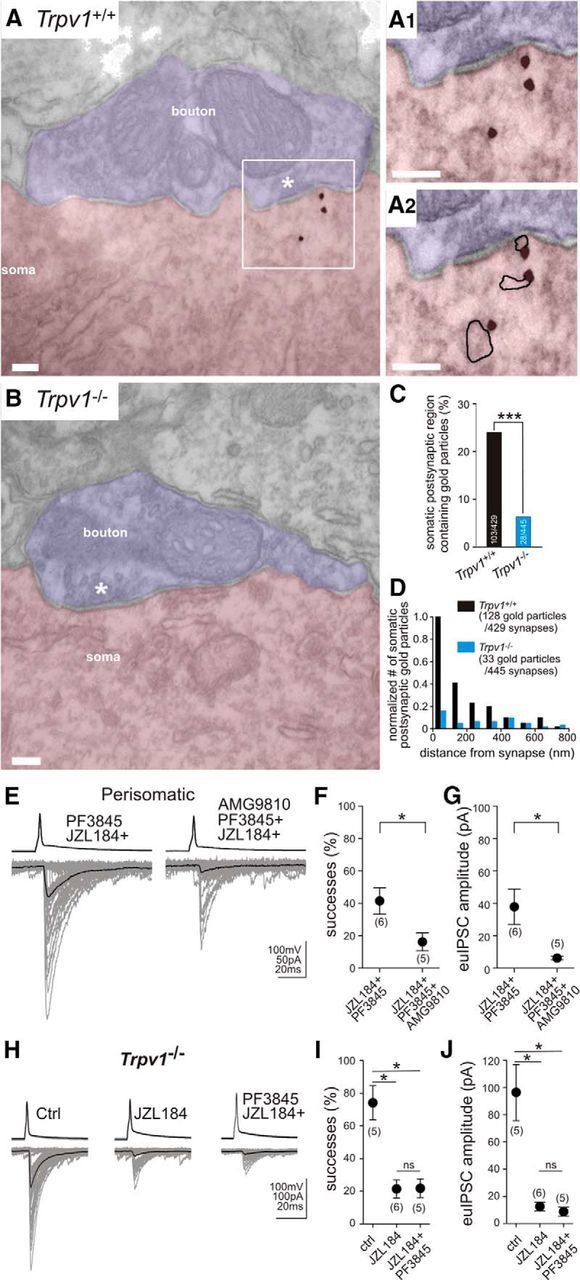
TRPV1 is present postsynaptically at perisomatic GABAergic synapses and regulates synaptic strength at CB1-expressing perisomatic GABAergic synapses. A, At the electron microscopic level, immunogold staining revealed that TRPV1 channels are located postsynaptically at perisomatic GABAergic synapses (labeled by asterisk) formed on the somata of CA1 pyramidal neurons in wild-type mice. A1, A2, Higher-magnification electron micrographs show that gold particles representing the position of TRPV1 were often attached to putative intracellular membrane cisternae (highlighted with black line in A2). B, In contrast, postsynaptic immunogold particles were usually not located at symmetrical GABAergic synapses obtained from TRPV1 knock-out mice. C, Summary bar graph illustrates that the proportion of randomly selected GABAergic synapses forming symmetrical synapses on the cell bodies of CA1 pyramidal neurons that contained immunogold particles in close proximity to the synapse was significantly higher on sections derived from wild-type mouse hippocampus compared with that obtained from TRPV1 knock-out mice. D, To assess the subsynaptic distribution of TRPV1 channels, immunogold particle distribution was further analyzed by measuring distance of each gold particle from the postsynaptic membrane. Notably, a sharply biased distribution of gold particles toward the postsynaptic membrane was observed. Altogether, 68% of all gold particles were found in the vicinity of the postsynaptic plasma membrane (<200 nm) and six times more gold particles were found in wild-type versus TRPV1 knock-out animals in the first two 100 nm bins. E, In accordance with these anatomical findings, coapplication of the selective TRPV1 antagonist AMG9810 (1 μm; 40 min) could efficiently rescue the effect of the FAAH inhibitor on 2-AG-mediated tonic inhibition of GABA release. F, G, Both the ratio of successful postsynaptic events and the amplitude of euIPSCs decreased significantly when AMG9810 was applied together with JZL184 and PF3845 compared with those when JZL184 and PF3845 were applied alone. H–J, MGL blockade with JZL184 (100 nm) strongly reduced success rate and euIPSCs compared with vehicle treatment (ctrl) in TRPV1 knock-out animals. However, the antagonistic effect of FAAH inhibition (seen in F and G in wild-type animals) was completely absent in TRPV1 knock-out mice (H). Importantly, when JZL184 (100 nm) was applied alone or together with PF3845 (1 μm), the success rate of action potentials evoking a postsynaptic IPSC and the amplitude of euIPSCs did not differ between the two conditions. The number of individual experiments is shown in parentheses. Error bars represent SEM. *p < 0.05, ***p < 0.001; ns, not significant; Ctrl, control.
The precise subcellular localization of the nonselective cation channel TRPV1 has also functional significance. Therefore, we first measured the distance of individual gold particles from the synapse. In total, 68% of gold particles were found adjacent to symmetrical synapses (within 200 nm), which was significantly different from the background distribution observed in knock-out animals (Fig. 8D; Kolmogorov–Smirnov test, p < 0.001; n = 128 and 33 gold particles from two wild-type and two knock-out mice, respectively). Surprisingly, the majority of gold particles representing the subcellular position of TRPV1 was found to be attached to intracellular membrane cisternae (Fig. 8A1,A2) instead of a plasma membrane localization (55 ± 6 vs 18 ± 4%). The subcellular compartment of the remaining 27 ± 2% of gold particles could not be identified unequivocally in the absence of clearly outlined nearby membranes, but these particles were all away from the plasma membrane. Considering that TRPV1 is an integral membrane channel and that those gold particles that were found close to the plasma membrane may also have belonged to intracellular membrane segments that were not identifiable, these data suggest that a surprisingly high proportion of TRPV1 is present on intracellular membrane compartments positioned under the postsynaptic plasma membrane of GABAergic synapses.
Endovanilloid signaling acting on postsynaptic TRPV1 channels regulates tonic 2-AG signaling on presynaptic CB1 receptors
Our electron microscopic findings revealed that the subcellular distribution of TRPV1 is similar to the compartmentalized subcellular localization of FAAH on intracellular membrane cisternae (Gulyás et al., 2004). Therefore, we tested the hypothesis that TRPV1 may be responsible for the functional interaction of FAAH inhibition with tonic 2-AG-mediated endocannabinoid signaling by using pharmacological and genetic tools. Paired recordings from perisomatically projecting interneurons and pyramidal neurons (Fig. 8E) revealed that the specific TRPV1 inhibitor AMG9810 [(E)-3-(4-t-butylphenyl)-N-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)acrylamide] (Gavva et al., 2005) completely rescued the effect of the FAAH inhibitor PF3845 on tonic 2-AG signaling. When AMG9810 (1 μm) was applied together with JZL184 (100 nm) and PF3845 (1 μm), both the success rate of action potentials evoking a postsynaptic IPSC (Fig. 8F; JZL184 + PF3845, 41 ± 8%, n = 6 pairs; JZL184 + PF3845 + AMG9810, 16 ± 6%, n = 5 pairs; unpaired t test, p < 0.05) and the amplitude of euIPSCs (Fig. 8G; JZL184 + PF3845, 37.9 ± 11.0 pA, n = 6 pairs; JZL184 + PF3845 + AMG9810, 6.2 ± 1.0 pA, n = 5 pairs; unpaired t test, p < 0.05) were decreased significantly. Accordingly, the proportion of successful events and the amplitude of euIPSCs were similar in the presence of the MGL inhibitor alone and when JZL184 was applied together with PF3845 and AMG9810 (unpaired t test, p > 0.05; n = 10 and 5 pairs). As a control, to exclude the possibility that the inhibitory effect of AMG9810 on the FAAH inhibitor PF3845-induced decrease in tonic 2-AG signaling is confounded by some unexpected off-target effects of AMG9810, we also tested whether AMG9810 attenuates GABA release from CB1-positive perisomatic interneurons in a CB1-dependent or CB1-independent manner. Our paired recordings revealed that pretreatment with AMG9810 (1 μm), in neither the absence nor the presence of the CB1 receptor antagonist AM251 (10 μm), affected the proportion of action potentials evoking a postsynaptic IPSC (vehicle, 60 ± 7%, n = 9 pairs; AMG9810, 65 ± 8%, n = 8 pairs; unpaired t test, p > 0.05; AM251, 92 ± 5%, n = 3 pairs; AMG9810 + AM251, 93 ± 2%, n = 3 pairs; AM251, 92 ± 5%, n = 3 pairs; unpaired t test, p > 0.5) and the amplitude of euIPSCs (vehicle, 53.1 ± 10 pA, n = 9 pairs; AMG9810, 66.4 ± 16 pA, n = 8 pairs; unpaired t test, p > 0.05; AM251, 160.4 ± 22.8 pA, n = 3 pairs; AMG9810 + AM251, 150.8 ± 15.5 pA, n = 3 pairs; unpaired t test, p > 0.5). In accordance with the above electron microscopic observation that the level of TRPV1 channels are under detection threshold at dendritic synapses, preincubation of AMG9810 alone did not have any apparent effect on the proportion of successful postsynaptic events (vehicle, 68 ± 7%, n = 6 pairs; AMG9810, 69 ± 3%, n = 4 pairs; unpaired t test, p = 0.86) and the amplitude of euIPSCs (vehicle, 33.5 ± 7.3 pA, n = 6 pairs; AMG9810, 37.3 ± 4.2 pA, n = 4 pairs; unpaired t test, p = 0.66) at dendritic connections. Furthermore, application of AM251 (10 μm) did not unmask persistent CB1 activity in slices pretreated with AMG9810 (successes: 73 ± 4%, n = 4 pairs, paired t test, p = 0.3; euIPSCs amplitudes: 34.4 ± 2.5 pA, n = 4 pairs, paired t test, p = 0.57). Thus, these control experiments indicate that the observed inhibition of the FAAH inhibitor-induced decrease in tonic 2-AG signaling by AMG9810 was mediated directly by TRPV1 blockade and that this interaction occurs selectively at perisomatic synapses.
Finally, to corroborate the involvement of TRPV1 in the functional interaction between FAAH activity and MGL activity, paired patch-clamp recordings and pharmacological treatments were performed in acute hippocampal slices obtained from TRPV1 knock-out mice. Notably, MGL blockade robustly reduced success rate and euIPSCs in both wild-type and TRPV1 knock-out animals (Figs. 3B–D, 8H–J), but the antagonistic effect of FAAH inhibition in wild-type animals (Fig. 7A–C) was completely absent in TRPV1 knock-out mice (Fig. 8H–J). When JZL184 (100 nm) was applied alone or together with PF3845 (1 μm), both the success rate of action potentials evoking a postsynaptic IPSC (Fig. 8I; JZL184, 21 ± 6%, n = 5 pairs; JZL184 + PF3845, 22 ± 6%, n = 5 pairs) and the amplitude of euIPSCs (Fig. 8J; JZL184, 12.6 ± 3.2 pA, n = 5 pairs; JZL184 + PF3845, 8.7 ± 3.3 pA, n = 5 pairs) were decreased significantly compared with control values obtained in TRPV1 knock-out mice (Fig. 8I; successes: 74 ± 11%, n = 5 pairs, Tukey–Kramer test, p < 0.05 for both comparisons; Fig. 8J; euIPSC amplitudes: 96.4 ± 20.8 pA, n = 5 pairs, Tukey–Kramer test, p < 0.05 for both comparisons). Data obtained by using the TRPV1 antagonist or the TRPV1 knock-out mice to interfere with tonic 2-AG signaling were highly similar (p > 0.5 for both parameters).
Collectively, these findings provide direct evidence that highly compartmentalized TRPV1 channels are concentrated on postsynaptic intracellular membranes at hippocampal perisomatic, but not at dendritic, GABAergic synapses. Moreover, the data show that TRPV1 activation by endovanilloid signaling regulates the magnitude of tonic 2-AG signaling at perisomatic GABAergic synapses.
Discussion
The qualitative diversity and quantitative variability of synaptic transmission are critically determined by neuromodulatory signaling (Marder, 2012). Persistent CB1 receptor activity represents one of the major modulatory mechanisms to establish synaptic strength (Losonczy et al., 2004). Our present findings indicate the following: (1) constitutively active CB1 receptors set GABA release probability selectively at perisomatic synapses in the hippocampus; (2) 2-AG is the primary mediator of ligand-driven tonic endocannabinoid signaling, but it is tightly controlled by presynaptic MGL activity; (3) a major regulatory mechanism calibrating the homosynaptic 2-AG-mediated endocannabinoid tone is endovanilloid signaling, which requires the activation of TRPV1 channels predominantly located on postsynaptic intracellular stores at GABAergic synapses; and (4) endovanilloid regulation of the 2-AG-mediated tone is controlled by FAAH. Together, these findings demonstrate a striking spatial segregation and functional division of labor of distinct molecular components of the endocannabinoid system in the tonic control of neurotransmitter release.
Persistent CB1 activity at hippocampal GABAergic synapses
Although the mechanistic details and functional importance of phasic endocannabinoid signaling have received widespread attention (Cachope, 2012; Castillo et al., 2012), the molecular mechanisms and significance of tonic endocannabinoid signaling are much less understood. Tonic signaling represents a major mode of operation for several other neuromodulatory systems, with tonic adenosine and ambient GABA signaling being two major examples (Brickley and Mody, 2012; Dias et al., 2013), suggesting that a deeper understanding of tonic cannabinoid control on neurotransmitter release may also lead to important insights. Therefore, our primary goal was to delineate how tonic endocannabinoid signaling is organized at hippocampal GABAergic synapses. Because various physiological or pathophysiological processes modulate endocannabinoid-mediated synaptic plasticity (Iremonger et al., 2013; Melis et al., 2014), our secondary goal was to identify new regulatory mechanisms that can fine-tune the extent of tonic endocannabinoid signaling at inhibitory synapses.
In the present study, the data revealed that AM251, an inverse agonist of CB1 receptors, could readily increase GABAergic neurotransmission. In contrast, NESS0327, a neutral antagonist, did not have an effect on tonic cannabinoid signaling, although it could block exogenous and endogenous ligand-activated CB1 signaling. This intriguing finding raises the possibility that a certain proportion of the highly abundant CB1 receptors on perisomatic boutons can take up a constitutively active conformation and thereby persistently block nearby voltage-gated calcium channels and GABA release (Szabó et al., 2014a). Indeed, biophysical modeling and mutational analysis identified several active conformations of CB1 receptors that trigger distinct downstream signaling mechanisms with different kinetics and determine subcellular receptor targeting (D'Antona et al., 2006; Simon et al., 2013; Gyombolai et al., 2015). In light of previous evidence that CB1-mediated inhibition of GABA release depends on the activity pattern of the presynaptic neuron (Losonczy et al., 2004; Földy et al., 2006) together with the emerging therapeutic significance of CB1 inverse agonism (Pertwee, 2005; Tam et al., 2012; Meye et al., 2013), it will be pivotal to determine in the future the extent of constitutive CB1 activity at other specific circuit locations and during distinct physiological or pathophysiological states.
Tonic 2-AG signaling is controlled by MGL activity at hippocampal GABAergic synapses
An alternative mechanism leading to persistent CB1 activity relies on tonic endocannabinoid mobilization. Indeed, somatodendritic CB1 receptors on hippocampal neurons are activated tonically by 2-AG (Leterrier et al., 2006; Thibault et al., 2013; Ladarre et al., 2014). Thus, although it is conceivable that the allosteric conformational change induced by NESS0327 docking prevents the extracellular 2-AG from reaching its intramembrane binding pocket on CB1 receptors in the presynaptic membrane, the possibility that NESS0327 fails to displace prebound 2-AG also cannot be fully excluded. Our finding that 2-AG is generated tonically in acute slice preparations indicates that the latter scenario is possible and has also general implication for synaptic research performed in acute slice preparations because of the widespread distribution of presynaptic CB1 receptors throughout the brain (Katona and Freund, 2012).
Our parallel lipid measurements and electrophysiological recordings revealed that MGL activity limits 2-AG levels in the hippocampus and controls how postsynaptically mobilized 2-AG affects GABA release. In contrast, anandamide and FAAH did not have direct effects on GABAergic synapses. Generally, these findings are consistent with the anatomical distribution of the molecular constituents of the endocannabinoid system. FAAH is found somatodendritically throughout the brain (Egertová et al., 1998; Gulyás et al., 2004); hence, it is segregated spatially from the presynaptic effector site of tonic endocannabinoid signaling. MGL is distributed ubiquitously within axon terminals, which is the ideal subcellular position to terminate 2-AG effects at presynaptic CB1 receptors (Gulyás et al., 2004; Ludányi et al., 2008; Uchigashima et al., 2011, Horváth et al., 2014). Because downregulation of MGL levels is associated with various physiological conditions and pathological situations (Sumislawski et al., 2011; Kallendrusch et al., 2012; Serrano et al., 2012), our results also highlight the importance to identify those mechanisms by which the presynaptic neuron can adjust MGL activity.
Synapse specificity of tonic cannabinoid signaling
Our previous findings uncovered tonic endocannabinoid signaling at perisomatic, but not dendritic, synaptic connections (Lee et al., 2010). Present observations now extend these findings by showing that neither tonic MGL nor tonic FAAH activity affects GABAergic transmission, despite the presence of functionally active CB1 receptors on dendritically projecting interneuron boutons. One simple explanation is that cannabinoid signaling is quantitatively different between perisomatic and dendritic axon terminals because of the distinct nanoscale architecture of the two terminals (Dudok et al., 2015). Indeed, we found that a CB1 receptor agonist more strongly inhibits GABA release from perisomatically projecting interneurons. An alternative explanation could be the cell-type-specific expression of cannabinoid receptor-interacting proteins, which bind to the C terminus of CB1 and block constitutive receptor activity (Niehaus et al., 2007; Smith et al., 2015).
The synapse specificity also provides clues as to why tonic endocannabinoid signaling may sometimes appear absent, when electrical stimulation of a heterogeneous mixture of fiber types is used to evoke compound IPSCs (Pan et al., 2011). Such a paradigm was shown to require the genetic inactivation of MGL to uncover the presence of endocannabinoid tone (Pan et al., 2011). Similarly, the absence of effect after FAAH blockade does not exclude that anandamide conducts tonic endocannabinoid signaling under specific physiological or pathophysiological conditions, such as during chronic inhibition of neuronal firing (Kim and Alger, 2010). Considering that postsynaptic neurons exploit tonic endocannabinoid signaling in an activity- and cell-type-specific manner (Losonczy et al., 2004; Hentges et al., 2005; Neu et al., 2007; Oliet et al., 2007; Lee et al., 2010; Huang and Woolley, 2012), our findings highlight the importance of focusing anatomical and physiological analyses on identified circuit locations to elucidate how cannabinoid signaling regulates circuit dynamics.
Synaptic division of labor between anandamide and 2-AG signaling
A major unresolved subject of endocannabinoid research is why the brain uses two different endocannabinoid substances (Piomelli, 2014). Both anandamide and 2-AG are bona fide CB1 ligands. Activation of presynaptic CB1 receptors inhibits voltage-gated calcium channels, thereby reducing neurotransmitter release (Twitchell et al., 1997; Sullivan, 1999; Hoffman and Lupica, 2000). Interestingly, both endocannabinoids can additionally activate TRPV1 channels (Zygmunt et al., 1999, 2013) and mediate long-term depression via postsynaptic AMPA receptor internalization (Chávez et al., 2010; Grueter et al., 2010; Puente et al., 2011).
Besides these independent synaptic functions of endocannabinoid and endovanilloid signaling, we found that these two pathways antagonistically interact to adjust synaptic strength at hippocampal inhibitory synapses. Endovanilloid signaling was reported to control 2-AG-mediated short-term plasticity evoked by acute stimulation of mGlu5 glutamate receptors or M1 muscarinic acetylcholine receptors in the striatum (Maccarrone et al., 2008; Musella et al., 2010). The model proposed by Maccarrone et al. (2008) also suggested that increased anandamide levels activate TRPV1 channels, which then leads to an inhibition of the 2-AG-synthesizing enzyme DGL-α. Consequently, mGlu5-triggered increase in striatal 2-AG levels and phasic endocannabinoid signaling were impaired after FAAH inhibition (Maccarrone et al., 2008). Our findings are entirely consistent with this model by showing the following: (1) selective FAAH blockade elevates anandamide levels but attenuates increased hippocampal 2-AG levels induced by MGL inhibition; (2) the latter effect is associated with a weakened tonic 2-AG signaling; (3) reduced 2-AG signaling after FAAH blockade can be rescued by using a potent TRPV1 antagonist or in TRPV1 knock-out mice; and (4) TRPV1 is present postsynaptically at GABAergic synapses.
The mechanistic similarity of how endovanilloid signaling controls endocannabinoid signaling in different brain areas suggests that this functional interaction may be a more general phenomenon than hitherto thought. Emerging evidence indicates that TRPV1 activation has opposite behavioral function with respect to CB1 signaling in several cognitive processes, including hippocampus-dependent learning and anxiety (Marsch et al., 2007; Li et al., 2008, Moreira et al., 2012; You et al., 2012). Importantly, human FAAH variants that cause reduced enzyme activity are also associated with decreased anxiety-related behaviors and attenuated trauma in posttraumatic stress disorder patients (Pardini et al., 2012; Dincheva et al., 2015). Thus, uncovering the functional interaction of these two signaling systems in relation to specific behavioral processes will also help to understand the neurobiological mechanisms underlying the potential of FAAH inhibitors as novel anxiolytics (Gunduz-Cinar et al., 2013).
Footnotes
This work was supported by Hungarian Academy of Sciences Momentum Program LP-54/2013, European Research Council Grant 243153 (I.K.), National Institute of Health Grant NS35915 (I.S.), and NASA Grants NNX10AD59G and NNX15AI22G (C.L.L., I.S.). Super-resolution microscopy was funded by Hungarian Academy of Sciences Equipment Grant IF-22/2012. I.K. is a recipient of a Wellcome Trust International Senior Research Fellowship. M.O. was funded by Minority Science Program National Institutes of Health/Maximizing Access to Research Careers Grant GM-69337. We are grateful to B. Pintér, G. Goda, E. Tischler, R. Zhu, C. Lozova, and K. Ding for excellent technical assistance and Profs. H. Bradshaw and O. Manzoni for advice on lipid and physiology experiments. We thank the Nikon Microscopy Center at the Institute of Experimental Medicine, Nikon Austria, Auro-Science Consulting, and the confocal facility of the Optical Biology Shared Resource of the Cancer Center Support Grant CA-62203 at the University of California, Irvine for kindly providing microscopy support.
The authors declare no conflict of interest.
This is an Open Access article distributed under the terms of the Creative Commons Attribution License Creative Commons Attribution 4.0 International, which permits unrestricted use, distribution and reproduction in any medium provided that the original work is properly attributed.
References
- Ahn K, Johnson DS, Mileni M, Beidler D, Long JZ, McKinney MK, Weerapana E, Sadagopan N, Liimatta M, Smith SE, Lazerwith S, Stiff C, Kamtekar S, Bhattacharya K, Zhang Y, Swaney S, Van Becelaere K, Stevens RC, Cravatt BF. Discovery and caharacterization of a highly selective FAAH inhibitor that reduces inflammatory pain. Chem Biol. 2009;16:411–420. doi: 10.1016/j.chembiol.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alger BE. Endocannabinoids at the synapse a decade after the dies mirabilis (29 March 2001): what we still do not know. J Physiol. 2012;590:2203–2212. doi: 10.1113/jphysiol.2011.220855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali AB, Todorova M. Asynchronous release of GABA via tonic cannabinoid receptor activation at identified interneuron synapses in rat CA1. Eur J Neurosci. 2010;31:1196–1207. doi: 10.1111/j.1460-9568.2010.07165.x. [DOI] [PubMed] [Google Scholar]
- Azad SC, Monory K, Marsicano G, Cravatt BF, Lutz B, Zieglgänsberger W, Rammes G. Circuitry for associative plasticity in the amygdala involves endocannabinoid signaling. J Neurosci. 2004;24:9953–9961. doi: 10.1523/JNEUROSCI.2134-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisogno T, Howell F, Williams G, Minassi A, Cascio MG, Ligresti A, Matias I, Schiano-Moriello A, Paul P, Williams EJ, Gangadharan U, Hobbs C, Di Marzo V, Doherty P. Cloning of the first sn1-DAG lipases points to the spatial and temporal regulation of endocannabinoid signaling in the brain. J Cell Biol. 2003;163:463–468. doi: 10.1083/jcb.200305129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankman JL, Cravatt BF. Chemical probes of endocannabinoid metabolism. Pharmacol Rev. 2013;65:849–871. doi: 10.1124/pr.112.006387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankman JL, Simon GM, Cravatt BF. A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem Biol. 2007;14:1347–1356. doi: 10.1016/j.chembiol.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouaboula M, Perrachon S, Milligan L, Canat X, Rinaldi-Carmona M, Portier M, Barth F, Calandra B, Pecceu F, Lupker J, Maffrand JP, Le Fur G, Casellas P. A selective inverse agonist for central cannabinoid receptor inhibits mitogen-activated protein kinase activation stimulated by insulin or insulin-like growth factor 1. Evidence for a new model of receptor/ligand interactions. J Biol Chem. 1997;272:22330–22339. doi: 10.1074/jbc.272.35.22330. [DOI] [PubMed] [Google Scholar]
- Branco T, Staras K. The probability of neurotransmitter release: variability and feedback control at single synapses. Nat Rev Neurosci. 2009;10:373–383. doi: 10.1038/nrn2634. [DOI] [PubMed] [Google Scholar]
- Brickley SG, Mody I. Extrasynaptic GABA(A) receptors: their function in the CNS and implications for disease. Neuron. 2012;73:23–34. doi: 10.1016/j.neuron.2011.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cachope R. Functional diversity on synaptic plasticity mediated by endocannabinoids. Philos Trans R Soc Lond B Biol Sci. 2012;367:3242–3253. doi: 10.1098/rstb.2011.0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo PE, Younts TJ, Chávez AE, Hashimotodani Y. Endocannabinoid signaling and synaptic function. Neuron. 2012;76:70–81. doi: 10.1016/j.neuron.2012.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chávez AE, Chiu CQ, Castillo PE. TRPV1 activation by endogenous anandamide triggers postsynaptic long-term depression in dentate gyrus. Nat Neurosci. 2010;13:1511–1518. doi: 10.1038/nn.2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chávez AE, Hernández VM, Rodenas-Ruano A, Chan CS, Castillo PE. Compartment-specific modulation of GABAergic synaptic transmission by TRPV1 channels in the dentate gyrus. J Neurosci. 2014;34:16621–16629. doi: 10.1523/JNEUROSCI.3635-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Ratzliff A, Hilgenberg L, Gulyás A, Freund TF, Smith M, Dinh TP, Piomelli D, Mackie K, Soltesz I. Long-term plasticity of endocannabinoid signaling induced by developmental febrile seizures. Neuron. 2003;39:599–611. doi: 10.1016/S0896-6273(03)00499-9. [DOI] [PubMed] [Google Scholar]
- Chen K, Neu A, Howard AL, Földy C, Echegoyen J, Hilgenberg L, Smith M, Mackie K, Soltesz I. Prevention of plasticity of endocannabinoid signaling inhibits persistent limbic hyperexcitability caused by developmental seizures. J Neurosci. 2007;27:46–58. doi: 10.1523/JNEUROSCI.3966-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cope DW, Maccaferri G, Márton LF, Roberts JD, Cobden PM, Somogyi P. Cholecystokinin-immunopositive basket and Schaffer collateral-associated interneurones target different domains of pyramidal cells in the CA1 area of the rat hippocampus. Neuroscience. 2002;109:63–80. doi: 10.1016/S0306-4522(01)00440-7. [DOI] [PubMed] [Google Scholar]
- Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature. 1996;384:83–87. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- Cristino L, Starowicz K, De Petrocellis L, Morishita J, Ueda N, Guglielmotti V, Di Marzo V. Immunohistochemical localization of anabolic and catabolic enzymes for anandamide and other putative endovanilloids in the hippocampus and cerebellar cortex of the mouse brain. Neuroscience. 2008;151:955–968. doi: 10.1016/j.neuroscience.2007.11.047. [DOI] [PubMed] [Google Scholar]
- Dani A, Huang B, Bergan J, Dulac C, Zhuang X. Superresolution imaging of chemical synapses in the brain. Neuron. 2010;68:843–856. doi: 10.1016/j.neuron.2010.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Antona AM, Ahn KH, Kendall DA. Mutations of CB1 T210 produce active and inactive receptor forms: correlations with ligand affinity, receptor stability, and cellular localization. Biochemistry. 2006;45:5606–5617. doi: 10.1021/bi060067k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias RB, Rombo DM, Ribeiro JA, Henley JM, Sebastião AM. Adenosine: setting the stage for plasticity. Trends Neurosci. 2013;36:248–257. doi: 10.1016/j.tins.2012.12.003. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, De Petrocellis L. Why do cannabinoid receptors have more than one endogenous ligand? Philos Trans R Soc Lond B Biol Sci. 2012;367:3216–3228. doi: 10.1098/rstb.2011.0382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dincheva I, Drysdale AT, Hartley CA, Johnson DC, Jing D, King EC, Ra S, Gray JM, Yang R, DeGruccio AM, Huang C, Cravatt BF, Glatt CE, Hill MN, Casey BJ, Lee FS. FAAH genetic variation enhances fronto-amygdala function in mouse and human. Nat Commun. 2015;6:6395. doi: 10.1038/ncomms7395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinh TP, Carpenter D, Leslie FM, Freund TF, Katona I, Sensi SL, Kathuria S, Piomelli D. Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc Natl Acad Sci U S A. 2002;99:10819–10824. doi: 10.1073/pnas.152334899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudok B, Barna L, Ledri M, Szabó SI, Szabadits E, Pintér B, Woodhams SG, Henstridge CM, Balla GY, Nyilas R, Varga C, Lee SH, Matolcsi M, Cervenak J, Kacskovics I, Watanabe M, Sagheddu C, Melis M, Pistis M, Soltesz I, Katona I. Cell-specific STORM super-resolution imaging reveals nanoscale organization of cannabinoid signaling. Nat Neurosci. 2015;18:75–86. doi: 10.1038/nn.3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvorzhak A, Semtner M, Faber DS, Grantyn R. Tonic mGluR5/CB1-dependent suppression of inhibition as a pathophysiological hallmark in the striatum of mice carrying a mutant form of huntingtin. J Physiol. 2013;591:1145–1166. doi: 10.1113/jphysiol.2012.241018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egertová M, Giang DK, Cravatt BF, Elphick MR. A new perspective on cannabinoid signalling: complementary localization of fatty acid amide hydrolase and the CB1 receptor in rat brain. Proc Biol Sci. 1998;265:2081–2085. doi: 10.1098/rspb.1998.0543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Földy C, Neu A, Jones MV, Soltesz I. Presynaptic, activity-dependent modulation of cannabinoid type 1 receptor-mediated inhibition of GABA release. J Neurosci. 2006;26:1465–1469. doi: 10.1523/JNEUROSCI.4587-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Földy C, Malenka RC, Südhof TC. Autism-associated neuroligin-3 mutations commonly disrupt tonic endocannabinoid signaling. Neuron. 2013;78:498–509. doi: 10.1016/j.neuron.2013.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukudome Y, Ohno-Shosaku T, Matsui M, Omori Y, Fukaya M, Tsubokawa H, Taketo MM, Watanabe M, Manabe T, Kano M. Two distinct classes of muscarinic action on hippocampal inhibitory synapses: M2-mediated direct suppression and M1/M3-mediated indirect suppression through endocannabinoid signalling. Eur J Neurosci. 2004;19:2682–2692. doi: 10.1111/j.0953-816X.2004.03384.x. [DOI] [PubMed] [Google Scholar]
- Gavva NR, Tamir R, Qu Y, Klionsky L, Zhang TJ, Immke D, Wang J, Zhu D, Vanderah TW, Porreca F, Doherty EM, Norman MH, Wild KD, Bannon AW, Louis JC, Treanor JJ. AMG 9810 [(E)-3-(4-t-butylphenyl)-N-(2,3-dihydrobenzo[b][1,4] dioxin-6-yl)acrylamide], a novel vanilloid receptor 1 (TRPV1) antagonist with antihyperalgesic properties. J Pharmacol Exp Ther. 2005;313:474–484. doi: 10.1124/jpet.104.079855. [DOI] [PubMed] [Google Scholar]
- Grueter BA, Brasnjo G, Malenka RC. Postsynaptic TRPV1 triggers cell type-specific long-term depression in the nucleus accumbens. Nat Neurosci. 2010;13:1519–1525. doi: 10.1038/nn.2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulyas AI, Cravatt BF, Bracey MH, Dinh TP, Piomelli D, Boscia F, Freund TF. Segregation of two endocannabinoid-hydrolyzing enzymes into pre- and postsynaptic compartments in the rat hippocampus, cerebellum and amygdala. Eur J Neurosci. 2004;20:441–458. doi: 10.1111/j.1460-9568.2004.03428.x. [DOI] [PubMed] [Google Scholar]
- Gunduz-Cinar O, Hill MN, McEwen BS, Holmes A. Amygdala FAAH and anandamide: mediating protection and recovery from stress. Trends Pharmacol Sci. 2013;34:637–644. doi: 10.1016/j.tips.2013.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyombolai P, Tóth AD, Tímár D, Turu G, Hunyady L. Mutations in the “DRY” motif of the CB1 cannabinoid receptor result in biased receptor variants. J Mol Endocrinol. 2015;54:75–89. doi: 10.1530/JME-14-0219. [DOI] [PubMed] [Google Scholar]
- Hanlon EC, Tasali E, Leproult R, Stuhr KL, Doncheck E, de Wit H, Hillard CJ, Van Cauter E. Circadian rhythm of circulating levels of the endocannabinoid 2-arachidonoylglycerol. J Clin Endocrinol Metab. 2015;100:220–226. doi: 10.1210/jc.2014-3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimotodani Y, Ohno-Shosaku T, Kano M. Presynaptic monoacylglycerol lipase activity determines basal endocannabinoid tone and terminates retrograde endocannabinoid signaling in the hippocampus. J Neurosci. 2007;27:1211–1219. doi: 10.1523/JNEUROSCI.4159-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentges ST, Low MJ, Williams JT. Differential regulation of synaptic inputs by constitutively released endocannabinoids and exogenous cannabinoids. J Neurosci. 2005;25:9746–9751. doi: 10.1523/JNEUROSCI.2769-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillard CJ. Biochemistry and pharmacology of the endocannabinoids arachidonylethanolamide and 2-arachidonylglycerol. Prostaglandins Other Lipid Mediat. 2000;61:3–18. doi: 10.1016/S0090-6980(00)00051-4. [DOI] [PubMed] [Google Scholar]
- Hoffman AF, Lupica CR. Mechanisms of cannabinoid inhibition of GABA(A) synaptic transmission in the hippocampus. J Neurosci. 2000;20:2470–2479. doi: 10.1523/JNEUROSCI.20-07-02470.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horváth E, Woodhams SG, Nyilas R, Henstridge CM, Kano M, Sakimura K, Watanabe M, Katona I. Heterogeneous presynaptic distribution of monoacylglycerol lipase, a multipotent regulator of nociceptive circuits in the mouse spinal cord. Eur J Neurosci. 2014;39:419–434. doi: 10.1111/ejn.12470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang GZ, Woolley CS. Estradiol acutely suppresses inhibition in the hippocampus through a sex-specific endocannabinoid and mGluR-dependent mechanism. Neuron. 2012;74:801–808. doi: 10.1016/j.neuron.2012.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iremonger KJ, Wamsteeker Cusulin JI, Bains JS. Changing the tune: plasticity and adaptation of retrograde signals. Trends Neurosci. 2013;36:471–479. doi: 10.1016/j.tins.2013.04.007. [DOI] [PubMed] [Google Scholar]
- Kallendrusch S, Hobusch C, Ehrlich A, Nowicki M, Ziebell S, Bechmann I, Geisslinger G, Koch M, Dehghani F. Intrinsic up-regulation of 2-AG favors an area specific neuronal survival in different in vitro models of neuronal damage. PLoS One. 2012;7:e51208. doi: 10.1371/journal.pone.0051208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kathuria S, Gaetani S, Fegley D, Valiño F, Duranti A, Tontini A, Mor M, Tarzia G, La Rana G, Calignano A, Giustino A, Tattoli M, Palmery M, Cuomo V, Piomelli D. Modulation of anxiety through blockade of anandamide hydrolysis. Nat Med. 2003;9:76–81. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- Katona I, Freund TF. Multiple functions of endocannabinoid signaling in the brain. Annu Rev Neurosci. 2012;35:529–558. doi: 10.1146/annurev-neuro-062111-150420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katona I, Sperlágh B, Sík A, Käfalvi A, Vizi ES, Mackie K, Freund TF. Presynaptically located CB1 cannabinoid receptors regulate GABA release from axon terminals of specific hippocampal interneurons. J Neurosci. 1999;19:4544–4558. doi: 10.1523/JNEUROSCI.19-11-04544.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katona I, Urbán GM, Wallace M, Ledent C, Jung KM, Piomelli D, Mackie K, Freund TF. Molecular composition of the endocannabinoid system at glutamatergic synapses. J Neurosci. 2006;26:5628–5637. doi: 10.1523/JNEUROSCI.0309-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T. Principles: receptor theory in pharmacology. Trends Pharmacol Sci. 2004;25:186–192. doi: 10.1016/j.tips.2004.02.012. [DOI] [PubMed] [Google Scholar]
- Khlaifia A, Farah H, Gackiere F, Tell F. Anandamide, cannabinoid type 1 receptor, and NMDA receptor activation mediate non-Hebbian presynaptically expressed long-term depression at the first central synapse for visceral afferent fibers. J Neurosci. 2013;33:12627–12637. doi: 10.1523/JNEUROSCI.1028-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Alger BE. Reduction in endocannabinoid tone is a homeostatic mechanism for specific inhibitory synapses. Nat Neurosci. 2010;13:592–600. doi: 10.1038/nn.2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klausberger T, Somogyi P. Neuronal diversity and temporal dynamics: the unity of hippocampal circuit operations. Science. 2008;321:53–57. doi: 10.1126/science.1149381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladarre D, Roland AB, Biedzinski S, Ricobaraza A, Lenkei Z. Polarized cellular patterns of endocannabinoid production and detection shape cannabinoid signaling in neurons. Front Cell Neurosci. 2014;8:426. doi: 10.3389/fncel.2014.00426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Földy C, Soltesz I. Distinct endocannabinoid control of GABA release at perisomatic and dendritic synapses in the hippocampus. J Neurosci. 2010;30:7993–8000. doi: 10.1523/JNEUROSCI.6238-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Marchionni I, Bezaire M, Varga C, Danielson N, Lovett-Barron M, Losonczy A, Soltesz I. Parvalbumin-positive basket cells differentiate among hippocampal pyramidal cells. Neuron. 2014;82:1129–1144. doi: 10.1016/j.neuron.2014.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenkey N, Kirizs T, Holderith N, Máté Z, Szabó G, Vizi ES, Hájos N, Nusser Z. Tonic endocannabinoid-mediated modulation of GABA release is independent of the CB1 content of axon terminals. Nat Commun. 2015;6:6557. doi: 10.1038/ncomms7557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner TN, Kreitzer AC. RGS4 is required for dopaminergic control of striatal LTD and susceptibility to parkinsonian motor deficits. Neuron. 2012;73:347–359. doi: 10.1016/j.neuron.2011.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leterrier C, Lainé J, Darmon M, Boudin H, Rossier J, Lenkei Z. Constitutive activation drives compartment-selective endocytosis and axonal targeting of type 1 cannabinoid receptors. J Neurosci. 2006;26:3141–3153. doi: 10.1523/JNEUROSCI.5437-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HB, Mao RR, Zhang JC, Yang Y, Cao J, Xu L. Antistress effect of TRPV1 channel on synaptic plasticity and spatial memory. Biol Psychiatry. 2008;64:286–292. doi: 10.1016/j.biopsych.2008.02.020. [DOI] [PubMed] [Google Scholar]
- Liedhegner ES, Sasman A, Hillard CJ. Brain region-specific changes in N-acylethanolamine contents with time of day. J Neurochem. 2014;128:491–506. doi: 10.1111/jnc.12495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JZ, Nomura DK, Vann RE, Walentiny DM, Booker L, Jin X, Burston JJ, Sim-Selley LJ, Lichtman AH, Wiley JL, Cravatt BF. Dual blockade of FAAH and MAGL identifies behavioral processes regulated by endocannabinoid crosstalk in vivo. Proc Natl Acad Sci U S A. 2009a;106:20270–20275. doi: 10.1073/pnas.0909411106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JZ, Li W, Booker L, Burston JJ, Kinsey SG, Schlosburg JE, Pavón FJ, Serrano AM, Selley DE, Parsons LH, Lichtman AH, Cravatt BF. Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat Chem Biol. 2009b;5:37–44. doi: 10.1038/nchembio.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losonczy A, Biró AA, Nusser Z. Persistently active cannabinoid receptors mute a subpopulation of hippocampal interneurons. Proc Natl Acad Sci U S A. 2004;101:1362–1367. doi: 10.1073/pnas.0304752101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovinger DM. Presynaptic modulation by endocannabinoids. Handb Exp Pharmacol. 2008;184:435–477. doi: 10.1007/978-3-540-74805-2_14. [DOI] [PubMed] [Google Scholar]
- Ludányi A, Eross L, Czirjak S, Vajda J, Halász P, Watanabe M, Palkovits M, Maglóczky Z, Freund TF, Katona I. Downregulation of the CB1 cannabinoid receptor and related molecular elements of the endocannabinoid system in epileptic human hippocampus. J Neurosci. 2008;28:2976–2990. doi: 10.1523/JNEUROSCI.4465-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccarrone M, Rossi S, Bari M, De Chiara V, Fezza F, Musella A, Gasperi V, Prosperetti C, Bernardi G, Finazzi-Agrò A, Cravatt BF, Centonze D. Anandamide inhibits metabolism and physiological actions of 2-arachidonoylglycerol in the striatum. Nat Neurosci. 2008;11:152–159. doi: 10.1038/nn2042. [DOI] [PubMed] [Google Scholar]
- Marder E. Neuromodulation of neuronal circuits: back to the future. Neuron. 2012;76:1–11. doi: 10.1016/j.neuron.2012.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsch R, Foeller E, Rammes G, Bunck M, Kössl M, Holsboer F, Zieglgänsberger W, Landgraf R, Lutz B, Wotjak CT. Reduced anxiety, conditioned fear, and hippocampal long-term potentiation in transient receptor potential vanilloid type 1 receptor-deficient mice. J Neurosci. 2007;27:832–839. doi: 10.1523/JNEUROSCI.3303-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathur BN, Tanahira C, Tamamaki N, Lovinger DM. Voltage drives diverse endocannabinoid signals to mediate striatal microcircuit-specific plasticity. Nat Neurosci. 2013;16:1275–1283. doi: 10.1038/nn.3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mato S, Pazos A, Valdizán EM. Cannabinoid receptor antagonism and inverse agonism in response to SR141716A on cAMP production in human and rat brain. Eur J Pharmacol. 2002;443:43–46. doi: 10.1016/S0014-2999(02)01575-3. [DOI] [PubMed] [Google Scholar]
- Melis M, Greco B, Tonini R. Interplay between synaptic endocannabinoid signaling and metaplasticity in neuronal circuit function and dysfunction. Eur J Neurosci. 2014;39:1189–1201. doi: 10.1111/ejn.12501. [DOI] [PubMed] [Google Scholar]
- Meye FJ, Trezza V, Vanderschuren LJ, Ramakers GM, Adan RA. Neutral antagonism at the cannabinoid 1 receptor: a safer treatment for obesity. Mol Psychiatry. 2013;18:1294–1301. doi: 10.1038/mp.2012.145. [DOI] [PubMed] [Google Scholar]
- Moreira FA, Aguiar DC, Terzian AL, Guimarães FS, Wotjak CT. Cannabinoid type 1 receptors and transient receptor potential vanilloid type 1 channels in fear and anxiety-two sides of one coin? Neuroscience. 2012;204:186–192. doi: 10.1016/j.neuroscience.2011.08.046. [DOI] [PubMed] [Google Scholar]
- Musella A, De Chiara V, Rossi S, Cavasinni F, Castelli M, Cantarella C, Mataluni G, Bernardi G, Centonze D. Transient receptor potential vanilloid 1 channels control acetylcholine/2-arachidonoylglicerol coupling in the striatum. Neuroscience. 2010;167:864–871. doi: 10.1016/j.neuroscience.2010.02.058. [DOI] [PubMed] [Google Scholar]
- Neu A, Földy C, Soltesz I. Postsynaptic origin of CB1-dependent tonic inhibition of GABA release at cholecystokinin-positive basket cell to pyramidal cell synapses in the CA1 region of the rat hippocampus. J Physiol. 2007;578:233–247. doi: 10.1113/jphysiol.2006.115691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niehaus JL, Liu Y, Wallis KT, Egertová M, Bhartur SG, Mukhopadhyay S, Shi S, He H, Selley DE, Howlett AC, Elphick MR, Lewis DL. CB1 cannabinoid receptor activity is modulated by the cannabinoid receptor interacting protein CRIP 1a. Mol Pharmacol. 2007;72:1557–1566. doi: 10.1124/mol.107.039263. [DOI] [PubMed] [Google Scholar]
- Nissen W, Szabo A, Somogyi J, Somogyi P, Lamsa KP. Cell type-specific long-term plasticity at glutamatergic synapses onto hippocampal interneurons expressing either parvalbumin or CB1 cannabinoid receptor. J Neurosci. 2010;30:1337–1347. doi: 10.1523/JNEUROSCI.3481-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyíri G, Cserép C, Szabadits E, Mackie K, Freund TF. CB1 cannabinoid receptors are enriched in the perisynaptic annulus and on preterminal segments of hippocampal GABAergic axons. Neuroscience. 2005;136:811–822. doi: 10.1016/j.neuroscience.2005.01.026. [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Kano M. Endocannabinoid-mediated retrograde modulation of synaptic transmission. Curr Opin Neurobiol. 2014;29:1–8. doi: 10.1016/j.conb.2014.03.017. [DOI] [PubMed] [Google Scholar]
- Oliet SH, Baimoukhametova DV, Piet R, Bains JS. Retrograde regulation of GABA transmission by the tonic release of oxytocin and endocannabinoids governs postsynaptic firing. J Neurosci. 2007;27:1325–1333. doi: 10.1523/JNEUROSCI.2676-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan B, Wang W, Zhong P, Blankman JL, Cravatt BF, Liu QS. Alterations of endocannabinoid signaling, synaptic plasticity, learning, and memory in monoacylglycerol lipase knock-out mice. J Neurosci. 2011;31:13420–13430. doi: 10.1523/JNEUROSCI.2075-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X, Ikeda SR, Lewis DL. SR 141716A acts as an inverse agonist to increase neuronal voltage-dependent Ca2+ currents by reversal of tonic CB1 cannabinoid receptor activity. Mol Pharmacol. 1998;54:1064–1072. doi: 10.1124/mol.54.6.1064. [DOI] [PubMed] [Google Scholar]
- Pardini M, Krueger F, Koenigs M, Raymont V, Hodgkinson C, Zoubak S, Goldman D, Grafman J. Fatty-acid amide hydrolase polymorphisms and post-traumatic stress disorder after penetrating brain injury. Transl Psychiatry. 2012;2:e75. doi: 10.1038/tp.2012.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawelzik H, Hughes DI, Thomson AM. Physiological and morphological diversity of immunocytochemically defined parvalbumin- and cholecystokinin-positive interneurones in CA1 of the adult rat hippocampus. J Comp Neurol. 2002;443:346–367. doi: 10.1002/cne.10118. [DOI] [PubMed] [Google Scholar]
- Pertwee RG. Inverse agonism and neutral antagonism at cannabinoid CB1 receptors. Life Sci. 2005;76:1307–1324. doi: 10.1016/j.lfs.2004.10.025. [DOI] [PubMed] [Google Scholar]
- Piomelli D. More surprises lying ahead. The endocannabinoids keep us guessing. Neuropharmacology. 2014;76:228–234. doi: 10.1016/j.neuropharm.2013.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puente N, Cui Y, Lassalle O, Lafourcade M, Georges F, Venance L, Grandes P, Manzoni OJ. Polymodal activation of the endocannabinoid system in the extended amygdala. Nat Neurosci. 2011;14:1542–1547. doi: 10.1038/nn.2974. [DOI] [PubMed] [Google Scholar]
- Puente N, Reguero L, Elezgarai I, Canduela MJ, Mendizabal-Zubiaga J, Ramos-Uriarte A, Fernández-Espejo E, Grandes P. The transient receptor potential vanilloid-1 is localized at excitatory synapses in the mouse dentate gyrus. Brain Struct Funct. 2015;220:1187–1194. doi: 10.1007/s00429-014-0711-2. [DOI] [PubMed] [Google Scholar]
- Roberto M, Cruz M, Bajo M, Siggins GR, Parsons LH, Schweitzer P. The endocannabinoid system tonically regulates inhibitory transmission and depresses the effect of ethanol in central amygdala. Neuropsychopharmacology. 2010;35:1962–1972. doi: 10.1038/npp.2010.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiu S, Pinna GA, Marchese G, Mussinu JM, Saba P, Tambaro S, Casti P, Vargiu R, Pani L. Synthesis and characterization of NESS 0327: a novel putative antagonist of the CB1 cannabinoid receptor. J Pharmacol Exp Ther. 2003;306:363–370. doi: 10.1124/jpet.103.049924. [DOI] [PubMed] [Google Scholar]
- Rust MJ, Bates M, Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM) Nat Methods. 2006;3:793–795. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlosburg JE, Blankman JL, Long JZ, Nomura DK, Pan B, Kinsey SG, Nguyen PT, Ramesh D, Booker L, Burston JJ, Thomas EA, Selley DE, Sim-Selley LJ, Liu QS, Lichtman AH, Cravatt BF. Chronic monoacylglycerol lipase blockade causes functional antagonism of the endocannabinoid system. Nat Neurosci. 2010;13:1113–1119. doi: 10.1038/nn.2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano A, Rivera P, Pavon FJ, Decara J, Suárez J, Rodriguez de Fonseca F, Parsons LH. Differential effects of single versus repeated alcohol withdrawal on the expression of endocannabinoid system-related genes in the rat amygdala. Alcohol Clin Exp Res. 2012;36:984–994. doi: 10.1111/j.1530-0277.2011.01686.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon AC, Loverdo C, Gaffuri AL, Urbanski M, Ladarre D, Carrel D, Rivals I, Leterrier C, Benichou O, Dournaud P, Szabo B, Voituriez R, Lenkei Z. Activation-dependent plasticity of polarized GPCR distribution on the neuronal surface. J Mol Cell Biol. 2013;5:250–265. doi: 10.1093/jmcb/mjt014. [DOI] [PubMed] [Google Scholar]
- Smith TH, Blume LC, Straiker A, Cox JO, David BG, McVoy JR, Sayers KW, Poklis JL, Abdullah RA, Egertová M, Chen CK, Mackie K, Elphick MR, Howlett AC, Selley DE. Cannabinoid receptor-interacting protein 1a modulates CB1 receptor signaling and regulation. Mol Pharmacol. 2015;87:747–765. doi: 10.1124/mol.114.096495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stella N, Schweitzer P, Piomelli D. A second endogenous cannabinoid that modulates long-term potentiation. Nature. 1997;388:773–778. doi: 10.1038/42015. [DOI] [PubMed] [Google Scholar]
- Straiker A, Mackie K. Cannabinoid signaling in inhibitory autaptic hippocampal neurons. Neuroscience. 2009;163:190–201. doi: 10.1016/j.neuroscience.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan JM. Mechanisms of cannabinoid-receptor-mediated inhibition of synaptic transmission in cultured hippocampal pyramidal neurons. J Neurophysiol. 1999;82:1286–1294. doi: 10.1152/jn.1999.82.3.1286. [DOI] [PubMed] [Google Scholar]
- Sumislawski JJ, Ramikie TS, Patel S. Reversible gating of endocannabinoid plasticity in the amygdala by chronic stress: a potential role for monoacylglycerol lipase inhibition in the prevention of stress-induced behavioral adaptation. Neuropsychopharmacology. 2011;36:2750–2761. doi: 10.1038/npp.2011.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabó GG, Lenkey N, Holderith N, Andrási T, Nusser Z, Hájos N. Presynaptic calcium channel inhibition underlies CB1 cannabinoid receptor-mediated suppression of GABA release. J Neurosci. 2014a;34:7958–7963. doi: 10.1523/JNEUROSCI.0247-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabó GG, Papp OI, Máté Z, Szabó G, Hájos N. Anatomically heterogeneous populations of CB1 cannabinoid receptor-expressing interneurons in the CA3 region of the hippocampus show homogeneous input-output characteristics. Hippocampus. 2014b;24:1506–1523. doi: 10.1002/hipo.22330. [DOI] [PubMed] [Google Scholar]
- Takács VT, Szõnyi A, Freund TF, Nyiri G, Gulyás AI. Quantitative ultrastructural analysis of basket and axo-axonic cell terminals in the mouse hippocampus. Brain Struct Funct. 2015;220:919–940. doi: 10.1007/s00429-013-0692-6. [DOI] [PubMed] [Google Scholar]
- Tam J, Cinar R, Liu J, Godlewski G, Wesley D, Jourdan T, Szanda G, Mukhopadhyay B, Chedester L, Liow JS, Innis RB, Cheng K, Rice KC, Deschamps JR, Chorvat RJ, McElroy JF, Kunos G. Peripheral cannabinoid-1 receptor inverse agonism reduces obesity by reversing leptin resistance. Cell Metab. 2012;16:167–179. doi: 10.1016/j.cmet.2012.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibault K, Carrel D, Bonnard D, Gallatz K, Simon A, Biard M, Pezet S, Palkovits M, Lenkei Z. Activation-dependent subcellular distribution patterns of CB1 cannabinoid receptors in the rat forebrain. Cereb Cortex. 2013;23:2581–2591. doi: 10.1093/cercor/bhs240. [DOI] [PubMed] [Google Scholar]
- Twitchell W, Brown S, Mackie K. Cannabinoids inhibit N- and P/Q-type calcium channels in cultured rat hippocampal neurons. J Neurophysiol. 1997;78:43–50. doi: 10.1152/jn.1997.78.1.43. [DOI] [PubMed] [Google Scholar]
- Uchigashima M, Yamazaki M, Yamasaki M, Tanimura A, Sakimura K, Kano M, Watanabe M. Molecular and morphological configuration for 2-arachidonoylglycerol-mediated retrograde signaling at mossy cell-granule cell synapses in the dentate gyrus. J Neurosci. 2011;31:7700–7714. doi: 10.1523/JNEUROSCI.5665-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughn LK, Denning G, Stuhr KL, de Wit H, Hill MN, Hillard CJ. Endocannabinoid signalling: has it got rhythm? Br J Pharmacol. 2010;160:530–543. doi: 10.1111/j.1476-5381.2010.00790.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vida I, Halasy K, Szinyei C, Somogyi P, Buhl EH. Unitary IPSPs evoked by interneurons at the stratum radiatum-stratum lacunosum-moleculare border in the CA1 area of the rat hippocampus in vitro. J Physiol. 1998;506:755–773. doi: 10.1111/j.1469-7793.1998.755bv.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T, Fukaya M, Uchigashima M, Miura E, Kamiya H, Kano M, Watanabe M. Localization of diacylglycerol lipase-α around postsynaptic spine suggests close proximity between production site of an endocannabinoid, 2-arachidonoyl-glycerol, and presynaptic cannabinoid CB1 receptor. J Neurosci. 2006;26:4740–4751. doi: 10.1523/JNEUROSCI.0054-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You IJ, Jung YH, Kim MJ, Kwon SH, Hong SI, Lee SY, Jang CG. Alterations in the emotional and memory behavioral phenotypes of transient receptor potential vanilloid type 1-deficient mice are mediated by changes in expression of 5-HT1A, GABA(A), and NMDA receptors. Neuropharmacology. 2012;62:1034–1043. doi: 10.1016/j.neuropharm.2011.10.013. [DOI] [PubMed] [Google Scholar]
- Zhu PJ, Lovinger DM. Retrograde endocannabinoid signaling in a postsynaptic neuron/synaptic bouton preparation from basolateral amygdala. J Neurosci. 2005;25:6199–6207. doi: 10.1523/JNEUROSCI.1148-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zygmunt PM, Petersson J, Andersson DA, Chuang H, Sørgård M, Di Marzo V, Julius D, Högestätt ED. Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature. 1999;400:452–457. doi: 10.1038/22761. [DOI] [PubMed] [Google Scholar]
- Zygmunt PM, Ermund A, Movahed P, Andersson DA, Simonsen C, Jönsson BA, Blomgren A, Birnir B, Bevan S, Eschalier A, Mallet C, Gomis A, Högestätt ED. Monoacylglycerols activate TRPV1—a link between phospholipase C and TRPV1. PLoS One. 2013;8:e81618. doi: 10.1371/journal.pone.0081618. [DOI] [PMC free article] [PubMed] [Google Scholar]