

Abstract
Sustained neuroinflammation mediated by resident microglia is recognized as a key pathophysiological contributor to many neurodegenerative diseases, including Parkinson's disease (PD), but the key molecular signaling events regulating persistent microglial activation have yet to be clearly defined. In the present study, we examined the role of Fyn, a non-receptor tyrosine kinase, in microglial activation and neuroinflammatory mechanisms in cell culture and animal models of PD. The well-characterized inflammogens LPS and TNFα rapidly activated Fyn kinase in microglia. Immunocytochemical studies revealed that activated Fyn preferentially localized to the microglial plasma membrane periphery and the nucleus. Furthermore, activated Fyn phosphorylated PKCδ at tyrosine residue 311, contributing to an inflammogen-induced increase in its kinase activity. Notably, the Fyn-PKCδ signaling axis further activated the LPS- and TNFα-induced MAP kinase phosphorylation and activation of the NFκB pathway, implying that Fyn is a major upstream regulator of proinflammatory signaling. Functional studies in microglia isolated from wild-type (Fyn+/+) and Fyn knock-out (Fyn−/−) mice revealed that Fyn is required for proinflammatory responses, including cytokine release as well as iNOS activation. Interestingly, a prolonged inflammatory insult induced Fyn transcript and protein expression, indicating that Fyn is upregulated during chronic inflammatory conditions. Importantly, in vivo studies using MPTP, LPS, or 6-OHDA models revealed a greater attenuation of neuroinflammatory responses in Fyn−/− and PKCδ −/− mice compared with wild-type mice. Collectively, our data demonstrate that Fyn is a major upstream signaling mediator of microglial neuroinflammatory processes in PD.
SIGNIFICANCE STATEMENT Parkinson's disease (PD) is a complex multifactorial disease characterized by the progressive loss of midbrain dopamine neurons. Sustained microglia-mediated neuroinflammation has been recognized as a major pathophysiological contributor to chronic degenerative processes in PD; however, the key molecular signaling mechanisms underlying microglial activation are not entirely clear. Herein, we identified a novel role for the non-receptor tyrosine kinase Fyn in regulating neuroinflammatory responses in microglia. Our data clearly suggest that the Fyn-PKCδ signaling axis acts as a major upstream signaling mediator of the sustained neuroinflammatory processes in cell culture and animal models of PD. Our finding has important clinical significance to PD because it identifies Fyn as a potential translational target for intervention of progressive neurodegenerative processes in PD.
Keywords: Fyn, kinase, microglia, neuroinflammation, Parkinson's disease, phosphorylation
Introduction
Parkinson's disease (PD) is a highly prevalent neurodegenerative disorder and is mainly characterized by the loss of dopaminergic neurons in the substantia nigra (SN) of the ventral midbrain region. Extra nigral lesions and non-motor deficits have recently been recognized (Chaudhuri et al., 2006; Bohnen et al., 2014). Although the etiopathogenesis of PD is not known, both environmental insults and genetic defects have been implicated in its onset. Mutations in seven disparate genes have been linked to Parkinsonism, which clinically resembles PD with varying onset and disease progression. Additionally, 19 other genes have been hypothesized to have a disease-causing role (Puschmann, 2013).
The pathophysiology of PD is complex and multifactorial, with mitochondrial dysfunction, oxidative stress, apoptosis and proteasomal dysfunction being identified among others as potential disease mechanisms underlying nigrostriatal dopaminergic neuronal degeneration (Przedborski, 2005; Jenner and Olanow, 2006; Olanow, 2007; Levy et al., 2009). Recently, a wealth of data from cell culture, animal models and postmortem analyses of human PD brains have established chronic, sustained microglia-mediated neuroinflammation as being a major event in the delayed and progressive loss of dopaminergic neurons within the SN (Imamura et al., 2003; Block et al., 2007; Glass et al., 2010; Tansey and Goldberg, 2010). As the macrophagic cells of the CNS, microglia compose a major component of the brain's innate immune system. Under “normal” physiological conditions, they produce anti-inflammatory and neurotrophic factors to promote neuronal survival and plasticity (Carson, 2002). However, when they encounter a potential pathogen, a dead or dying neuron, or neurotoxic stress, they switch to an “activated” phenotype, producing proinflammatory cytokines and chemokines, reactive nitrogen species and reactive oxygen species. Activated microglia may also directly contribute to cell death by phagocytizing dopaminergic neurons (Barcia et al., 2012; Virgone-Carlotta et al., 2013). Thus, the pathophysiology of PD is accompanied by a sustained proinflammatory microglial response that contributes to neuron death, thereby exacerbating disease progression.
Fyn, a member of the Src family of kinases, is a non-receptor tyrosine kinase expressed in the brain. The kinase has been shown to play a role in amyloid-mediated apoptosis in cortical neurons (Lambert et al., 1998), astrocyte migration (Dey et al., 2008), and oligodendrocyte differentiation (Sperber et al., 2001). In the peripheral immune system, Fyn plays a role in mast cell and B-/T-cell activation (Palacios and Weiss, 2004; Gomez et al., 2005). Fyn was shown to mediate proinflammatory mediator production in mast cells, macrophages, basophils, as well as in natural killer cells (Rajasekaran et al., 2013). Fyn was shown to be activated following fibrillar β-amyloid peptide engagement of its receptor CD36, contributing to activation and migration of primary murine peritoneal macrophages and microglia (Moore et al., 2002; Stuart et al., 2007), and in BV2 microglial cells stimulated with the neurotoxic fragment of prion protein (Kouadir et al., 2012). Recently, we have identified a proapoptotic Fyn/PKCδ-mediated signaling pathway that contributes to oxidative stress-induced cell death in dopaminergic neurons (Kaul et al., 2005; Saminathan et al., 2011). However, the role of Fyn in microglial activation and neuroinflammation has never been studied in PD. Therefore, we sought to characterize the role of the Fyn-PKCδ signaling pathway in microglial activation and neuroinflammation in cell culture and animal models of PD. The results from these comprehensive studies reveal that Fyn kinase plays a key role in microglial activation and sustained neuroinflammation in the nigral dopaminergic system.
Materials and Methods
Chemicals and reagents.
DMEM/F-12, ascorbic acid, RPMI, FBS, l-glutamine, Hoechst nuclear stain, penicillin, streptomycin, and other cell culture reagents were purchased from Invitrogen. Recombinant tumor necrosis factor alpha (TNFα) was purchased from Peprotech, and lipopolysaccharide (LPS) (Escherichia coli 0111:B4, endotoxin content 6.6000000 EU/mg), 6-hydroxydopamine (6-OHDA), and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) were purchased from Sigma. The mouse Fyn antibody was purchased from Thermo Scientific. Antibodies for rabbit Fyn, PKCδ, p-Y311 PKCδ, IκBα, Lamin-B, NOS2 (iNOS), and mouse tubulin were purchased from Santa Cruz Biotechnology. Antibodies against rabbit p-Src family kinase Y416 (p-Y416 SFK), native p65, p-p38 MAP kinase, native p38 MAP kinase, p-p44/42 MAP kinase (p-ERK), and native p44/42 MAP kinase (ERK) were purchased from Cell Signaling Technology. The gp91phox antibody was purchased from BD Biosciences. The mouse GFAP antibody was purchased from Millipore. The TH antibody was purchased from Millipore. Mouse M2 FLAG and β-actin antibodies as well as the rabbit β-actin antibody were purchased from Sigma. Rabbit and goat Iba-1 antibodies were purchased from Wako Chemicals and Abcam, respectively. The goat TNFα antibody was purchased from R&D Systems. 32P-ATP was purchased from PerkinElmer and the histone substrate from Sigma. The Bradford protein assay kit was purchased from Bio-Rad Laboratories. The dual-luciferase reporter construct containing 3.1 kb human Fyn promoter fragment was provided by Dr. Joya Chandra (University of Texas M. D. Anderson Cancer Center, Houston, TX). FLAG-tagged human wild-type (WT) Fyn and Y417A mutant Fyn constructs were obtained as described previously (Kaspar and Jaiswal, 2011).
Animal studies.
The Fyn−/− and PKCδ−/− mice used in these studies were bred in our animal facility. Fyn−/− mice were originally obtained from Dr. Dorit Ron's laboratory at the University of California, San Francisco, and are available from The Jackson Laboratory (stock #002271). PKCδ−/− mice were obtained originally from Dr. Keiichi Nakayama's laboratory (Division of Cell Biology, Department of Molecular and Cellular Biology, Medical Institute of Bioregulation, Kyushu University, Fukuoka, Japan). Wild-type (Fyn+/+ and PKCδ+/+), PKCδ−/−, and Fyn−/− mice were housed under standard conditions of constant temperature (22 ± 1°C), humidity (relative, 30%), and a 12 h light cycle with food and water provided ad libitum. Six- to eight-week-old male mice were used for all studies. The well-characterized acute MPTP mouse model of PD (Wu et al., 2003; Przedborski et al., 2004; Kim et al., 2007; Hu et al., 2008) was primarily used for neuroinflammation studies. The mice from the MPTP treatment group received four intraperitoneal injections of MPTP-HCl (18 mg/kg free-base) dissolved in saline at 2 h intervals. Mice were killed 24 h after the last injection. The nigral neuroinflammatory response was also studied using the systemic LPS injection model (Qin et al., 2007), which induces chronic neuroinflammation and progressive dopaminergic degeneration in mice. A single injection of LPS (5 mg/kg, i.p.) was delivered to wild-type, Fyn−/−, and PKCδ−/− mice. Mice were killed 3 h later. Control groups for both MPTP and LPS received equivolume injections of saline. We injected 2 μl of 6-OHDA, diluted at a concentration of 5 μg/μl in 0.02% ascorbic acid, into the left striatum (0.2 μl/min) using the Angle 2 stereotaxic apparatus (Leica Biosystems). The coordinates relative to bregma were as follows: 0.7 mm anteroposterior, 2 mm lateral, and 2.4 mm ventral. The contralateral side was either not injected or injected with 2 μl of 0.02% ascorbic acid diluted in sterile PBS as a negative control. All animal procedures were approved by the Iowa State University Institutional Animal Care and Use Committee.
Primary microglial cultures and treatments.
Primary microglial cultures were prepared from wild-type, Fyn−/−, and PKCδ−/− postnatal day 1 (P1) mouse pups as described previously (Gordon et al., 2011). Briefly, mouse brains were harvested, meninges removed, and then placed in DMEM-F12 supplemented with 10% heat-inactivated FBS, 50 IU/ml penicillin, 50 μg/ml streptomycin, 2 mm l-glutamine, 100 μm nonessential amino acids, and 2 mm sodium pyruvate. Brain tissues were then incubated in 0.25% trypsin-EDTA for 30 min with gentle agitation. The trypsin reaction was stopped by adding double the volume of DMEM/F12 complete medium and then washing brain tissues three times. Tissues were then triturated gently to prepare a single-cell suspension, which was then passed through a 70 μm nylon mesh cell strainer to remove tissue debris and aggregates. The cell suspension was then made up in DMEM/F12 complete medium and seeded into T-75 flasks, which were incubated in humidified 5% CO2 at 37°C. The medium was changed after 5–6 d, and the mixed glial cells were grown to confluence. Microglial cells were separated from confluent mixed glial cultures by differential adherence and magnetic separation to >97% purity and then were allowed to recover for 48 h after plating. Primary microglia were treated in DMEM/F12 complete medium containing 2% FBS. For signaling experiments, the protocol used by Stuart (2007) was used with a small modification. For this, the primary microglial cells were kept in 2% DMEM/F12 complete medium for 5 h at 37°C before treatment. The microglial cells were treated with 100–200 ng/ml LPS and 10–30 ng/ml TNFα for durations sampled at prespecified time points. We selected the LPS doses used in this study based on previous studies in which stimulation of cultured primary mouse microglia with 100 and 200 ng/ml LPS resulted in significant microglial activation (Haynes et al., 2006; Crotti et al., 2014; Lee et al., 2014).
siRNAs and transfections of microglia.
Transient transfections of primary microglia with Fyn promoter reporter were performed using Lipofectamine LTX and Plus Reagent according to the manufacturer's protocol. Primary microglia were plated at 0.75 × 106 cells/well in 12-well plates 1 d before transfection. We transiently transfected 3 μg of Fyn promoter construct. Cells were treated 24 h after transfection with or without 200 ng/ml of LPS for 12 h and then lysed. Luciferase activity was measured using a Dual-luciferase assay kit (Promega) on a Synergy 2 multimode microplate reader (BioTek Instruments). Firefly luciferase luminescence values were used to normalize Renilla luciferase luminescence values. The predesigned, on-target plus SMART pool Fyn siRNA (a combination of four siRNAs, catalog #LQ-040112-00-0002) and scrambled siRNA (catalog #D-001210-03-05) were purchased from Dharmacon. We performed siRNA transfections in primary mouse microglial cells with Lipofectamine 3000 reagent according to the manufacturer's protocol. Briefly, primary microglia were plated at 2 × 106 cells/well in 6-well plates 1 d before transfection. For each well, 300 pmol of Fyn siRNA pool (75 pmol each) or an equal amount of scrambled siRNA mixed with 5 μl of Lipofectamine 3000 was added to the cells. Seventy-two hours after the initial transfection, cells were analyzed by Western blotting to confirm the extent of Fyn knockdown or treated with LPS (200 ng/ml) for 24 h further, after which cytokine content was analyzed by Luminex bioassay.
Transfection of BV2 microglia with WT Fyn-FLAG, Y417A Fyn-FLAG, and Empty vector pcDNA3.1 constructs was performed using the Amaxa Nucleofector kit (Lonza). Briefly, BV2 cells were resuspended in transfection buffer (solution 1: 400 μm ATP-disodium, Sigma A7699; 600 μm MgCl2-6H2O in water; solution 2: 100 μm KH2PO4, 20 μm NaHCO3, 5 μm glucose in water) to a final concentration of 3 × 106 cells per 100 μl and mixed with the respective vector; 5 μg of vector DNA was used per transfection.
Immunohistochemistry and immunofluorescence studies.
Immunohistochemistry was performed on sections from the SN and other brain regions of interest as described previously (Jin et al., 2011b; Ghosh et al., 2013). Briefly, mice were anesthetized with a mixture of 100 mg/kg ketamine and 10 mg/kg xylazine and then perfused transcardially with freshly prepared 4% PFA. Extracted brains were postfixed in 4% PFA for 48 h, and 30 μm sections were cut using a freezing microtome (Leica Microsystems). Antigen retrieval was performed in citrate buffer (10 mm sodium citrate, pH 8.5) for 30 min at 90°C. Sections were then washed several times in PBS and blocked with PBS containing 2% BSA, 0.2% Triton X-100, and 0.05% Tween 20 for 1 h at room temperature. Sections were then incubated with primary antibodies overnight at 4°C and washed 7 times in PBS on a Belly Dancer shaker (SPI Supplies). The sections were incubated with Alexa dye-conjugated secondary antibodies for 75 min at room temperature, and their cell nuclei were stained with Hoechst dye. Sections were mounted on slides using Prolong antifade gold mounting medium (Invitrogen) according to the manufacturer's instructions. Samples were visualized using an inverted fluorescence microscope (Nikon TE-2000U), and images were captured using a Spot digital camera (Diagnostic Instruments).
Immunofluorescence studies in primary microglia were performed according to previously published protocols with some modifications (Gordon et al., 2011). Briefly, microglial cells were grown on poly-d-lysine-coated coverslips and treated 48 h later. At the end of treatments, cells were fixed with 4% PFA, washed in PBS, and incubated in blocking buffer (PBS containing 2% BSA, 0.5% Triton X-100 and 0.05% Tween 20) for 1 h at room temperature. The coverslips were then incubated overnight at 4°C with respective primary antibodies diluted in PBS containing 2% BSA. Samples were then washed several times in PBS and incubated with Alexa-488 and -555 dye-conjugated secondary antibodies. The nuclei were labeled with Hoechst stain (10 μg/ml), and coverslips were mounted with Fluoromount medium (Sigma-Aldrich) on glass slides for visualization. Quantification of the number of microglial/astroglial cells obtained after separation was accomplished using JACoP, a downloadable ImageJ plugin from Fabrice P. Cordelières and Susanne Bolte. Original Hoechst or antibody TIFF files were converted into 8-bit black-and-white images, and a colocalization image was generated. Counting of Hoechst-positive and Iba-1+Hoechst-positive cells was done using the cell counter function of the default “Analyze” plugin in ImageJ.
Confocal imaging and Z-stack image acquisition and analysis.
Confocal imaging was performed at the Iowa State University Microscopy Facility, using a Leica DMIRE2 confocal microscope with the 63× and 43× oil objectives and Leica Confocal Software. One optical series covered 11–13 optical slices of 0.5 μm thickness each. Microglial neuronal contact identification and quantification were performed by counting the number of colocalizations of the two markers, with TH marked red by anti-555 and Iba-1 marked green by anti-488 using the methodology described by Barcia et al. (2012). The Imaris software was used to analyze the Z-stack images for contact identification. The surface reconstruction wizard in the Imaris software was used to make 3D reconstructions of stacks for easier viewing of microglial-dopaminergic contacts and surface topology.
qRT-PCR.
RNA isolation from primary microglial cells and brain tissue samples was performed using the Absolutely RNA Miniprep kit, and then 1 μg total of isolated RNA was used for reverse transcription with the AffinityScript qPCR cDNA synthesis system (Agilent Technologies) according to the manufacturer's instructions. Quantitative SYBR Green PCR assays for gene expression were performed using the RT2 SYBR Green Master Mix with prevalidated primers (SA Biosciences). Catalog numbers of the primers were as follows: Fyn, PPM04015A; pro-IL1β, PPM03109E; and TNFα, PPM03113G. The mouse 18S rRNA gene (catalog #PPM57735E) was used as the housekeeping gene for normalization. For each primer, the amount of template providing maximum efficiency without inhibiting the PCR was determined during initial optimization experiments. For all experiments, dissociation curves were generated to ensure that a single peak was obtained at the right melting temperature without nonspecific amplicons. The fold change in gene expression was determined by the ΔΔCt method using the threshold cycle (Ct) value for the housekeeping gene and the respective target gene of interest in each sample.
Western blotting.
Brain tissue and microglial cell lysates were prepared using modified RIPA buffer and were normalized for equal amounts of protein using the Bradford protein assay kit. Equal amounts of protein (12–25 μg for cell lysates and 30–40 μg for tissue lysates) were loaded for each sample and separated on either 12% or 15% SDS-PAGE gels depending on the molecular weight of the target protein. After separation, proteins were transferred to a nitrocellulose membrane, and the nonspecific binding sites were blocked for 1 h using a blocking buffer specifically formulated for fluorescent Western blotting (Rockland Immunochemicals). Membranes were then probed with the respective primary antibodies for 3 h at room temperature or overnight at 4°C. After incubation, the membranes were washed 7 times with PBS containing 0.05% Tween 20, and then secondary IR-680-conjugated anti-mouse (1:10,000, goat anti-mouse, Invitrogen) and IR-800-conjugated anti-rabbit (1:10,000, goat anti-rabbit, Rockland) were used for antibody detection with the Odyssey IR imaging system (LiCor). Antibodies for β-actin and tubulin were used as loading controls.
Coimmunoprecipitation studies.
We adopted an immunoprecipitation protocol with slight modifications from Gao et al. (2011). Cell lysates were prepared in TNE buffer (10 mm Tris-HCl at pH 7.5, 1% Nonidet P-40, 0.15 M NaCl, 1 mm EDTA, and 1:100 protease inhibitor mixture) and centrifuged at 17,400 × g for 40 min at 4°C. The supernatant protein concentration was measured and normalized between samples. Approximately 20 μl of the sample containing 50 μg protein was used as input. For immunoprecipitation analysis, 1 mg of protein in 400 μl TNE buffer was used. Next, 10 μl (2 μg) of Fyn rabbit polyclonal antibody was added to the lysates, and the samples were set on an orbital shaker overnight at 4°C. The next day, protein G Sepharose beads were spun down at 17,400 × g for 5 min, and the ethanol supernatant was replaced with an equal volume of the lysis buffer. The Protein G Sepharose slurry was washed once, and 50 μl was added to each sample. The samples were set on an orbital shaker overnight at 4°C. Protein G beads were collected by centrifugation at 2000 × g for 5 min and were washed four times with TNE buffer. The bound proteins were eluted by boiling in 2× protein-loading dye for 5 min. Immunoblots were performed on 12% SDS-PAGE gels as described for Western blotting.
Nuclear and cytoplasmic fractionation.
Nuclear and cytoplasmic fractions were performed using the NE-PER kit (Thermo Scientific) as previously described (Jin et al., 2011a, 2014). Briefly, 5 × 106 cells were treated with LPS or TNFα for 15 min. CER1 reagent (200 μl) was used for each sample to extract the cytoplasmic fraction, and 50 μl of NER reagent was used to extract the nuclear fraction. Tubulin or β-actin was used as a cytosolic fraction marker. Lamin B was used as a nuclear fraction marker.
Fyn kinase assays.
Cell pellets were washed with ice-cold PBS and resuspended in lysis buffer (25 mm HEPES at pH 7.5, 20 mm β-glycerophosphate, 0.1 mm sodium orthovanadate, 0.1% Triton X-100, 0.3 M NaCl, 1.5 mm MgCl2, 0.2 mm EDTA, 0.5 mm dithiothreitol, 10 mm NaF, and 4 μg/ml each of aprotinin and leupeptin) (Kaul et al., 2005). Next, 50 μg of crude protein was incubated with 150 mm Fyn kinase substrate (Biomol), 100 mCi of [γ-32P] ATP, Src-Mn-ATP mixture, and Src reaction buffer (Millipore) for 10 min at 30°C with agitation. To precipitate the Fyn kinase substrate peptide, 20 μl of 40% trichloroacetic acid was added. Then 25 μl of the mixture was spotted onto a P81 phosphocellulose square; and 5 min after spotting, the squares were washed five times in 0.75% phosphoric acid in PBS with a final wash step in acetone. The squares were transferred into a scintillation vial, and the counts per minute were read in a liquid scintillation system after adding 5 ml of scintillation mixture to each vial.
PKCδ kinase assays.
PKCδ IP kinase activity assays were performed as described previously (Anantharam et al., 2002; Latchoumycandane et al., 2011; Harischandra et al., 2014) with some modifications for microglial cells. Briefly, primary microglial cells were collected after treatments, washed in ice-cold PBS, and resuspended in a mild RIPA lysis buffer containing protease and phosphatase inhibitor mixture (Pierce Biotechnology). The cells were placed on ice for 20 min to allow for complete lysis and then centrifuged at 16,200 × g for 45 min. The supernatant protein concentration was determined using the Bradford protein assay kit. Samples were normalized to a uniform total protein concentration of 2 μg/ml, and then 200 μg of total protein in a 250 μl reaction volume was immunoprecipitated overnight at 4°C using 5 μg of PKCδ antibody. The next day, protein A agarose beads (Sigma-Aldrich) were incubated for 1 h at room temperature. The protein A-bound antibody complexes were collected and washed 3 times in 2× kinase assay buffer (40 mm Tris, pH 7.4, 20 mm MgCl2, 20 μm ATP, and 2.5 mm CaCl2), and then resuspended in the same buffer. The kinase reaction was started by adding 40 μl of the reaction buffer containing 0.4 mg of histone H1, 50 μg/ml phosphatidylserine, 4 μm dioleoylglycerol, and 10 μCi of [γ-32P] ATP at 3000 Ci/mm to the immunoprecipitated samples. The samples were then incubated for 10 min at 30°C, and the kinase reaction was stopped by adding 2× SDS loading buffer and boiling for 5 min. Proteins were separated on a 15% SDS-PAGE gel, and the phosphorylated histone bands were imaged using a Fujifilm FLA 5000 imager. Image analysis and band quantification were performed using ImageJ.
Nitric oxide detection.
Nitric oxide production by primary microglia was measured indirectly by quantification of nitrite in the supernatant using the Griess reagent (Sigma-Aldrich). Microglia were plated in poly-d-lysine-coated 96-well plates at 1 × 105 cells/well. Cells were treated with 100 ng/ml of LPS for 24 h; and after 100 μl of supernatant was collected from each well, an equal volume of the Griess reagent was added. The samples were incubated on a plate shaker at room temperature for 15 min until a stable color was obtained. The absorbance at 540 nm was measured using the Synergy 2 multimode microplate reader, and the nitrite concentration was determined from a sodium nitrite standard curve.
Multiplex cytokine Luminex immunoassays.
Primary microglia obtained from wild-type, PKCδ−/−, and Fyn−/− mice were seeded in poly-d-lysine-coated 96-well plates at 1 × 105 cells/well. The cells were treated for 24 h with 100–200 ng/ml LPS or 10 ng/ml TNFα. After treatment, 50 μl of supernatant from each well was collected and frozen at −80°C. The levels of cytokines and chemokines in the supernatants were determined using the Luminex bead-based immunoassay platform (Vignali 2000) and prevalidated multiplex kits (Milliplex mouse cytokine panel, Millipore) according to the manufacturer's instructions.
Diaminobenzidine immunostaining and grading of microglial morphology.
Diaminobenzidine (DAB) immunostaining was performed on striatal and nigral sections as described previously (Ghosh et al., 2010). Briefly, mice were perfused with 4% PFA, and brains were postfixed with PFA for 48 h before storage in 30% sucrose. Fixed brains were embedded in OCT compound (Tissue-Tek) and stored frozen at −80°C until the frozen blocks were sliced into 30 μm coronal sections using a cryostat. Sections were probed with the primary antibodies overnight at 4°C and then incubated with biotinylated anti-rabbit secondary antibody. The sections were then treated with avidin peroxidase (Vectastain ABC Elite kit). The DAB reagent was used for producing the brown colored stain. Grading of microglial morphology was performed as described previously (Lastres-Becker et al., 2012). For microglial grading, images were sharpened in ImageJ so the morphology could be more clearly visualized. The cell counter function in the “Analyze” plugin was used to count the number of Type A-D microglia in the ventral midbrain sections.
Data analysis.
Data analysis was performed using Prism 4.0 (GraphPad Software). The data were initially analyzed using one-way ANOVA and Bonferroni's post-test to compare the means of treatment groups. Differences of p < 0.05 were considered statistically significant. Student's t test was used when comparing two groups.
Results
Fyn and PKCδ are differentially expressed in primary astrocytes and microglia
Primary mouse microglia were prepared as described in our recent publication using a magnetic separation method, which enables us to obtain a high-yield pure fraction of microglia from mixed glial cultures (Gordon et al., 2011). Iba-1 and GFAP immunocytochemistry confirmed that the microglial fraction obtained after magnetic separation was devoid of astrocytes (Fig. 1A). Quantification of Hoechst colocalized Iba-1-positive microglia and GFAP-positive astrocytes using the ImageJ plugin JACoP revealed a microglial population that was ∼97% pure after separation (Fig. 1B,C). Immunoblotting analysis revealed that microglia-enriched fractions expressed significantly more Fyn (60 kDa) and PKCδ (76 kDa) than did astrocyte-enriched (microglia-depleted) fractions (Fig. 1D,E). The differential expression of both Fyn and PKCδ in microglia compared with astrocytes prompted us to study the roles these proteins may play in microglial proinflammatory signaling.
Figure 1.

Fyn and PKCδ are differentially expressed in primary astrocytes and microglia. A, Representative image from immunocytochemical analysis for the microglial marker Iba-1 and the astrocytic marker GFAP on both the magnetically purified and pour-off fractions of cells obtained after separation revealed almost no astrocytic contamination in the samples. Scale bar, 200 μm. B, The colocalization image of Hoechst (nuclear stain) and Iba-1 images in 6 random image fields were obtained using the ImageJ plugin JACoP. C, The number of Hoechst-positive and colocalization-positive cells were counted using ImageJ. The magnetically purified samples were >97% positive for microglial cells. D, E, Western blot analyses of the magnetically separated cells revealed that the microglial fraction expressed higher amounts of the non-receptor Src kinase Fyn and the serine threonine kinase PKCδ than did the astrocyte-rich pour-off fraction. *p < 0.05. **p < 0.01.
Fyn kinase is rapidly activated in microglial cells and in the ventral midbrain following inflammogen stimulation
Our initial experiment to determine whether the non-receptor tyrosine kinase Fyn plays a role in regulating neuroinflammatory responses in PD was performed in BV2 microglial cells, which are widely used in vitro models of neuroinflammation (Henn et al., 2009; Gao et al., 2011; Kim et al., 2013b). We treated BV2 cells with 1 μg/ml LPS for 10–60 min and measured Fyn activity using an in vitro kinase assay (Saminathan et al., 2011). A kinase reaction mixture containing 32P-ATP and a Fyn-specific peptide substrate were added to whole-cell lysates. LPS stimulation of BV2 microglia rapidly induced Fyn activity as early as 10 min after LPS stimulation (Fig. 2A), and maximal activity was attained 30 min after LPS stimulation. In addition to the Fyn kinase activity assay, we also determined the phosphorylation status of the Y416 residue in its activation loop domain by using the phospho Y416 Src family kinase (p-Y416 SFK) antibody, which recognizes activated Src family kinases. This antibody has been used extensively to demonstrate Fyn kinase activation (Wake et al., 2011; Kouadir et al., 2012; Larson et al., 2012; Um et al., 2012). Our immunoblotting analysis of LPS-treated BV2 lysates using the p-Y416 SFK antibody revealed LPS-induced SFK activation (Fig. 2B). To further confirm inflammogen-induced Fyn activation in BV2 microglia, we transiently transfected BV2 cells with FLAG-tagged WT-Fyn and Y417A-Fyn (activation loop mutant) constructs. We then performed immunoprecipitation studies in LPS-treated transfected BV2 cells. We pulled down Fyn from FLAG-tagged WT-Fyn and Y417A-Fyn transfected, LPS-treated BV2 cells and immunoblotted for p-Y416 SFK levels. A strong p-Y416 SFK signal was detected in the LPS-treated WT-Fyn-FLAG-transfected cells, but not in the LPS-treated Y417A-Fyn-transfected cells (Fig. 2C,D).
Figure 2.

Fyn kinase is rapidly activated in microglial cells and in the ventral midbrain following inflammogen stimulation. A, Fyn kinase assay shows that Fyn activity was highly induced in BV2 microglia treated with 1 μg/ml LPS for 10, 15, and 30 min. *p < 0.05. **p < 0.01. B, Immunoblots showing a concomitant rise in p-Y416 SFK levels in BV2 cell lysates after LPS treatment. C, D, Immunoprecipitation studies revealed that WT Fyn, but not activation loop tyrosine-mutant Fyn (Y417A Fyn), when overexpressed in BV2 microglia, was activated following LPS stimulation. E, F, Treatment of (E) primary microglia with LPS and TNFα (F) for 15 and 30 min increased p-Y416 SFK levels in primary microglia obtained from Fyn+/+, but not Fyn−/− mice, identifying Fyn as the primary Src family kinase that was activated by inflammogen stimulation. G, Pretreatment of primary microglia with the TLR-signaling antagonist IAXO-101 or the TNFα receptor decoy etanercept abolished Fyn activation by LPS or TNFα stimulation (p44/42 phosphorylation used as marker for early microglial activation). H, Immunocytochemistry of LPS-treated WT primary microglia showing that activated Fyn expression greatly increased and was localized preferentially to the membrane periphery of the microglial cell. Scale bar, 20 μm. I, Immunoblots of ventral midbrain lysates showed that peripheral administration of the inflammogen LPS (5 mg/kg) increased p-Y416 SFK levels in Fyn+/+, but not in Fyn−/− ventral midbrain tissues.
Next, we extended our studies to primary microglia derived from both wild-type and Fyn-deficient (Fyn−/−) mice. These were treated with 200 ng/ml LPS for 0–30 min. In line with the analyses of BV2 cells, stimulation of the primary microglia from Fyn+/+ mice rapidly increased the levels of p-Y416 SFK (Fig. 2E). Interestingly, p-Y416 SFK was not detected in LPS-treated Fyn−/− microglia, suggesting that LPS preferentially induces Fyn phosphorylation in microglia over other Src family kinases. Treatment of wild-type and Fyn-deficient microglia with TNFα also yielded similar results. Both 10 ng/ml and 30 ng/ml TNFα treatments induced similar levels of p-Y416 SFK in wild-type, but not in Fyn-deficient microglia (Fig. 2F). Pretreatment of wild-type microglia with either the Toll-like receptor (TLR) antagonist IAXO-101 or the TNFα signaling antagonist etanercept significantly attenuated both LPS- and TNFα-mediated Fyn activation, respectively (Fig. 2G). We also examined subcellular localization of activated Fyn following LPS stimulation. The Iba-1/p-Y416 SFK double-immunocytochemical analysis showed that LPS treatment dramatically increased p-Y416 Fyn levels in WT primary microglia (Fig. 2H). Active Fyn seems to be preferentially expressed at the periphery of the microglia, possibly allowing it to become activated quickly in response to a proinflammatory stimulus. Additionally, activated Fyn was also found in the nucleus of LPS-treated primary microglia. Next, we wanted to confirm that LPS treatment would activate Fyn in the SN of mice. Knowing that a single intraperitoneal LPS injection elicits microglial cell activation in the SN (Qin et al., 2007), we challenged Fyn+/+ and Fyn−/− mice with 5 mg/kg LPS or sterile PBS vehicle intraperitoneally for 3 h. Immunoblot analysis of ventral midbrain lysates revealed that LPS significantly increased p-Y416 SFK levels in wild-type compared with saline control, whereas LPS failed to increase p-Y416 SFK levels in the Fyn−/− ventral midbrain lysates (Fig. 2I). These studies indicate that stimulating microglia with inflammatory stimuli rapidly activates Fyn kinase in both cell culture and animal models of neuroinflammation.
Fyn contributes to LPS- and TNFα-induced tyrosine phosphorylation and activation of PKCδ in primary microglia
It has been shown that Src family kinases, including Fyn, phosphorylate PKCδ at residue Y311 in platelets and in immortalized dopaminergic neuronal cells (Steinberg, 2004; Saminathan et al., 2011). Therefore, we investigated whether Fyn-PKCδ signaling regulates microglial proinflammatory responses using primary microglia cultures from wild-type, Fyn−/−, and PKCδ−/− mice. Stimulation with LPS induced a rapid and time-dependent increase in p-Y311 PKCδ in wild-type microglia. In contrast, LPS failed to increase Y311 phosphorylation of PKCδ in the Fyn−/− microglia (Fig. 3A,B). Similarly, TNFα stimulation of microglia also increased PKCδ Y311 phosphorylation in wild-type, but not in Fyn-deficient primary microglia (Fig. 3C,D). As expected, immunoblot analysis did not detect any LPS-induced phosphorylation of Y311 PKCδ in PKCδ−/− microglia (Fig. 3A). To confirm further that Fyn mediates the activation of PKCδ in activated microglia, we measured PKCδ kinase activity in wild-type and Fyn−/− microglia. An in vitro PKCδ kinase assay showed that LPS rapidly increased PKCδ kinase activity in wild-type microglia; however, LPS-induced PKCδ kinase activity was significantly less in Fyn−/− microglia (Fig. 3E). To further confirm the Fyn-PKCδ interaction, we performed coimmunoprecipitation studies in BV2 cells transfected with the WT-Fyn-FLAG construct. As shown in Figure 3F, G, coimmunoprecipitation analysis of WT-Fyn-FLAG-transfected lysates revealed that Fyn and PKCδ interact during LPS stimulation. Together with the PKCδ kinase activity results, these data reveal that Fyn kinase mediates LPS- and TNFα-induced activation of PKCδ in primary microglia.
Figure 3.

Fyn contributes to LPS- and TNFα-induced tyrosine phosphorylation and activation of PKCδ in primary microglia. Western blot analysis revealed that stimulation of microglia with LPS (A, B) and TNFα (C, D) induced a time-dependent increase in p-Y311 PKCδ levels in wild-type but not Fyn−/− microglia. *p < 0.05. **p < 0.01. E, LPS-induced PKCδ kinase activity was reduced in Fyn−/− microglial lysates in contrast to wild-type lysates, as measured by PKCδ kinase assay. F, G, Coimmunoprecipitation studies showed that LPS stimulation elicited a physical interaction between Fyn and PKCδ in WT Fyn-transfected BV2 microglial cells.
The Fyn-PKCδ signaling axis mediates MAP kinase activation in microglial cells
We next examined whether the Fyn-PKCδ signaling axis plays a role in mediating activation of the MAP kinase pathway, a key hallmark of neuroinflammatory signaling in microglia. MAP kinases are important regulators of proinflammatory cytokine synthesis in microglial cells (Koistinaho and Koistinaho, 2002; Tansey and Goldberg, 2010). For this purpose, we treated wild-type, Fyn−/−, and PKCδ−/− microglia with LPS for 15, 30, and 45 min each and determined MAPK activation. The LPS treatment significantly increased the phosphorylation of p38 and p44/42 (ERK) kinases in wild-type microglia (Fig. 4A,B), with LPS-induced phosphorylation peaking at 15 min and decreasing thereafter. In contrast, LPS-induced phosphorylation of p38 and p44/42 was significantly reduced in Fyn−/− and PKCδ−/− primary microglia. Similar results were obtained with TNFα treatment of wild-type, Fyn−/−, and PKCδ−/− microglia (Fig. 4C,D). These results suggest that Fyn-PKCδ signaling is an important upstream regulator of MAP kinases in microglia during both LPS and TNFα stimulation.
Figure 4.

The Fyn-PKCδ signaling axis mediates MAP kinase activation in microglial cells. A, B, Immunoblot analysis demonstrated diminished LPS-induced p38 and p44/42 (ERK) phosphorylation in Fyn−/− and PKCδ−/− microglia. *p < 0.05. ***p < 0.001. C, D, Diminished TNFα-induced p38 and p44/42 (ERK) phosphorylation in Fyn−/− and PKCδ−/− microglia. *p < 0.05.
Fyn contributes to inflammogen-mediated NFκB pathway activation in microglial cells
Proinflammatory signaling mediated by both LPS and TNFα converges at the NFκB pathway. Activation of NFκB signaling during the proinflammatory process is characterized by the phosphorylation and subsequent degradation of the inhibitory protein IκBα, after which the NFκB p65-p50 heterodimer enters the nucleus, leading to the transcription of various proinflammatory genes (Hayden and Ghosh, 2004). To elucidate whether Fyn mediates the nuclear translocation and activation of NFκB signaling in activated microglia, primary microglia obtained from wild-type and Fyn−/− microglia were treated with LPS for 15–45 min. Whole-cell lysates were prepared and probed for IκBα. LPS treatment induced a greater degradation of IκBα in wild-type microglia than in Fyn−/− microglia at the 15 min time point, followed by the resynthesis of IκBα 30 and 45 min after stimulation in the WT cells. Resynthesis of IκBα in Fyn−/− microglia was almost completely abrogated, indicating diminished NFκB activation (Fig. 5A,B). Next, we investigated the role of Fyn in the nuclear translocation of the p65 component of the NFκB complex in response to LPS and TNFα treatments. Nuclear and cytoplasmic fractions were prepared from WT and Fyn−/− microglia treated with LPS or TNFα for 15 min before being assessed for p65 content. Immunoblotting revealed lesser nuclear translocation of p65 in LPS- and TNFα-treated Fyn−/− microglia than in wild-type microglia (Fig. 5C,D). These results were further supported by Iba-1/p65 double-immunocytochemistry showing strong LPS-induced nuclear translocation of p65 in wild-type, but not in the Fyn−/− microglia (Fig. 5E). Together, these results clearly suggest that Fyn kinase regulates NFκB activation in microglial cells.
Figure 5.

Fyn contributes to inflammogen-mediated NFκB pathway activation in microglial cells. A, B, Immunoblot analyses of whole-cell lysates of wild-type and Fyn−/− microglia treated with LPS for 15–45 min revealed reduced IκBα degradation in Fyn−/− microglia at 15 min, and attenuated IκBα resynthesis at 30 and 45 min. *p < 0.05. **p < 0.01. C, D, Cytosolic and nuclear fractionation of LPS- and TNFα-treated wild-type and Fyn−/− microglia revealed diminished nuclear translocation of the p65 subunit of the NFκB complex in the Fyn−/− microglia. **p < 0.01. ***p < 0.001. E, Immunocytochemistry also showed reduced nuclear p65 in LPS-treated Fyn−/− microglia. Scale bar, 50 μm.
LPS- or TNFα-induced proinflammatory cytokine production is suppressed in Fyn/PKCδ-deficient microglia
Next, we determined whether Fyn-PKCδ signaling axis regulates microglia-mediated proinflammatory mediator production. After treating wild-type, PKCδ−/−, and Fyn−/− microglial cultures with LPS or TNFα, we used multiplexed immunoassays to quantify inflammogen-induced cytokine secretion. We observed significant production of the cytokines IL-6, IL-12p70, and TNFα from wild-type microglia treated with LPS (Fig. 6A). However, the production of these cytokines was significantly dampened in Fyn- and PKCδ-deficient microglia, providing evidence for the hypothesis that attenuated proinflammatory signaling in Fyn−/− and PKCδ−/− microglia suppresses proinflammatory mediator production. When we knocked down Fyn expression in wild-type primary microglia by Fyn-specific siRNA (Fig. 6B), diminished amounts of the proinflammatory cytokines IL-6 and TNFα were produced in response to LPS treatment (Fig. 6C). Next, treatment of wild-type and Fyn−/− microglia with TNFα yielded similar results, with the Fyn−/− microglia showing reduced IL-6 and TNFα production (Fig. 6D). Western blot analysis also demonstrated that Fyn-deficient microglia produced less TNFα relative to wild-type microglia (Fig. 6E). To further confirm the role of Fyn in proinflammatory cytokine production, we expressed Fyn wild-type (WT-Fyn-FLAG) or activation loop mutant (kinase-deficient Fyn kinase, Y417A Fyn-FLAG) in BV2 microglial cells (Fig. 6F). Following the transfection, BV2 cells transfected WT-Fyn-FLAG, Y417A Fyn-FLAG, or empty vector constructs were treated with 1 μg/ml LPS for 24 h. Luminex immunoassay of cell supernatants revealed that overexpressing wild-type Fyn augmented proinflammatory cytokine release, whereas overexpressing the activation loop Y417A Fyn mutant suppressed the production of IL-6 and IL-12 (Fig. 6F,G).
Figure 6.
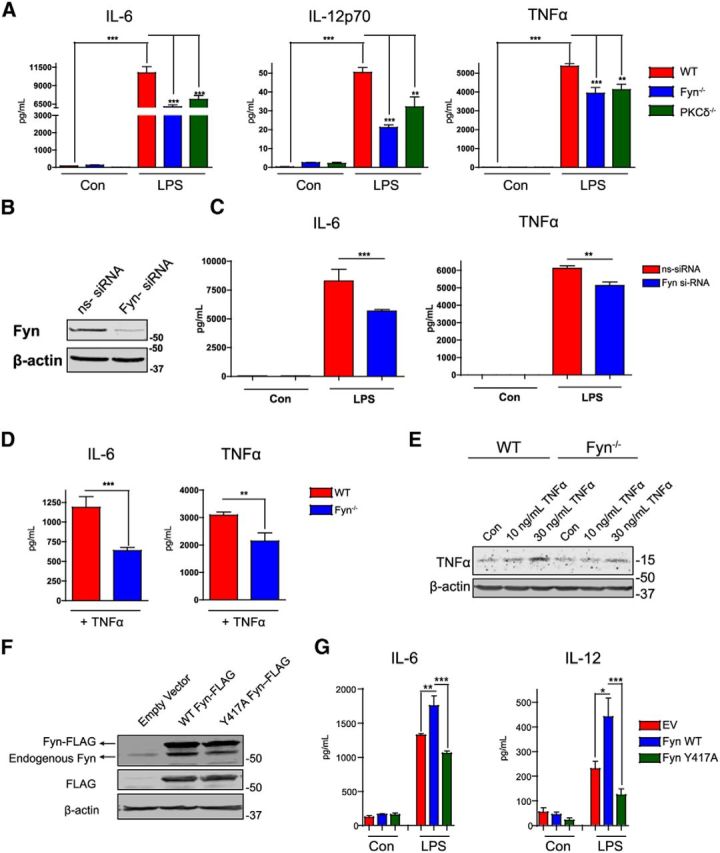
LPS- or TNFα-induced proinflammatory cytokine production is suppressed in Fyn/PKCδ-deficient microglia. A, Luminex analyses of supernatants from LPS-treated wild-type, PKCδ−/−, and Fyn−/− microglia revealed reduced secretion of the proinflammatory cytokines IL-6, IL-12, and TNFα. **p < 0.01. ***p < 0.001. B, Wild-type primary microglia were transfected with nontargeting and Fyn-specific siRNA for 72 h. Knockdown of Fyn was evaluated by Western blot. C, Fyn-depleted microglia demonstrated diminished IL-6 and TNFα secretion in response to LPS stimulation. **p < 0.01. ***p < 0.001. D, TNFα stimulation of Fyn−/− microglia reduced IL-6 and TNFα production in contrast to wild-type microglia. **p < 0.01. ***p < 0.001. E, Immunoblots showing reduced TNFα levels in Fyn-deficient microglia after TNFα stimulation in contrast to wild-type microglia. F, G, Overexpressing the FLAG-tagged activation loop tyrosine mutant of Fyn in BV2 microglia attenuated IL-6 and IL-12 production when the cells were treated with LPS, as shown by Luminex cytokine analysis. *p < 0.05. **p < 0.01. ***p < 0.001.
Fyn/PKCδ regulates the induction of neuroinflammatory markers iNOS and gp91phox in microglia during LPS stimulation
We further assessed whether Fyn alters the induction of iNOS and gp91phox, which are key proinflammatory responses of microglial activation following LPS treatment. Treatment with LPS induced a stronger nitrite response from wild-type microglia than from Fyn−/− microglia (Fig. 7A). This was further confirmed by immunostaining and immunoblotting for iNOS, the enzyme that mediates nitrite production. There was a greater induction of iNOS in Fyn wild-type microglia relative to Fyn−/− microglia (Fig. 7B–D). We also determined the expression of other key neuroinflammatory markers, including gp91phox and Iba-1, in response to LPS stimulation. We, as well as other groups, have previously shown increased expression of the NADPH oxidase component gp91phox and Iba-1 following LPS stimulation of primary microglia (Gao et al., 2011; Gordon et al., 2011). Western blot analysis revealed that LPS increased expression of both gp91phox and Iba-1 in wild-type, but not in Fyn−/− or PKCδ−/− microglia (Fig. 7E,F). Collectively, these data indicate that Fyn-PKCδ signaling plays a major proinflammatory role in microglial cells.
Figure 7.
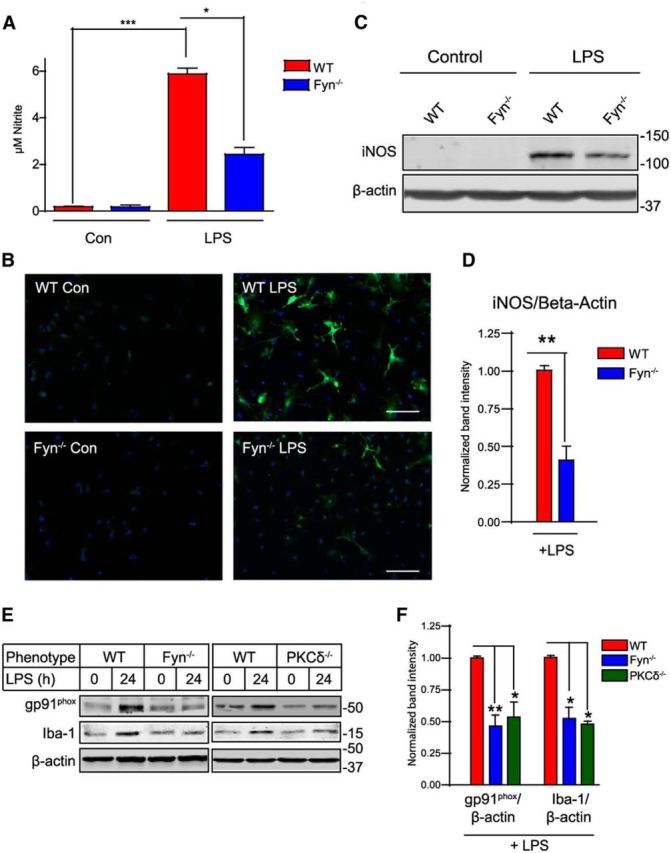
Fyn plays a role in LPS-induced iNOS expression, nitrite production, and neuroinflammatory marker expression. A, Griess nitrite measurement assay demonstrated that LPS-induced nitrite production was reduced in Fyn−/− microglia. *p < 0.05. ***p < 0.001. B–D, Diminished iNOS expression in LPS-treated Fyn−/− microglia. **p < 0.01. Scale bar, 100 μm. E, F, Reduced gp91phox and Iba-1 expression in LPS-treated Fyn−/− and PKCδ−/− microglia, as shown by immunoblotting analysis. *p < 0.05. **p < 0.01.
Fyn−/− and PKCδ−/− mice are resistant to LPS- and MPTP-induced neuroinflammatory responses
To extend our findings from isolated primary microglia to in vivo animal models of neuroinflammation, we first used the LPS model, which has previously been used to evoke neuroinflammatory responses in vivo (Choi et al., 2007; Qin et al., 2007). Wild-type (PKCδ+/+ and Fyn+/+), PKCδ−/−, and Fyn−/− mice were injected with 5 mg/kg LPS or PBS and were killed 3 h later. Striatal mRNA contents of the proinflammatory cytokines pro-IL-1β and TNFα were determined by qRT-PCR. The levels of cytokine induction were almost identical in both wild-type groups, and we thus pooled the results. Systemic LPS administration strongly increased the levels of pro-IL-1β and TNFα transcripts in wild-type striata, but not in Fyn−/− and PKCδ−/− striata (Fig. 8A). To further establish the role of Fyn relevant to PD-associated neuroinflammation, we used the well-known Parkinsonian toxicant MPTP. We subjected wild-type and Fyn−/− mice to an acute MPTP regimen (4 × 18 mg/kg, 2 h apart) and collected their brains for immunohistochemical analysis 24 h after the final MPTP injection. This acute MPTP model has been widely adopted for studying the neuroinflammatory response in the nigrostriatal pathway because maximal microglial activation occurs 24–48 h after the MPTP challenge (Wu et al., 2002, 2003; Sriram et al., 2006; Hirsch and Hunot, 2009). Following the MPTP challenge, successive 30 μm ventral midbrain sections from Fyn+/+ and Fyn−/− mice were stained for the microglial marker Iba-1, and then microglial morphology was quantified using a recently well-established morphometric rating scale as discussed by others (Lastres-Becker et al., 2012). Representations of Type A-D microglial phenotype are provided in Figure 8B. Treating Fyn+/+ mice with the acute MPTP regimen increased Iba-1 expression and discernibly shifted microglial morphology from its typical ramified state to its more amoeboid, activated morphology. After MPTP administration, significantly fewer Type A and more Type B and C microglia were observed in the Fyn+/+ SN, but this shift in microglial morphology was not apparent in the Fyn−/− mice (Fig. 8C,D). We also determined the induction of the NADPH oxidase component gp91phox in MPTP animal model of neuroinflammation. Immunoblotting analysis revealed that MPTP increased expression of gp91phox in WT but not in Fyn−/− ventral midbrain tissues (Fig. 8E,F). Overall, these results confirm that our in vitro data translate well to animal models of neuroinflammation.
Figure 8.
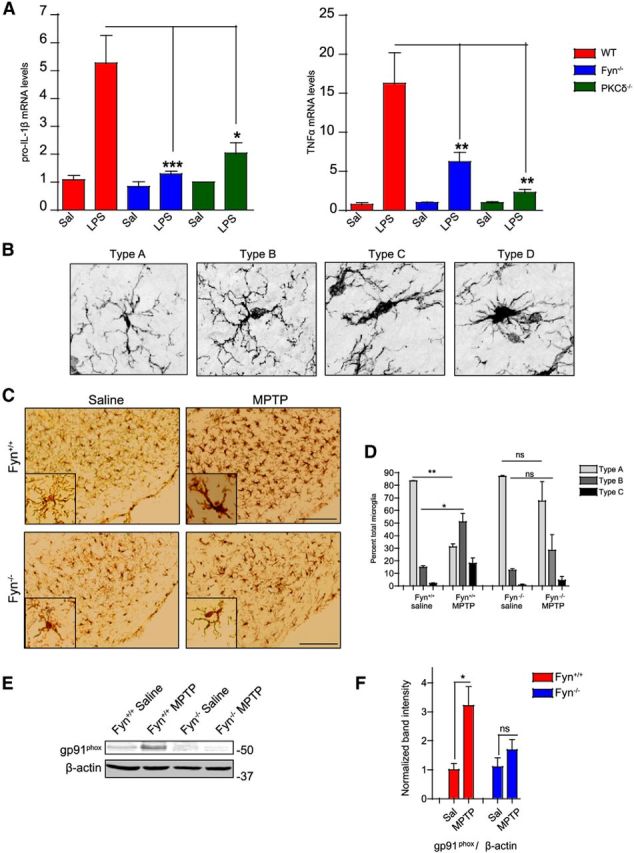
Fyn−/− and PKCδ−/− mice are resistant to LPS- and MPTP-induced neuroinflammatory responses. A, Wild-type, PKCδ−/−, and Fyn−/− mice were injected intraperitoneally with 5 mg/kg LPS for 3 h. Striatal cytokine mRNA levels, assessed by qRT-PCR, showed significantly reduced induction of pro-IL-1β and TNFα mRNA levels in PKCδ−/− and Fyn−/− mice in contrast to wild-type mice. *p < 0.05. **p < 0.01. ***p < 0.001. B, The transitional stages of microglial activation, from ramified (inactivated, Type A) to amoeboid (activated, Types B-D), are shown by representative images. C, D, Iba-1-DAB immunohistochemistry in MPTP-injected Fyn−/− and wild-type ventral midbrain sections demonstrated nigral microgliosis, assessed by quantification of microglial morphology, in the wild-type, but not the Fyn−/− sections. Scale bar, 75 μm. *p < 0.05. **p < 0.01. ns, Not significant. E, F, Fyn−/− mice showed diminished induction of the proinflammatory marker gp91phox in ventral midbrain lysates following the acute MPTP regimen. *p < 0.05.
Fyn−/− and PKCδ−/− mice are protected against 6-OHDA-induced nigrostriatal dopaminergic neuronal deficits and microgliosis
The 6-OHDA mouse model has recently been shown to elicit a neuroinflammatory response and neurodegeneration in the nigrostriatal dopaminergic system (Stott and Barker, 2014). While studying the role of Fyn in dopamine D1 receptor agonist-induced redistribution of NMDA receptor subunits, it was serendipitously discovered that Fyn−/− mice were remarkably resistant to 6-OHDA-induced behavioral deficits and striatal TH loss (Dunah et al., 2004). Fyn+/+ and Fyn−/− mice, injected unilaterally with 6-OHDA (Fig. 9B), were killed 9 d after treatment because mice at this treatment stage concurrently exhibit fewer striatal dopaminergic terminals, significantly fewer TH-positive cells in the SN, and microgliosis within the SN (Stott and Barker, 2014). Fyn−/− mice were more resistant to 6-OHDA-induced striatal nerve terminal degeneration relative to Fyn+/+ mice (Fig. 9A,C). We also show in our studies that 6-OHDA induced massive gliosis coupled with dopaminergic neuronal loss (Fig. 9D). However, Fyn−/− mice show both greater survival of nigral dopaminergic neurons and a diminished neuroinflammatory microglial response.
Figure 9.

Fyn−/− mice are protected against 6-OHDA-induced nigrostriatal dopaminergic neuronal deficits and microgliosis. A, TH-DAB immunohistochemistry in 6-OHDA-injected Fyn−/− and wild-type mouse striatal sections. Scale bar, 1000 μm. B, Schematic diagram of a coronal section through the mouse striatum at the level of the injection. C, Significant preservation of 6-OHDA-induced degeneration of dopaminergic terminals is seen in the Fyn−/− mice in contrast to wild-type mice. **p < 0.01. ***p < 0.001. D, Immunofluorescence staining of 6-OHDA-injected Fyn−/− and wild-type ventral midbrain sections reveals diminished microgliosis and concomitant nigral neuroprotection in Fyn−/− mice after 6-OHDA administration, in contrast to the massive microgliosis and nigral dopaminergic neuronal death observed in the wild-type mice. Scale bar, 200 μm.
In the next set of in vivo experiments, we checked whether PKCδ−/− mice were also resistant to 6-OHDA-induced nigral microgliosis and dopaminergic neuronal loss. PKCδ+/+ and PKCδ−/− mice were injected unilaterally with 6-OHDA for 9 d, and DAB-TH immunostaining was performed on striatal sections as described above (Fig. 10B). Similar to Fyn−/− mice, PKCδ−/− mice showed reduced striatal TH loss following 6-OHDA treatment (Fig. 10A,C). We also assessed nigral microgliosis by double-staining ventral midbrain sections for TH and Iba-1. As shown in Figure 10D, E, PKCδ+/+ mice showed less TH-positive neuronal staining in the SN along with significantly more microgliosis on the ipsilateral side than on the contralateral side; however, the PKCδ−/− mice showed a marked resistance to 6-OHDA-induced nigral TH loss as well as microgliosis. Thus, results from both Fyn and PKCδ knock-out models of 6-OHDA neurotoxicity confirm the role of the Fyn-PKCδ signaling axis in a neuroinflammatory response in the nigrostriatal dopaminergic system.
Figure 10.
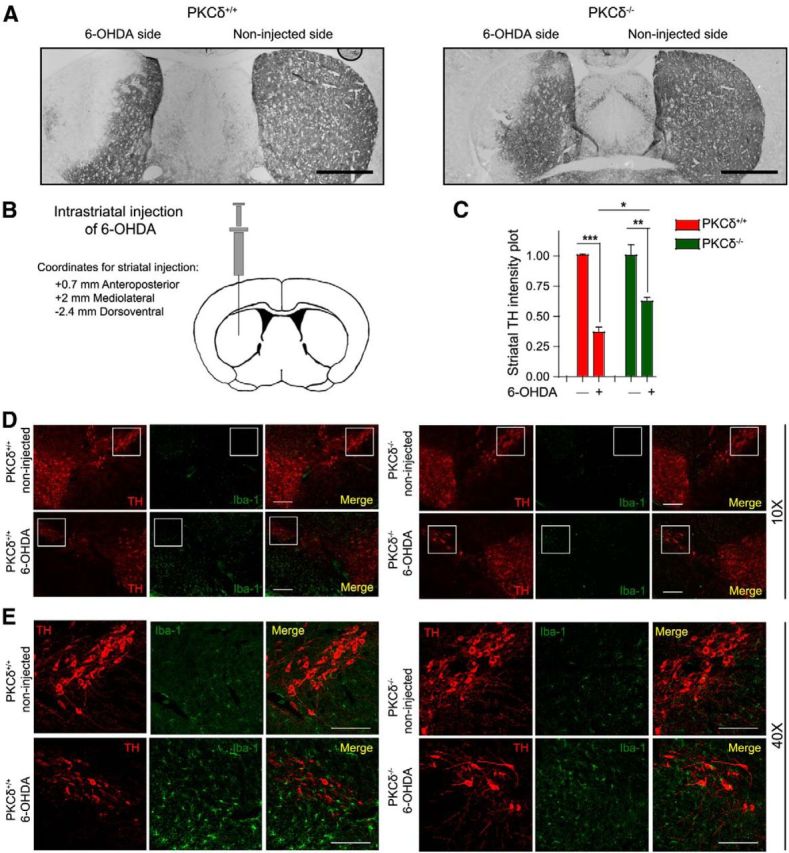
PKCδ−/− mice are resistant to 6-OHDA-induced nigrostriatal dopaminergic neuronal deficits and microgliosis. A, TH-DAB immunohistochemistry in 6-OHDA-injected PKCδ−/− and wild-type mouse striatal sections. Scale bar, 1000 μm. B, Schematic diagram of a coronal section through the mouse striatum at the level of the injection. C, Significant preservation of dopaminergic terminals is seen in the 6-OHDA-treated PKCδ−/− mice in contrast to wild-type mice. *p < 0.05. **p < 0.01. ***p < 0.001. D, Immunofluorescence staining of 6-OHDA-injected PKCδ−/− and wild-type ventral midbrain sections reveals reduced nigral TH degeneration and microgliosis in PKCδ−/− mice after 6-OHDA administration, in contrast to the wild-type mice. Scale bar, 200 μm. E, High-magnification image of 6-OHDA-injected PKCδ−/− and wild-type ventral midbrain sections. Scale bar, 50 μm.
Diminished 6-OHDA-induced glial neuronal contact formation in the Fyn−/− SN
Recently, it was demonstrated that treating mice with MPTP rapidly increased the number of microglial-neuronal appositions, termed gliapses (Barcia et al., 2012). These contacts preceded neuronal phagocytosis by the microglia. Similar appositions between microglia and dopaminergic neurons were demonstrated in the 6-OHDA model, with evidence suggesting that microglial cells actually phagocytized neurons (Virgone-Carlotta et al., 2013), which has been postulated to occur if the neurons are dysfunctional. Our confocal high-magnification Z-stack image analysis (Imaris software) revealed a sharply increased number of microglial-neuronal contacts formed in the Fyn+/+ SN after 6-OHDA treatment as indicated in Fig. 11A, B, F (arrowhead). The 3D reconstructions of the respective stacks demonstrating contacts between dopaminergic neurons and microglia are shown adjacent to the original images (Fig. 11B). The number of gliapses per SN dopaminergic neuron was dramatically reduced in the 6-OHDA-injected Fyn−/− mice (Fig. 11C,D,F). Typical contacts formed between microglial processes and dopaminergic neuronal cell bodies (termed Process-Body, or Pr-B contacts), and those formed between the microglial cell body and the dopaminergic neuronal cell body (Body-Body, or B-B contacts), are shown in Figure 11E. Image analysis involving optical slices through the Z-plane allowed us to both easily count gliapses and visualize actual engulfment events. Representative (Fig. 11B,D) gliapses between a dopaminergic neuron and a microglial cell in the SN of 6-OHDA-injected Fyn+/+ and Fyn−/− mice reveal a conspicuous reduction in the number of gliapses per neuron. Collectively, our confocal imaging results demonstrate that Fyn plays a key role in activation of microglial morphological changes in vivo during inflammatory insults in nigrostriatal system.
Figure 11.
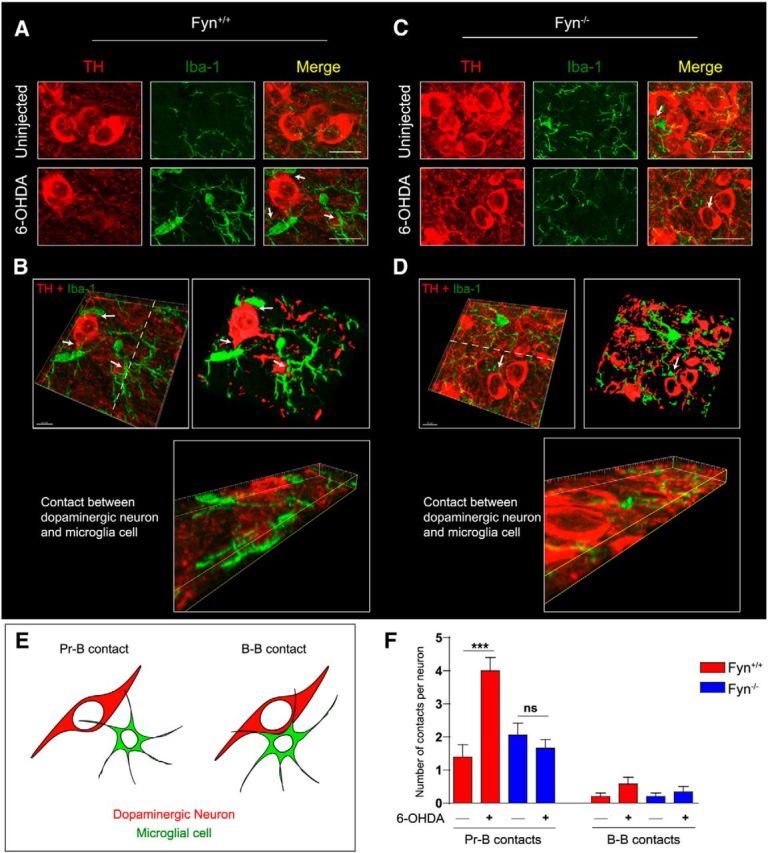
Diminished 6-OHDA-induced glial-neuronal contact (gliapse) formation in the Fyn−/− SN. A, C, Confocal Z-stack maximum projection image analysis of ventral midbrain sections reveals a strongly increased number of microglial-neuronal contacts and appositions upon 6-OHDA treatment of Fyn+/+ but not Fyn−/− mice. Scale bar, 12 μm. B, D, Confocal Z-stack images were rotated and optically sectioned along the Z-plane using Imaris software, allowing easy visualization of gliapse formation. Scale bar, 10 μm. E, Diagrams of Process-Body (Pr-B) and Body-Body (B-B) gliapses formed between dopaminergic neurons and microglia. F, Fyn ventral midbrain sections revealed significantly fewer gliapses formed per dopaminergic neuron in the SN. ***p < 0.001. ns, Not significant.
Prolonged inflammogen stimulation causes Fyn induction upon microglial activation
Thus far, our results demonstrated that short-term treatment of microglial cells with LPS and TNFα brings about an increase in Fyn activity, but not its expression. Strikingly, we discovered that prolonged treatment (12–24 h) of microglia with LPS or TNFα actually resulted in increased Fyn expression, evidenced by Western blot and immunocytochemistry (Fig. 12A–C). To confirm whether this is really due to induction of Fyn protein or increased protein stability, we performed qRT-PCR for Fyn mRNA expression in control and LPS-treated microglial cells. The result showed that treatment of microglia with LPS for 12 h brought about an increase in Fyn transcript levels (Fig. 12D). We also evaluated the effects of prolonged LPS treatment on Fyn promoter activity. For this, we transiently transfected primary microglia with a dual-luciferase Fyn reporter construct containing the 3.1 kb Fyn promoter fragment. LPS treatment significantly increased Fyn promoter activity (Fig. 12E), indicating strongly that Fyn is transcriptionally induced in microglial cells after prolonged inflammogen administration. To further examine whether LPS upregulates Fyn mRNA expression, we injected wild-type mice with a single dose of LPS (5 mg/kg, i.p.) and evaluated the Fyn mRNA expression by qRT-PCR analysis. As shown in Figure 12F, administration of LPS also induced Fyn transcript levels in the striatum. Together, these data suggest that prolonged LPS exposure induces Fyn gene upregulation in microglia, indicating that Fyn may have a sustained role in chronic neuroinflammatory processes.
Figure 12.

Prolonged inflammogen stimulation induces Fyn upon microglial activation. A, B, Stimulation of primary microglia with LPS for 12 h and TNFα for 24 h increased Fyn expression, as evidenced by Western blotting. *p < 0.05. C, Immunohistochemistry analysis of Fyn expression. Scale bar, 20 μm. D, qRT-PCR analysis of Fyn mRNA levels in LPS-stimulated primary microglia and BV2 microglia revealed induction of Fyn at the message level. *p < 0.05. **p < 0.01. E, Induction of Fyn promoter activity in primary microglia following LPS activation of wild-type primary microglia. *p < 0.05. F, Increased striatal Fyn mRNA levels were seen in the Fyn+/+ mice injected intraperitoneally with LPS (5 mg/kg) for 12 h, as assessed by qRT-PCR. **p < 0.01.
Discussion
Evidence from experimental models and human PD postmortem studies strongly implicates the microglia-mediated inflammatory response as a major driver in the progression of PD; however, the key upstream cell signaling mechanisms that govern the neuroinflammatory processes have yet to be elucidated. Our results obtained from both cell culture and animal models provide novel insight into the role of the Fyn-PKCδ signaling cascade in regulating microglia-mediated neuroinflammation as related to PD pathogenesis. We have demonstrated dual regulation of proneuroinflammatory responses in microglia involving post-translational tyrosine phosphorylation of Fyn at its activation loop during the early stages of an inflammatory insult as well as transcriptional upregulation of Fyn upon prolonged exposure to proinflammatory stimuli. We have also shown that Fyn serves as a major upstream signaling molecule that works in concert with PKCδ to influence MAP kinase downstream and the NFκB proinflammatory cascade. Collectively, our study provides novel and significant insight into the proinflammatory function of Fyn-PKCδ signaling in PD models; and to the best of our knowledge, we are the first to discern this key signaling cascade that is relevant to microglia-mediated neuroinflammation in the nigrostriatal dopaminergic system.
We demonstrate that both the tyrosine kinase Fyn and the serine/threonine kinase PKCδ are differentially expressed in microglia and astrocytes (Fig. 1). No prior comparative data are available on Fyn and PKCδ expression in primary microglia. Although the roles of Src family kinases in TLR signaling are being identified, most studies have used peripheral immune and nonimmune cells to determine Src kinase signaling. For example, multiple Src family kinases were activated by LPS in human lung microvascular endothelial cells (Gong et al., 2008). The activation of Src kinases mediated by TLR agonists depends on CD14, TLR2, and TLR4 (Reed-Geaghan et al., 2009), and Fyn has been shown to be associated with TLR2 in TLR2-overexpressing HEK293 cells (Finberg et al., 2012). Peritoneal macrophages have often been used as putative substitutes for brain microglia; Fyn contributes to CD36-mediated signaling responses upon Aβ1–42 stimulation of macrophages (Moore et al., 2002). Of note, the authors reported unaltered LPS-induced MAP kinase activation in Fyn−/− peritoneal macrophages compared with WT macrophages. These apparent discrepancies may be attributed to the inherent differences between the microglial and macrophage gene expression profiles (Hickman et al., 2013). Many studies have used the p-Y416 Src family kinase antibody as a direct indicator of Fyn activation, without using immunoprecipitation or Fyn−/− primary microglia as confirmatory tools to establish Fyn activation. In the present study, we demonstrate that Fyn is rapidly activated in primary microglia within 15–30 min of exposure to inflammogens (Fig. 2A–F). Immunoprecipitation studies and experiments with Fyn−/− microglia clearly confirmed that Fyn kinase is specifically activated during LPS and TNFα stimulation. LPS and TNFα activate microglia/macrophages via TLR4 and TNFα receptor 1 (TNFR1) signaling, respectively (Olson and Miller, 2004; Parameswaran and Patial, 2010). Importantly, our study reveals that Fyn is a common signaling conduit in both TLR- and TNFR1-mediated signaling because the TLR antagonist IAXO-101 and the TNFα signaling antagonist etanercept attenuated Fyn activation (Fig. 2G). Immunocytochemistry analysis revealed that activated Fyn primarily localized to the microglial cell membrane. Although the functional relevance of this localization is not presently known, it is possible that movement of activated Fyn to the microglial membrane may regulate cell migration and cytokine release. Our results with the LPS mouse model provide in vivo evidence for rapid Fyn activation in the ventral midbrain region during inflammatory insults (Fig. 2I).
Our group has previously shown that PKCδ kinase proteolytic activation promotes oxidative stress-induced proapoptotic signaling pathways in dopaminergic neuronal cells (Kaul et al., 2003; Zhang et al., 2007; Jin et al., 2011a, b). Recently, it was demonstrated that PKCδ is proteolytically cleaved by caspase-3 in LPS-treated BV2 cells (Burguillos et al., 2011). In the present study, we demonstrate that activated Fyn associates with PKCδ to phosphorylate the Y311 site, resulting in increased PKCδ kinase activity (Fig. 3). To the best of our knowledge, we are the first group to show the assembly of the Fyn-PKCδ signaling complex in microglial cells during proinflammatory conditions.
MAP kinase activation is necessary for cytokine production in various immune cell types, including microglia (El Benna et al., 1996; Koistinaho and Koistinaho, 2002). We demonstrate that Fyn-PKCδ signaling contributes to MAP kinase phosphorylation during microglial activation. Both LPS and TNFα stimulations rapidly activated the p38 and ERK MAP kinases in WT, but to a significantly lesser extent in the Fyn−/− and PKCδ−/− microglia (Fig. 4), indicating that Fyn-PKCδ signaling lies upstream of MAP kinase in microglia. Given that p38 is a prominent MAP kinase associated with the inflammatory cascade, our results suggest that Fyn and PKCδ are key upstream regulators of the proinflammatory function of this kinase. The downstream events of MAP kinase activation include NFκB signaling, which plays a cardinal role in eliciting proinflammatory responses in microglia. Selective inhibition of NFκB signaling has also proved beneficial in vitro as well as in an experimental mouse model of PD (Ghosh et al., 2007). We show here that IκBα degradation and p65-NFκB nuclear translocation were diminished in Fyn−/− microglia stimulated with LPS or TNFα (Fig. 5), lending credence to the hypothesis that upstream Fyn signaling contributes to NFκB pathway activation in microglia. To our knowledge, the role of Fyn signaling in NFκB-mediated proinflammatory signaling in microglia has never been explored. Fyn has been shown to contribute to anaphylaxis inducer DNP36-HSA mediated NFκB activation in mast cells (Gomez et al., 2005). More recently, Fyn was shown to mediate the nuclear translocation of p65-NFκB downstream of NKG2D and CD137 activation in natural killer cells, using a signaling mechanism dependent on adhesion- and degranulation-promoting adaptor protein (ADAP) (Rajasekaran et al., 2013). This signaling pathway is almost certainly distinct from the Fyn-dependent microglial activation pathway, evidenced by the fact that ADAP−/− microglia display unaltered proinflammatory responses (Engelmann et al., 2013).
Classical activation of microglia by TLR and TNFR1 agonists produces proinflammatory cytokines and chemokines, which mediate the downstream effects of microglial activation. Recently, we showed that TNFα directly induces dopaminergic neuronal apoptosis (Gordon et al., 2012). In our present study, the induction of the cytokines IL-6, IL-12, and TNFα was all diminished in Fyn−/− and PKCδ−/− microglia compared with wild-type microglia (Fig. 6A). Consistently, genetic knockdown of Fyn via siRNA also resulted in diminished LPS-induced proinflammatory cytokine secretion (Fig. 6B,C). TNFα-mediated production of IL-6 and TNFα was also diminished in Fyn-deficient microglia (Fig. 6D,E). Overexpressing the Fyn Y417A activation loop kinase-deficient mutant construct in BV2 microglial cells also diminished LPS-stimulated cytokine production, implicating that the phosphorylation of tyrosine 417 is critical to the proinflammatory function of Fyn (Fig. 6F,G). Furthermore, we showed that the LPS-induced expression of iNOS and secretion of nitrite were significantly attenuated in the Fyn−/− microglia (Fig. 7A–D). We and several other groups have reported increased expression of the NADPH oxidase component gp91phox, as well as the microglial marker Iba-1 following proinflammatory stimulation of microglia (Gao et al., 2011; Gordon et al., 2011). We demonstrate herein that prolonged LPS stimulation brought about the induction of these neuroinflammatory markers in wild-type, but not in Fyn−/− and PKCδ−/− microglia (Fig. 7E,F).
We extended our in vitro studies to well-characterized animal models of neuroinflammation, wherein a single intraperitoneal injection of LPS in mice increases TNFα in the brain to levels that remain elevated long after serum TNFα levels have returned to normal (Qin et al., 2007). We used this model system to check for LPS-induced striatal proinflammatory cytokine induction in wild-type, Fyn−/−, and PKCδ−/− mice. Strikingly, a single injection of LPS strongly increased WT striatal TNFα and pro-IL-1β mRNA levels; however, the induction of these cytokines was greatly diminished in Fyn−/− and PKCδ−/− striata (Fig. 8A). In addition to the LPS model, we also determined the pro-inflammatory role of Fyn in the well-studied acute MPTP model of neuroinflammation. MPTP induced reactive microgliosis and increased gp91phox expression in the nigra of WT mice, but not in Fyn−/− mice (Fig. 8E,F). Interestingly, a quiescent ramified state of microglial morphology was observed in MPTP-treated Fyn−/− mice, whereas more amoeboid activated microglia were noted in Fyn wild-type mice (Fig. 8C,D). In addition to the MPTP model, we further used the 6-OHDA-induced selective dopaminergic lesion model to validate that ablating Fyn or PKCδ confers resistance to nigrostriatal dopaminergic degeneration and microgliosis (Figs. 9, 10). Together, our results indicate that the Fyn-PKCδ signaling axis plays an important role in mediating the proinflammatory response in both cell culture and animal models of neuroinflammation.
Recent imaging studies have demonstrated the formation of glial-neuronal contacts, called gliapses, formed between dopaminergic neurons and microglia that precede neuron loss in the MPTP model (Barcia et al., 2012, 2013). To determine whether Fyn plays a role in microglial-dopaminergic neuron contact formation, we adopted the 6-OHDA mouse model. The formation of gliapses was described recently in the 6-OHDA model (Virgone-Carlotta et al., 2013). Our results from high-magnification confocal analysis revealed that the formation of gliapses was almost completely blocked in 6-OHDA-injected Fyn−/− mice (Fig. 11). The reduced number of gliapses correlated well with reduced dopaminergic neuronal loss following 6-OHDA administration to the Fyn−/− mice. Lastly, prolonged stimulation of microglial cells with inflammogens strongly elicited an induction in Fyn kinase expression levels (Fig. 12). The aggregated form of α-synuclein, the primary component of PD-associated Lewy bodies, can activate microglia by using CD36- and TLR2-dependent pathways (Su et al., 2008; Kim et al., 2013a). Studies are underway in our laboratory to demonstrate the role that Fyn plays in aggregated α-synuclein-induced neuroinflammatory events.
As summarized in Figure 13, we demonstrate that Fyn activation plays an upstream regulatory role in eliciting proinflammatory signaling following both acute and chronic states of microglia stimulation. We arrived at this conclusion based on various lines of experimental evidence from cell culture, primary culture, and in vivo models using both Fyn and PKCδ knock-out mice. Our mechanistic studies revealed that Fyn serves as a major upstream regulator of proinflammatory signaling involving PKCδ, MAP kinase, and the NFκB pathways. Thus, Fyn could be exploited as a potential signaling node in the development of novel antineuroinflammatory drug candidates for treating PD and other related neurodegenerative diseases with associated microglia-mediated proinflammatory processes.
Figure 13.

Proposed scheme of Fyn-mediated neuroinflammatory signaling pathway in microglia. LPS and TNFα bind to their receptors, TLR4 and TNFR1, respectively, leading to early Fyn activation. Fyn then phosphorylates and activates PKCδ, which leads to the downstream activation of the MAP kinase and NFκB pathways. The p65 component of the NFκB complex enters the nucleus and binds to the promoter of various proinflammatory cytokine genes. Fyn is also upregulated to sustain the heightened inflammatory response during prolonged stimulation of microglia, possibly contributing to progressive neurodegeneration in PD.
Footnotes
This work was supported by National Institutes of Health Grants NS088206, ES10586, and NS78247, the W. Eugene and Linda Lloyd Endowed Chair to A.G.K., and the Dean Endowed Professorship to A.K. We thank Gary Zenitsky for assistance in preparing the manuscript.
A.G.K. and V.A. are shareholders of PK Biosciences Corporation (Ames, Iowa), which is interested in translating mechanistic studies into therapies targeting kinases, including Fyn. The remaining authors have no conflicts of interest.
References
- Anantharam V, Kitazawa M, Wagner J, Kaul S, Kanthasamy AG. Caspase-3-dependent proteolytic cleavage of protein kinase Cδ is essential for oxidative stress-mediated dopaminergic cell death after exposure to methylcyclopentadienyl manganese tricarbonyl. J Neurosci. 2002;22:1738–1751. doi: 10.1523/JNEUROSCI.22-05-01738.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcia C, Ros CM, Annese V, Carrillo-de Sauvage MA, Ros-Bernal F, Gomez A, Yuste JE, Campuzano CM, de Pablos V, Fernandez-Villalba E, Herrero MT. ROCK/Cdc42-mediated microglial motility and gliapse formation lead to phagocytosis of degenerating dopaminergic neurons in vivo. Sci Rep. 2012;2:809. doi: 10.1038/srep00809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcia C, Ros CM, Ros-Bernal F, Gómez A, Annese V, Carrillo-de Sauvage MA, Yuste JE, Campuzano CM, de Pablos V, Fernández-Villalba E, Herrero MT. Persistent phagocytic characteristics of microglia in the substantia nigra of long-term parkinsonian macaques. J Neuroimmunol. 2013;261:60–66. doi: 10.1016/j.jneuroim.2013.05.001. [DOI] [PubMed] [Google Scholar]
- Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- Bohnen NI, Frey KA, Studenski S, Kotagal V, Koeppe RA, Constantine GM, Scott PJ, Albin RL, Müller ML. Extra-nigral pathological conditions are common in Parkinson's disease with freezing of gait: an in vivo positron emission tomography study. Mov Disord. 2014;29:1118–1124. doi: 10.1002/mds.25929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burguillos MA, Deierborg T, Kavanagh E, Persson A, Hajji N, Garcia-Quintanilla A, Cano J, Brundin P, Englund E, Venero JL, Joseph B. Caspase signalling controls microglia activation and neurotoxicity. Nature. 2011;472:319–324. doi: 10.1038/nature09788. [DOI] [PubMed] [Google Scholar]
- Carson MJ. Microglia as liaisons between the immune and central nervous systems: functional implications for multiple sclerosis. Glia. 2002;40:218–231. doi: 10.1002/glia.10145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri KR, Healy DG, Schapira AH. Non-motor symptoms of Parkinson's disease: diagnosis and management. Lancet Neurol. 2006;5:235–245. doi: 10.1016/S1474-4422(06)70373-8. [DOI] [PubMed] [Google Scholar]
- Choi HB, Ryu JK, Kim SU, McLarnon JG. Modulation of the purinergic P2X7 receptor attenuates lipopolysaccharide-mediated microglial activation and neuronal damage in inflamed brain. J Neurosci. 2007;27:4957–4968. doi: 10.1523/JNEUROSCI.5417-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotti A, Benner C, Kerman BE, Gosselin D, Lagier-Tourenne C, Zuccato C, Cattaneo E, Gage FH, Cleveland DW, Glass CK. Mutant Huntingtin promotes autonomous microglia activation via myeloid lineage-determining factors. Nat Neurosci. 2014;17:513–521. doi: 10.1038/nn.3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey N, Crosswell HE, De P, Parsons R, Peng Q, Su JD, Durden DL. The protein phosphatase activity of PTEN regulates SRC family kinases and controls glioma migration. Cancer Res. 2008;68:1862–1871. doi: 10.1158/0008-5472.CAN-07-1182. [DOI] [PubMed] [Google Scholar]
- Dunah AW, Sirianni AC, Fienberg AA, Bastia E, Schwarzschild MA, Standaert DG. Dopamine D1-dependent trafficking of striatal N-methyl-d-aspartate glutamate receptors requires Fyn protein tyrosine kinase but not DARPP-32. Mol Pharmacol. 2004;65:121–129. doi: 10.1124/mol.65.1.121. [DOI] [PubMed] [Google Scholar]
- El Benna J, Faust RP, Johnson JL, Babior BM. Phosphorylation of the respiratory burst oxidase subunit p47phox as determined by two-dimensional phosphopeptide mapping: phosphorylation by protein kinase C, protein kinase A, and a mitogen-activated protein kinase. J Biol Chem. 1996;271:6374–6378. doi: 10.1074/jbc.271.11.6374. [DOI] [PubMed] [Google Scholar]
- Engelmann S, Togni M, Thielitz A, Reichardt P, Kliche S, Reinhold D, Schraven B, Reinhold A. T cell-independent modulation of experimental autoimmune encephalomyelitis in ADAP-deficient mice. J Immunol. 2013;191:4950–4959. doi: 10.4049/jimmunol.1203340. [DOI] [PubMed] [Google Scholar]
- Finberg RW, Yim C, Yan J, Cao LC, Mandell L, Kurt-Jones EA. Phosphorylated Toll-like receptor 2 interacts with Fyn and cross talks with the phosphorylation-independent TLR2-signaling pathway. Open Immunol J. 2012;5:36–45. doi: 10.2174/1874226201205010036. [DOI] [Google Scholar]
- Gao HM, Zhou H, Zhang F, Wilson BC, Kam W, Hong JS. HMGB1 acts on microglia Mac1 to mediate chronic neuroinflammation that drives progressive neurodegeneration. J Neurosci. 2011;31:1081–1092. doi: 10.1523/JNEUROSCI.3732-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Roy A, Liu X, Kordower JH, Mufson EJ, Hartley DM, Ghosh S, Mosley RL, Gendelman HE, Pahan K. Selective inhibition of NF-kappaB activation prevents dopaminergic neuronal loss in a mouse model of Parkinson's disease. Proc Natl Acad Sci U S A. 2007;104:18754–18759. doi: 10.1073/pnas.0704908104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Chandran K, Kalivendi SV, Joseph J, Antholine WE, Hillard CJ, Kanthasamy A, Kalyanaraman B. Neuroprotection by a mitochondria-targeted drug in a Parkinson's disease model. Free Radic Biol Med. 2010;49:1674–1684. doi: 10.1016/j.freeradbiomed.2010.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Saminathan H, Kanthasamy A, Anantharam V, Jin H, Sondarva G, Harischandra DS, Qian Z, Rana A, Kanthasamy AG. The peptidyl-prolyl isomerase Pin1 up-regulation and proapoptotic function in dopaminergic neurons: relevance to the pathogenesis of Parkinson disease. J Biol Chem. 2013;288:21955–21971. doi: 10.1074/jbc.M112.444224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140:918–934. doi: 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez G, Gonzalez-Espinosa C, Odom S, Baez G, Cid ME, Ryan JJ, Rivera J. Impaired FcϵRI-dependent gene expression and defective eicosanoid and cytokine production as a consequence of Fyn deficiency in mast cells. J Immunol. 2005;175:7602–7610. doi: 10.4049/jimmunol.175.11.7602. [DOI] [PubMed] [Google Scholar]
- Gong P, Angelini DJ, Yang S, Xia G, Cross AS, Mann D, Bannerman DD, Vogel SN, Goldblum SE. TLR4 signaling is coupled to SRC family kinase activation, tyrosine phosphorylation of zonula adherens proteins, and opening of the paracellular pathway in human lung microvascular endothelia. J Biol Chem. 2008;283:13437–13449. doi: 10.1074/jbc.M707986200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon R, Hogan CE, Neal ML, Anantharam V, Kanthasamy AG, Kanthasamy A. A simple magnetic separation method for high-yield isolation of pure primary microglia. J Neurosci Methods. 2011;194:287–296. doi: 10.1016/j.jneumeth.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon R, Anantharam V, Kanthasamy AG, Kanthasamy A. Proteolytic activation of proapoptotic kinase protein kinase Cδ by tumor necrosis factor alpha death receptor signaling in dopaminergic neurons during neuroinflammation. J Neuroinflammation. 2012;9:82. doi: 10.1186/1742-2094-9-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harischandra DS, Kondru N, Martin DP, Kanthasamy A, Jin H, Anantharam V, Kanthasamy AG. Role of proteolytic activation of protein kinase Cδ in the pathogenesis of prion disease. Prion. 2014;8:143–153. doi: 10.4161/pri.28369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- Haynes SE, Hollopeter G, Yang G, Kurpius D, Dailey ME, Gan WB, Julius D. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat Neurosci. 2006;9:1512–1519. doi: 10.1038/nn1805. [DOI] [PubMed] [Google Scholar]
- Henn A, Lund S, Hedtjärn M, Schrattenholz A, Pörzgen P, Leist M. The suitability of BV2 cells as alternative model system for primary microglia cultures or for animal experiments examining brain inflammation. ALTEX. 2009;26:83–94. doi: 10.14573/altex.2009.2.83. [DOI] [PubMed] [Google Scholar]
- Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang LC, Means TK, El Khoury J. The microglial sensome revealed by direct RNA sequencing. Nat Neurosci. 2013;16:1896–1905. doi: 10.1038/nn.3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch EC, Hunot S. Neuroinflammation in Parkinson's disease: a target for neuroprotection? Lancet Neurol. 2009;8:382–397. doi: 10.1016/S1474-4422(09)70062-6. [DOI] [PubMed] [Google Scholar]
- Hu X, Zhang D, Pang H, Caudle WM, Li Y, Gao H, Liu Y, Qian L, Wilson B, Di Monte DA, Ali SF, Zhang J, Block ML, Hong JS. Macrophage antigen complex-1 mediates reactive microgliosis and progressive dopaminergic neurodegeneration in the MPTP model of Parkinson's disease. J Immunol. 2008;181:7194–7204. doi: 10.4049/jimmunol.181.10.7194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura K, Hishikawa N, Sawada M, Nagatsu T, Yoshida M, Hashizume Y. Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson's disease brains. Acta Neuropathol. 2003;106:518–526. doi: 10.1007/s00401-003-0766-2. [DOI] [PubMed] [Google Scholar]
- Jenner P, Olanow CW. The pathogenesis of cell death in Parkinson's disease. Neurology. 2006;66:S24–S36. doi: 10.1212/WNL.66.10_suppl_4.S24. [DOI] [PubMed] [Google Scholar]
- Jin H, Kanthasamy A, Anantharam V, Rana A, Kanthasamy AG. Transcriptional regulation of pro-apoptotic protein kinase Cδ: implications for oxidative stress-induced neuronal cell death. J Biol Chem. 2011a;286:19840–19859. doi: 10.1074/jbc.M110.203687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin H, Kanthasamy A, Ghosh A, Yang Y, Anantharam V, Kanthasamy AG. α-Synuclein negatively regulates protein kinase Cδ expression to suppress apoptosis in dopaminergic neurons by reducing p300 histone acetyltransferase activity. J Neurosci. 2011b;31:2035–2051. doi: 10.1523/JNEUROSCI.5634-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin H, Kanthasamy A, Harischandra DS, Kondru N, Ghosh A, Panicker N, Anantharam V, Rana A, Kanthasamy AG. Histone hyperacetylation up-regulates protein kinase Cδ in dopaminergic neurons to induce cell death: relevance to epigenetic mechanisms of neurodegeneration in Parkinson disease. J Biol Chem. 2014;289:34743–34767. doi: 10.1074/jbc.M114.576702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaspar JW, Jaiswal AK. Tyrosine phosphorylation controls nuclear export of Fyn, allowing Nrf2 activation of cytoprotective gene expression. FASEB J. 2011;25:1076–1087. doi: 10.1096/fj.10-171553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul S, Kanthasamy A, Kitazawa M, Anantharam V, Kanthasamy AG. Caspase-3 dependent proteolytic activation of protein kinase Cδ mediates and regulates 1-methyl-4-phenylpyridinium (MPP+)-induced apoptotic cell death in dopaminergic cells: relevance to oxidative stress in dopaminergic degeneration. Eur J Neurosci. 2003;18:1387–1401. doi: 10.1046/j.1460-9568.2003.02864.x. [DOI] [PubMed] [Google Scholar]
- Kaul S, Anantharam V, Yang Y, Choi CJ, Kanthasamy A, Kanthasamy AG. Tyrosine phosphorylation regulates the proteolytic activation of protein kinase Cδ in dopaminergic neuronal cells. J Biol Chem. 2005;280:28721–28730. doi: 10.1074/jbc.M501092200. [DOI] [PubMed] [Google Scholar]
- Kim C, Ho DH, Suk JE, You S, Michael S, Kang J, Joong Lee S, Masliah E, Hwang D, Lee HJ, Lee SJ. Neuron-released oligomeric alpha-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun. 2013a;4:1562. doi: 10.1038/ncomms2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Choi DJ, Jeong HK, Kim J, Kim DW, Choi SY, Park SM, Suh YH, Jou I, Joe EH. DJ-1 facilitates the interaction between STAT1 and its phosphatase, SHP-1, in brain microglia and astrocytes: a novel anti-inflammatory function of DJ-1. Neurobiol Dis. 2013b;60:1–10. doi: 10.1016/j.nbd.2013.08.007. [DOI] [PubMed] [Google Scholar]
- Kim YS, Choi DH, Block ML, Lorenzl S, Yang L, Kim YJ, Sugama S, Cho BP, Hwang O, Browne SE, Kim SY, Hong JS, Beal MF, Joh TH. A pivotal role of matrix metalloproteinase-3 activity in dopaminergic neuronal degeneration via microglial activation. FASEB J. 2007;21:179–187. doi: 10.1096/fj.06-5865com. [DOI] [PubMed] [Google Scholar]
- Koistinaho M, Koistinaho J. Role of p38 and p44/42 mitogen-activated protein kinases in microglia. Glia. 2002;40:175–183. doi: 10.1002/glia.10151. [DOI] [PubMed] [Google Scholar]
- Kouadir M, Yang L, Tan R, Shi F, Lu Y, Zhang S, Yin X, Zhou X, Zhao D. CD36 participates in PrP106–126-induced activation of microglia. PLoS One. 2012;7:e30756. doi: 10.1371/journal.pone.0030756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Ab1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson M, Sherman MA, Amar F, Nuvolone M, Schneider JA, Bennett DA, Aguzzi A, Lesné SE. The complex PrP(c)-Fyn couples human oligomeric Abeta with pathological tau changes in Alzheimer's disease. J Neurosci. 2012;32:16857–16871. doi: 10.1523/JNEUROSCI.1858-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Lastres-Becker I, Ulusoy A, Innamorato NG, Sahin G, Rábano A, Kirik D, Cuadrado A. α-Synuclein expression and Nrf2 deficiency cooperate to aggravate protein aggregation, neuronal death and inflammation in early-stage Parkinson's disease. Hum Mol Genet. 2012;21:3173–3192. doi: 10.1093/hmg/dds143. [DOI] [PubMed] [Google Scholar]
- Latchoumycandane C, Anantharam V, Jin H, Kanthasamy A, Kanthasamy A. Dopaminergic neurotoxicant 6-OHDA induces oxidative damage through proteolytic activation of PKCδ in cell culture and animal models of Parkinson's disease. Toxicol Appl Pharmacol. 2011;256:314–323. doi: 10.1016/j.taap.2011.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Nam Y, Koo JY, Lim D, Park J, Ock J, Kim J, Suk K, Park SB. A small molecule binding HMGB1 and HMGB2 inhibits microglia-mediated neuroinflammation. Nat Chem Biol. 2014;10:1055–1060. doi: 10.1038/nchembio.1669. [DOI] [PubMed] [Google Scholar]
- Levy OA, Malagelada C, Greene LA. Cell death pathways in Parkinson's disease: proximal triggers, distal effectors, and final steps. Apoptosis. 2009;14:478–500. doi: 10.1007/s10495-008-0309-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore KJ, El Khoury J, Medeiros LA, Terada K, Geula C, Luster AD, Freeman MW. A CD36-initiated signaling cascade mediates inflammatory effects of beta-amyloid. J Biol Chem. 2002;277:47373–47379. doi: 10.1074/jbc.M208788200. [DOI] [PubMed] [Google Scholar]
- Olanow CW. The pathogenesis of cell death in Parkinson's disease—2007. Mov Disord. 2007;22(Suppl 17):S335–S342. doi: 10.1002/mds.21675. [DOI] [PubMed] [Google Scholar]
- Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol. 2004;173:3916–3924. doi: 10.4049/jimmunol.173.6.3916. [DOI] [PubMed] [Google Scholar]
- Palacios EH, Weiss A. Function of the Src-family kinases, Lck and Fyn, in T-cell development and activation. Oncogene. 2004;23:7990–8000. doi: 10.1038/sj.onc.1208074. [DOI] [PubMed] [Google Scholar]
- Parameswaran N, Patial S. Tumor necrosis factor-α signaling in macrophages. Crit Rev Eukaryot Gene Expr. 2010;20:87–103. doi: 10.1615/CritRevEukarGeneExpr.v20.i2.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przedborski S. Pathogenesis of nigral cell death in Parkinson's disease. Parkinsonism Relat Disord. 2005;11(Suppl 1):S3–S7. doi: 10.1016/j.parkreldis.2004.10.012. [DOI] [PubMed] [Google Scholar]
- Przedborski S, Tieu K, Perier C, Vila M. MPTP as a mitochondrial neurotoxic model of Parkinson's disease. J Bioenerg Biomembr. 2004;36:375–379. doi: 10.1023/B:JOBB.0000041771.66775.d5. [DOI] [PubMed] [Google Scholar]
- Puschmann A. Monogenic Parkinson's disease and parkinsonism: clinical phenotypes and frequencies of known mutations. Parkinsonism Relat Disord. 2013;19:407–415. doi: 10.1016/j.parkreldis.2013.01.020. [DOI] [PubMed] [Google Scholar]
- Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS, Knapp DJ, Crews FT. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia. 2007;55:453–462. doi: 10.1002/glia.20467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajasekaran K, Kumar P, Schuldt KM, Peterson EJ, Vanhaesebroeck B, Dixit V, Thakar MS, Malarkannan S. Signaling by Fyn-ADAP via the Carma1-Bcl-10-MAP3K7 signalosome exclusively regulates inflammatory cytokine production in NK cells. Nat Immunol. 2013;14:1127–1136. doi: 10.1038/ni.2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed-Geaghan EG, Savage JC, Hise AG, Landreth GE. CD14 and toll-like receptors 2 and 4 are required for fibrillar Aβ-stimulated microglial activation. J Neurosci. 2009;29:11982–11992. doi: 10.1523/JNEUROSCI.3158-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saminathan H, Asaithambi A, Anantharam V, Kanthasamy AG, Kanthasamy A. Environmental neurotoxic pesticide dieldrin activates a non receptor tyrosine kinase to promote PKCδ-mediated dopaminergic apoptosis in a dopaminergic neuronal cell model. Neurotoxicology. 2011;32:567–577. doi: 10.1016/j.neuro.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperber BR, Boyle-Walsh EA, Engleka MJ, Gadue P, Peterson AC, Stein PL, Scherer SS, McMorris FA. A unique role for Fyn in CNS myelination. J Neurosci. 2001;21:2039–2047. doi: 10.1523/JNEUROSCI.21-06-02039.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriram K, Matheson JM, Benkovic SA, Miller DB, Luster MI, O'Callaghan JP. Deficiency of TNF receptors suppresses microglial activation and alters the susceptibility of brain regions to MPTP-induced neurotoxicity: role of TNF-alpha. FASEB J. 2006;20:670–682. doi: 10.1096/fj.05-5106com. [DOI] [PubMed] [Google Scholar]
- Steinberg SF. Distinctive activation mechanisms and functions for protein kinase Cδ. Biochem J. 2004;384:449–459. doi: 10.1042/BJ20040704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stott SR, Barker RA. Time course of dopamine neuron loss and glial response in the 6-OHDA striatal mouse model of Parkinson's disease. Eur J Neurosci. 2014;39:1042–1056. doi: 10.1111/ejn.12459. [DOI] [PubMed] [Google Scholar]
- Stuart LM, Bell SA, Stewart CR, Silver JM, Richard J, Goss JL, Tseng AA, Zhang A, El Khoury JB, Moore KJ. CD36 signals to the actin cytoskeleton and regulates microglial migration via a p130Cas complex. J Biol Chem. 2007;282:27392–27401. doi: 10.1074/jbc.M702887200. [DOI] [PubMed] [Google Scholar]
- Su X, Maguire-Zeiss KA, Giuliano R, Prifti L, Venkatesh K, Federoff HJ. Synuclein activates microglia in a model of Parkinson's disease. Neurobiol Aging. 2008;29:1690–1701. doi: 10.1016/j.neurobiolaging.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tansey MG, Goldberg MS. Neuroinflammation in Parkinson's disease: its role in neuronal death and implications for therapeutic intervention. Neurobiol Dis. 2010;37:510–518. doi: 10.1016/j.nbd.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Um JW, Nygaard HB, Heiss JK, Kostylev MA, Stagi M, Vortmeyer A, Wisniewski T, Gunther EC, Strittmatter SM. Alzheimer amyloid-beta oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat Neurosci. 2012;15:1227–1235. doi: 10.1038/nn.3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virgone-Carlotta A, Uhlrich J, Akram MN, Ressnikoff D, Chrétien F, Domenget C, Gherardi R, Despars G, Jurdic P, Honnorat J, Nataf S, Touret M. Mapping and kinetics of microglia/neuron cell-to-cell contacts in the 6-OHDA murine model of Parkinson's disease. Glia. 2013;61:1645–1658. doi: 10.1002/glia.22546. [DOI] [PubMed] [Google Scholar]
- Wake H, Lee PR, Fields RD. Control of local protein synthesis and initial events in myelination by action potentials. Science. 2011;333:1647–1651. doi: 10.1126/science.1206998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu DC, Jackson-Lewis V, Vila M, Tieu K, Teismann P, Vadseth C, Choi DK, Ischiropoulos H, Przedborski S. Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J Neurosci. 2002;22:1763–1771. doi: 10.1523/JNEUROSCI.22-05-01763.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu DC, Teismann P, Tieu K, Vila M, Jackson-Lewis V, Ischiropoulos H, Przedborski S. NADPH oxidase mediates oxidative stress in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson's disease. Proc Natl Acad Sci U S A. 2003;100:6145–6150. doi: 10.1073/pnas.0937239100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Anantharam V, Kanthasamy A, Kanthasamy AG. Neuroprotective effect of protein kinase C δinhibitor rottlerin in cell culture and animal models of Parkinson's disease. J Pharmacol Exp Ther. 2007;322:913–922. doi: 10.1124/jpet.107.124669. [DOI] [PubMed] [Google Scholar]