

Abstract
Back pain is common in the general population, but only a subgroup of back pain patients develops a disabling chronic pain state. The reasons for this are incompletely understood, but recent evidence implies that both preexisting and pain-related variations in the structure and function of the nervous system may contribute significantly to the development of chronic pain. Here, we addressed the role of striatal dopamine (DA) D2/D3 receptor (D2/D3R) function in chronic non-neuropathic back pain (CNBP) by comparing CNBP patients and healthy controls using PET and the D2/D3R-selective radioligand [11C]raclopride. D2/D3R availability was measured at baseline and during a pain challenge, yielding in vivo measures of receptor availability (binding potential, BPND) and DA release (change in BPND from baseline to activated state). At baseline, CNBP patients demonstrated reductions in D2/D3R BPND in the ventral striatum compared with controls. These reductions were associated with greater positive affect scores and pain tolerance measures. The reductions in D2/D3R BPND were also correlated with μ-opioid receptor BPND and pain-induced endogenous opioid system activation in the amygdala, further associated with measures of positive affect, the affective component of back pain and pain tolerance. During the pain challenge, lower magnitudes of DA release, and therefore D2/D3R activation, were also found in the ventral striatum in the CNBP sample compared with controls. Our results show that CNBP is associated with adaptations in ventral striatal D2/D3R function, which, together with endogenous opioid system function, contribute to the sensory and affective-motivational features of CNBP.
SIGNIFICANCE STATEMENT The neural systems that underlie chronic pain remain poorly understood. Here, using PET, we provide insight into the molecular mechanisms that regulate sensory and affective dimensions of pain in chronic back pain patients. We found that patients with back pain have alterations in brain dopamine function that are associated with measures of pain sensitivity and affective state, but also with brain endogenous opioid system functional measures. These findings suggest that brain dopamine–opioid interactions are involved in the pathophysiology of chronic pain, which has potential therapeutic implications. Our results may also help to explain individual variation in susceptibility to opioid medication misuse and eventual addiction in the context of chronic pain.
Keywords: amygdala, chronic pain, dopamine, endogenous opioid, striatum
Introduction
The neural mechanisms underpinning interindividual variability in the development and clinical course of chronic back pain and associated symptoms are poorly understood. Recently, it has been shown that one of the critical brain regions associated with the development of chronic back pain is the striatum, an area that participates in the formation of coherent behavioral responses by integrating sensorimotor, emotional, and motivational functions (Haber, 2003; Baliki et al., 2010; Baliki et al., 2012). Striatal function is strongly regulated by dopaminergic projections from the midbrain, the activity of which has been linked to pain control mechanisms, including endogenous opioid, μ-opioid receptor (MOR)-mediated antinociception (Morgan and Franklin, 1990; Gear et al., 1999; Schmidt et al., 2002). The effects of striatal dopamine (DA) on pain are thought to be mediated by the DA D-2 receptor (D2R) because striatal administration of selective D2R agonists reduced, and D2R antagonists enhanced, pain responses in animal models of persistent pain (Morgan and Franklin, 1991; Magnusson and Fisher, 2000; Taylor et al., 2003). Furthermore, the development of an experimental chronic pain state in animal models has been found to parallel reductions in both D2R expression and excitatory drive in D2R-expressing neurons in the nucleus accumbens (Chang et al., 2014; Schwartz et al., 2014).
PET imaging studies in healthy humans using [11C]raclopride, a radioligand selective for D2Rs and D3Rs, have shown that striatal D2/D3R availability (binding potential, BPND) at baseline is positively correlated with pain sensitivity (Hagelberg et al., 2002), whereas activation of this neurotransmitter system, assessed as acute reductions in BPND during painful stimulation, is associated with greater pain report in the dorsal striatum and with negative affect and fear ratings in the nucleus accumbens (Scott et al., 2006). In chronic pain patients, increases in baseline striatal D2/D3R BPND were found in orofacial pain (Hagelberg et al., 2003a; Hagelberg et al., 2003b), whereas fibromyalgia (FM) was associated with reductions in baseline D2/D3R BPND (Wood et al., 2007). The study by Wood et al. (2007) also found that, whereas painful stimulation led to significant activation of D2/D3R-mediated neurotransmission in healthy controls (HCs), less activation was observed in the FM sample, suggesting a blunted response of this neurotransmitter system in FM.
Here, we investigated whether patients with chronic non-neuropathic back pain (CNBP) display alterations in striatal D2/D3R BPND and pain-induced activation of DA neurotransmission. We hypothesized that CNBP would be associated with reductions in baseline D2/D3R BPND and in the activation of D2/D3R-mediated neurotransmission during experimental pain in a manner similar to patients with FM, a disorder that is also characterized by chronic musculoskeletal pain but that is typically more widespread (Wood et al., 2007). In addition, based on the known interactions between DA and opioid systems (Chen et al., 1993; Steiner and Gerfen, 1998; Soderman and Unterwald, 2009) and the specific role of MOR neurotransmission in the regulation of clinical pain in CNBP patients (Martikainen et al., 2013), we also investigated whether brain MOR measures at baseline and during painful stimulation are associated with D2/D3R-mediated neurotransmission in CNBP.
Materials and Methods
Subjects.
We compared 16 CNBP patients (9 males and 7 females, age 35 ± 11 years, mean ± SD) with 16 age- and sex-matched HC subjects (9 males and 7 females, age 35 ± 10 years). The patients were part of a larger sample of CNBP patients from which the patient sample of our earlier study was also derived (Martikainen et al., 2013). All participants were right-handed nonsmokers who drank <10 units of alcohol per week, did not perform physical exercise in excess than 1 h/d, and had no history of recreational drug use. The HC subjects had no current or past history of medical illness and were not taking any regular medications.
The CNBP patients were recruited from a university-based pain clinic with the following main inclusion criteria: current average back or neck pain intensity between 3 and 8 on a 0 to 10 visual analog rating scale (VAS) with 0 representing no pain and 10 representing the greatest pain intensity imaginable, pain duration of at least 1 year, no current or previous opioid use within the past year, and no history of psychiatric disease with the exception of mild depressive symptoms not meeting the diagnostic criteria for major depression. The back pain was clinically characterized as nonspecific and patients with spinal cord injury, nonspinal etiology, or radicular symptoms were not included in the study. The pain was limited to the low back in seven patients, to the neck area in three patients, and to larger areas in the back and neck in six patients. The most intense pain was localized in the lower back region in 12/16 patients. CNBP patients had a median pain duration of 6 years (interquartile range: 5), with average pain intensity of 49 ± 21 units on a 0 to 100 VAS scale. The pain varied from 57 ± 19 at worst to 35 ± 18 at best. At the time of PET imaging, back pain intensity and pain unpleasantness were rated at 48 ± 23 and 53 ± 20 (0 to 100 VAS), respectively.
To include CNBP patients representative of those encountered in routine practice, the use of nonopioid analgesic medications was allowed during the study. The most common analgesic medications used by the CNBP patients were nonsteroidal anti-inflammatory drugs and acetaminophen (5/16), muscle relaxants (3/16), selective serotonin reuptake inhibitors (2/16), and gabapentin/pregabalin (2/16). Duloxetine (a serotonin-norepinephrine reuptake inhibitor) was used by one patient and topiramate (an anticonvulsant) was also used by one patient.
All participants provided written informed consent before entering the study. The study protocol was approved by the Institutional Review Board and the Radioactive Drug Research Committee at the University of Michigan.
Neuroimaging methods.
PET scans were acquired with a Siemens HR+ scanner in 3D mode with septa retracted and scatter correction. The reconstructed full width at half-maximum (FWHM) resolution was 5.5 mm in-plane and 5.0 mm axially. Each participant was positioned comfortably in the PET scanner gantry and an antecubital intravenous line was placed in the right arm. Movement during the scan was minimized by using a light forehead restraint. [11C]raclopride was synthesized at high specific activity by the reaction of O-desmethyl raclopride with [11C]methyltriflate. Fifty percent of the [11C]raclopride dose was administered as a bolus and the remaining 50% was administered by continuous infusion for the remainder of the study. The total dose was 16 ± 0.6 mCi (592 ± 24 MBq). Under these conditions, equilibrium conditions are achieved 35 min after tracer administration (Carson et al., 1997). Twenty-eight frames of images were acquired over 90 min with an increasing duration (30 s up to 10 min). The HC subjects and CNBP patients underwent a 90 min PET scan with [11C]raclopride, which included two experimental conditions: a control condition with nonpainful isotonic saline infusion (baseline) and a pain condition with noxious hypertonic 5% saline.
PET images were reconstructed using iterative algorithms (brain mode; Fourier rebinning with ordered subsets-expectation maximization, four iterations, 16 subsets; no smoothing) into a 128 × 128 pixel matrix in a 28.8 cm diameter field of view (FOV). Attenuation correction was performed through a 6 min transmission scan (68Ge source) obtained before the PET study and with iterative reconstruction of the blank/transmission data, which was followed by segmentation of the attenuation image. Before analysis, small head motions were corrected by an automated computer algorithm and the images were coregistered with the same software (Minoshima et al., 1993). Time points were decay corrected during reconstruction of the PET data.
The [11C]raclopride image data were transformed on a voxel-by-voxel basis into two sets of parametric maps: (1) a tracer transport measure (K1 ratio) and (2) a receptor-related measure at equilibrium (distribution volume ratio at equilibrium, DVReq). The DVReq measure was calculated using the ratio of brain activity to activity in the cerebellum (Carson et al., 1997; Watabe et al., 2000) using values obtained from 35–45 min (isotonic infusion) and 60–80 min (hypertonic infusion) after tracer administration. DVReq-1 (f2Bmax/Kd) is the receptor-related measure (D2/D3R availability in vivo or nondisplaceable binding potential, BPND). Bmax is the concentration of receptors and Kd their affinity. The term f2 refers to the concentration of free radiotracer in the extracellular fluid and is considered to represent a constant and very small value. [11C]raclopride BPND reflects binding exclusively to D2 receptors in the dorsal striatum, whereas in the ventral striatum, one-third of the [11C]raclopride BPND may be contributed by D3Rs (Seeman et al., 2006).
Of the 16 CNBP patients who participated in the [11C]raclopride scans, 15 also participated in the PET scans performed with [11C]carfentanil, a radiotracer selective for MORs. [11C]carfentanil PET scans were acquired with the same scanner using identical methods for data acquisition, image reconstruction, and processing. [11C]carfentanil was synthesized at high specific activity by the reaction of [11C]methyliodide and a nonmethyl precursor (Jewett, 2001); 15 ± 1.0 mCi (555 ± 37 MBq) were administered during the scan, with a mass of carfentanil injected of < 0.03 μg/kg per scan. The CNBP patients underwent two 90 min PET scans with [11C]carfentanil, one scan without any intervention for baseline MOR BPND assessment and another for the measurement of MOR activation during painful hypertonic saline infusion, with a preceding nonpainful isotonic saline infusion as a control condition, as described previously (Martikainen et al., 2013). Scan order was randomized and counterbalanced.
[11C]carfentanil PET data were transformed into a tracer transport measure (K1 ratio) and a receptor-related measure (distribution volume ratio, DVR) using modified Logan graphical analysis (Logan et al., 1996). The occipital cortex, an area devoid of MORs, was used as the reference region. The slope of the Logan plot was used for the estimation of the DVR (f2Bmax/Kd + 1; as with [11C]raclopride, f2Bmax/Kd is the measure of receptor availability, MOR BPND).
Anatomical T1-weighted MRI data were acquired on a 3 T scanner (Signa LX; General Electric) using axial 3D inversion recovery-prepared fast-spoiled gradient-recalled acquisition [repetition time (TR) = 9.2 ms, echo time (TE) = 1.9 ms, inversion time = 500 ms, flip angle (FA) = 15°, bandwidth = 16 kHz, number of excitations = 1, 256 × 256 matrix, FOV = 25/26 cm, number of contiguous images = 154, isotropic voxel size = 1 mm3].
MR and PET images were coregistered to each other and to the Montreal Neurological Institute (MNI) stereotactic atlas orientation using Statistical Parametric Mapping software (SPM8; Wellcome Trust Centre for Neuroimaging, London, UK; http://www.fil.ion.ucl.ac.uk/spm/). The D2/D3R and MOR BPND maps were normalized with the deformation field obtained from the normalization of the MR images to the MNI atlas orientation using VBM8 toolbox (http://dbm.neuro.uni-jena.de/vbm/). The accuracy of the coregistration and normalization algorithms was confirmed for each subject individually by comparing the transformed images with each other and with the MNI atlas template. Statistical parametric maps of group differences were generated with SPM8. No global normalization was applied to the data, so the calculations presented are based on absolute BPND estimates. Only regions with specific D2/D3R and MOR BPND were included in the analyses (i.e., voxels with BPND values >0.1). A 6 mm FWHM Gaussian filter was applied to each PET scan to compensate for small residual anatomic variations across subjects and to improve signal-to-noise ratios.
Infusion paradigm.
The infusion paradigm during the PET scan consisted of a nonpainful control condition and a pain condition. In the pain condition, moderate muscle pain was maintained for 20 min after radiotracer administration (45–65 min after the start of the scan) by the infusion of a small amount of 5% hypertonic saline into the left masseter muscle via a computer-controlled closed-loop system (Zhang et al., 1993; Stohler and Kowalski, 1999). This method of sustained painful stimulation during the PET scan allows the determination of acute changes in D2/D3R and MOR BPND associated with the activation of DA and endogenous opioid release, respectively, which are related to the individual assessments of the pain experience (Zubieta et al., 2001; Scott et al., 2006). The feedback mechanism ensures that the pain intensity is standardized across individual subjects and subject groups (Stohler and Kowalski, 1999).
In this study, a computer-controlled pump injected an average volume of 2.1 ± 1.0 ml into the left masseter muscle. The pain level was measured every 15 s by a computer version of a manual 0 to 100 VAS, with the lower and upper bound of the scale marked with numbers 0 and 100, representing the range from “no pain” to “the highest pain intensity imaginable.” Based on the VAS pain reports, the individual infusion requirements were modeled continuously and updated with the aim of keeping the present pain intensity scores at a target of 40 VAS units. The actual average VAS rating in the experiment over the whole subject group was 32 ± 11 units. The potential biases that might be induced by the VAS measurements during PET imaging were taken into account by placing the computer screen and pointing the device in a way that minimized movement during pain reporting and by having an identical rating task during the control condition.
The control condition was an otherwise identical infusion with nonpainful 0.9% isotonic saline administered 5–25 min after the start of the scan. The isotonic saline was infused at the same rate as the average hypertonic infusion and was applied in the right masseter muscle. The volunteers were informed of the experimental conditions, but not of their order or laterality. The first condition of the experiment was always the control condition, which was followed by the pain challenge condition to avoid carryover effects (Scott et al., 2006; Scott et al., 2007). In the data analysis, the magnitude of D2/D3R and MOR system activations during pain (reflecting processes associated with neurotransmitter release, such as competition between radiotracer and endogenous neurotransmitter and potentially neurotransmitter-induced receptor internalization) were defined as the change (reductions) in BPND (ΔBPND) during the hypertonic infusion compared with their respective baselines (Scott et al., 2006; Scott et al., 2008).
Before the scan and after the experimental conditions, subjects completed the Positive and Negative Affect Schedule (PANAS) (Watson and Clark, 1999) and the Profile of Mood States (POMS) questionnaires (McNair et al., 1992). Also before the scan and after the pain challenge, the CNBP patients rated their back pain using a 0–100 VAS of the pain intensity and unpleasantness and the McGill Pain Questionnaire (MPQ) (Melzack and Torgerson, 1971), which uses weighted word descriptors of pain. Separate MPQ and VAS estimates of pain were collected after the pain challenge, rating the experience of experimental (masseter muscle) pain. These measures, together with the momentary pain intensity ratings acquired every 15 s for the 20 min challenge, provided the measures of the individual pain experience during the experimental muscle pain. Individual sustained pain tolerance was measured as the total volume of hypertonic saline (in milliliters) infused to maintain the pain intensity during the experimental period and pain sensitivity was measured as the ratio of average VAS ratings acquired every 15 s divided by the total volume of hypertonic saline infused to maintain pain at the target level. To complement these measures of affective state and sensory and affective-motivational dimensions of pain, information on pain duration was used along with the measures of clinical pain to estimate the total burden of pain at the individual level. Together, these measures, albeit overlapping in some cases, provide profiles of effects on clinical and experimental pain and affective state. These measures were included in the analyses without correction for multiple comparisons to ensure the consistency of the findings and for completeness.
We studied the function of brain DA D2/D3R and MOR systems, not only at baseline (in absence of painful stimulation), but also during experimental pain to gain insight into the functional responses of these neurotransmitter systems during increases in pain signal, mimicking acute exacerbation in clinical pain. Considering the evidence that CNBP is associated with alterations in pain control (Giesecke et al., 2004; O'Neill et al., 2007; Martikainen et al., 2013), painful stimulation might have a differential effect on neurotransmitter activation in HCs and CNBP patients and therefore bias the PET measurements. To prevent the potential impact of altered function of pain systems in the CNBP sample, experimental pain was delivered to an area that was free of spontaneous pain and sensory alterations in all subjects, both in HCs and CNBP patients (the masseter muscle). Further, the individual pain level was standardized across individuals and subject samples, which ensured that all subjects had a similar pain experience during the painful stimulation regardless of individual pain sensitivity.
In the CNBP sample, however, the experimental painful stimulation could theoretically interact with clinical pain by heterosegmental pain-inhibitory mechanisms or attentional modulation (Sowman et al., 2011; Ladouceur et al., 2012). The possible differential impact of painful stimulation in the subject samples could therefore be reflected in the DA responses of the ventral striatum, which is involved in the valuation of emotionally valenced stimuli in relation to expected outcomes also in the setting of chronic pain (Baliki et al., 2010). Nevertheless, in our sample, there was no evidence of any significant improvements in back pain or positive and negative affect ratings during the pain challenge (VAS intensity and unpleasantness scores, p > 0.3; MPQ total scores, p > 0.1; PANAS negative affect scores, p > 0.7). On the contrary, positive affect ratings, as measured with the PANAS, showed significant reductions during painful stimulation in the CNBP sample (t15 = 3.3, p = 0.005) and in HCs (t15 = 4.5, p = 0.0004).
An additional potential source of bias related to the infusion paradigm is that the isotonic infusion always preceded the hypertonic infusion. This was modeled in a manner similar to that described for pharmacological challenges with [11C]raclopride using full equilibrium conditions for this radiotracer (Carson et al., 1997). The order of the infusions was fixed to prevent carryover effects of pain-induced neurotransmitter release and receptor activation on the baseline receptor BPND, as shown previously for intervals <60 min between conditions (Scott et al., 2007). Although the nonrandomized sequence of experimental conditions might still theoretically create a small systematic error in the analysis of neurotransmitter release measures, it is highly unlikely that such error would have a significant impact on the main objective of this study, group effects in striatal DA function.
Data analysis.
We examined the effects of CNBP on receptor BPND by applying a general linear model (GLM) on a voxel-by-voxel basis using SPM8 and MATLAB (The MathWorks). Baseline D2/D3R BPND was compared between patients with CNBP and age- and sex-matched HC subjects using an independent-samples t test. Paired t test was used to assess the sustained pain-induced effects on striatal D2/D3R BPND, which were defined as the change in the D2/D3R BPND measure from the control condition (baseline) to painful hypertonic saline condition. Group × condition interactions in pain-induced D2/D3R activation were determined using a mixed-model ANOVA, with the diagnostic group (HC/CNBP) as the between-subject factor and the change in D2/D3R BPND as the within-subject dependent variable. For all analyses, age was included as a covariate of no interest, whereas for mixed-model ANOVA, we also included the average VAS pain ratings during the hypertonic saline infusion as a covariate. Significant effects were detected using a statistical threshold that controls a type I error rate at p < 0.05 (false discovery rate corrected for multiple comparisons); for regions with specific hypotheses, statistical significance was set at p < 0.005. The numerical values for D2/D3R BPND were extracted from the image data by averaging the values of voxels contained in the area in which significant effects were obtained. Statistical analyses of associations between D2/D3R BPND and clinical measures were performed using SPSS version 19 at p < 0.05. No correction for multiple comparisons was used for these correlations.
To determine whether alterations in D2/D3R functional measures in CNBP would be associated with endogenous opioid-MOR function, a whole-brain voxel-by-voxel regression analysis was performed in SPM8. In this analysis, values for baseline D2/D3R BPND and change in D2/D3R BPND during pain in regions showing significant alterations in the CNBP sample were used as regressors and images of baseline MOR BPND and pain-induced MOR activation (subtraction images representing ΔBPND, i.e., baseline MOR BPND − MOR BPND during painful stimulation) were used as dependent variables. Age and sex were included as covariates of no interest. Our hypothesis was that D2/D3R BPND would be associated with MOR functional measures in the thalamus and amygdala based on the findings of our previous study (Martikainen et al., 2013).
Results
A summary of significant [11C]raclopride PET neuroimaging findings and their relationship to behavioral measures and MOR function is shown in Table 1.
Table 1.
Summary of [11C]raclopride PET neuroimaging findings and their relationship to behavioral measures and MOR function
| Main effects | Region; x, y, z coordinates; Z score; cluster size; BPND change | Correlations |
|||
|---|---|---|---|---|---|
| Measure | CNBP | HC | All subjects | ||
| Reductions in baseline D2/D3R BPND in the CNBP sample | Right ventral striatum, (4,16, 0), Z = 3.7, 400 mm3, 21% | Positive affect at baseline (PANAS) | r = −0.59; p = 0.02* | r = −0.17; p = 0.54 | r = −0.25; p = 0.16 |
| Total volume of hypertonic saline infused (mL) | r = −0.66; p = 0.007** | r = −0.10; p = 0.71 | r = −0.20; p = 0.28 | ||
| Pain sensitivity (VAS/total volume) | r = 0.54; p = 0.048* | r = 0.32; p = 0.25 | r = 0.39; p = 0.04* | ||
| Baseline MOR BPND in the right amygdala, (24, −2, −24), Z = 2.6, 1472 mm3 | r = 0.65; p = 0.009** | ||||
| Baseline MOR BPND in the left amygdala, (−22, −6, −24), Z = 2.6, 592 mm3 | r = 0.55; p = 0.03* | ||||
| Pain-induced MOR activation in the left amygdala, (−20, −6, −20), Z = 3.4, 912 mm3 | r = −0.57; p = 0.03* | ||||
| Pain-induced D2/D3R activation across all subjects (HC, CNBP) | Left caudate nucleus, (−18, 20, 2), Z = 4.8, 1600 mm3, 6% | Pain unpleasantness (VAS) | r = −0.35; p = 0.25 | r = 0.65; p = 0.02* | r = −0.04; p = 0.87 |
| Left putamen, (−30, 4, 4), Z = 4.7, 648 mm3, 6% | |||||
| Right nucleus accumbens, (16, 12, −4), Z = 4.1, 2072 mm3, 5% | |||||
| Reductions in pain-induced D2/D3R activation in the CNBP sample | Right ventral striatum, (6, 14, 2), Z = 3.4, 328 mm3, 130% | ||||
*p < 0.05;
**p < 0.01.
Baseline striatal D2/D3R BPND in chronic back pain
CNBP patients displayed significant reductions in baseline D2/D3R BPND in the right ventral striatum compared with their control sample, with a peak effect localized in the area of ventromedial caudate nucleus/nucleus accumbens [peak MNI coordinates (x, y, z) at (4, 16, 0), cluster size = 400 mm3, Z = 3.7, mean BPND reduction = 21%; Fig. 1]. No significant effects were found in the opposite contrast (HC < CNBP). We did not find any associations between the D2/D3R BPND in the right ventral striatum and clinical pain duration (r = 0.12, p = 0.65) or back pain intensity at the time of PET imaging as measured with MPQ or VAS (p > 0.3). A significant negative correlation was found between D2/D3R BPND in the ventral striatum and positive affect ratings as measured by PANAS both at baseline and after the pain challenge (baseline: r = −0.59, p = 0.02; pain: r = −0.55, p = 0.03). The ratings of negative affect were not associated with D2/D3R BPND (PANAS; p > 0.3). Consistent with those findings, the vigor (positive valence) score of POMS (POMS-vigor) was negatively correlated with D2/D3R BPND (baseline: r = −0.50, p = 0.049; pain: r = −0.52, p = 0.04); however, the total mood disturbance score of POMS (POMS-TMD) (negative valence) was not correlated with D2/D3R BPND (p > 0.3). The D2/D3R BPND in the ventral striatum was also negatively correlated with the total volume of hypertonic saline infusion required to achieve the target pain intensity (r = −0.66, p = 0.007) and positively with the ratio of VAS intensity ratings acquired every 15 s over the infusion volume (r = 0.54, p = 0.048), measures of sustained pain tolerance and sensitivity, respectively.
Figure 1.

Levels of striatal D2/D3R BPND in CNBP patients and HCs. Significant reductions in D2/D3R BPND, compared with HC sample, were observed at baseline in the right ventral striatum. A, Top, Significant region in the ventral striatum and plots of average D2/D3R BPND values for HC subjects (empty circles) and CNBP patients (filled circles). The D2/D3R BPND in this region was negatively correlated with positive affect and pain tolerance measures in CNBP patients (B, bottom).
Activation of striatal D2/D3R-mediated neurotransmission during sustained experimental pain
Consistent with the adaptive pain maintenance system used, no significant differences were observed between the HC and CNBP samples in their average 0–100 VAS ratings acquired every 15 s during the experimental pain challenge (HC: 33 ± 9, CNBP: 32 ± 12; t30 = 0.26, p = 0.80) or experimental pain total MPQ scores (HC: 26 ± 9, CNBP: 22 ± 13; t30 = 1.0, p = 0.32). No statistically significant (p < 0.05) differences were found between the HC and CNBP samples in the indices of pain tolerance and sensitivity, the total volume of hypertonic saline infused (HC: 2.4 ± 1.1 ml, CNBP: 1.9 ± 0.9 ml; t29 = 1.3, p = 0.24), and the average VAS over the total volume of hypertonic saline infused (HC: 21 ± 21, CNBP: 23 ± 20; t29 = 0.28, p = 0.95). There were no significant differences in the affective state measures between HC and CNBP samples before scanning (PANAS positive: F(1,21) = 0.001, p = 0.98; PANAS negative: F(1,21) = 0.78, p = 0.39; POMS-vigor: F(1,21) = 3.9, p = 0.06; POMS-TMD: F(1,21) = 3.2, p = 0.09), after the nonpainful isotonic saline infusion (PANAS positive: F(1,21) = 0.21, p = 0.65; PANAS negative: F(1,21) = 1.2, p = 0.28; POMS-vigor: F(1,21) = 0.51, p = 0.48; POMS-TMD: F(1,21) = 0.79, p = 0.38), or after the pain challenge (PANAS positive: F(1,21) = 0.10, p = 0.75; PANAS negative: F(1,21) = 1.5, p = 0.24; POMS-vigor: F(1,21) = 0.32, p = 0.58; POMS-TMD: F(1,21) = 0.84, p = 0.37).
Significant pain-induced activation of DA D2/D3R neurotransmission (ΔBPND from baseline to pain) was observed bilaterally in the striatum across the two volunteer groups, with significant peaks in the head of the left caudate nucleus [(−18, 20, 2), 1600 mm3, Z = 4.8, mean BPND reduction 6%], left putamen [(−30, 4, 4), 648 mm3, Z = 4.7, mean reduction 6%], and right nucleus accumbens [(16, 12, −4), 2072 mm3, Z = 4.1, mean reduction 5%] (Fig. 2A). The peak in right caudate nucleus (14, 18, 6) showed a trend to statistical significance after correction for multiple comparisons (Z = 4.0; p = 0.06).
Figure 2.

Sustained experimental muscle pain-induced changes in D2/D3R BPND (DA activation) in the striatum, in HC subjects and CNBP patients. In the entire sample, significant D2/D3R activation was observed bilaterally in the ventral and dorsal striatum, with peak effects localized in the left caudate, left putamen, and right nucleus accumbens (A). A significant group × condition interaction in D2/D3R activation was observed in the right ventral striatum, with peak in the right ventral caudate (B). Illustrated on the right are plots of average regional D2/D3R activation in the areas showing significant D2/D3R activation in the entire sample (A) and in the area that showed a significant group × condition interaction (B).
In the HC group, pain-induced D2/D3R activation in the left caudate nucleus was positively correlated with the ratings of experimental pain unpleasantness (r = 0.65, p = 0.02), but not pain intensity or MPQ scores (p > 0.1). No significant associations were found between PANAS scores and striatal D2/D3R activation in these regions (p > 0.2). In the CNBP sample, no associations were found between striatal D2/D3R activation and ratings of clinical and experimental pain or affective state (p > 0.2).
A mixed-model ANOVA was used to examine significant group × condition interactions in experimental pain-induced D2/D3R activation, with the hypothesis that they would be colocalized with the baseline D2/D3R BPND changes in the ventral striatum. This analysis found a significant region in the right ventral striatum in which CNBP patients demonstrated less DA release (ΔBPND) than HC subjects [(6, 14, 2), 328 mm3, Z = 3.4, 130% differential between activation levels; Fig. 2B]. No significant effects were obtained in any other region or for the opposite contrast (HC < CNBP).
No associations were observed between theexperimental pain-induced D2/D3R system activation in this region and reductions in baseline D2/D3R BPND in the same region in the CNBP sample (p > 0.5). In addition, no associations were found between pain-induced ventral striatal D2/D3R system activation and measures of back pain (MPQ: p > 0.2), affect ratings (PANAS positive, negative; POMS-vigor, POMS-TMD: p > 0.2) or measures of pain sensitivity and tolerance (p > 0.6).
Associations between ventral striatal D2/D3R function and endogenous opioid system functional measures
In a previous study, we found that CNBP was associated with increases in baseline MOR BPND in the thalamus bilaterally and reductions in endogenous opioid-MOR activation during painful stimulation in the thalamus bilaterally and the left amygdala (Martikainen et al., 2013). Given the known interactions between the DA and the endogenous opioid systems (Chen et al., 1993; Steiner and Gerfen, 1998; Soderman and Unterwald, 2009), we investigated whether reductions in ventral striatal D2/D3R BPND in CNBP would be associated with alterations in MOR BPND at baseline and MOR system activation during pain.
Using the baseline D2/D3R BPND values in CNBP as regressors, a whole-brain voxel-by-voxel analysis found a significant positive correlation between ventral striatal D2/D3R BPND and baseline MOR BPND in the amygdala bilaterally [right: (24, −2, −24), 1472 mm3, Z = 2.6; left: (−22, −6, −24), 592 mm3, Z = 2.6; Fig. 3A,B]. Baseline D2/D3R BPND in the ventral striatum was in turn negatively correlated with the activation of MOR-mediated neurotransmission during the pain challenge in the left amygdala [(−20, −6, −20), 912 mm3, Z = 3.4; Fig. 3C]. No significant associations were found among pain-induced D2/D3R system activation, baseline MOR BPND, or MOR system activation during pain.
Figure 3.
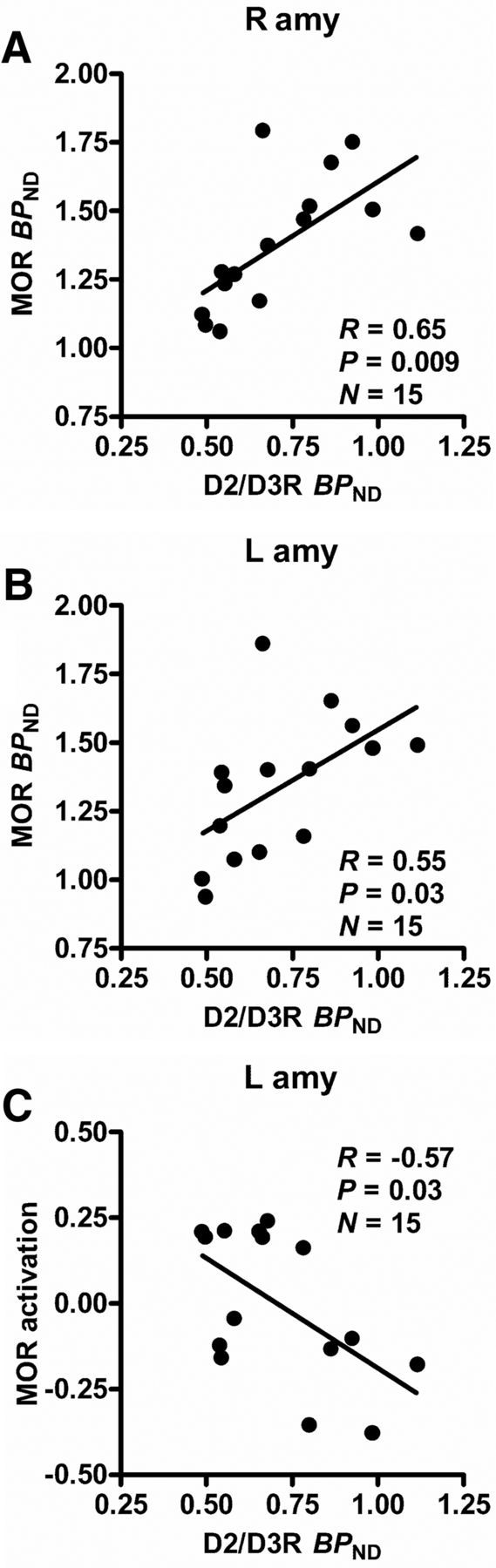
Associations between baseline D2/D3R BPND in the right ventral striatum and MOR functional measures in the amygdala in CNBP patients. In patients with CNBP, significant associations were found between baseline D2/D3R BPND in the right ventral striatum and baseline MOR BPND in the right (A) and left (B) amygdala and MOR activation during experimental pain in the left amygdala (C). The r and p-values were calculated using data extracted from the significant regions in the voxel-by-voxel regression analyses.
The baseline MOR BPND in the right amygdala was negatively correlated with positive affect as measured by PANAS (r = −0.52, p = 0.049) and the baseline MOR BPND in the amygdala bilaterally was negatively correlated with POMS-vigor score (right: r = −0.57, p = 0.03; left: r = −0.56, p = 0.03) and with the volume of hypertonic saline infused during the pain challenge (right: r = −0.66, p = 0.01; left: r = −0.67, p = 0.008), but not with average VAS over the volume infused, a measure of pain sensitivity (p > 0.4). The MOR activation in the left amygdala was negatively correlated with back pain MPQ pain affect subscale scores (r = −0.68, p = 0.006), whereas negative correlations with other measures of back pain (VAS and MPQ) were short of significance (p > 0.07).
Discussion
We found reductions in baseline D2/D3R BPND in the right ventral striatum in CNBP patients compared with HCs. These reductions were positively associated with measures of positive affect and pain tolerance and negatively with pain sensitivity. During experimental pain, both subject groups showed striatal D2/D3R activation, although this response was more variable in the CNBP sample. A group effect for experimental pain-induced D2/D3R system activation was also found in the right ventral striatum, where significant reductions in activation were found in CNBP patients compared with HCs. We further determined the associations between the ventral striatal D2/D3R system measures and brain MOR system function in CNBP. In these analyses, we found that ventral striatal D2/D3R BPND was positively correlated with baseline MOR BPND in the bilateral amygdala and negatively with MOR activation during experimental pain in the left amygdala. MOR system function in the amygdala was also associated with measures of positive affect, the affective quality of the clinical pain, and experimental pain tolerance.
Significant alterations in D2/D3R neurochemical measures in the CNBP group were localized in the ventral striatum, a region where DA coordinates incentive motivational behavior and response to salient stimuli, including pain (Altier and Stewart, 1999; Horvitz, 2000). Pain activates the dopaminergic system in animal models (Abercrombie et al., 1989; Rougé-Pont et al., 1993; Schmidt et al., 2002; Brischoux et al., 2009) and these data have been corroborated in healthy humans by the observation that experimentally induced increases in pain signal lead to activation of striatal DA D2/D3R neurotransmission as measured using [11C]raclopride and PET (Scott et al., 2006; Wood et al., 2007). The D2/D3R system activation during experimental pain was associated with enhanced ratings of sensory and affective qualities of pain in the dorsal striatum and with emotional responses in the nucleus accumbens, indicating that striatal D2/D3R responses to pain reflect both the somatosensory aspects and emotional saliency of the stimulus depending on their location (Scott et al., 2006).
Consistent with the above evidence showing that the striatal D2/D3Rs are involved in both sensory and affective aspects of the individual pain experience, we found significant relationships between the reductions in baseline D2/D3R BPND and the maintenance of positive affect, as well as greater pain tolerance and lesser pain sensitivity in the CNBP group. Regarding positive affect, our results are consistent with earlier research showing that positive affect is associated with DA system function (Burgdorf and Panksepp, 2006; Salamone and Correa, 2012); however, as for pain sensitivity and DA, the evidence from experimental studies in humans has been ambiguous: in healthy subjects, pain sensitivity and tolerance have been associated with genetic polymorphisms related to DA system function (Treister et al., 2009; Jääskeläinen et al., 2014), whereas no change was found in these measures after experimental reductions in brain DA (Becker et al., 2013; Tiemann et al., 2014). Nonetheless, specifically regarding striatal DA D2/D3R function, [11C]raclopride PET studies have found significant positive correlations between baseline striatal D2/D3R BPND and pain sensitivity in HCs (Hagelberg et al., 2002; Scott et al., 2006) and in patients with FM, who showed significant positive correlations between striatal D2/D3R BPND and measures of both experimental pain sensitivity and widespread tenderness (Wood et al., 2007). Together, these previous studies and the present data suggest that the interindividual variability in baseline striatal D2/D3R function may be an important factor in the development and persistence of generalized hyperalgesia in chronic musculoskeletal pain, which is a frequent finding not only in FM, but also in chronic back pain (Giesecke et al., 2004; O'Neill et al., 2007). Considering that both high positive affect and low pain sensitivity have been shown to be associated with increased resilience to pain and negative affective states (Clauw et al., 1999; Zautra et al., 2005; Strand et al., 2006; Tang et al., 2008) and improved long-term outcomes (Edwards et al., 2003; Von Korff and Miglioretti, 2005) in chronic pain patients, the observed reductions in striatal D2/D3R BPND may represent an adaptive process that increases the individual ability to cope with persistent pain and its associated physical and psychological stress, thereby influencing clinical outcomes.
Earlier studies in chronic pain patients have found reductions in baseline striatal D2/D3R BPND in patients with FM (Wood et al., 2007), whereas patients with orofacial pain presented with increases in baseline D2/D3R BPND compared with HCs (Hagelberg et al., 2003a; Hagelberg et al., 2003b). The existing data thus suggest that, whereas the dopaminergic system may display differential alterations across chronic pain conditions, similar changes in baseline striatal D2/D3R measures exist in patients with CNBP and FM. Experimental studies in animals have found evidence for increases in striatal DA and reductions in striatal D2R expression in persistent pain states (Austin et al., 2010; Chang et al., 2014); however, it is presently unknown whether the presence of chronic pain in CNBP and FM is also associated with tonic increases in striatal DA whereby reductions in baseline D2/D3R availability and experimental pain-induced DA release would serve an adaptive role (Chen et al., 1993).
During sustained experimental pain, we found significant striatal D2/D3R activation in both subject groups, although the CNBP group displayed more variable and generally lower responses than HCs, which is consistent with data in FM patients (Wood et al., 2007). A significant group effect in the magnitude of DA D2/D3R activation was observed in the right ventral striatum, where significant reductions in D2/D3R activation were found in the CNBP sample compared with HCs, whereas more regionally widespread reductions had been observed in FM (Wood et al., 2007). In the HC sample, pain-induced D2/D3R system activation was correlated with the perceived unpleasantness of the pain, as described previously (Scott et al., 2006; Wood et al., 2007), whereas in the CNBP sample, no such association was detected, again as also observed in FM patients (Wood et al., 2007). Together, these data appear to reflect a dysregulated dopaminergic response to increases in pain signal in CNBP in a manner that parallels, but is more regionally localized than, data previously reported for FM.
Our previous work compared MOR measures in CNBP patients with matched HCs and found increases in baseline MOR BPND in the thalamus bilaterally and reductions in endogenous opioid-MOR system activation during sustained experimental pain in the thalamus and left amygdala (Martikainen et al., 2013). Because of the reciprocal influences between the opioidergic and dopaminergic systems, in particular, the effects of chronic DA D2R stimulation on enkephalinergic neurotransmission and MORs in the indirect mesolimbic pathway (Steiner and Gerfen, 1998; Soderman and Unterwald, 2009), we hypothesized that associations between D2/D3R and MOR function would also be observed in CNBP. Consistent with this hypothesis, we found that baseline D2/D3R BPND in the ventral striatum was associated with MOR measures in the amygdala that were further related to pain tolerance, positive affect, and back pain. These results suggest that deficits in ventral striatal DA induce compensatory changes in MOR neurotransmission in the amygdala, the activation of which has been shown to suppress sensory and affective dimensions of pain, thereby influencing CNBP clinical presentations (Zubieta et al., 2001; Martikainen et al., 2013). In agreement with this, and in persistent back pain samples, neural activity in the ventral striatum has been shown to be significantly correlated with amygdala and medial prefrontal cortex activity (Baliki et al., 2010). Projections from the ventral striatum reach both midbrain and ventral pallidum and the latter, through the mediodorsal thalamus, projects further to a number of brain areas involved in pain representation and affective regulation, including the amygdala, but also the insular and cingulate cortices, which are known to contribute significantly to the integration and response to the experience of pain (Coghill et al., 1999; Zubieta et al., 2001; Haber, 2011). Functional interactions between the ventral striatum and amygdala may also engage direct amygdalostriatal projections and potentially also projections from the amygdala to the dopaminergic neurons in the midbrain (Kelley et al., 1982; Fudge and Haber, 2000; Phillips et al., 2003).
In summary, our data suggest that pain sensitivity and positive affect in the context of CNBP are regulated by D2/D3R function in the ventral striatum and MOR function in the amygdala. The results help to explain interindividual variability in the presentation of CNBP in both sensory and affective-motivational dimensions and point to DA–opioid interactions as an important CNS mechanism involved in persistent pain pathophysiology and, potentially, risk for the development of persistent pain syndromes. These are also neurotransmitter systems implicated in the action of opioid medications, with reductions in baseline D2/D3R BPND and DA function in the striatum having been described in association with opioid and other drug dependence (Volkow et al., 2007), raising the possibility that DA–opioid mechanisms and their interactions may also be implicated in the risk for opioid medication misuse and eventual addiction. Future research appears warranted to determine the relationship of DA dysfunction and DA–endogenous opioid interactions with treatment outcomes in persistent pain conditions, as well as the involvement of these mechanisms in risk for opioid dependence in chronic pain samples.
Footnotes
This work was supported by the National Institute on Drug Abuse–National Institutes of Health (Grants R01 DA 022520 and R01 027494) and the Phil F. Jenkins Foundation. E.B.N. was supported in part by the National Institutes of Health (Grants 5T32EY017878 and 5T32DA007281). We thank nurses Kathleen Singer and Laurie Carr for patient screening and recruitment and the nuclear medicine technologists at the Positron Emission Tomography Center at the University of Michigan for the assistance in PET data acquisition and reconstruction.
The authors declare no competing financial interests.
References
- Abercrombie ED, Keefe KA, DiFrischia DS, Zigmond MJ. Differential effect of stress on in vivo dopamine release in striatum, nucleus accumbens, and medial frontal cortex. J Neurochem. 1989;52:1655–1658. doi: 10.1111/j.1471-4159.1989.tb09224.x. [DOI] [PubMed] [Google Scholar]
- Altier N, Stewart J. The role of dopamine in the nucleus accumbens in analgesia. Life Sci. 1999;65:2269–2287. doi: 10.1016/S0024-3205(99)00298-2. [DOI] [PubMed] [Google Scholar]
- Austin PJ, Beyer K, Bembrick AL, Keay KA. Peripheral nerve injury differentially regulates dopaminergic pathways in the nucleus accumbens of rats with either ‘pain alone’ or ‘pain and disability’. Neuroscience. 2010;171:329–343. doi: 10.1016/j.neuroscience.2010.08.040. [DOI] [PubMed] [Google Scholar]
- Baliki MN, Geha PY, Fields HL, Apkarian AV. Predicting value of pain and analgesia: nucleus accumbens response to noxious stimuli changes in the presence of chronic pain. Neuron. 2010;66:149–160. doi: 10.1016/j.neuron.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baliki MN, Petre B, Torbey S, Herrmann KM, Huang L, Schnitzer TJ, Fields HL, Apkarian AV. Corticostriatal functional connectivity predicts transition to chronic back pain. Nat Neurosci. 2012;15:1117–1119. doi: 10.1038/nn.3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker S, Ceko M, Louis-Foster M, Elfassy NM, Leyton M, Shir Y, Schweinhardt P. Dopamine and pain sensitivity: neither sulpiride nor acute phenylalanine and tyrosine depletion have effects on thermal pain sensations in healthy volunteers. PLoS One. 2013;8:e80766. doi: 10.1371/journal.pone.0080766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brischoux F, Chakraborty S, Brierley DI, Ungless MA. Phasic excitation of dopamine neurons in ventral VTA by noxious stimuli. Proc Natl Acad Sci U S A. 2009;106:4894–4899. doi: 10.1073/pnas.0811507106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgdorf J, Panksepp J. The neurobiology of positive emotions. Neurosci Biobehav Rev. 2006;30:173–187. doi: 10.1016/j.neubiorev.2005.06.001. [DOI] [PubMed] [Google Scholar]
- Carson RE, Breier A, de Bartolomeis A, Saunders RC, Su TP, Schmall B, Der MG, Pickar D, Eckelman WC. Quantification of amphetamine-induced changes in [11C]raclopride binding with continuous infusion. J Cereb Blood Flow Metab. 1997;17:437–447. doi: 10.1097/00004647-199704000-00009. [DOI] [PubMed] [Google Scholar]
- Chang PC, Pollema-Mays SL, Centeno MV, Procissi D, Contini M, Baria AT, Martina M, Apkarian AV. Role of nucleus accumbens in neuropathic pain: linked multi-scale evidence in the rat transitioning to neuropathic pain. Pain. 2014;155:1128–1139. doi: 10.1016/j.pain.2014.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JF, Aloyo VJ, Weiss B. Continuous treatment with the D2 dopamine receptor agonist quinpirole decreases D2 dopamine receptors, D2 dopamine receptor messenger RNA and proenkephalin messenger RNA, and increases mu opioid receptors in mouse striatum. Neuroscience. 1993;54:669–680. doi: 10.1016/0306-4522(93)90238-B. [DOI] [PubMed] [Google Scholar]
- Clauw DJ, Williams D, Lauerman W, Dahlman M, Aslami A, Nachemson AL, Kobrine AI, Wiesel SW. Pain sensitivity as a correlate of clinical status in individuals with chronic low back pain. Spine. 1999;24:2035–2041. doi: 10.1097/00007632-199910010-00013. [DOI] [PubMed] [Google Scholar]
- Coghill RC, Sang CN, Maisog JM, Iadarola MJ. Pain intensity processing within the human brain: a bilateral, distributed mechanism. J Neurophysiol. 1999;82:1934–1943. doi: 10.1152/jn.1999.82.4.1934. [DOI] [PubMed] [Google Scholar]
- Edwards RR, Doleys DM, Lowery D, Fillingim RB. Pain tolerance as a predictor of outcome following multidisciplinary treatment for chronic pain: differential effects as a function of sex. Pain. 2003;106:419–426. doi: 10.1016/j.pain.2003.09.004. [DOI] [PubMed] [Google Scholar]
- Fudge JL, Haber SN. The central nucleus of the amygdala projection to dopamine subpopulations in primates. Neuroscience. 2000;97:479–494. doi: 10.1016/S0306-4522(00)00092-0. [DOI] [PubMed] [Google Scholar]
- Gear RW, Aley KO, Levine JD. Pain-induced analgesia mediated by mesolimbic reward circuits. J Neurosci. 1999;19:7175–7181. doi: 10.1523/JNEUROSCI.19-16-07175.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giesecke T, Gracely RH, Grant MA, Nachemson A, Petzke F, Williams DA, Clauw DJ. Evidence of augmented central pain processing in idiopathic chronic low back pain. Arthritis Rheum. 2004;50:613–623. doi: 10.1002/art.20063. [DOI] [PubMed] [Google Scholar]
- Haber SN. The primate basal ganglia: parallel and integrative networks. J Chem Neuroanat. 2003;26:317–330. doi: 10.1016/j.jchemneu.2003.10.003. [DOI] [PubMed] [Google Scholar]
- Haber SN. Neuroanatomy of reward: a view from the ventral striatum. In: Gottfried JA, editor. Neurobiology of sensation and reward. Boca Raton, FL: CRC; 2011. [PubMed] [Google Scholar]
- Hagelberg N, Martikainen IK, Mansikka H, Hinkka S, Någren K, Hietala J, Scheinin H, Pertovaara A. Dopamine D2 receptor binding in the human brain is associated with the response to painful stimulation and pain modulatory capacity. Pain. 2002;99:273–279. doi: 10.1016/S0304-3959(02)00121-5. [DOI] [PubMed] [Google Scholar]
- Hagelberg N, Forssell H, Rinne JO, Scheinin H, Taiminen T, Aalto S, Luutonen S, Någren K, Jääskeläinen S. Striatal dopamine D1 and D2 receptors in burning mouth syndrome. Pain. 2003a;101:149–154. doi: 10.1016/S0304-3959(02)00323-8. [DOI] [PubMed] [Google Scholar]
- Hagelberg N, Forssell H, Aalto S, Rinne JO, Scheinin H, Taiminen T, Någren K, Eskola O, Jääskeläinen SK. Altered dopamine D2 receptor binding in atypical facial pain. Pain. 2003b;106:43–48. doi: 10.1016/S0304-3959(03)00275-6. [DOI] [PubMed] [Google Scholar]
- Horvitz JC. Mesolimbocortical and nigrostriatal dopamine responses to salient non-reward events. Neuroscience. 2000;96:651–656. doi: 10.1016/S0306-4522(00)00019-1. [DOI] [PubMed] [Google Scholar]
- Jääskeläinen SK, Lindholm P, Valmunen T, Pesonen U, Taiminen T, Virtanen A, Lamusuo S, Forssell H, Hagelberg N, Hietala J, Pertovaara A. Variation in the dopamine D2 receptor gene plays a key role in human pain and its modulation by transcranial magnetic stimulation. Pain. 2014;155:2180–2187. doi: 10.1016/j.pain.2014.08.029. [DOI] [PubMed] [Google Scholar]
- Jewett DM. A simple synthesis of [11C]carfentanil using an extraction disk instead of HPLC. Nucl Med Biol. 2001;28:733–734. doi: 10.1016/S0969-8051(01)00226-8. [DOI] [PubMed] [Google Scholar]
- Kelley AE, Domesick VB, Nauta WJ. The amygdalostriatal projection in the rat–an anatomical study by anterograde and retrograde tracing methods. Neuroscience. 1982;7:615–630. doi: 10.1016/0306-4522(82)90067-7. [DOI] [PubMed] [Google Scholar]
- Ladouceur A, Tessier J, Provencher B, Rainville P, Piché M. Top-down attentional modulation of analgesia induced by heterotopic noxious counterstimulation. Pain. 2012;153:1755–1762. doi: 10.1016/j.pain.2012.05.019. [DOI] [PubMed] [Google Scholar]
- Logan J, Fowler JS, Volkow ND, Wang GJ, Ding YS, Alexoff DL. Distribution volume ratios without blood sampling from graphical analysis of PET data. J Cereb Blood Flow Metab. 1996;16:834–840. doi: 10.1097/00004647-199609000-00008. [DOI] [PubMed] [Google Scholar]
- Magnusson JE, Fisher K. The involvement of dopamine in nociception: the role of D(1) and D(2) receptors in the dorsolateral striatum. Brain Res. 2000;855:260–266. doi: 10.1016/S0006-8993(99)02396-3. [DOI] [PubMed] [Google Scholar]
- Martikainen IK, Peciña M, Love TM, Nuechterlein EB, Cummiford CM, Green CR, Harris RE, Stohler CS, Zubieta JK. Alterations in endogenous opioid functional measures in chronic back pain. J Neurosci. 2013;33:14729–14737. doi: 10.1523/JNEUROSCI.1400-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNair D, Lorr M, Droppleman L. EdITS manual for the profile of mood states. San Diego: Educational and Industrial Testing Service; 1992. [Google Scholar]
- Melzack R, Torgerson WS. On the language of pain. Anesthesiology. 1971;34:50–59. doi: 10.1097/00000542-197101000-00017. [DOI] [PubMed] [Google Scholar]
- Minoshima S, Koeppe RA, Mintun MA, Berger KL, Taylor SF, Frey KA, Kuhl DE. Automated detection of the intercommissural line for stereotactic localization of functional brain images. J Nucl Med. 1993;34:322–329. [PubMed] [Google Scholar]
- Morgan MJ, Franklin KB. 6-Hydroxydopamine lesions of the ventral tegmentum abolish D-amphetamine and morphine analgesia in the formalin test but not in the tail flick test. Brain Res. 1990;519:144–149. doi: 10.1016/0006-8993(90)90072-J. [DOI] [PubMed] [Google Scholar]
- Morgan MJ, Franklin KB. Dopamine receptor subtypes and formalin test analgesia. Pharmacol Biochem Behav. 1991;40:317–322. doi: 10.1016/0091-3057(91)90560-O. [DOI] [PubMed] [Google Scholar]
- O'Neill S, Manniche C, Graven-Nielsen T, Arendt-Nielsen L. Generalized deep-tissue hyperalgesia in patients with chronic low-back pain. Eur J Pain. 2007;11:415–420. doi: 10.1016/j.ejpain.2006.05.009. [DOI] [PubMed] [Google Scholar]
- Phillips AG, Ahn S, Howland JG. Amygdalar control of the mesocorticolimbic dopamine system: parallel pathways to motivated behavior. Neurosci Biobehav Rev. 2003;27:543–554. doi: 10.1016/j.neubiorev.2003.09.002. [DOI] [PubMed] [Google Scholar]
- Rougé-Pont F, Piazza PV, Kharouby M, Le Moal M, Simon H. Higher and longer stress-induced increase in dopamine concentrations in the nucleus accumbens of animals predisposed to amphetamine self-administration. A microdialysis study. Brain Res. 1993;602:169–174. doi: 10.1016/0006-8993(93)90260-T. [DOI] [PubMed] [Google Scholar]
- Salamone JD, Correa M. The mysterious motivational functions of mesolimbic dopamine. Neuron. 2012;76:470–485. doi: 10.1016/j.neuron.2012.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt BL, Tambeli CH, Barletta J, Luo L, Green P, Levine JD, Gear RW. Altered nucleus accumbens circuitry mediates pain-induced antinociception in morphine-tolerant rats. J Neurosci. 2002;22:6773–6780. doi: 10.1523/JNEUROSCI.22-15-06773.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz N, Temkin P, Jurado S, Lim BK, Heifets BD, Polepalli JS, Malenka RC. Chronic pain. Decreased motivation during chronic pain requires long-term depression in the nucleus accumbens. Science. 2014;345:535–542. doi: 10.1126/science.1253994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott DJ, Heitzeg MM, Koeppe RA, Stohler CS, Zubieta JK. Variations in the human pain stress experience mediated by ventral and dorsal basal ganglia dopamine activity. J Neurosci. 2006;26:10789–10795. doi: 10.1523/JNEUROSCI.2577-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott DJ, Stohler CS, Koeppe RA, Zubieta JK. Time-course of change in [11C]carfentanil and [11C]raclopride binding potential after a nonpharmacological challenge. Synapse. 2007;61:707–714. doi: 10.1002/syn.20404. [DOI] [PubMed] [Google Scholar]
- Scott DJ, Stohler CS, Egnatuk CM, Wang H, Koeppe RA, Zubieta JK. Placebo and nocebo effects are defined by opposite opioid and dopaminergic responses. Arch Gen Psychiatry. 2008;65:220–231. doi: 10.1001/archgenpsychiatry.2007.34. [DOI] [PubMed] [Google Scholar]
- Seeman P, Wilson A, Gmeiner P, Kapur S. Dopamine D2 and D3 receptors in human putamen, caudate nucleus, and globus pallidus. Synapse. 2006;60:205–211. doi: 10.1002/syn.20298. [DOI] [PubMed] [Google Scholar]
- Soderman AR, Unterwald EM. Cocaine-induced mu opioid receptor occupancy within the striatum is mediated by dopamine D2 receptors. Brain Res. 2009;1296:63–71. doi: 10.1016/j.brainres.2009.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowman PF, Wang K, Svensson P, Arendt-Nielsen L. Diffuse noxious inhibitory control evoked by tonic craniofacial pain in humans. Eur J Pain. 2011;15:139–145. doi: 10.1016/j.ejpain.2010.06.009. [DOI] [PubMed] [Google Scholar]
- Steiner H, Gerfen CR. Role of dynorphin and enkephalin in the regulation of striatal output pathways and behavior. Exp Brain Res. 1998;123:60–76. doi: 10.1007/s002210050545. [DOI] [PubMed] [Google Scholar]
- Stohler CS, Kowalski CJ. Spatial and temporal summation of sensory and affective dimensions of deep somatic pain. Pain. 1999;79:165–173. doi: 10.1016/S0304-3959(98)00171-7. [DOI] [PubMed] [Google Scholar]
- Strand EB, Zautra AJ, Thoresen M, Ødegård S, Uhlig T, Finset A. Positive affect as a factor of resilience in the pain-negative affect relationship in patients with rheumatoid arthritis. J Psychosom Res. 2006;60:477–484. doi: 10.1016/j.jpsychores.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Tang NK, Salkovskis PM, Hodges A, Wright KJ, Hanna M, Hester J. Effects of mood on pain responses and pain tolerance: an experimental study in chronic back pain patients. Pain. 2008;138:392–401. doi: 10.1016/j.pain.2008.01.018. [DOI] [PubMed] [Google Scholar]
- Taylor BK, Joshi C, Uppal H. Stimulation of dopamine D2 receptors in the nucleus accumbens inhibits inflammatory pain. Brain Res. 2003;987:135–143. doi: 10.1016/S0006-8993(03)03318-3. [DOI] [PubMed] [Google Scholar]
- Tiemann L, Heitmann H, Schulz E, Baumkötter J, Ploner M. Dopamine precursor depletion influences pain affect rather than pain sensation. PLoS One. 2014;9:e96167. doi: 10.1371/journal.pone.0096167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treister R, Pud D, Ebstein RP, Laiba E, Gershon E, Haddad M, Eisenberg E. Associations between polymorphisms in dopamine neurotransmitter pathway genes and pain response in healthy humans. Pain. 2009;147:187–193. doi: 10.1016/j.pain.2009.09.001. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Fowler JS, Wang GJ, Swanson JM, Telang F. Dopamine in drug abuse and addiction: results of imaging studies and treatment implications. Arch Neurol. 2007;64:1575–1579. doi: 10.1001/archneur.64.11.1575. [DOI] [PubMed] [Google Scholar]
- Von Korff M, Miglioretti DL. A prognostic approach to defining chronic pain. Pain. 2005;117:304–313. doi: 10.1016/j.pain.2005.06.017. [DOI] [PubMed] [Google Scholar]
- Watabe H, Endres CJ, Breier A, Schmall B, Eckelman WC, Carson RE. Measurement of dopamine release with continuous infusion of [11C]raclopride: optimization and signal-to-noise considerations. J Nucl Med. 2000;41:522–530. [PubMed] [Google Scholar]
- Watson D, Clark LA. Manual for the positive and negative affect schedule–expanded form. Iowa City, IA: The University of Iowa; 1999. [Google Scholar]
- Wood PB, Schweinhardt P, Jaeger E, Dagher A, Hakyemez H, Rabiner EA, Bushnell MC, Chizh BA. Fibromyalgia patients show an abnormal dopamine response to pain. Eur J Neurosci. 2007;25:3576–3582. doi: 10.1111/j.1460-9568.2007.05623.x. [DOI] [PubMed] [Google Scholar]
- Zautra AJ, Johnson LM, Davis MC. Positive affect as a source of resilience for women in chronic pain. J Consult Clin Psychol. 2005;73:212–220. doi: 10.1037/0022-006X.73.2.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Ashton-Miller JA, Stohler CS. A closed-loop system for maintaining constant experimental muscle pain in man. IEEE Trans Biomed Eng. 1993;40:344–352. doi: 10.1109/10.222327. [DOI] [PubMed] [Google Scholar]
- Zubieta JK, Smith YR, Bueller JA, Xu Y, Kilbourn MR, Jewett DM, Meyer CR, Koeppe RA, Stohler CS. Regional mu opioid receptor regulation of sensory and affective dimensions of pain. Science. 2001;293:311–315. doi: 10.1126/science.1060952. [DOI] [PubMed] [Google Scholar]