

Abstract
Spinal cord injury (SCI) activates macrophages, endowing them with both reparative and pathological functions. The mechanisms responsible for these divergent functions are unknown but are likely controlled through stochastic activation of different macrophage receptor subtypes. Various danger-associated molecular patterns released from dying cells in the injured spinal cord likely activate distinct subtypes of macrophage pattern recognition receptors, including bacterial toll-like receptors (TLRs) and fungal C-type lectin receptors (e.g., dectin-1). To determine the in vivo consequences of activating these receptors, ligands specific for TLR2 or dectin-1 were microinjected, alone or in combination, into intact spinal cord. Both ligands elicit a florid macrophage reaction; however, only dectin-1 activation causes macrophage-mediated demyelination and axonal injury. Coactivating TLR2 reduced the injurious effects of dectin-1 activation. When injected into traumatically injured spinal cord, TLR2 agonists enhance the endogenous macrophage reaction while conferring neuroprotection. Indeed, dieback of axons was reduced, leading to smaller lesion volumes at the peak of the macrophage response. Moreover, the density of NG2+ cells expressing vimentin increased in and near lesions that were enriched with TLR2-activated macrophages. In dectin-1-null mutant (knock-out) mice, dieback of corticospinal tract axons also is reduced after SCI. Collectively, these data support the hypothesis that the ability of macrophages to create an axon growth-permissive microenvironment or cause neurotoxicity is receptor dependent and it may be possible to exploit this functional dichotomy to enhance CNS repair.
SIGNIFICANCE STATEMENT There is a growing appreciation that macrophages exert diverse functions in the injured and diseased CNS. Indeed, both macrophage-mediated repair and macrophage-mediated injury occur, and often these effector functions are elicited simultaneously. Understanding the mechanisms governing the reparative and pathological properties of activated macrophages is at the forefront of neuroscience research. In this report, using in vitro and in vivo models of relevance to traumatic spinal cord injury (SCI), new data indicate that stochastic activation of toll-like and c-type lectin receptors on macrophages causes neuroprotection or neurotoxicity, respectively. Although this manuscript focuses on SCI, these two innate immune receptor subtypes are also involved in developmental processes and become activated in macrophages that respond to various neurological diseases.
Keywords: dieback, microglia, neuroinflammation, regeneration, TLR2, zymosan
Introduction
Spinal cord injury (SCI) elicits a neuroinflammatory reaction dominated by microglia and monocyte-derived macrophages. Together, these CNS macrophages work to restore tissue homeostasis; however, in doing so they also cause additional injury to neurons and glia. The mechanisms responsible for these divergent effects are not known but likely involve stochastic activation of different innate immune receptors.
Pattern-recognition receptors (PRRs) detect molecular sequences that were first discovered on bacteria and viruses (Bianchi, 2007). Emerging data indicate that similar molecular sequences are released from injured or dying mammalian cells and both microglia and macrophages recognize these as “damage-associated molecular patterns” or DAMPs. Common DAMPs include heat-shock proteins, high-mobility group box 1 protein, and mRNA, and each activates a range of PRRs, including toll-like receptors (TLRs) and C-type lectin receptors (CLRs). In the CNS, activation of microglia or macrophage PRRs can cause gliogenesis or regeneration of injured axons; however, such reparative effects are often accompanied by secondary demyelination and neurotoxicity (Kigerl et al., 2007; Schonberg et al., 2007; Gensel et al., 2009; Kigerl and Popovich, 2009; Stirling et al., 2014). These divergent effects could be explained by convergent signaling elicited by individual DAMPs acting on discrete PRRs or by the unique effects of distinct DAMP–PRR interactions. Insight into the functional consequences of activating macrophages via multiple PRRs can be gained from experiments using zymosan, a glucan polysaccharide found on yeast.
Zymosan binds to both TLR2 and dectin-1, a CLR (Lien et al., 1999; Gantner et al., 2003; Brown, 2006). When injected into brain or spinal cord, zymosan elicits macrophage-mediated killing of neurons while also creating a microenvironment that supports axon regeneration (Fitch et al., 1999; Yin et al., 2003, 2006, 2009; Okada et al., 2005; Steinmetz et al., 2005; Gensel et al., 2009; Benowitz and Popovich, 2011). In this manuscript, new data show that the neurotoxic effects of zymosan-activated macrophages (ZAMs) are due to CLR activation. Selective activation of dectin-1 significantly increases the magnitude of axonal injury and tissue pathology compared with zymosan alone and dieback of corticospinal tract (CST) axons is reduced in dectin-1 knock-out (KO) mice. Conversely, selective TLR2 activation causes florid inflammation but without causing marked neuropathology. Importantly, it is possible to attenuate the injurious effects of endogenous macrophages responding to SCI by injecting TLR2-selective agonists into the injury site; augmenting TLR2 activation reduces dieback (retraction) of injured CNS axons and promotes the accumulation of axon growth-permissive molecules. Collectively, these data implicate different macrophage PRRs in injury and repair processes after CNS injury and identify new molecular targets for achieving cell-specific immune modulation.
Materials and Methods
Animals.
Adult female Sprague Dawley rats (200–225 g; Harlan) or C57BL/6 female Clec7a−/− (dectin-1 KO) mice (provided by Dr. Gordon Brown at University of Cape Town, South Africa; Taylor et al., 2007) and wild-type (WT) littermates were used for individual experiments. Cells for in vitro analysis were prepared from adult C57BL/6 WT male and female mice (8–12 weeks; Jackson Laboratory). All procedures were performed in accordance with the guidelines and protocols of the Office of Responsible Research Practices and with approval of the Institutional Animal Care and Use Committees at The Ohio State University or the University of Kentucky.
Spinal cord injuries.
Rat dorsal column crush (DCC) injuries were performed at C8 as previously described (Horn et al., 2008). Briefly, rats were anesthetized with intraperitoneally injected ketamine (80 mg/kg) and xylazine (50 mg/kg), and muscle and skin layers were dissected to expose the gap between the T1 and T2 spinal processes. The ligament between the spinal processes was removed to expose the C8 cervical spinal level. Bilateral, pin-prick-size durotomies were made with microscissors 0.75 mm lateral to midline to allow insertion of jeweler's forceps (Dumont #55) through the dura and into the arachnoid mater, pia mater, and then spinal cord to a depth of 1 mm. Forceps were then retracted and Dumont #3 jeweler forceps were inserted into the spinal cord through the same hole to a depth of 1 mm. Then a 1.5-mm-wide DCC lesion was created by squeezing the forceps together three times, holding maximal pressure for 10 s each time. For mice, a complete crush injury was performed at the T9 spinal level. After performing a laminectomy, tips of a Dumont #5 jeweler's forceps were inserted bilaterally along the sides of the exposed spinal cord until both tips touch the ventral surface of the spinal canal. To cause injury, forceps were closed completely, held in place for 2 s, and then released. Complete lesions were verified by observation of white-matter clearing (without damage to the dura).
Intraspinal macrophage activation.
Rats were anesthetized with intraperitoneally injected ketamine (80 mg/kg) and xylazine (50 mg/kg) and a T8/T9 laminectomy was performed as described previously (Popovich et al., 2002; Gensel et al., 2009). For macrophage activation after DCC, animals were anesthetized (as above) 2 d after injury and the injury site re-exposed. Microinjections were performed via calibrated pressure microinjection (PV800 Pneumatic PicoPump, David Kopf Instruments) using pulled glass micropipettes (PD-5 Micropipette Puller, Narishige). Pipettes were beveled to an external diameter of 30–80 mm (Beveller 1300 M-C, World Precision Instruments) and were sterilized overnight under UV light. To target the lateral white matter in the thoracic cord, stereotaxic injections were made 900 mm lateral to midline and 800 mm deep (Schonberg et al., 2007). For macrophage activation in the crush site, stereotaxic injections were made on midline 500 mm deep into the center of the injury. Macrophages were activated through 100 nl intraspinal microinjections of zymosan (10 mg/ml; Sigma-Aldrich, catalog #Z4250), depleted zymosan (10 mg/ml; InvivoGen, catalog #tlrl-dzn), 1 μl microinjections of PAM2CSK4 (1 mg/ml; InvivoGen, catalog #tlrl-pm2s), and PBS (vehicle) at a rate of 300 nl/min.
Axon labeling.
Two days before killing rats via perfusion, while under anesthetic, the dorsal column axons were labeled unilaterally with Texas Red-conjugated 3000 molecular weight (MW) dextran (Invitrogen, catalog #D-3328). The right sciatic nerve was exposed and crushed 3–3.5 cm from the knee (4–5 mm from bifurcation of nerve) with Dumont #3 forceps for 10 s and repeated two additional times as described previously (Horn et al., 2008; Ikeda et al., 2008). Via a 32-gauge Hamilton syringe, 1.5–2 μl of 3000 MW dextran 10% in sterile water was injected into the crush site. To trace CST axons in mice, 1 μl (total) of biotinylated dextran amine (BDA; 10%) was injected into sensorimotor cortex at three sites using a NanoJect system (anterior–posterior coordinates from bregma: −0.1, 2 mm; 1.5, 2 mm; and 1.5, 1.5 mm; all at a depth of 0.6 mm into cortex). BDA injections were completed at 2 weeks postinjury, a time when CST axons are undergoing axon dieback and activated macrophages are evident in the degenerating CST (Wang et al., 2009).
After SCI or intraspinal microinjection, muscle layers were sutured with 4-0 nylon suture and the skin was closed with surgical staples. Postoperatively, animals received 5 ml of subcutaneous saline, were kept warm on a heating plate during recovery from anesthesia, and allowed access to food and water ad libitum.
Tissue preparation, histology, and immunohistochemistry.
At various times after microinjection, dorsal column injury, or spinal cord crush injury, animals were terminally anesthetized and transcardially perfused with 0.1 PBS and then 4% paraformaldehyde. Spinal cords were removed, postfixed for 2 h, and then rinsed in 0.2 m phosphate buffer and stored overnight at 4°C. Tissues were cryoprotected by immersing in 30% sucrose over several days, then were blocked and embedded in Tissue-Tek OCT (Fisher), and then frozen on dry ice. Frozen spinal cord blocks were sectioned on a cryostat in either 10-μm-thick frontal or 15–20-μm-thick longitudinal (from dorsal to ventral) sections. Tissues were labeled using standard indirect immunofluorescence techniques. Primary antibodies used were OX42 (1:2000; Serotec); Neurofilament-1 (NF-1; 1:1000; Aves); GFAP [1:20,000; Polysciences; or 1:2000; Wako (for mouse immunohistochemistry)]; vimentin (1:500; Aves); Texas Red (1:200; Life Technologies); ED-1 (1:1000; Serotec); β-amyloid precursor protein (APP; 1:1000; Invitrogen); tomato lectin (TomL; 1:1000; Sigma-Aldrich); protein kinase Cγ (PKCγ; 1:200; Santa Cruz Biotechnology); NG2 (1:1000; United States Biological). Secondary Alexafluor (AF; 1:1000)-conjugated antibodies AF488, AF546, AF633 were used to visualize primary antibody labeling. DAPI (Sigma-Aldrich) was used to label cell nuclei. Sections were stained for macrophages/microglia (OX-42; 1:4000; mouse AbD Serotec) or axons (NF200; 1:5000; rabbit, Millipore Bioscience Research Reagents) using peroxidase-based immunohistochemistry or eriochrome cyanine for myelin. Images were captured using a Zeiss 510 Meta (Carl Zeiss) confocal microscope, a Nikon Eclipse Ti laser-scanning confocal microscope, or an Axioplan 2 imaging (Carl Zeiss) microscope.
Analysis.
Sensory axon retraction from the center of the DCC site (identified by the forceps tracks and maximum width of gliosis), CST retraction from the lesion edge (identified by GFAP labeling), and rostral–caudal lesion lengths were quantified using an Axioplan 2 imaging microscope (Zeiss) with a motorized X-Y stage controlled by MCID image quantification software (Imaging Research). As described previously (Horn et al., 2008), the distance of Texas Red-labeled axon retraction was determined for three sections per animal spaced 200 μm apart and starting 200 μm below the dorsal surface of the spinal cord. As performed previously, the measurements from all sections from all animals in a group were averaged to determine the average distance of retraction per time point (Horn et al., 2008). In rat DCC injuries, lesion length was defined as the widest area of pathology/gliosis per section (Stirling et al., 2014). Retraction of dorsal CST axons in mice was measured in one section per mouse at a depth of ∼280 μm.
Lesion and macrophage volume estimates were calculated using the Cavalieri method on digitized images at 0.1 mm intervals centered on the injection site (Donnelly et al., 2011). Proportional areas of macrophage, NF, and vimentin, were quantified similarly to techniques used previously to quantify macrophage activation (Donnelly et al., 2009; Gensel et al., 2009). Briefly, the density of labeling above background was quantified using threshold-based measures within predefined areas on tissue sections. The MCID, MetaMorph (Molecular Devices), or ImageJ image quantification software programs were used to determine proportional area measurements. For quantification of axon density within areas of activated macrophages, manual counts of NF axons were normalized to the area of activated, TomL+ macrophages. Then the average of two adjacent sections (100 μm apart) was used for each animal. For vimentin, uniform areas were sampled from regions centered on BDA-labeled axons. The MetaMorph colocalization plug-in was used to analyze colocalization of vimentin with BDA-labeled axons.
Cell culture.
Bone marrow-derived macrophages (BMDMs) were isolated from the leg bones of C57BL/6 mice as described previously (Longbrake et al., 2007). Briefly, BMDMs were obtained from bilateral femurs and tibias using aseptic techniques. Marrow cores were flushed into sterile tubes using syringes fit with 26 gauge needles and filled with Roswell Park Memorial Institute (RPMI) 1640 (or DMEM) plus 10% FBS. Cells were triturated 3–5× and then red blood cells were lysed in lysis buffer (0.15 m NH4Cl, 10 mm KHCO3, and 0.1 mm Na2EDTA, pH 7.4). Cells were washed once in media, and then plated and cultured in differentiation media: DMEM or RPMI 1640 supplemented with 1% penicillin/streptomycin, 1% HEPES, 0.001% β-mercaptoethanol, 10% FBS, and 20% supernatant from sL929 cells. The sL929 (which contains macrophage colony stimulating factor) is needed to promote differentiation of bone marrow cells into macrophages with phenotypically similar properties as CNS macrophages (7–10 d; Burgess et al., 1985; Longbrake et al., 2007).
Macrophage-conditioned media (MCM) was generated as described by Gensel et al. (2009). After 6 d in culture, BMDMs were cold-shocked (eliminates need for trypsin) with media for 5 min and then removed from flasks using a cell scraper (BD Falcon, catalog #353086). Cells were replated overnight at 1 × 106 cells/ml in differentiation media without sL929 supernatant. The following day, media was replaced with Neurobasal A (NBA) media supplemented with 2% B27, 1% Glutamax, and 1% penicillin-streptomycin containing zymosan (1.25 mg/ml or 100 μg/ml), PAM2CSK4 (200 ng/ml), depleted zymosan (1.25 mg/ml or 100 μg/ml), or fresh NBA. Macrophages were activated for 8 h at 37°C in 5% CO2, after which the MCM was collected, centrifuged (1200 rpm, 5 min), and then filtered (0.22 μm low-protein binding filter; Millipore) to remove the zymosan/stimulant particles and cell debris. All MCM was stored at −20°C.
Dorsal root ganglion (DRG) neurons were isolated from terminally anesthetized adult WT C57BL/6 mice. DRG cultures were prepared on glass coverslips as described previously (Gensel et al., 2009, 2010). Cell death was determined by plating cells directly into MCM (1:1 MCM/NBA-B27 base media) at 4000 cells/coverslip or plating at 400 cells/coverslip and treating on the third day in vitro with MCM for 48 h. Five days after plating, neurons were fixed with 2% paraformadehyde and then labeled with β-tubulin III (1:2000; Sigma-Aldrich) and AF546 and DAPI (Sigma-Aldrich). The density of β-tubulin III labeling was determined by randomly sampling digitized images of DRG cells. Numbers of DRG neurons were estimated by unbiased counting of β-tubulin III+ neurons in randomized fields covering ∼45% of each coverslip (∼125 fields per coverslip; Gensel et al., 2009; Kigerl et al., 2009).
Statistical analysis.
All data were analyzed with the investigator blind to experimental conditions. All experimental data are representative of ≥2 independent replicate experiments. Specific replicates and animal numbers are provided in the legends that accompany each dataset. Data were analyzed using ANOVA followed by Dunnett's or Tukey's test for multiple comparisons. Independent-samples t tests were also used to compare two groups separately. All p ≤ 0.05 were considered significant. Unless noted otherwise, all data represent the mean ± SEM. All analyses were performed using Prism 5.0/6.0 (GraphPad Software). Figures were prepared using Adobe Photoshop CS5/6 (Adobe Systems) and Prism 5.0/6.0.
Results
Selective activation of TLR2 or dectin-1 elicits distinct macrophage functions in vivo and in vitro
Zymosan activates both TLR-2 and dectin-1 (CLR) receptors found on microglia and macrophages. In brain and spinal cord, ZAMs cause profound axonal injury, glial cell loss, and demyelination, but these cells also can enhance axon regeneration (Fitch et al., 1999; Popovich et al., 2002; Yin et al., 2003; Steinmetz et al., 2005; Okada et al., 2006; Schonberg et al., 2007; Gensel et al., 2009). To clarify the seemingly conflicting effects of coactivating macrophage TLR2 and CLRs, receptor-selective agonists were injected into intact rat thoracic spinal cord. PAM2CSK4 is a TLR2-selective agonist, whereas removing the TLR2-stimulating component of zymosan allows selective activation of dectin-1. Depleted zymosan (see Materials and Methods) and heat-killed Saccharomyces cerevisiae are both selective dectin-1 agonists (Ikeda et al., 2008) and, when injected into intact spinal cord, both CLR agonists produced similar results. Accordingly, only depleted zymosan data are described and are designated as “CLR” in all figures.
To account for possible differences in the magnitude of macrophage activation caused by each agonist, lesion volumes were normalized to macrophage volumes (presented as the lesion/macrophage ratio). Consistent with previous data from our laboratory, coactivating TLR2 and CLRs with zymosan caused axonal injury and demyelination (Popovich et al., 2002; Schonberg et al., 2007; Gensel et al., 2009). Overall, ∼75% of the area occupied by ZAMs was devoid of axons or myelin (Fig. 1A,D,G,H,K). However, ZAMs also were colocalized with intact axons and myelin (Fig. 1H,K). In contrast, activating only dectin-1 caused a similarly robust macrophage reaction but all areas occupied by macrophages were devoid of axons and myelin and regions of pathology extended beyond the inflammatory focus (ratio, >1.0; Fig. 1B,E,G,I,L). Intraspinal injection of the TLR2 agonist PAM2CSK4, also elicits a florid macrophage response but with minimal axon or myelin pathology (Fig. 1C,F,G,J,M).
Figure 1.

Intraspinal injections of dectin-1 (CLR) and TLR2 receptor agonists cause variable degrees of axonal injury and demyelination. Cross-sections of rat thoracic spinal cord 3 d after microinjecting TLR2 or dectin-1 (CLR) agonists. A–F, All agonists activate macrophages (OX42, brown, A–C) but with varying degrees of myelin (eriochrome cyanine, blue, D–F) and axon (NF, brown, D–F) pathology. G, Normalizing the area of axon/myelin pathology to area occupied by round OX42+ macrophages reveals marked differences in macrophage effector potential when activating these receptors, alone or in combination. A–G, Pathology is minimal when TLR2 is activated alone (PAM2CSK4, 1 μg in 1.0 μl). Conversely, activating CLR (D-ZYM, 1 μg in 0.1 μl) causes significantly more pathology when compared with the effects of coactivating TLR2 and CLR (zymosan, 1 μg in 0.1 μl; ANOVA p < 0.0001; n = 3 rats/group, macrophage and lesion volumes were quantified for each subject using the Cavalieri method). H–M, Confocal projections reveal differences in axon preservation (H–J, NF) after activating TLR2 or CLR, alone and in combination. Only when macrophages are activated via TLR2 alone (J, M) do axons persist within the inflammatory foci (K–M, right of the dotted line). In contrast, few axons remain when macrophages are activated via dectin-1 (CLR), alone or in combination with TLR2 (H, I, K, L). The most significant pathology is evident after dectin-1 (CLR) activation; axon loss is evident beyond the dense core of activated macrophages (I, L, left of dotted line). N, Quantification of axon density in areas colocalized with activated macrophages reveals significant preservation of axons only when macrophages are activated by TLR2. Data are representative of three independent experiments (n = 3/rats/group/experiment). *p < 0.05; ***p < 0.001; ****p < 0.0001 versus other conditions. Scale bars: A–F, 50 μm; H–M, 20 μm.
In an independent experiment, axon density was quantified within regions occupied by activated macrophages (Fig. 1H–N). Activating dectin-1 alone increased axonal injury >5-fold when compared with selective activation of TLR2 (Fig. 1N). Coactivation of TLR2 and dectin-1 consistently caused pathology that was intermediate between activation of TLR2 or dectin-1 alone (Fig. 1G,N). These data indicate that TLR2 activation might mitigate the pathologic effects of activating dectin-1.
Indeed, activation of TLR-2 resulted in the least amount of axonal damage. Three-dimensional confocal reconstructions of axons in the longitudinal plane confirmed the unique relationship between TLR2-activated macrophages and spared (or sprouting) axons. More intact axon profiles were found projecting through and around TLR2-activated macrophages than macrophages activated via dectin-1 (CLR) or dectin-1 plus TLR2 (Fig. 2A–F). Within foci of macrophages activated via both TLR2 and dectin-1 or dectin-1 alone, either no neurofilament labeling was present or axons were dystrophic or fragmented (Fig. 2A–D). The autofluorescence caused by depleted zymosan particles (Fig. 2C) obscures clear visualization of dystrophic axons in each condition. Accordingly, adjacent sections were immunolabeled to reveal β-APP+ axon profiles. β-APP is a sensitive measure of axon injury (Pierce et al., 1996). More APP+ axons colocalized with macrophages activated via dectin-1, either alone or in combination with TLR2 (Fig. 2G–L). Together, data in Figures 1 and 2 indicate that the neurotoxic effects of activated macrophages are receptor dependent; activating dectin-1, a CLR, elicits pathological effector functions, whereas TLR2-activated macrophages do not. Also, TLR2 activation may inhibit the pathologic effects of activating dectin-1.
Figure 2.
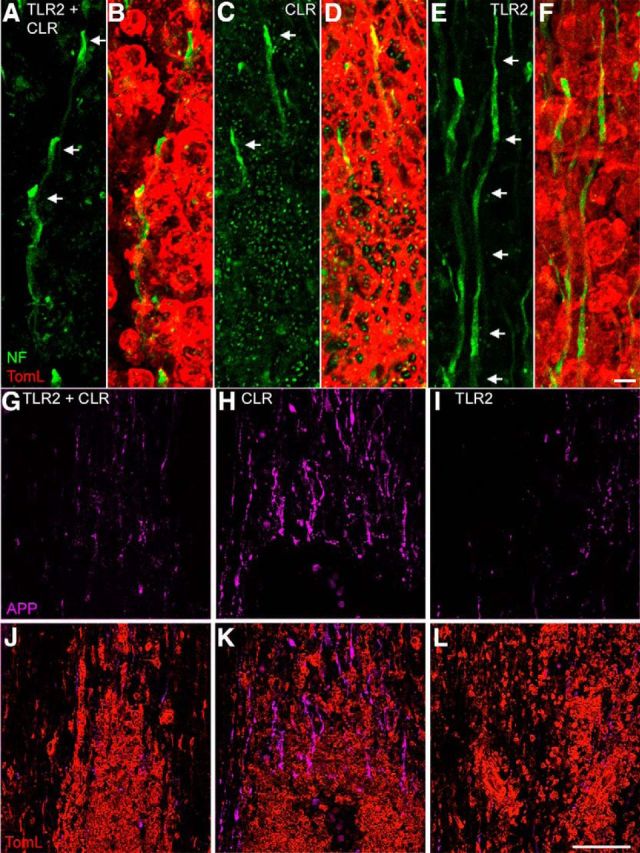
Selective activation of macrophage dectin-1 (CLR), but not TLR2, causes axonal degeneration. Representative longitudinal thoracic spinal cord sections 3 d after microinjecting TLR2 and dectin-1 agonists. A–D, Axons in proximity to macrophages activated via CLR plus TLR2 (A, B, TLR2 + CLR) or CLR (C, D) were not continuous and had dystrophic endings (arrows; note that green “dots” in C and D are not fragmented axons; this is autofluorescence caused by depleted zymosan particles). E, F, In contrast, intact axons coursed through areas occupied by TLR2-activated macrophages (arrows indicate continuous axon). G–L, Confocal projections reveal accumulation of APP (purple) in injured axons. All agonists elicit comparable magnitudes of macrophage activation (B, D, E, red); however, a notable increase in APP staining is evident only after injecting a dectin-1 agonist (H, K, compare with G, J and I, L). Scale bars: A–F, 3 μm; G–L, 100 μm.
Since cells other than macrophages (and microglia) can express TLRs (Kigerl and Popovich, 2009), we next tested whether the in vivo effects of the dectin-1 and/or TLR2 agonists were caused by direct activation of microglia/macrophages. Accordingly, in vitro assays were used. Specifically, media conditioned by macrophages (MCM) activated with zymosan (TLR2 plus CLR) or selective dectin-1 (CLR) or TLR2 agonists were compared with media from unstimulated macrophages. All MCM was overlaid onto cultured primary adult DRG neurons.
When compared with MCM from unstimulated macrophages, which kills ∼10–20% of neurons in culture, TLR2 MCM was neuroprotective. Greater numbers of DRG neurons were present after exposure to TLR2 MCM and axons were protected from spontaneous degeneration (Fig. 3A,B,E). Conversely, activating dectin-1 (CLR) caused significant neurotoxicity (Fig. 3A,B,D). Coactivating TLR2 with dectin-1 partially reversed the neurotoxic effects of activating dectin-1 alone, but axon pathology persisted (Fig. 3A–C).
Figure 3.

Dectin-1 (CLR)-activated macrophages cause toxicity and axonal degeneration. MCM from macrophages activated via CLR plus TLR2 (TLR2+CLR, zymosan) or CLR alone (depleted zymosan) cause neurotoxicity and axonal degeneration, whereas MCM from TLR2-activated macrophages is neuroprotective or enhances axon growth. A, B, Total numbers of DRG neurons (A; normalized to DRG neurons incubated with unstimulated MCM; ANOVA, p = 0.0055; *p < 0.05 vs other conditions) and overall density of β-tubulin staining (B; ANOVA, p = 0.0028, *p = 0.01 vs CLR) were quantified as indices of neuron survival and axon degeneration, respectively. DRG neurons were plated at low density (400 cells per coverslip) and then, beginning on day 3 in vitro, were stimulated with MCM for 2 d (A). C–F, Representative images of adult DRG neurons cultured for 5 d in the presence of different types of MCM (quantified in B). Data are representative of three independent experiments. Dotted line in A and B represents unstimulated MCM. Scale bar, 50 μm.
Delayed delivery of a TLR2 agonist reprograms the injurious effects of the endogenous macrophage response caused by SCI
Data in Figures 1–3 support the hypothesis that activating dectin-1 causes macrophages to release neurotoxic factors while TLR2 activation stimulates the release of neuroprotective or growth-promoting factors. After SCI, macrophages actively promote axonal dieback (Popovich et al., 1999; Horn et al., 2008; Evans et al., 2014). To test whether these injurious effects of the endogenous macrophage response can be “reprogrammed,” a TLR2-selective agonist, Pam2CSK4, was microinjected into the injury site at 2 d postinjury (dpi), a time preceding the onset of monocyte recruitment and macrophage-mediated dieback (Horn et al., 2008). Two days before killing rats, a subset of dorsal column sensory axons was labeled by injecting fluorescent dextran into the sciatic nerve (Horn et al., 2008). Two or 6 d after intraspinal injection of vehicle or Pam2CSK4 (corresponding with 4 or 8 dpi, respectively), spinal cords were removed and the distances between ends of the labeled fibers and the lesion center were quantified (Horn et al., 2008; Busch et al., 2009).
Consistent with a previous report (Horn et al., 2008), significant axonal dieback occurred 4–8 dpi in control SCI rats with axons retracting ∼0.6–0.8 mm during this time interval (Fig. 4A,C,D,E). Conversely, in rats injected with a TLR2 agonist, axon dieback was reduced ∼40 and ∼15% at 4 and 8 dpi, respectively (Fig. 4B,C,F,G). Notably, within areas where axon dieback was reduced, TLR2 activation enhanced the endogenous macrophage response >2-fold (Figs. 4D–G, 5). Consistent with the observation that TLR2-activated macrophages attenuate axonal dieback, lesion size also was reduced ∼30% at 4 dpi (1.9 ± 0.2 mm) versus vehicle (3.0 ± 0.2 mm; Fig. 5F).
Figure 4.

TLR2-activated macrophages reduce axon dieback in vivo after SCI. A, B, Longitudinal spinal cord sections 4 d after DCC injury and 2 d after intraspinal injection of vehicle (A) or TLR2 agonist (Pam2CSK4; B). Dotted line indicates the center of the crush lesion. Inverted fluorescent images of BDA-labeled axons are presented below labeled sections in A and B. C, Significantly less axon dieback (arrows) occurs when TLR2 agonist (Pam2CSK4) is microinjected compared with vehicle injections. *p < 0.05, ***p < 0.001 (n = 3/group). D–G, High-powered confocal montages of labeled axons in proximity to activated macrophages. Note the increase in macrophage (green, OX42) and axon (red, BDA-labeled) density closer to the lesion center (*) when lesion contains TLR2-activated macrophages (F, G) compared with spinal cord injected with vehicle (D, E). Data are representative of two independent experiments (n = 3/group in each experiment). Scale bars: A, B, 500 μm; D–G, 212 μm.
Figure 5.

Intraspinal injection of TLR2 ligand elicits a florid macrophage response and reduces lesion size after SCI. A, B, D, E, Representative confocal montages of longitudinal spinal cord sections 4 d after DCC and 2 d after intraspinal injection (1 μl) of vehicle (A, B) or Pam2CSK4, a selective TLR2 agonist (D, E). Note the marked increase in the density of macrophages (green, OX42, quantified in C) in or near the injury site in spinal cords injected with TLR2 agonist (E) versus vehicle (B). The rostral–caudal lesion length (lesion identified by dotted line) is significantly reduced in spinal cords enriched with TLR2-activated macrophages (A vs D, quantified in F). Images are representative of spinal cords obtained in two independent experiments. Individual animal data are presented in Figure 4F (n = 6–9 rats/group). **p < 0.01. Scale bar, 200 μm.
Published data indicate that vimentin-positive glial progenitor cells provide a stabilizing substrate that protects dystrophic axons from macrophage-mediated dieback (Busch et al., 2010; Filous et al., 2014). To determine whether activating macrophages via TLR2 was associated with changes in this stabilizing substrate, we examined vimentin immunoreactivity in proximity to labeled axons. The density of vimentin labeling was significantly increased after injecting the TLR2 agonist into the injured spinal cord (∼1.5× vs vehicle; Fig. 6A–G). Also, more axons were closely apposed to vimentin-positive cells in the TLR2 condition (Fig. 6H–N). Orthogonal confocal projections revealed that cells positive for vimentin double-labeled with NG2 (Fig. 6O–Q). Neither ED-1+ macrophages (Fig. 6R–T) or GFAP+ astrocytes (data not shown) colabeled with NG2. Since published data indicate that vimentin in NG2+ glial progenitor cells can protect axons from dieback (Busch et al., 2010; Filous et al., 2014), these data suggest that TLR2-activated macrophages may limit dieback in part by creating an axon-stabilizing substrate at or near the lesion.
Figure 6.

TLR2-activated macrophages create a microenvironment capable of stabilizing dystrophic axons after SCI. Confocal montages of longitudinal spinal cord sections 4 dpi. A–F, Vimentin expression is increased 2 d after intraspinal injection (1 μl) of TLR2 agonist Pam2CSK4 (D–F) compared with vehicle injection (A–C). Edge of the lesion center in A and D labeled with asterisks. G, The density of vimentin labeling in proximity to labeled axons is significantly increased in spinal cords injected with the TLR2 agonist (**p < 0.01, n = 3/group). H–M, High-powered confocal projections show more axons closely associated with vimentin-positive cells (arrows) in spinal cords injected with TLR2 agonist. Arrowheads depict axons that are not associated with vimentin. N, Significantly more axons were apposed to vimentin-positive cells following injection of the TLR2 agonist (***p < 0.001, n = 3/group). O–T, Confocal images reveal vimentin colocalization with NG2-positive (O–Q) but not ED-1-positive (R–T) cells (z-plane to the right and bottom of each image). Data are representative of two independent experiments. Scale bars: A–F, 100 μm; H–M, 50 μm; O–T, 10 μm.
Dectin-1-mediated activation of macrophages contributes to dieback of CST axons after SCI
To test the hypothesis that dectin-1 activation elicits destructive effector functions in endogenous macrophages leading to axon dieback, we analyzed the magnitude of CST retraction from the injury site in dectin-1 KO or WT mice subjected to a complete crush injury of the thoracic spinal cord (Taylor et al., 2007). CST axon dieback was significantly reduced in dectin-1 KO mice compared with WT littermate control mice (p < 0.005; Fig. 7A–C). Neither lesion size nor the magnitude of macrophage accumulation was different between KO and WT animals (data not shown). Complete injury to the dorsal CST was confirmed by immunolabeling for PKCγ below the level of injury (Lieu et al., 2013; Fig. 7D).
Figure 7.

Genetic deletion of dectin-1 limits dieback of CST axons after SCI. A, B, Horizontal spinal cord sections revealing BDA-labeled dorsal CST axons (inverted) from individual dectin-1 KO (A) or littermate WT control mice (B) 5 weeks after complete crush injury (3 weeks after BDA labeling). Dotted line indicates the boundary of GFAP+ glial scar at a depth of ∼280 μm (middle of dorsal CST) from the dorsal surface of the spinal cord. C, Quantification of distance of BDA-labeled CST fibers from the lesion border reveals that CST axon dieback is significantly reduced in dectin-1 KO mice (vs WT controls). D, Cross-sections of mouse spinal cord caudal to the injury stained with anti-PKCγ antibody. Normal (intact) CST axons are PKCγ+. Absence of PKCγ labeling confirms complete injury to dorsal CST axons (dotted line) after SCI. **p = 0.003 (n = 7 dectin-1 KO mice; n = 5 WT mice). Scale bar, 200 μm. Data are the combined results of two independent experiments.
Discussion
Macrophages exert reparative and degradative effects on injured tissue. Several variables likely contribute to this functional heterogeneity, including the size/shape of the ligand, the polarization state of responding macrophages, and the stochastic activation of diverse macrophage receptors in both spared and lesioned tissue (Gantner et al., 2005; Rosas et al., 2008; Goodridge et al., 2011; Stirling et al., 2014). These variables have not been thoroughly studied in the context of traumatic SCI. In this report, we attempted to define how two different families of macrophage PRRs—the TLRs and CLRs—influence macrophage effector functions in the spinal cord. Expression of each of these receptor families increases after SCI, providing a molecular mechanism by which macrophages can bind endogenous proteins and nucleic acids that are released or that become newly synthesized by dead or damaged cells (Horn et al., 2008; Chen et al., 2013; Kigerl et al., 2014). Data in this report show that activating intraspinal macrophages via the CLR dectin-1, either in isolation or together with TLR2, causes pathology. Conversely, TLR2-activated macrophages confer neuroprotection and are able to attenuate pathology caused by dectin-1 activation.
The unique functional properties associated with these different PRRs endow macrophages with the ability to distinguish between bound/embedded stimuli and stimuli that are shed from the same microbe or damaged/stressed cell. TLRs are activated by low concentrations of soluble ligands, whereas CLRs are nonopsonic phagocytic receptors. CLRs sense carbohydrate moieties in pathogens as well as glycosylated proteins and DAMPs derived from apoptotic and necrotic cells (Gardai et al., 2006; Matsunaga and Moody, 2009; Miyake et al., 2010; Donnelly et al., 2011). In the injured CNS, this type of dual recognition scheme would prioritize host defense at or near the injury site where entry of microbes or other pathogens is likely, while allowing macrophage subpopulations in penumbra or spared tissues to respond to soluble DAMPs that are newly expressed by injured neurons or glia.
Previously, our laboratory implicated TLR2 signaling in the induction of a reparative macrophage phenotype after SCI. Indeed, locomotor recovery is impaired and lesion pathology is exacerbated in TLR2 KO mice (Kigerl et al., 2007; Donnelly et al., 2009; Gensel et al., 2009). Independent reports showing that regenerative axon growth or neuroprotection can be achieved by injecting TLR2 agonists into the eye or an ex vivo SCI preparation further supports a protective role for TLR2 signaling (Longbrake et al., 2007; Hauk et al., 2010; Stirling et al., 2014). Data in the present report show that delayed (2 dpi) intraspinal injection of a TLR2 agonist enhances the endogenous macrophage response without exacerbating lesion pathology. In fact, axon dieback and secondary lesion expansion were significantly reduced. Associated with these changes was an increase in the number of vimentin-positive NG2 glia. Previous data indicate that these cells create a permissive or axon-stabilizing substrate that can protect dystrophic axons from macrophage-mediated dieback (Burgess et al., 1985; Longbrake et al., 2007; Busch et al., 2010). Collectively, these data indicate that a broad therapeutic window exists for manipulating macrophage effector functions and that TLR2 may be a novel target for therapeutic development. Also, the in vivo neuroprotective effects of delayed macrophage activation via TLR2 may be as effective or more effective than systemic macrophage depletion (Popovich et al., 1999; Horn et al., 2008; Gensel et al., 2009). Other TLRs, including TLR4 and TLR3, also may be therapeutic targets after SCI (Gensel et al., 2009, 2010; Kigerl and Popovich, 2009). Systemic injections of LPS, a TLR4 agonist, augment intraspinal macrophage recruitment while reducing tissue damage and increasing axonal growth/sprouting after SCI. These neuroprotective effects were associated with modest improvements in functional recovery (Guth et al., 1994; Gensel et al., 2009; Popovich et al., 2012). TLR3 ligands can increase production of neurotrophic factors and anti-inflammatory cytokines (Kigerl et al., 2009; Bsibsi et al., 2012); however, these effects may not be macrophage specific and direct injection of TLR ligands can adversely affect axon growth and plasticity (Popovich et al., 2002; Cameron et al., 2007; Schonberg et al., 2007; Gensel et al., 2009, 2012).
In contrast to the effects of activating TLR2, no benefit was associated with intraspinal activation of dectin-1. When injected into intact spinal cord, dectin-1 agonists caused axon loss and demyelination, whereas axon dieback was reduced in mice deficient in dectin-1. Like TLRs, dectin-1 signaling activates both nuclear factor κB and MAPK cascades; however, dectin-1 also may impart unique and presumably pathological effector functions (e.g., respiratory burst, phagocytosis) in macrophages through the immunoreceptor tyrosine-based activation motif in its cytoplasmic domain that recruits the tyrosine kinase Syk (Yin et al., 2003; Okada et al., 2005; Steinmetz et al., 2005; Brown, 2006; Gensel et al., 2009).
Although unique functional attributes are associated with activating TLR2 or dectin-1 in the intact spinal cord, after SCI these receptors are coexpressed by macrophages and it is likely that simultaneous activation of these receptors occurs in the dynamic lesion microenvironment. How intraspinal macrophages respond to zymosan is instructive in this context since zymosan activates both TLR2 and dectin-1. Published data indicate that, like macrophages that become activated by SCI, zymosan-activated macrophages can kill neurons but they also create a microenvironment that supports axon regeneration (Popovich et al., 2002; Yin et al., 2006; Schonberg et al., 2007; Gensel et al., 2009). Our current data indicate that the effects of CLR ligation, alone or in synergy with TLR2, predominate at the injury site and the net effect of engaging both TLRs and nonopsonic CLRs does more harm than good. Indeed, when injected into injured spinal cord, zymosan failed to induce axon growth or neuroprotection (McPhail et al., 2004; Ward et al., 2014). Also, Kroner et al. showed that synergistic signaling induced by phagocytosis and cytokines released downstream of TLR activation potentiate the development of a destructive inflammatory macrophage phenotype (Kigerl and Popovich, 2009; Kroner et al., 2014). Those authors also found that the default pathway for macrophage polarization after SCI is not necessarily destructive. Myelin phagocytosis, which is prominent in the lesion early after SCI, can trigger anti-inflammatory or resolution signaling in macrophages. However, the signaling required for induction of an anti-inflammatory macrophage phenotype is subjugated by other signaling cascades initiated by iron phagocytosis and inflammatory cytokines (e.g., TNFa; Kroner et al., 2014). These latter signals predominate in the lesion core and likely squelch signaling induced by myelin. Even if iron-dependent and TNF-dependent signaling were reduced, the possibility remains that impaired phagocytosis of myelin (“frustrated phagocytosis”) would continue to fuel destructive inflammation via overactivation of dectin-1 or other CLRs (Rosas et al., 2008). Our current data indicate that blocking CLRs or augmenting specific TLR signaling pathways could be a novel way to reduce inflammatory macrophage-mediated injury after SCI.
Even when the same receptor is activated on different cells, ligand dimorphism and the magnitude of receptor cross-linking could promote polarization of distinct macrophage phenotypes. As an example, solubilized β-glucans do not effectively activate dectin-1, whereas particulate/bound β-glucans form “phagocytic synapses” that fully activate dectin-1 signaling pathways (Goodridge et al., 2011). A practical application of this divergent recognition by dectin-1 was described for fungal (yeast) infections. Reproductive budding in yeast leaves scars where β-glucans are exposed. This form of bound β-glucan is a potent dectin-1 agonist. Conversely, in its filamentous form, Candida albicans does not activate dectin-1 because β-glucans are not accessible (Gantner et al., 2005). An analogy in the injured spinal cord might be structural or intracellular chaperone proteins that are normally sequestered in intact cells but then are exposed as cells become stressed or die. If bound to dying cells or released and sequestered in the surrounding extracellular matrix, these ligands could preferentially activate dectin-1 or other CLRs. Other soluble DAMPs could diffuse throughout the lesion and spared tissue and activate macrophages via TLRs without concurrent CLR ligation. In this context, recent data show that dissolving chondroitin sulfate proteoglycan-enriched glial scars, a reaction that likely solubilizes various PRR ligands, elicits a reparative macrophage phenotype (Bartus et al., 2014; Didangelos et al., 2014).
While the present data are an oversimplification of the complex and stochastic nature by which macrophages become activated after SCI, they highlight the need for future studies that consider how the lesion microenvironment affects the expression of other macrophage receptors. In addition to TLRs and CLRs, macrophages express scavenger receptors, integrins, chemokine and cytokine receptors, and various cytosolic proteins [e.g., nucleotide-binding oligomerization domains (NODs) and NAcht leucine-rich-repeat proteins (NALPs)] capable of regulating intracellular signaling that will ultimately determine macrophage phenotype (Taylor et al., 2005).
Footnotes
This work was supported by the University of Kentucky and the Spinal Cord and Brain Injury Research Center, The Craig H. Neilsen Foundation (J.C.G.), the American Academy of Neurology (K.A.B.), National Institute of Neurological Disorders and Stroke (NINDS) NS037846 (P.G.P.), NINDS P30-NS045758 (Ohio State University), NINDS NS051220 P30 (University of Kentucky), and The Ray W. Poppleton Endowment (P.G.P.). We also thank Kyeong (Rachel) Jang (supported through a summer fellowship from the American Physiological Society), Linda Simmerman, Dr. Gordon Brown (contributed dectin-1-null mutant mice), and Brian Brautigam for their help with this work.
The authors declare no competing financial interests.
References
- Bartus K, James ND, Didangelos A, Bosch KD, Verhaagen J, Yáñez-Muñoz RJ, Rogers JH, Schneider BL, Muir EM, Bradbury EJ. Large-scale chondroitin sulfate proteoglycan digestion with chondroitinase gene therapy leads to reduced pathology and modulates macrophage phenotype following spinal cord contusion injury. J Neurosci. 2014;34:4822–4836. doi: 10.1523/JNEUROSCI.4369-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benowitz LI, Popovich PG. Inflammation and axon regeneration. Curr Opin Neurol. 2011;24:577–583. doi: 10.1097/WCO.0b013e32834c208d. [DOI] [PubMed] [Google Scholar]
- Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81:1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- Brown GD. Dectin-1: a signalling non-TLR pattern-recognition receptor. Nat Rev Immunol. 2006;6:33–43. doi: 10.1038/nri1745. [DOI] [PubMed] [Google Scholar]
- Bsibsi M, Nomden A, van Noort JM, Baron W. Toll-like receptors 2 and 3 agonists differentially affect oligodendrocyte survival, differentiation, and myelin membrane formation. J Neurosci Res. 2012;90:388–398. doi: 10.1002/jnr.22767. [DOI] [PubMed] [Google Scholar]
- Burgess AW, Metcalf D, Kozka IJ, Simpson RJ, Vairo G, Hamilton JA, Nice EC. Purification of two forms of colony-stimulating factor from mouse L-cell-conditioned medium. J Biol Chem. 1985;260:16004–16011. [PubMed] [Google Scholar]
- Busch SA, Horn KP, Silver DJ, Silver J. Overcoming macrophage-mediated axonal dieback following CNS injury. J Neurosci. 2009;29:9967–9976. doi: 10.1523/JNEUROSCI.1151-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch SA, Horn KP, Cuascut FX, Hawthorne AL, Bai L, Miller RH, Silver J. Adult NG2+ cells are permissive to neurite outgrowth and stabilize sensory axons during macrophage-induced axonal dieback after spinal cord injury. J Neurosci. 2010;30:255–265. doi: 10.1523/JNEUROSCI.3705-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron JS, Alexopoulou L, Sloane JA, DiBernardo AB, Ma Y, Kosaras B, Flavell R, Strittmatter SM, Volpe J, Sidman R, Vartanian T. Toll-like receptor 3 is a potent negative regulator of axonal growth in mammals. J Neurosci. 2007;27:13033–13041. doi: 10.1523/JNEUROSCI.4290-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Deng S, Lu H, Zheng Y, Yang G, Kim D, Cao Q, Wu JQ. RNA-Seq characterization of spinal cord injury transcriptome in acute/subacute phases: a resource for understanding the pathology at the systems level. PLoS One. 2013;8:e72567. doi: 10.1371/journal.pone.0072567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didangelos A, Iberl M, Vinsland E, Bartus K, Bradbury EJ. Regulation of IL-10 by chondroitinase ABC promotes a distinct immune response following spinal cord injury. J Neurosci. 2014;34:16424–16432. doi: 10.1523/JNEUROSCI.2927-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly DJ, Gensel JC, Ankeny DP, van Rooijen N, Popovich PG. An efficient and reproducible method for quantifying macrophages in different experimental models of central nervous system pathology. J Neurosci Methods. 2009;181:36–44. doi: 10.1016/j.jneumeth.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly DJ, Longbrake EE, Shawler TM, Kigerl KA, Lai W, Tovar CA, Ransohoff RM, Popovich PG. Deficient CX3CR1 signaling promotes recovery after mouse spinal cord injury by limiting the recruitment and activation of Ly6Clo/iNOS+ macrophages. J Neurosci. 2011;31:9910–9922. doi: 10.1523/JNEUROSCI.2114-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans TA, Barkauskas DS, Myers JT, Hare EG, You JQ, Ransohoff RM, Huang AY, Silver J. High-resolution intravital imaging reveals that blood-derived macrophages but not resident microglia facilitate secondary axonal dieback in traumatic spinal cord injury. Exp Neurol. 2014;254:109–120. doi: 10.1016/j.expneurol.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filous AR, Tran A, Howell CJ, Busch SA, Evans TA, Stallcup WB, Kang SH, Bergles DE, Lee SI, Levine JM, Silver J. Entrapment via synaptic-like connections between NG2 proteoglycan+ cells and dystrophic axons in the lesion plays a role in regeneration failure after spinal cord injury. J Neurosci. 2014;34:16369–16384. doi: 10.1523/JNEUROSCI.1309-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitch MT, Doller C, Combs CK, Landreth GE, Silver J. Cellular and molecular mechanisms of glial scarring and progressive cavitation: in vivo and in vitro analysis of inflammation-induced secondary injury after CNS trauma. J Neurosci. 1999;19:8182–8198. doi: 10.1523/JNEUROSCI.19-19-08182.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantner BN, Simmons RM, Canavera SJ, Akira S, Underhill DM. Collaborative induction of inflammatory responses by dectin-1 and toll-like receptor 2. J Exp Med. 2003;197:1107–1117. doi: 10.1084/jem.20021787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantner BN, Simmons RM, Underhill DM. Dectin-1 mediates macrophage recognition of Candida albicans yeast but not filaments. EMBO J. 2005;24:1277–1286. doi: 10.1038/sj.emboj.7600594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardai SJ, Bratton DL, Ogden CA, Henson PM. Recognition ligands on apoptotic cells: a perspective. J Leukoc Biol. 2006;79:896–903. doi: 10.1189/jlb.1005550. [DOI] [PubMed] [Google Scholar]
- Gensel JC, Nakamura S, Guan Z, van Rooijen N, Ankeny DP, Popovich PG. Macrophages promote axon regeneration with concurrent neurotoxicity. J Neurosci. 2009;29:3956–3968. doi: 10.1523/JNEUROSCI.3992-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gensel JC, Schonberg DL, Alexander JK, McTigue DM, Popovich PG. Semi-automated Sholl analysis for quantifying changes in growth and differentiation of neurons and glia. J Neurosci Methods. 2010;190:71–79. doi: 10.1016/j.jneumeth.2010.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gensel JC, Kigerl KA, Mandrekar-Colucci SS, Gaudet AD, Popovich PG. Achieving CNS axon regeneration by manipulating convergent neuro-immune signaling. Cell Tissue Res. 2012;349:201–213. doi: 10.1007/s00441-012-1425-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodridge HS, Reyes CN, Becker CA, Katsumoto TR, Ma J, Wolf AJ, Bose N, Chan AS, Magee AS, Danielson ME, Weiss A, Vasilakos JP, Underhill DM. Activation of the innate immune receptor dectin-1 upon formation of a “phagocytic synapse.”. Nature. 2011;472:471–475. doi: 10.1038/nature10071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guth L, Zhang Z, DiProspero NA, Joubin K, Fitch MT. Spinal cord injury in the rat: treatment with bacterial lipopolysaccharide and indomethacin enhances cellular repair and locomotor function. Exp Neurol. 1994;126:76–87. doi: 10.1006/exnr.1994.1043. [DOI] [PubMed] [Google Scholar]
- Hauk TG, Leibinger M, Müller A, Andreadaki A, Knippschild U, Fischer D. Stimulation of axon regeneration in the mature optic nerve by intravitreal application of the toll-like receptor 2 agonist Pam3Cys. Invest Ophthalmol Vis Sci. 2010;51:459–464. doi: 10.1167/iovs.09-4203. [DOI] [PubMed] [Google Scholar]
- Horn KP, Busch SA, Hawthorne AL, van Rooijen N, Silver J. Another barrier to regeneration in the CNS: activated macrophages induce extensive retraction of dystrophic axons through direct physical interactions. J Neurosci. 2008;28:9330–9341. doi: 10.1523/JNEUROSCI.2488-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda Y, Adachi Y, Ishii T, Miura N, Tamura H, Ohno N. Dissociation of toll-like receptor 2-mediated innate immune response to zymosan by organic solvent-treatment without loss of dectin-1 reactivity. Biol Pharm Bull. 2008;31:13–18. doi: 10.1248/bpb.31.13. [DOI] [PubMed] [Google Scholar]
- Kigerl KA, Popovich PG. Toll-like receptors in spinal cord injury. Curr Top Microbiol Immunol. 2009;336:121–136. doi: 10.1007/978-3-642-00549-7_7. [DOI] [PubMed] [Google Scholar]
- Kigerl KA, Lai W, Rivest S, Hart RP, Satoskar AR, Popovich PG. Toll-like receptor (TLR)-2 and TLR-4 regulate inflammation, gliosis, and myelin sparing after spinal cord injury. J Neurochem. 2007;102:37–50. doi: 10.1111/j.1471-4159.2007.04524.x. [DOI] [PubMed] [Google Scholar]
- Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci. 2009;29:13435–13444. doi: 10.1523/JNEUROSCI.3257-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kigerl KA, de Rivero Vaccari JP, Dietrich WD, Popovich PG, Keane RW. Pattern recognition receptors and central nervous system repair. Exp Neurol. 2014;258:5–16. doi: 10.1016/j.expneurol.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroner A, Greenhalgh AD, Zarruk JG, Passos Dos Santos R, Gaestel M, David S. TNF and increased intracellular iron alter macrophage polarization to a detrimental M1 phenotype in the injured spinal cord. Neuron. 2014;83:1098–1116. doi: 10.1016/j.neuron.2014.07.027. [DOI] [PubMed] [Google Scholar]
- Lien E, Sellati TJ, Yoshimura A, Flo TH, Rawadi G, Finberg RW, Carroll JD, Espevik T, Ingalls RR, Radolf JD, Golenbock DT. Toll-like receptor 2 functions as a pattern recognition receptor for diverse bacterial products. J Biol Chem. 1999;274:33419–33425. doi: 10.1074/jbc.274.47.33419. [DOI] [PubMed] [Google Scholar]
- Lieu A, Tenorio G, Kerr BJ. Protein kinase C gamma (PKCγ) as a novel marker to assess the functional status of the corticospinal tract in experimental autoimmune encephalomyelitis (EAE) J Neuroimmunol. 2013;256:43–48. doi: 10.1016/j.jneuroim.2013.01.003. [DOI] [PubMed] [Google Scholar]
- Longbrake EE, Lai W, Ankeny DP, Popovich PG. Characterization and modeling of monocyte-derived macrophages after spinal cord injury. J Neurochem. 2007;102:1083–1094. doi: 10.1111/j.1471-4159.2007.04617.x. [DOI] [PubMed] [Google Scholar]
- Matsunaga I, Moody DB. Mincle is a long sought receptor for mycobacterial cord factor. J Exp Med. 2009;206:2865–2868. doi: 10.1084/jem.20092533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPhail LT, Stirling DP, Tetzlaff W, Kwiecien JM, Ramer MS. The contribution of activated phagocytes and myelin degeneration to axonal retraction/dieback following spinal cord injury. Eur J Neurosci. 2004;20:1984–1994. doi: 10.1111/j.1460-9568.2004.03662.x. [DOI] [PubMed] [Google Scholar]
- Miyake Y, Ishikawa E, Ishikawa T, Yamasaki S. Self and nonself recognition through C-type lectin receptor, Mincle. Self Nonself. 2010;1:310–313. doi: 10.4161/self.1.4.13736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada S, Nakamura M, Katoh H, Miyao T, Shimazaki T, Ishii K, Yamane J, Yoshimura A, Iwamoto Y, Toyama Y, Okano H. Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat Med. 2006;12:829–834. doi: 10.1038/nm1425. [DOI] [PubMed] [Google Scholar]
- Okada T, Ichikawa M, Tokita Y, Horie H, Saito K, Yoshida J, Watanabe M. Intravitreal macrophage activation enables cat retinal ganglion cells to regenerate injured axons into the mature optic nerve. Exp Neurol. 2005;196:153–163. doi: 10.1016/j.expneurol.2005.07.015. [DOI] [PubMed] [Google Scholar]
- Pierce JE, Trojanowski JQ, Graham DI, Smith DH, McIntosh TK. Immunohistochemical characterization of alterations in the distribution of amyloid precursor proteins and beta-amyloid peptide after experimental brain injury in the rat. J Neurosci. 1996;16:1083–1090. doi: 10.1523/JNEUROSCI.16-03-01083.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovich PG, Guan Z, Wei P, Huitinga I, van Rooijen N, Stokes BT. Depletion of hematogenous macrophages promotes partial hindlimb recovery and neuroanatomical repair after experimental spinal cord injury. Exp Neurol. 1999;158:351–365. doi: 10.1006/exnr.1999.7118. [DOI] [PubMed] [Google Scholar]
- Popovich PG, Guan Z, McGaughy V, Fisher L, Hickey WF, Basso DM. The neuropathological and behavioral consequences of intraspinal microglial/macrophage activation. J Neuropathol Exp Neurol. 2002;61:623–633. doi: 10.1093/jnen/61.7.623. [DOI] [PubMed] [Google Scholar]
- Popovich PG, Tovar CA, Wei P, Fisher L, Jakeman LB, Basso DM. A reassessment of a classic neuroprotective combination therapy for spinal cord injured rats: LPS/pregnenolone/indomethacin. Exp Neurol. 2012;233:677–685. doi: 10.1016/j.expneurol.2011.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas M, Liddiard K, Kimberg M, Faro-Trindade I, McDonald JU, Williams DL, Brown GD, Taylor PR. The induction of inflammation by dectin-1 in vivo is dependent on myeloid cell programming and the progression of phagocytosis. J Immunol. 2008;181:3549–3557. doi: 10.4049/jimmunol.181.5.3549. [DOI] [PubMed] [Google Scholar]
- Schonberg DL, Popovich PG, McTigue DM. Oligodendrocyte generation is differentially influenced by toll-like receptor (TLR) 2 and TLR4-mediated intraspinal macrophage activation. J Neuropathol Exp Neurol. 2007;66:1124–1135. doi: 10.1097/nen.0b013e31815c2530. [DOI] [PubMed] [Google Scholar]
- Steinmetz MP, Horn KP, Tom VJ, Miller JH, Busch SA, Nair D, Silver DJ, Silver J. Chronic enhancement of the intrinsic growth capacity of sensory neurons combined with the degradation of inhibitory proteoglycans allows functional regeneration of sensory axons through the dorsal root entry zone in the mammalian spinal cord. J Neurosci. 2005;25:8066–8076. doi: 10.1523/JNEUROSCI.2111-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirling DP, Cummins K, Mishra M, Teo W, Yong VW, Stys P. Toll-like receptor 2-mediated alternative activation of microglia is protective after spinal cord injury. Brain. 2014;137:707–723. doi: 10.1093/brain/awt341. [DOI] [PubMed] [Google Scholar]
- Taylor PR, Martinez-Pomares L, Stacey M, Lin HH, Brown GD, Gordon S. Macrophage receptors and immune recognition. Annu Rev Immunol. 2005;23:901–944. doi: 10.1146/annurev.immunol.23.021704.115816. [DOI] [PubMed] [Google Scholar]
- Taylor PR, Tsoni SV, Willment JA, Dennehy KM, Rosas M, Findon H, Haynes K, Steele C, Botto M, Gordon S, Brown GD. Dectin-1 is required for beta-glucan recognition and control of fungal infection. Nat Immunol. 2007;8:31–38. doi: 10.1038/ni1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Hu B, Wong WM, Lu P, Wu W, Xu XM. Glial and axonal responses in areas of Wallerian degeneration of the corticospinal and dorsal ascending tracts after spinal cord dorsal funiculotomy. Neuropathology. 2009;29:230–241. doi: 10.1111/j.1440-1789.2008.00969.x. [DOI] [PubMed] [Google Scholar]
- Ward RE, Huang W, Kostusiak M, Pallier PN, Michael-Titus AT, Priestley JV. A characterization of white matter pathology following spinal cord compression injury in the rat. Neuroscience. 2014;260:227–239. doi: 10.1016/j.neuroscience.2013.12.024. [DOI] [PubMed] [Google Scholar]
- Yin Y, Cui Q, Li Y, Irwin N, Fischer D, Harvey AR, Benowitz LI. Macrophage-derived factors stimulate optic nerve regeneration. J Neurosci. 2003;23:2284–2293. doi: 10.1523/JNEUROSCI.23-06-02284.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y, Henzl MT, Lorber B, Nakazawa T, Thomas TT, Jiang F, Langer R, Benowitz LI. Oncomodulin is a macrophage-derived signal for axon regeneration in retinal ganglion cells. Nat Neurosci. 2006;9:843–852. doi: 10.1038/nn1701. [DOI] [PubMed] [Google Scholar]
- Yin Y, Cui Q, Gilbert HY, Yang Y, Yang Z, Berlinicke C, Li Z, Zaverucha-do-Valle C, He H, Petkova V, Zack DJ, Benowitz LI. Oncomodulin links inflammation to optic nerve regeneration. Proc Natl Acad Sci U S A. 2009;106:19587–19592. doi: 10.1073/pnas.0907085106. [DOI] [PMC free article] [PubMed] [Google Scholar]