

Abstract
Cardinal is an R package for statistical analysis of mass spectrometry-based imaging (MSI) experiments of biological samples such as tissues. Cardinal supports both Matrix-Assisted Laser Desorption/Ionization (MALDI) and Desorption Electrospray Ionization-based MSI workflows, and experiments with multiple tissues and complex designs. The main analytical functionalities include (1) image segmentation, which partitions a tissue into regions of homogeneous chemical composition, selects the number of segments and the subset of informative ions, and characterizes the associated uncertainty and (2) image classification, which assigns locations on the tissue to pre-defined classes, selects the subset of informative ions, and estimates the resulting classification error by (cross-) validation. The statistical methods are based on mixture modeling and regularization.
Contact: o.vitek@neu.edu
Availability and implementation: The code, the documentation, and examples are available open-source at www.cardinalmsi.org under the Artistic-2.0 license. The package is available at www.bioconductor.org.
1 Introduction
Mass spectrometry-based imaging (MSI) experiments characterize the chemical composition of biological samples (such as tissues) or non-biological samples at spatial resolution (Watrous et al., 2011). The experiments repeatedly acquire mass spectra at gridded locations on a tissue. Two typical goals of statistical analysis of MSI are image segmentation, which partitions a tissue into regions of homogeneous spectral profiles, and image classification, which assigns locations of the tissue to pre-defined classes based on their spectral profiles. However, achieving these goals is often quite difficult due to the large and complex nature of the datasets, and due to the biological and technical variation in intensities of spectral features. Statistical inference is key for distinguishing the systematic signals in the spectra from noise. The availability of statistical methods and software for MSI experiments is currently limited.
We introduce Cardinal, an open-source R-based software package for processing and visualization of mass spectra, and for statistical segmentation and classification of the resulting images. Cardinal differs from other publicly available software tools such as BioMap, DataCube Explorer and MSiReader in its emphasis on statistical modeling and inference. It differs from commercial tools such as SCiLS Lab (SCiLS), flexImaging (Bruker), HDI (Waters) and TissueView (AB Sciex) in being open-source. It differs from the existing R packages for mass spectrometry such as MALDIquant and MSnbase in being designed specifically for MSI.
2 Description
2.1 Applicability and requirements
Cardinal is applicable to experiments aiming at segmentation and classification of a single tissue, or multiple tissues collected across biological subjects. It is applicable to Desorption Electrospray Ionization workflows, and to Matrix-Assisted Laser Desorption/Ionization (MALDI) workflows analyzing either intact or in situ-digested proteins. Cardinal has been tested on raw MS1 spectra from Thermo LTQ linear ion trap, ABSciex TOF/TOF and Bruker Autoflex MALDI-TOF instruments with resolving powers ranging from 1000 to 22 000. Cardinal is compatible with Windows, Mac and Linux operating systems. The size of the input dataset must be such that it can be loaded entirely into computer memory. Cardinal runs optimally when the available memory is twice the size of the dataset.
2.2 Data import, processing and visualization
Cardinal supports input data in the imzML format (Schramm et al., 2012), and the Analyze7.5 format. Free converters to imzML are available for most other formats at www.imzml.org, and the converted imzML input data can be read into Cardinal.
Cardinal implements a complete set of common spectral processing methods (Yang et al., 2009), including normalization (e.g. using total ion current), baseline correction (e.g. using median interpolation), peak detection [e.g. using LIMPIC (Mantini et al., 2007)] and peak alignment (e.g. using mean spectrum).
Cardinal visualizes mass spectra, molecular ion images and results of the statistical analyses. The images are optimized with contrast enhancement and smoothing. The plots can be conditioned on experimental metadata (such as the type of the tissue), and viewed separately using a grid layout with multiple conditions, or jointly in a superposition.
2.3 Functionalities for statistical analysis
For image segmentation, Cardinal implements several existing methods, e.g. principle component analysis and Spatially-Aware (SA) and Spatially Aware Structurally Adaptive (SASA) segmentation (Alexandrov and Kobarg, 2011). Cardinal also introduces a novel method, called Spatial Shrunken Centroids, for model-based unsupervised image segmentation (Fig. 1). It combines the spatial distance from SA and SASA with the mixture modeling and regularization from Nearest Shrunken Centroids (Tibshirani et al., 2003). The mixture modeling allows us to estimate the probability that a location on the tissue belongs to a particular segment. Statistical regularization allows us to automatically select the spectral features that define each segments, as well as the total number of segments.
Fig. 1.
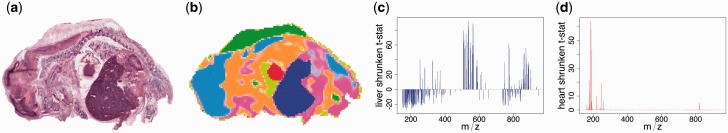
Unsupervised model-based segmentation of a cross-section of a pig fetus. (a) Optical image of a hematoxylin & eosin-stained tissue highlights its morphology, e.g. the brain (left), the heart (center) and the liver (dark region below the heart). (b) Joint segmentation of five adjacent tissue sections from 28 016 non-background pixels and 10 200 mass features. Cardinal detected 298 peaks during peak picking. The segmentation with Spatial Shrunken Centroids and Spatially Aware distance selected 11 tissue segments. (c) The t-statistics quantified the relative importance of the peaks in the liver. Ninety-two peaks were systematically enriched and 153 were systematically absent, as compared with the mean spectrum. (d) As in (c), but for the heart segment. Only 23 peaks were systematically enriched in the heart, and none were systematically absent. Similar analyses can be performed in a supervised manner for image classification
For image classification, Cardinal implements partial least squares discriminant analysis and orthogonal projections to latent structures discriminant analysis (Dill et al., 2010). Cardinal also introduces a novel Spatial Shrunken Centroids for model-based image classification, which utilizes the same principles as the model-based image segmentation but works in a supervised manner. For all the methods, Cardinal automates the estimation of classification error rate by (cross-)validation.
2.4 Implementation and performance
Cardinal employs efficient data structures to store the data and the metadata, and optimized methods for data manipulation. As the result, Cardinal can be used with any dataset that fits in the computer memory. For example, the dataset in Figure 1 with 28 016 pixels was 2.2 GB before the peak picking, and the processed version was 63.7 MB after the peak picking. Computation of the first 20 principal components took 86.9 sec on the raw data and 4.3 s on the picked peaks on a MacBook Pro with a 2.6 GHz Intel Core i7 and 16 GB memory. The segmentation with Spatial Shrunken Centroids on the picked peaks took 241 s (shortest) to 827 s (longest), depending on the initial values of regularization parameters and the number of clusters, on the same computer.
Cardinal facilitates the development of new functionalities, and the interoperability with other software. For example, raw mass spectra can be stored as either a R matrix or any matrix-like object, such as a sparse matrix. Most of the processing methods use an extendable framework pixelApply, similar to the apply family of methods in R. The ResultSet data structure allows the developers to store the results of any analyses, and directly access the Cardinal’s plotting capabilities. Cardinal also has functions for simulating mass spectra, to assist method testing. It is publicly available at www.bioconductor.org
3 Conclusions
Cardinal is a general, flexible, open-source tool for the analysis of MSI experiments. It can be used by researchers with and without background in R and computing. For experimenters, Cardinal provides a full toolchain for multiple workflows, with emphasis on multivariate statistical modeling, inference and model-based visualization. For developers, Cardinal provides a foundation for designing and implementing new methods of computational and statistical analysis of MSI experiments. Users can find more support through the Google Group, accessible through the project website.
Acknowledgements
This work was supported by the NSF Graduate Research Fellowship No. 103049 to K.B., by the Carver Fellowship, Purdue University, to A.H., and by the NSF CAREER award DBI-1054826 to O.V. The authors thank R. Graham Cooks for invaluable discussions and the data.
Conflict of Interest: none declared.
References
- Alexandrov T., Kobarg J.H. (2011) Efficient spatial segmentation of large imaging mass spectrometry datasets with spatially aware clustering. Bioinformatics, 27, i230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dill A.L., et al. (2010) Multivariate statistical differentiation of renal cell carcinomas based on lipidomic analysis by ambient ionization imaging mass spectrometry. Anal. Bioanal. Chem., 398, 2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantini D., et al. (2007) LIMPIC: a computational method for the separation of protein MALDI-TOF-MS signals from noise. BMC Bioinf., 8, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramm T., et al. (2012) imzML – A common data format for the flexible exchange and processing of mass spectrometry imaging data. J. Proteom., 75, 5106. [DOI] [PubMed] [Google Scholar]
- Tibshirani R., et al. (2003) Class prediction by nearest shrunken with applications to DNA microarrays. Stat. Sci., 18, 104. [Google Scholar]
- Watrous J.D., et al. (2011) The evolving field of imaging mass spectrometry and its impact on future biological research. J.Mass Spectrom., 46, 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C., et al. (2009) Comparison of public peak detection algorithms for MALDI mass spectrometry data analysis. BMC Bioinf., 10, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]