

Summary
Centromeres are essential chromosomal structures that mediate accurate chromosome segregation during cell division. Centromeres are specified epigenetically by the heritable incorporation of the centromeric histone H3 variant, CENP-A. While many of the primary factors that mediate centromeric deposition of CENP-A are known, the chromatin and DNA requirements in this process have remained elusive. Here, we uncover a role for transcription in Drosophila CENP-A deposition. Using an inducible ectopic centromere system that uncouples CENP-A deposition from endogenous centromere function and cell-cycle progression, we demonstrate that CENP-A assembly by its loading factor, CAL1, requires RNAPII-mediated transcription of the underlying DNA. This transcription depends on the novel CAL1 binding partner FACT, but not on CENP-A incorporation. Our work establishes RNAPII passage as a key step in chaperone-mediated CENP-A chromatin establishment and propagation.
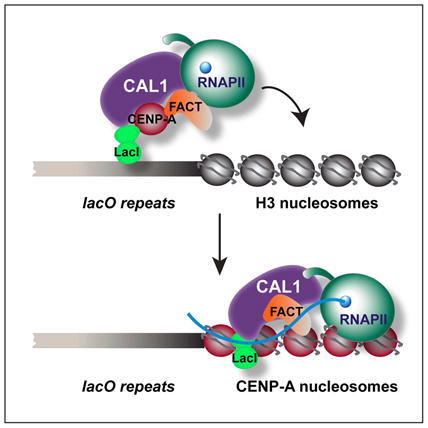
Graphical Abstract
Introduction
Accurate chromosome segregation during cell division is dependent upon the correct assembly and propagation of a distinct region of the chromosome known as the centromere. The centromere forms the structural basis for the assembly of the kinetochore, a multi-protein complex to which spindle microtubules attach during mitosis and meiosis. In most eukaryotes, the position of the centromere is defined epigenetically through the heritable incorporation of the histone H3 variant, CENP-A, which is both necessary and sufficient for centromere activity (De Rop et al., 2012).
Centromeric chromatin displays a conserved organization composed of interspersed blocks of CENP-A and H3 nucleosomes (Blower et al., 2002). During DNA replication in human cells, no new CENP-A deposition occurs (Jansen et al., 2007) and histone H3.3, and H3.1 are deposited as place-holders (Dunleavy et al., 2011). CENP-A deposition occurs during or after mitosis in Drosophila and humans, respectively (Hemmerich et al., 2008; Jansen et al., 2007; Mellone et al., 2011; Schuh et al., 2007) and is mediated by specialized histone chaperones known as Scm3 in fungi (Camahort et al., 2007; Pidoux et al., 2009; Stoler et al., 2007), HJURP in tetrapods (Barnhart et al., 2011; Bernad et al., 2011; Dunleavy et al., 2009; Foltz et al., 2009; Sanchez-Pulido et al., 2009; Shuaib et al., 2010), and CAL1 in flies (Chen et al., 2014). Each of these chaperones has been shown to selectively bind CENP-A, and not canonical H3, and to mediate the formation of CENP-A nucleosomes in vitro. However, how place-holder nucleosomes are reorganized to incorporate CENP-A/H4 tetramers is unknown. Additional histone chaperones have been found to either bind to CENP-A or contribute to proper CENP-A localization in vertebrate cells (Foltz et al., 2006; Okada et al., 2009; Perpelescu et al., 2009), but whether or not they are involved in this reorganization is unknown.
Mounting evidence points to a functional interplay between the transcription of centromeric repeats and centromere function across species. For instance, manipulation of a human artificial chromosome (HAC) revealed that targeting a transcriptional silencer to alpha-satellite repeats caused loss of CENP-A (Nakano et al., 2008). Remarkably, transcripts emanating from centromeric DNA have been identified in yeast, human, wallabies and plants (Carone et al., 2009; Chan et al., 2012; Choi et al., 2011; Ohkuni and Kitagawa, 2011; Quenet and Dalal, 2014; Topp et al., 2004)), and have been shown to be important for centromere integrity. However, the idea that specific RNAs play a role in centromere integrity is inconsistent with the notion that centromeres can form independently of centromeric DNA (Marshall et al., 2008). Additionally, the functional significance of transcription in the CENP-A assembly cascade remains poorly defined.
Here, we identify RNA Polymerase II (RNAPII)-dependent transcription as a key requirement for Drosophila CENP-A deposition. Using an inducible ectopic centromere system, which allows for the direct comparison of chromatin states in the presence or absence of active CENP-A deposition, we find that CENP-A assembly by its loading factor CAL1 is coupled with transcription of the underlying DNA. We identify FACT (Facilitates Chromatin Transcription (Orphanides et al., 1998)) as a central molecular player in this process, and show that its role in centromere integrity is that of driving DNA sequence-independent RNAPII transcription through centromeric chromatin via a direct interaction with CAL1. Thus, current models for centromere transcription must take into account the transcriptional requirements for CENP-A recruitment by its assembly factor.
Results
De novo CENP-A incorporation temporally coincides with transcription of the underlying DNA
Transcription of centromeric DNA has been described in several species (Chan and Wong, 2012), but whether or not it is directly linked to CENP-A deposition has remained elusive. One limitation of studying transcription at endogenous centromeres is the inability to precisely compare the same genomic locus in the presence and absence of active CENP-A deposition without interfering with cell-cycle progression or global transcription, which can result in reciprocal perturbation (Adolph et al., 1993; Whitfield et al., 2002). Furthermore, the endogenous CENP-A-bound DNA sequences of Drosophila are unknown, making the assessment of their transcription unfeasible. To overcome these limitations, we employed an ectopic centromere strategy based on the LacI/lacO system (Straight et al., 1996). This system utilizes a stably inserted lacO vector (10 kb of lacO repeats and 3kb of vector backbone inserted within one arm of chromosome 2 or 3 (Mendiburo et al., 2011)) coupled with the inducible expression of the Drosophila CENP-A chaperone CAL1 fused to the lac repressor, LacI, (CAL1-GFP-LacI; Figure 1A). A GFP-LacI protein is used as a negative control. LacO tethered CAL1-GFP-LacI induces the formation of fully functional and epigenetically propagated ectopic centromeres at the lacO site (Chen et al., 2014), allowing the direct comparison of the transcriptional status between lacO DNA with or without ongoing CENP-A incorporation.
Figure 1. CENP-A deposition at the ectopic lacO site is associated with transcription.

A) Experimental approach to determine if transcription is coupled with CENP-A deposition. The lacO vector is stably inserted in S2 cells and contains 256 lacO repeats (lacO array; blue bar), the bacterial Amp resistance gene (black arrow), and the yeast TRP1 gene (red arrow). Primer set 1.6 (arrow) is within the lacO vector backbone (lacOb). B) Experimental strategy used to follow ectopic CENP-A deposition and transcription from lacOb after induction of CAL1-GFP-LacI. C) Quantification of the presence (+) or absence (−) of CAL1-GFP-LacI and CENP-A foci at the lacO site during a time course. n=100 cells for each time point. D) qRT-PCR analysis of lacOb (black) and CAL1-GFP-LacI transcripts (blue) in induced CAL1-GFP-LacI cells at the indicated times. Error bars, SD of 3 technical replicates. E) qRT-PCR measuring lacOb transcription after 24h induction in cell lines: lacO only (no LacI), lacO with GFP-LacI (GFP), and lacO with CAL1-GFP-LacI (CAL1). Shown are the means ±SD of 3 experiments. p=0.01, unpaired t-test. F) Transcription from lacOb determined by qRT-PCR in CAL1-GFP-LacI cells (induced 24h) where the lacO plasmid is episomal. Error bars, 95% CI of 3 technical replicates. See also Figure S1.
CAL1-GFP-LacI is under the control of a metallothionein (MT) promoter, which can be induced by addition of CuSO4 to the growth medium. First, we investigated how long after induction of CAL1-GFP-LacI CENP-A foci become visible at the lacO site by immunofluorescence (IF) on metaphase chromosome spreads. In parallel, we assessed transcription from the lacO backbone (lacOb) by quantitative reverse-transcription PCR (qRT-PCR; primer set 1.6; Figure 1A–B; (Mendiburo et al., 2011)). Transcription of the lacO array portion of the vector could not be assessed by this method due to its repetitive nature. 2h after induction, 54% of metaphase spreads displayed CAL1-GFP-LacI foci on the lacO-containing chromosome arm, with ~65% of these foci also containing CENP-A, showing that the recruitment of CENP-A at the lacO site occurs soon after CAL1-GFP-LacI induction (Figure 1C). Strikingly, at the 2h time point, a 28-fold change in transcription from lacOb was detected by qRT-PCR. Furthermore, lacOb transcript abundance persisted throughout the remainder of the time course (Figure 1D). The levels of LacI and lacOb transcripts displayed a decrease at 8h post-induction that was also observed in an independent time course experiment (Figure S1A). This transient dip likely reflects the kinetics of induction of the MT promoter (Bunch et al., 1988) driving CAL1-GFP-LacI. Detection of nascent lacOb transcripts showed they are produced continuously, reaching a peak between 24–30h after induction (Figure S1B–C), a time when it is expected that most lacO cells have acquired CENP-A at the lacO site (Figure 1C). Thus, we reveal a striking correlation between transcription and CENP-A incorporation at the lacO site.
To ensure that transcription of lacOb is not due to the addition of CuSO4 or not somehow linked to the binding of the GFP-LacI fusion protein to the lacO array, we carried out qRT-PCR comparisons in the presence or absence of CuSO4 between the following cell lines: CAL1-GFP-LacI cells, GFP-LacI cells, and cells completely lacking any LacI transgene, yet still harboring the integrated lacO plasmid (lacO). These experiments showed that transcription from lacOb was only observed in induced CAL1-GFP-LacI cells (Figure 1E). No lacOb transcription was detected in induced cells containing CAL1-GFP-LacI or GFP-LacI without lacO (data not shown).
The expression of a control gene, actin, which is transcribed by RNAPII, was unaffected, suggesting that addition of CuSO4 does not cause non-specific transcriptional up-regulation (Figure S1D). An increase in transcription from lacOb was also observed when the lacO plasmid was introduced episomally, along with the CAL1-GFP-LacI plasmid, via transient transfection in S2 cells (Figure 1F). These transient transfections displayed low efficiency (~12%, as estimated by IF with anti-GFP antibodies), which resulted in less transcripts being detected by qRT-PCR compared to stable cells, nonetheless they demonstrate that transcription of lacOb occurs independently of its chromosomal insertion.
Transcription of lacOb correlates with CENP-A and RNAPII distribution
To gain more insight into the relationship between transcription and CENP-A occupancy across the lacO locus, we performed paired-end RNA-seq and ChIP-seq experiments. RNA-seq of induced CAL1-GFP-LacI cells revealed that 28.5 fragments per million (fpm) mapped to lacOb whereas only 0.45 fpm mapped to the lacO array itself, where CAL1 is tethered. This could be due to blockage of RNAP passage by LacI bound to lacO repeats (Jacob and Monod, 1961). Thus, upon CAL1-GFP-LacI induction, most of the transcription originates from lacOb sequences. Consistent with our qRT-PCR results (Figure 1D–F), induced GFP-LacI cells displayed fewer reads mapping to lacOb (12.8 fpm; p<0.05; Figure 2A).
Figure 2. Transcription of lacOb correlates with CENP-A and RNAPII distribution.

A) Coverage tracks of paired-end RNA-seq, from induced (24h) CAL1-GFP-LacI (top) and GFP-LacI cells (bottom), mapped to the lacO vector. The x axis represents the position along the vector, while the y axis represents the fragments per million reads (normalized to the sequencing depth of each library). p<0.05 (see supplementary experimental procedures). B) Coverage tracks of CENP-A and RNAPIIS2p paired-end ChIP-seq, from induced (24) and uninduced CAL1-GFP-LacI cells, mapped to the lacO vector. The x axis represents the position along the vector, while the y axis represents the fragments per million reads (normalized to the sequencing depth of each library). C) Schematic of the lacO vector as in Figure 1A. D) GFP ChIP-qPCR in CAL1-GFP-LacI uninduced and induced (24h). Error bars, 95% CI. p-value, unpaired t-test. E) Western blots with indicated antibodies of CAL1 IPs from chromatin extracts digested with either DNase or MNase. IN= input, M=mock (IP with beads only).
CENP-A ChIP-seq of induced CAL1-GFP-LacI cells revealed a preferential association of CENP-A with lacOb versus to the lacO array (4751.6 versus 2462.5 fpm; Figure 2B–C). Since GFP ChIP-qPCR showed that CAL1-GFP-LacI is also enriched at lacOb (Figure 2D), we conclude that CENP-A and CAL1-GFP-LacI spread to lacOb from the lacO array where CAL1-GFP-LacI is initially tethered.
RNAPII occupancy and low-level transcription has been observed at the central core of fission yeast centromeres (Catania et al., 2015), raising the possibility that RNAPII may mediate transcription of lacOb upon CAL1-GFP-LacI tethering. ChIP-seq with antibodies specific for the elongating form of RNAPII (RNAPIIS2p) showed a marked increase in RNAPIIS2p occupancy at the lacOb after CAL1-GFP-LacI induction (178.3 versus 75.5 fpm), suggesting that RNAPII mediates lacOb transcription. Furthermore, the distribution of RNAPIIS2p closely resembled that of CENP-A in induced cells, with low occupancy on the lacO array and higher on lacOb (32.3 versus 178.3 fpm; Figure 2B–C), consistent with a functional interplay between CENP-A assembly and transcriptional activity.
Low levels of CENP-A and RNAPIIS2p were observed in uninduced CAL1-GFP-LacI cells (Figure 2B), possibly due to leaky expression of CAL1-GFP-LacI, but these were found to be significantly lower than in induced samples (p<0.001 and q<0.001 for both CENP-A and RNAPIIS2p ChIPs, see supplementary experimental procedures, ChIP-seq and RNA-seq mapping).
Interestingly, immunoprecipitation (IP) of chromatin-associated CAL1 revealed a physical association between CAL1 and RNAPIIS2p (Figure 2E). This interaction and our ChIP-seq results indicate that CAL1 recruits RNAPIIS2p onto chromatin, which in turn stimulates transcription. Why only a subset of the RNAPIIS2p-associated sequences produced transcripts by RNA-seq remains unclear. It’s possible that some of these transcripts are unstable and cannot be detected by this type of assay.
Isolation of FACT, a novel CAL1 interactor
Having shown that targeting of CAL1 to lacO triggers accumulation of RNAPIIS2p and transcription, we sought to identify the key components of this process by isolating CAL1-interacting factors. We performed IPs of FLAG-tagged CAL1 (Chen et al., 2012) from chromatin-free (CF) and chromatin-associated (CA) cell extracts with anti-FLAG- M2 agarose beads, using Drosophila S2 cells (no FLAG tag) as a negative control. Mass spectrometric analysis (see Table S1 and data not shown) identified among the highest scoring unique hits Spt16 (called Dre4 in Drosophila), and SSRP1, the two subunits of the heterodimeric FACT complex (Orphanides et al., 1999). FACT allows the progression of the transcriptional machinery through chromatinized templates (Belotserkovskaya et al., 2003; Orphanides et al., 1999), by a mechanism involving nucleosome destabilization (Hondele and Ladurner, 2013; Hondele et al., 2013), and was found to be associated with human CENP-A (Foltz et al., 2006), and to be important for CENP-A localization in chickens (Okada et al., 2009). Thus, FACT is a strong candidate for mediating CENP-A deposition-coupled transcription. In order to confirm the association between CAL1 and the FACT complex, CF and CA fractions were prepared from S2 cells and CAL1 IPs were performed using anti-CAL1 antibodies. Dre4 and SSRP1 were present in both fractions (Figure 3A) and were associated with CAL1 in both cases (Figure 3B), confirming our proteomic results. Reciprocal IPs were performed using anti-FLAG antibodies to precipitate FLAG-Dre4 or FLAG-SSRP1, however, CAL1 was not detected (Figure S2A), suggesting that only a small fraction of FACT interacts with CAL1.
Figure 3. FACT interacts with CAL1 and localizes to the centromere in S2 cells.

A) Western blots of chromatin-free and (CF) chromatin-associated (CA) extracts from S2 cells with indicated antibodies. Tubulin and histone H3 antibodies are positive controls for their respective fractions. B) Western blots of IPs with anti-CAL1 antibodies from CF and CA extracts. Mock are IPs with rabbit IgGs. C) Direct interaction between in vitro translated 35S-methionine-labeled CAL1 with recombinant His::Dre4 or His::SSRP1 bound to Ni-NTA beads. His::MBP, negative control. D) IF with anti-SSRP1 or anti-Dre4 (green), anti-CENP-A (red), and anti-fibrillarin (blue) antibodies. DAPI shown in gray. Insets show 3× magnifications of boxed centromere. Bar 5μm. E) IF on metaphase chromosomes with anti-SSRP1 or anti-Dre4 (green), and anti-CENP-A (red) antibodies. DAPI shown in gray. Bar 1μm. See also Figure S2 and Table S1.
Next, we sought to determine if the association between CAL1, Dre4, and SSRP1 is direct, by analyzing protein-protein interactions between recombinant His-Dre4 or His-SSPR1 and in vitro translated 35S-methionine-labeled CAL1. The His-Dre4 and His-SSRP1 proteins heterodimerized in vitro (Figure S2B), suggesting that they are properly folded, and both pulled-down CAL1 (Figure 3C), demonstrating direct interaction.
FACT is involved in RNAPII (Belotserkovskaya et al., 2003; Krogan et al., 2002; LeRoy et al., 1998; Orphanides et al., 1998), RNAPI, and RNAPIII transcription (Birch et al., 2009); therefore, it is expected to be broadly distributed throughout chromatin. To determine if FACT displays any centromeric enrichment, we used IF with anti-Dre4 and anti-SSRP1 antibodies. After extraction with detergent pre-fixation, a treatment expected to remove loosely-chromatin bound proteins, FACT was enriched at interphase centromeres (identified by CENP-A staining) and at the nucleolus (identified by Fibrillarin staining; Figure 3D). Examination of FACT localization on metaphase chromosome spreads revealed an even more striking centromeric accumulation of Dre4 and SSRP1, demonstrating that during mitosis, when active deposition of newly synthesized CENP-A takes place (Mellone et al., 2011), FACT is more strongly associated with the centromere than with other regions of the genome (Figure 3E). These results were confirmed with epitope tagged SSRP1 and Dre4 (data not shown) and are consistent with a previous study in chicken DT-40 cells (Okada et al., 2009).
The transcription associated with CENP-A deposition requires FACT
Given that FACT enables RNAP progression, we next investigated whether FACT is required for the transcription we observed during CAL1-mediated CENP-A assembly at the lacO site. First, we investigated if CAL1-GFP-LacI recruits FACT to lacOb using ChIP-qPCR with anti-SSRP1 antibodies (Nakayama et al., 2007). CENP-A ChIPs were performed in parallel. CAL1-GFP-LacI or GFP-LacI (negative control) cells were induced for 24h. CENP-A and SSRP1 were both found enriched in induced CAL1-GFP-LacI cells (Figure 4A). We conclude that FACT is recruited by CAL1-GFP-LacI to lacOb.
Figure 4. FACT is required for CENP-A deposition-coupled transcription.

A) CENP-A and SSRP1 ChIP-qPCR in CAL1-GFP-LacI and GFP-LacI lacO cells. The graph shows the enrichment of induced (24h) relative to uninduced cells. Error bars, 95% CI of 3 technical replicates. Significant p-values (unpaired t-test) are shown. B) qRT-PCR lacOb transcripts in CAL1-GFP-LacI cells induced (24h) 6 days after the indicated RNAi treatments. p-values (unpaired t-test) are shown. C) qRT-PCR lacOb transcripts in control (purple) and SSRP1/Dre4 RNAi (blue) cells at the indicated times. Error bars, SD of 3 technical replicates. D) IF with anti-CENP-A (red) and anti-GFP (green) antibodies in lacO cells expressing full length CAL1-GFP-LacI (top) or CAL1Δ1-40-GFP-LacI (bottom). DAPI is shown in gray. Arrow points to the lacO site. Bar 1μm. E) Direct interaction between in vitro translated 35S-methionine-labelled CAL1Δ1-40 (35S-Δ1-40) with recombinant His::Dre4 (Dre4) or His::SSRP1 (SSRP1) bound to Ni-NTA beads. His::MBP (MBP) is a negative control. F) qRT-PCR of lacOb transcripts in induced cells (24h) transiently expressing full length (f.l.) CAL1-GFP-LacI or CAL1Δ1-40-GFP-LacI. Shown is mean ±SEM of 3 experiments p=0.68 (not significant; unpaired t-test).
We next measured lacOb transcription by qRT-PCR after induction of CAL1-GFP-LacI cells in which Dre4 and SSRP1 had been knocked-down by RNA interference (RNAi) for 6 days. Control cells displayed, on average, a 6.8 fold increase in lacOb transcript levels 24h after induction with CuSO4. In contrast, cells lacking FACT showed virtually no increase (Figure 4B). This result was also confirmed in a time course experiment, in which, after 6 days of FACT RNAi, CAL1-GFP-LacI was induced and qRT-PCR was performed on RNA extracted every 4h for 24h (Figure 4C). Together, these data demonstrate that FACT is required for the transcription observed upon CAL1 targeting.
CAL1-directed transcription could be a by-product of CENP-A incorporation or it could occur independently of CENP-A deposition through the recruitment of RNAPII and FACT onto chromatin. To distinguish between these two possibilities, we used a CAL1 mutation lacking a short Scm3-like domain ((Phansalkar et al., 2012); CAL1Δ1-40), which is defective in recruiting CENP-A to the lacO (Figure 4D; (Chen et al., 2014)). Importantly, CAL1Δ1-40 can interact directly with Dre4 and SSRP1 (Figure 4E). When we tethered CAL1Δ1-40-GFP-LacI to the lacO, we observed levels of lacOb transcription indistinguishable from those initiated by CAL1-GFP-LacI (Figure 4F). We conclude that lacOb transcription depends on CAL1 and FACT, but it does not require CENP-A incorporation.
FACT-mediated transcription is required for de novo CENP-A incorporation
CAL1 binds directly to FACT and recruits FACT and RNAPII to sites of CENP-A assembly. To determine if the absence of lacOb transcription caused by knock-down of FACT affects deposition of CENP-A at the lacO site, we depleted Dre4 or SSRP1 by RNAi for 5 days, induced CAL1-GFP-LacI for 24h, and performed IF with anti-CENP-A and anti-GFP antibodies on metaphase spreads (Figure 5A). Ectopic targeting of CAL1 via LacI/lacO leads to efficient de novo incorporation of CENP-A (Chen et al., 2014). In contrast, depletion of either SSRP1 or Dre4 resulted in a significant reduction in the percentage of CENP-A positive lacO sites (Figure 5B–C). Since FACT depletion did not affect the formation of the CENP-A/CENP-C/CAL1 complex ((Erhardt et al., 2008); Figure S3A), a defect in CENP-A incorporation is the most likely explanation for this reduction in ectopic CENP-A. Thus, these experiments demonstrate that efficient recruitment of CENP-A by CAL1 requires FACT and imply that CAL1 is not sufficient to assemble CENP-A into nucleosomes when chromatin is the substrate, as opposed to when naked DNA is the substrate (Chen et al., 2014).
Figure 5. FACT is required for the de novo incorporation of CENP-A at an ectopic locus.

A) Cartoon depicting the experimental strategy to assess de novo CENP-A recruitment in the absence of FACT. B) lacO CAL1-GFP-LacI cells were subjected to RNAi to deplete Dre4, SSRP1, or a control for 5 days followed by induction with CuSO4 for 24h. Metaphase chromosome spreads were stained with anti-GFP (green) and anti-CENP-A antibodies (red). DNA was stained with DAPI (blue). C) Graph showing the percentage of CAL1-GFP-LacI positive cells in which ectopic CENP-A signal was present or absent. Bars = 1μm. p<0.0001 (Chi-square). D) FACS profile of control, CAL1, and SSRP1/Dre4 (FACT) RNAi cells. See also Figure S3 and Table S2.
Given the ubiquitous role of FACT in DNA metabolism, we investigated possible pleiotropic effects that could account for the CENP-A incorporation defect seen after FACT RNAi. FACS analysis showed no change in the distribution of cells in G1, S or G2-M upon FACT RNAi (Figure 5D), suggesting that the CENP-A incorporation defect is not due to a cell cycle defect. Additionally, qRT-PCR analyses of cenp-a, cal1, or cenp-c transcripts (Figure S3B–C) and Western blot analyses from total protein extracts (Figure S3D) demonstrated that FACT depletion did not decrease the expression of these essential centromere genes. Similarly, expression of 8 hand-picked genes that are bound to Dre4 based on ChIP-chip data ((Kharchenko et al., 2011); Table S2), as well as that of the FACT-associated gene Hsp70 (Saunders et al., 2003), did not decrease upon FACT RNAi. This suggests that general transcription involves redundant chromatin remodeling activities, at least in Drosophila cultured cells. Altogether, these results demonstrate that FACT plays a specific function in CENP-A deposition.
Depletion of FACT causes defective CENP-A recruitment at endogenous centromeres
To determine if FACT is required for the recruitment of newly-synthesized CENP-A at endogenous centromeres, we performed quench-chase-pulse experiments in cells stably expressing SNAP-tagged CENP-A (Jansen et al., 2007; Mellone et al., 2011). FACT was knocked-down by simultaneous RNAi of Dre4 and SSRP1 for 6 days after which pre-existing SNAP-tagged CENP-A was irreversibly quenched using the BG-blocking agent (T0; quench). RNAi of CAL1 was used as a positive control. After a chase that lasted until cells had divided once, newly synthesized SNAP-tagged CENP-A was labeled using TMR* (T1; pulse) and cells were fixed and processed for IF (Figure 6A). Immediately after quenching SNAP-CENP-A, no TMR* labeled-CENP-A signal was observed, as expected, while labeling with an anti-CENP-A antibody showed that low levels of CENP-A were still present in both FACT and CAL1 RNAi (Figure 6B, T0). After one cell division, cells were incubated with TMR* to label newly synthesized SNAP-CENP-A. Newly synthesized SNAP-CENP-A was clearly visible at the centromeres of control cells (Figure 6B, T1, top). In contrast, there was a significant drop in the TMR*-CENP-A intensity levels of FACT RNAi cells, consistent with defective CENP-A recruitment (Figure 6B, T1, and 6C).
Figure 6. FACT is required for CENP-A recruitment at endogenous centromeres.

A) Diagram of the quench-chase-pulse experiment. B) IF with anti-CENP-A antibodies (green) in control and Dre4/SSRP1 RNAi SNAP-CENP-A cells pulsed with TMR* (red) immediately after BG-block (T0, left panel) or after having completed one cell division (T1, right panel). DAPI is shown in blue. Bars 5μm. C) Quantification of the signal intensity of TMR*-labeled CENP-A foci. Shown are the means ±SEM of 3 experiments (100 cells quantified per RNAi treatment). p<0.0001 for control versus SSRP1/Dre4 RNAi (unpaired t-test. D) The total CENP-A centromeric intensity for control cells, CAL1 RNAi cells, and SSRP/Dre4 RNAi (FACT) was quantified at T0 and T1. Shown is the mean change in CENP-A intensity at T1 relative to T0 ±SEM. n=3 experiments (150 cells each RNAi treatment). Unpaired t-test p-values are shown. E) IF with anti-CENP-A antibodies of S2 cells subjected to the indicated RNAi treatments. DNA is stained with DAPI. Bar 5μm. F) Scatter dot plot showing total centromeric CENP-A signal intensity per cell from the experiment in E. n=50 cells per condition. Unpaired t-test p-values are shown. See also Figure S4.
To determine if FACT is also required to retain pre-existing centromeric CENP-A through one cell division, we quantified the total centromeric CENP-A IF signal at T0 and T1. In control cells, retention and recruitment of CENP-A are intact; therefore, no change in total CENP-A intensity occurs over one cell division (T1/T0= 100%). In contrast, in cells lacking FACT, centromeric CENP-A signal displayed a decrease in intensity consistent with a loading defect (T1/T0= ~59%; Figure 6D; a ratio lower that 50% would be expected if the retention of pre-existing CENP-A were also affected). These results also explain why CENP-A is lost at a relatively slower rate in the absence of FACT (6 days): its loading is compromised, but its retention is not. In contrast, loss of CENP-A from the centromere in the absence of CAL1 is much more rapid (Figure 6D), consistent with the dual role of CAL1 in CENP-A loading and stabilization from degradation (Chen et al., 2014). Collectively, our data demonstrate that FACT is required for the centromeric recruitment of newly synthesized CENP-A at the centromere.
To examine if FACT depletion can lead to complete loss of CENP-A from centromeres, we knocked-down Dre4 or SSRP1, transfecting S2 cells twice with double stranded RNA over 6 days, and examined the intensity of centromeric CENP-A by IF. We observed a dramatic decrease in the intensity of CENP-A foci upon Dre4 or SSRP1 RNAi compared to control cells (Figure 6E–F), demonstrating that two consecutive RNAi lead to nearly-complete loss of CENP-A from centromeres. Consistent with defective CENP-A recruitment, we observed a significant increase in chromosome missegregation in mitosis in cells lacking FACT (Figure S4).
H3.1 and H3.3 accumulate within centromeric chromatin upon FACT RNAi
In human cells, histone H3 nucleosomes are deposited in S phase as temporary placeholders that need to be replaced by CENP-A in order to maintain centromere identity (Dunleavy et al., 2011). Whether CENP-A chaperones or other factors perform this exchange is unknown. Transcription at the centromere could mediate the eviction of placeholder H3 during CAL1-mediated CENP-A deposition, analogously to H3.3 deposition at active genes (Schwartz and Ahmad, 2005). To determine if, in the absence of FACT and transcription, histone H3.1 or H3.3 accumulate at centromeres, we depleted Dre4 (which causes loss of SSRP1 as well; Figure S5) in S2 cells transiently transfected with plasmids expressing V5 tagged H3.1 and H3.3, and inspected centromeric chromatin by IF on stretched chromatin fibers. In Dre4 depleted cells, the average length of continuous CENP-A fibers was about half that of control cells (Figure 7A) and the CENP-A signal became less contiguous, suggesting that CENP-A is lost throughout CENP-A centromeric chromatin stretches as well as from their edges. IF with anti-CENP-A and anti-V5 antibodies showed that H3.1 and H3.3 were present continuously across CENP-A fibers in control and in RNAi cells, indicating that upon loss of CENP-A no “gaps” were left at the centromere (Figure 7B). These results, which are consistent with a previous study that looked at centromeric fibers upon CENP-A depletion (Blower et al., 2002), suggest defective exchange between placeholders H3.1/H3.3 and CENP-A in the absence of FACT and transcription.
Figure 7. FACT depletion results in the accumulation of histone H3-containing nucleosomes within centromeric chromatin.

A) Quantification of CENP-A fiber length from 3 experiments (n=74 total fibers per condition). Shown are means ± SD. p<0.0001 for contr. vs each RNAi (unpaired t-test). B) IF on stretched chromatin fibers from control and Dre4 RNAi S2 cells expressing H3.1-V5 or H3.3-V5. V5 shown in red and CENP-A in green (n=10–17 fibers per condition). Bar 5μm. C) Model for the role of RNAPII transcription in centromere propagation. FACT is recruited to the centromere along with CAL1 and CENP-A/H4; here, it destabilizes H3-containing nucleosomes allowing the passage of RNAPII through chromatin (1). RNAPII transcribes through the region causing the eviction of H3/H4 tetramers (2) thereby allowing deposition of new CENP-A/H4 tetramers (3).
Collectively, our data suggest a model in which FACT is recruited to the centromere by interacting directly with CAL1 in a pre-nucleosomal complex. Once at the centromere, FACT destabilizes nucleosomes (Hondele and Ladurner, 2013; Hondele et al., 2013) allowing transcription through the region via RNAPII. Finally, transcription by RNAPII causes the eviction of place-holders H3.1 and H3.3 allowing the deposition of CENP-A by CAL1 (Figure 7C).
Discussion
The epigenetic maintenance of centromeres through faithful CENP-A deposition is a process crucial for genome stability. Much of the recent advances in understanding this process in metazoans have focused on the dissection of the specific proteins involved in CENP-A recruitment. In contrast, the roles of DNA and chromatin in CENP-A deposition have largely remained elusive. In this study, we have uncovered a key role for transcription in Drosophila CENP-A deposition and have identified FACT as a central player in this process. This mechanism of nucleosome reorganization- combining RNAPII passage with CENP-A/H3 exchange- is analogous to other paradigms seen during transcription and development. For example, FACT is recruited to specific genomic loci by the GAGA factor where it destabilizes nucleosomes allowing replacement of histone H3.1 with H3.3 by the chaperone HIRA, thereby modulating the expression of Hox genes (Nakayama et al., 2007; Shimojima et al., 2003).
To ensure the fidelity of centromere propagation, CENP-A chromatin must be replenished after each round of DNA replication. In human cells, newly synthesized CENP-A is recruited to centromeric chromatin along with newly synthesized histone H4, indicating that CENP-A and H4 form a sub-nucleosomal core that is assembled simultaneously (Bodor et al., 2013). As such, it is conceivable that CENP-A/H4 deposition involves the eviction of pre-existing H3/H4 tetramers.
To determine if CENP-A assembly is coupled to transcription, we used an inducible ectopic centromere system in Drosophila S2 cells. We discovered that a remarkable change in transcription occurs rapidly upon CAL1-GFP-LacI targeting at the lacO site. The same DNA that is transcribed is enriched in RNAPII, suggesting that this polymerase is the one mediating this transcription. The interaction between CAL1 and RNAPII supports this idea, although the involvement of additional RNAPs cannot be ruled out.
In order to characterize this phenomenon mechanistically, we biochemically isolated the novel CAL1 partner FACT and demonstrated that it is necessary for the transcription of the lacO site. Despite its function in global RNAP elongation, the depletion of FACT did not cause a decrease in expression of FACT-associated genes, suggesting a redundancy of mechanisms directing general transcription in Drosophila cells. In contrast, upon FACT RNAi, transcription at the lacO site was impaired, resulting in defective de novo CENP-A deposition, demonstrating a specific disruption of centromere chromatin assembly.
Surprisingly, we found that transcription at the lacO site is independent of CENP-A assembly, revealing that CENP-A chaperones can initiate local chromatin reorganization through the recruitment of FACT and RNAPII.
The discovery that chromatin poses a barrier to CENP-A deposition by its chaperone and the involvement of FACT-mediated transcription in overcoming this barrier are likely to be relevant to other complex eukaryotes. In budding yeast, FACT allows Phs1 to access misincorporated CENP-A/Cse4 nucleosomes allowing the ubiquitylation and subsequent degradation of CENP-A/Cse4 (Deyter and Biggins, 2014). However, our studies in Drosophila demonstrate that FACT is directly implicated in CENP-A deposition. The finding that FACT is required for CENP-A localization in chicken (Okada et al., 2009) and interacts with human CENP-A (Foltz et al., 2006) raises the possibility that the mechanism by which FACT promotes chromatin reorganization during CENP-A deposition by its chaperone may be conserved conserved from flies to vertebrates.
In budding yeast, FACT increases nucleosome accessibility to nucleases in the absence of H2A-H2B dimer displacement, suggesting that it can reorganize nucleosomes in a more open configuration, while maintaining their original composition (Xin et al., 2009). Consistent with this, the crystal structure and mutational analyses of Spt16/Dre4 showed that FACT allows a gradual invasion of the nucleosome, breaking strong octamer-DNA contacts and allowing the passage of polymerases (Hondele et al., 2013). Thus, FACT is likely to function as a nucleosome destabilizer (Hondele and Ladurner, 2013) allowing the passage of RNAPII, which in turn interacts with CAL1 (Figure 7C).
A question that remains unanswered is whether the transcripts produced during CENP-A deposition are simply a by-product of the ongoing chromatin reorganization or are necessary components of centromere structure and identity. Specific RNAs emanating from centromeres do appear to play a role in centromere/kinetochore integrity (Quenet and Dalal, 2014; Rosic et al., 2014; Topp et al., 2004). However, the sequence requirements of these RNAs remain poorly defined. Our work demonstrates a requirement for transcription in CENP-A deposition as a means to reorganize nucleosomes and suggests the dispensability of specific centromeric RNA sequences in this process. Either there is a generic, non-sequence specific role for RNA at the centromere, or specific sequences emanating from the centromere possess additional structural properties. Further work is needed to elucidate the functional relationship between CENP-A deposition-coupled transcription and structural centromeric RNAs.
Experimental procedures
Large-scale immunoprecipitation and mass spectrometry
FLAG-CAL1 complexes were purified from chromatin-free extracts generated from 2×109 S2 cells, as described previously (Chen et al., 2012; Mellone et al., 2011). FLAG-CAL1 complexes from chromatin-associated complexes were generated by homogenization, nuclear extraction, and digestion with benzonase. Extracts were added to anti-FLAG M2 beads (Sigma-Aldrich). After washing, complexes were eluted with FLAG peptide (Sigma-Aldrich) and sent off for mass spectrometric analysis (see supplementary experimental procedures for details).
Small-scale immunoprecipitations
Extracts from chromatin-free and chromatin-associated fractions were prepared from 108 cells, as described before (Mellone et al., 2011). Extracts were added to Dynabeads-protein A beads (Life Technologies) coupled with anti-CAL1 or anti-FLAG antibodies (Sigma-Aldrich) and incubated for 10 min at room temperature followed by a 30 min incubation at 4°C with rotation. Beads were washed three times with PBS-T (PBS; 0.1% Triton). 6% of the input and 50% of the IP was analyzed by 10% SDS-PAGE followed by Western blot. See supplementary experimental procedures for CAL1/RNAPII IPs.
In vitro protein binding assay
All steps were performed at room temperature. ~ 5μg of purified His::MBP (negative control), His::Dre4 or His::SSRP1 immobilized onto Ni-NTA agarose (Qiagen) were equilibrated in binding buffer containing 50mM HEPES pH 7.4, 150mM NaCl, 1mM MgCl2, 1mM EGTA, 0.1 % Triton X-100, 1× EDTA-free protease inhibitor cocktail (Roche), 20mM imidazole and 0.5mg/ml BSA, mixed with 35S-methionine-labelled proteins expressed by a coupled in vitro transcription translation system (IVTT) and incubated for 1h. Beads were then washed in binding buffer (without BSA), proteins were eluted by boiling in Laemmli sample buffer and subjected to SDS-PAGE followed by autoradiography. See also supplementary experimental procedures.
Cell culture and RNAi
Stable S2 cells containing an integrated lacO plasmid (pAFS52; (Straight et al., 1996)) were described before (Chen et al., 2014; Mendiburo et al., 2011). Additional stable S2 cells were generated by transfection with Cellfectin reagent (Life Technologies) and selection with 450μg/ml hygromycin. Stable lacO S2 cells were re-thawed after one month in culture due to loss of the lacO array over time. Transient transfections were performed by treating cells with FuGENE HD (Promega) for 2 days. Cells were induced with 0.5mM CuSO4 for 24h. Stable CAL1-GFP-LacI or GFP-LacI cells were induced with 0.5mM CuSO4 at 25°C for 1–48h or left uninduced. RNAi was performed using DOTAP and 10μg of dsRNA (see supplementary experimental procedures).
Total RNA extraction and qRT-PCR
Total RNA was isolated from 1×107 cells using TRI-reagent (Sigma-Aldrich). 10μg of RNA was treated with 1μl of Turbo DNase (Life Technologies) for 30 min at 37°C. RNA was reverse-transcribed using the iScript cDNA synthesis kit (Bio-Rad) and 2μl were used in qPCR using SYBR-green (Bio-Rad) on a CFX96 Real-Time system (Bio-Rad). Transcription from the lacOb (using primer pairs 1.6 or 3 (Mendiburo et al., 2011)) was normalized to uninduced samples. Values were calculated using the Pfaffl method (Pfaffl, 2001) with Rp49 (unaffected by FACT (Nakayama et al., 2007)) as a reference gene. Some variability in the fold increase in lacOb transcription between experiments was observed due to instability of the lacO array during cell culture over time. See supplementary experimental procedures for primer sequences. For RNA-seq, libraries were generated using the Tru-Seq kit (Illumina) and ran on a HiSeq. See supplementary experimental procedures for mapping information.
Total protein extraction and Western blotting
Total cell extracts were obtained from 1×106 cells resuspended in 15μl of RIPA buffer (150mM NaCl, 50mM Tris, pH8, 1% NP40, 0.1% SDS), incubated on ice for 10min, and digested with 1μl of benzonase (Novagen) for 20min at 37°C. Extracts were separated by 10% SDS-PAGE and transferred to nitrocellulose membranes. After 30 min incubation in blocking buffer (TBS, 0.1% Tween 20, 5% powder non-fat milk), membranes were incubated overnight at 4°C with anti-CAL1 (rabbit, 1:1000; from A. Straight), anti-CENP-A (rabbit, 1:1000; Active Motif), anti-FLAG (mouse, 1:1000; Sigma-Aldrich), anti-Dre4 and anti-SSRP1 (rabbit, 1:1000; from S. Hirose), and anti-RNAPIIS2p (mouse, 1:1000; Abcam); anti-CENP-C (guinea pig, 1:3000; (Mellone et al., 2011)); anti-tubulin (mouse, 1:1000; Sigma-Aldrich) or anti-histone H3 (rabbit, 1:5000; Abcam) were used as loading controls.
Immunofluorescence
IF on settled cells and metaphase spreads was performed as described (Chen et al., 2014). For pre-extraction with detergent, settled cells were immersed in 100μl of PBS-T for 5 min followed by addition of 11μl of 37% formaldehyde for 10min. Stretched chromatin fibers were performed essentially as described (Sullivan, 2010), using twice the amount of primary antibodies than conventional IF. Only extensively stretched fibers (DAPI nearly undetectable) were used for our analyses. The antibodies used were anti-CENP-A (chicken, 1:1000, (Blower and Karpen, 2001)), anti-GFP (rabbit-488 conjugated, 1:100, Life Technologies), anti-CENP-C (guinea pig, 1:1000, (Mellone et al., 2011)), anti-fibrillarin (mouse, 1:500; Cytoskeleton, Inc.), and anti-V5 (mouse, 1:50, Life Technologies). Secondary antibodies (Life Technologies Alexa-Fluor 488 or 546 conjugated, and Santa Cruz biotechnology CY5 conjugated; 1:500) were used as appropriate. Slides were mounted in Slowfade (Life Technologies) containing DAPI.
Quench-Pulse-Chase Assay
RNAi of both Dre4 and SSRP1 simultaneously was performed for 6 days in a 12-well plate. Quench-chase-pulse followed by IF was performed as described (Mellone et al., 2011) making sure the cells had divided once (~24h for control and 24–48h for FACT RNAi) between BG-block (quench) and TMR* labeling (chase).
Fluorescence Activated Cell Sorting
After 6 days of RNAi (Brown for control and SSRP1/Dre4 for FACT), 1×106 S2 cells were washed in PBS with 2% BSA then incubated in PBS containing 50μg/mL Propidium Iodide, 200μg/mL RNase A, and 0.1% Triton-X-100 for 15min at 25°C in the dark. Samples were analyzed on a BD FACSCaliber Flow Cytometer and analyzed using FloJo.
Imaging
All images were taken at 25°C on a Olympus fluorescence microscope (PersonalDV; Applied Precision) equipped with a 60×1.42 NA or a 100×1.40 NA oil immersion objective (Olympus) and a CoolSnap HQ2 camera (Photometrics), keeping exposure conditions constant between all samples. Images were acquired and deconvolved using softWoRx (Applied Precision), maintaining the scaling constant between samples, and saved as Photoshop files. Figures were assembled in Adobe Illustrator. See supplementary experimental procedures for image quantifications.
Chromatin immunoprecipitations
ChIPs were performed using the MAGnify kit (Life Technologies). 106 cells (~10μg DNA) was used for each IP and chromatin was sheared to fragments 100–300bp long. 1μl of anti-CENP-A (rabbit, Active Motif), anti-SSRP1 (Nakayama et al., 2007), anti-GFP (Abcam), or anti-RNAPIIS2p (Abcam) were coupled to 10μl beads for 2h and mixed with chromatin overnight at 4°C. Immunoprecipitated DNA was eluted in 50μl of elution buffer, and analyzed by qPCR. Normalization was performed using the formula: , where AE is the amplification efficiency calculated by the formula . The values obtained for induced were normalized by those for uninduced cells to calculate enrichment. For ChIP-seq, DNA from three independent ChIPs were pooled and made into libraries with the TruSeq ChIP kit (Illumina). Samples were ran on a MiSeq using Reagent Kit v3. See supplementary experimental procedures for mapping information.
Statistical methods
Standard error, standard deviation, and confidence intervals were calculated using Numbers (Apple). Unpaired t-test and Chi-square were performed in Prism (Graphpad). See supplementary experimental procedures for statistical analysis of next-gen sequencing data.
Supplementary Material
Highlights.
CENP-A deposition is coupled with transcription
CAL1 recruits RNAPII onto DNA during CENP-A deposition
CAL1 interacts directly with the histone chaperone FACT
FACT depletion causes loss of transcription and defective CENP-A deposition
Acknowledgments
We thank Bo Reese (CGI) for sequencing support, G. Karpen, S. Hirose, A. Straight and P. Heun for reagents, I. Cheeseman for critical reading of the manuscript, L. Core for suggestions and help with ChIP-chip data analysis, and the Drosophila RNAi Screening Center (DRSC) for resources. This work was supported by awards NSF 1024973 and NIH GM108829 to B.G.M. D.M.G. is supported by grants from Cancer Research UK (C3/A11431) and Medical Research Council (G1001696), Z.L. by a long-term FEBS fellowship, and R.J.O. by NSF award 1244146.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- Adolph S, Brusselbach S, Muller R. Inhibition of transcription blocks cell cycle progression of NIH3T3 fibroblasts specifically in G1. J Cell Sci. 1993;105 ( Pt 1):113–122. doi: 10.1242/jcs.105.1.113. [DOI] [PubMed] [Google Scholar]
- Barnhart MC, Kuich PH, Stellfox ME, Ward JA, Bassett EA, Black BE, Foltz DR. HJURP is a CENP-A chromatin assembly factor sufficient to form a functional de novo kinetochore. J Cell Biol. 2011;194:229–243. doi: 10.1083/jcb.201012017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belotserkovskaya R, Oh S, Bondarenko VA, Orphanides G, Studitsky VM, Reinberg D. FACT facilitates transcription-dependent nucleosome alteration. Science. 2003;301:1090–1093. doi: 10.1126/science.1085703. [DOI] [PubMed] [Google Scholar]
- Bernad R, Sanchez P, Rivera T, Rodriguez-Corsino M, Boyarchuk E, Vassias I, Ray-Gallet D, Arnaoutov A, Dasso M, Almouzni G, et al. Xenopus HJURP and condensin II are required for CENP-A assembly. J Cell Biol. 2011;192:569–582. doi: 10.1083/jcb.201005136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birch JL, Tan BC, Panov KI, Panova TB, Andersen JS, Owen-Hughes TA, Russell J, Lee SC, Zomerdijk JC. FACT facilitates chromatin transcription by RNA polymerases I and III. EMBO J. 2009;28:854–865. doi: 10.1038/emboj.2009.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blower MD, Karpen GH. The role of Drosophila CID in kinetochore formation, cell-cycle progression and heterochromatin interactions. Nat Cell Biol. 2001;3:730–739. doi: 10.1038/35087045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blower MD, Sullivan BA, Karpen GH. Conserved organization of centromeric chromatin in flies and humans. Dev Cell. 2002;2:319–330. doi: 10.1016/s1534-5807(02)00135-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodor DL, Valente LP, Mata JF, Black BE, Jansen LE. Assembly in G1 phase and long-term stability are unique intrinsic features of CENP-A nucleosomes. Mol Biol Cell. 2013;24:923–932. doi: 10.1091/mbc.E13-01-0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunch TA, Grinblat Y, Goldstein LS. Characterization and use of the Drosophila metallothionein promoter in cultured Drosophila melanogaster cells. Nucleic Acids Res. 1988;16:1043–1061. doi: 10.1093/nar/16.3.1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camahort R, Li B, Florens L, Swanson SK, Washburn MP, Gerton JL. Scm3 is essential to recruit the histone h3 variant cse4 to centromeres and to maintain a functional kinetochore. Mol Cell. 2007;26:853–865. doi: 10.1016/j.molcel.2007.05.013. [DOI] [PubMed] [Google Scholar]
- Carone DM, Longo MS, Ferreri GC, Hall L, Harris M, Shook N, Bulazel KV, Carone BR, Obergfell C, O'Neill MJ, et al. A new class of retroviral and satellite encoded small RNAs emanates from mammalian centromeres. Chromosoma. 2009;118:113–125. doi: 10.1007/s00412-008-0181-5. [DOI] [PubMed] [Google Scholar]
- Catania S, Pidoux AL, Allshire RC. Sequence features and transcriptional stalling within centromere DNA promote establishment of CENP-A chromatin. PLoS Genet. 2015;11:e1004986. doi: 10.1371/journal.pgen.1004986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan FL, Marshall OJ, Saffery R, Kim BW, Earle E, Choo KH, Wong LH. Active transcription and essential role of RNA polymerase II at the centromere during mitosis. Proc Natl Acad Sci U S A. 2012;109:1979–1984. doi: 10.1073/pnas.1108705109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan FL, Wong LH. Transcription in the maintenance of centromere chromatin identity. Nucleic Acids Res. 2012;40:11178–11188. doi: 10.1093/nar/gks921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CC, Dechassa ML, Bettini E, Ledoux MB, Belisario C, Heun P, Luger K, Mellone BG. CAL1 is the Drosophila CENP-A assembly factor. J Cell Biol. 2014;204:313–329. doi: 10.1083/jcb.201305036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CC, Greene E, Bowers SR, Mellone BG. A Role for the CAL1-Partner Modulo in Centromere Integrity and Accurate Chromosome Segregation in Drosophila. PLoS One. 2012;7:e45094. doi: 10.1371/journal.pone.0045094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi ES, Stralfors A, Castillo AG, Durand-Dubief M, Ekwall K, Allshire RC. Identification of noncoding transcripts from within CENP-A chromatin at fission yeast centromeres. The Journal of biological chemistry. 2011;286:23600–23607. doi: 10.1074/jbc.M111.228510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rop V, Padeganeh A, Maddox PS. CENP-A: the key player behind centromere identity, propagation, and kinetochore assembly. Chromosoma. 2012;121:527–538. doi: 10.1007/s00412-012-0386-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deyter GM, Biggins S. The FACT complex interacts with the E3 ubiquitin ligase Psh1 to prevent ectopic localization of CENP-A. Genes Dev. 2014;28:1815–1826. doi: 10.1101/gad.243113.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunleavy EM, Almouzni G, Karpen GH. H3.3 is deposited at centromeres in S phase as a placeholder for newly assembled CENP-A in G(1) phase. Nucleus. 2011;2:146–157. doi: 10.4161/nucl.2.2.15211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunleavy EM, Roche D, Tagami H, Lacoste N, Ray-Gallet D, Nakamura Y, Daigo Y, Nakatani Y, Almouzni-Pettinotti G. HJURP is a cell-cycle-dependent maintenance and deposition factor of CENP-A at centromeres. Cell. 2009;137:485–497. doi: 10.1016/j.cell.2009.02.040. [DOI] [PubMed] [Google Scholar]
- Erhardt S, Mellone BG, Betts CM, Zhang W, Karpen GH, Straight AF. Genome-wide analysis reveals a cell cycle-dependent mechanism controlling centromere propagation. J Cell Biol. 2008;183:805–818. doi: 10.1083/jcb.200806038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foltz DR, Jansen LE, Bailey AO, Yates JR, 3rd, Bassett EA, Wood S, Black BE, Cleveland DW. Centromere-specific assembly of CENP-a nucleosomes is mediated by HJURP. Cell. 2009;137:472–484. doi: 10.1016/j.cell.2009.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foltz DR, Jansen LE, Black BE, Bailey AO, Yates JR, 3rd, Cleveland DW. The human CENP-A centromeric nucleosome-associated complex. Nat Cell Biol. 2006;8:458–469. doi: 10.1038/ncb1397. [DOI] [PubMed] [Google Scholar]
- Hemmerich P, Weidtkamp-Peters S, Hoischen C, Schmiedeberg L, Erliandri I, Diekmann S. Dynamics of inner kinetochore assembly and maintenance in living cells. J Cell Biol. 2008;180:1101–1114. doi: 10.1083/jcb.200710052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hondele M, Ladurner AG. Catch me if you can: how the histone chaperone FACT capitalizes on nucleosome breathing. Nucleus. 2013;4:443–449. doi: 10.4161/nucl.27235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hondele M, Stuwe T, Hassler M, Halbach F, Bowman A, Zhang ET, Nijmeijer B, Kotthoff C, Rybin V, Amlacher S, et al. Structural basis of histone H2A-H2B recognition by the essential chaperone FACT. Nature. 2013;499:111–114. doi: 10.1038/nature12242. [DOI] [PubMed] [Google Scholar]
- Jacob F, Monod J. Genetic regulatory mechanisms in the synthesis of proteins. J Mol Biol. 1961;3:318–356. doi: 10.1016/s0022-2836(61)80072-7. [DOI] [PubMed] [Google Scholar]
- Jansen LE, Black BE, Foltz DR, Cleveland DW. Propagation of centromeric chromatin requires exit from mitosis. J Cell Biol. 2007;176:795–805. doi: 10.1083/jcb.200701066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharchenko PV, Alekseyenko AA, Schwartz YB, Minoda A, Riddle NC, Ernst J, Sabo PJ, Larschan E, Gorchakov AA, Gu T, et al. Comprehensive analysis of the chromatin landscape in Drosophila melanogaster. Nature. 2011;471:480–485. doi: 10.1038/nature09725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogan NJ, Kim M, Ahn SH, Zhong G, Kobor MS, Cagney G, Emili A, Shilatifard A, Buratowski S, Greenblatt JF. RNA polymerase II elongation factors of Saccharomyces cerevisiae: a targeted proteomics approach. Mol Cell Biol. 2002;22:6979–6992. doi: 10.1128/MCB.22.20.6979-6992.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeRoy G, Orphanides G, Lane WS, Reinberg D. Requirement of RSF and FACT for transcription of chromatin templates in vitro. Science. 1998;282:1900–1904. doi: 10.1126/science.282.5395.1900. [DOI] [PubMed] [Google Scholar]
- Marshall OJ, Chueh AC, Wong LH, Choo KH. Neocentromeres: new insights into centromere structure, disease development, and karyotype evolution. Am J Hum Genet. 2008;82:261–282. doi: 10.1016/j.ajhg.2007.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellone BG, Grive KJ, Shteyn V, Bowers SR, Oderberg I, Karpen GH. Assembly of Drosophila centromeric chromatin proteins during mitosis. PLoS Genet. 2011;7:e1002068. doi: 10.1371/journal.pgen.1002068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendiburo MJ, Padeken J, Fulop S, Schepers A, Heun P. Drosophila CENH3 is sufficient for centromere formation. Science. 2011;334:686–690. doi: 10.1126/science.1206880. [DOI] [PubMed] [Google Scholar]
- Nakano M, Cardinale S, Noskov VN, Gassmann R, Vagnarelli P, Kandels-Lewis S, Larionov V, Earnshaw WC, Masumoto H. Inactivation of a human kinetochore by specific targeting of chromatin modifiers. Dev Cell. 2008;14:507–522. doi: 10.1016/j.devcel.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama T, Nishioka K, Dong YX, Shimojima T, Hirose S. Drosophila GAGA factor directs histone H3.3 replacement that prevents the heterochromatin spreading. Genes Dev. 2007;21:552–561. doi: 10.1101/gad.1503407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkuni K, Kitagawa K. Endogenous transcription at the centromere facilitates centromere activity in budding yeast. Curr Biol. 2011;21:1695–1703. doi: 10.1016/j.cub.2011.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada M, Okawa K, Isobe T, Fukagawa T. CENP-H-containing complex facilitates centromere deposition of CENP-A in cooperation with FACT and CHD1. Mol Biol Cell. 2009;20:3986–3995. doi: 10.1091/mbc.E09-01-0065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orphanides G, LeRoy G, Chang CH, Luse DS, Reinberg D. FACT, a factor that facilitates transcript elongation through nucleosomes. Cell. 1998;92:105–116. doi: 10.1016/s0092-8674(00)80903-4. [DOI] [PubMed] [Google Scholar]
- Orphanides G, Wu WH, Lane WS, Hampsey M, Reinberg D. The chromatin-specific transcription elongation factor FACT comprises human SPT16 and SSRP1 proteins. Nature. 1999;400:284–288. doi: 10.1038/22350. [DOI] [PubMed] [Google Scholar]
- Perpelescu M, Nozaki N, Obuse C, Yang H, Yoda K. Active establishment of centromeric CENP-A chromatin by RSF complex. J Cell Biol. 2009;185:397–407. doi: 10.1083/jcb.200903088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phansalkar R, Lapierre P, Mellone BG. Evolutionary insights into the role of the essential centromere protein CAL1 in Drosophila. Chromosome Res. 2012;20:493–504. doi: 10.1007/s10577-012-9299-7. [DOI] [PubMed] [Google Scholar]
- Pidoux AL, Choi ES, Abbott JK, Liu X, Kagansky A, Castillo AG, Hamilton GL, Richardson W, Rappsilber J, He X, et al. Fission yeast Scm3: A CENP-A receptor required for integrity of subkinetochore chromatin. Mol Cell. 2009;33:299–311. doi: 10.1016/j.molcel.2009.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quenet D, Dalal Y. A long non-coding RNA is required for targeting centromeric protein A to the human centromere. eLife. 2014;3:e03254. doi: 10.7554/eLife.03254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosic S, Kohler F, Erhardt S. Repetitive centromeric satellite RNA is essential for kinetochore formation and cell division. J Cell Biol. 2014;207:335–349. doi: 10.1083/jcb.201404097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Pulido L, Pidoux AL, Ponting CP, Allshire RC. Common ancestry of the CENP-A chaperones Scm3 and HJURP. Cell. 2009;137:1173–1174. doi: 10.1016/j.cell.2009.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders A, Werner J, Andrulis ED, Nakayama T, Hirose S, Reinberg D, Lis JT. Tracking FACT and the RNA polymerase II elongation complex through chromatin in vivo. Science. 2003;301:1094–1096. doi: 10.1126/science.1085712. [DOI] [PubMed] [Google Scholar]
- Schuh M, Lehner CF, Heidmann S. Incorporation of Drosophila CID/CENP-A and CENP-C into centromeres during early embryonic anaphase. In Curr Biol. 2007:237–243. doi: 10.1016/j.cub.2006.11.051. [DOI] [PubMed] [Google Scholar]
- Schwartz BE, Ahmad K. Transcriptional activation triggers deposition and removal of the histone variant H3.3. Genes Dev. 2005;19:804–814. doi: 10.1101/gad.1259805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimojima T, Okada M, Nakayama T, Ueda H, Okawa K, Iwamatsu A, Handa H, Hirose S. Drosophila FACT contributes to Hox gene expression through physical and functional interactions with GAGA factor. Genes Dev. 2003;17:1605–1616. doi: 10.1101/gad.1086803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuaib M, Ouararhni K, Dimitrov S, Hamiche A. HJURP binds CENP-A via a highly conserved N-terminal domain and mediates its deposition at centromeres. Proc Natl Acad Sci U S A. 2010;107:1349–1354. doi: 10.1073/pnas.0913709107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoler S, Rogers K, Weitze S, Morey L, Fitzgerald-Hayes M, Baker RE. Scm3, an essential Saccharomyces cerevisiae centromere protein required for G2/M progression and Cse4 localization. Proc Natl Acad Sci U S A. 2007;104:10571–10576. doi: 10.1073/pnas.0703178104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straight AF, Belmont AS, Robinett CC, Murray AW. GFP tagging of budding yeast chromosomes reveals that protein-protein interactions can mediate sister chromatid cohesion. Curr Biol. 1996;6:1599–1608. doi: 10.1016/s0960-9822(02)70783-5. [DOI] [PubMed] [Google Scholar]
- Sullivan BA. Optical mapping of protein-DNA complexes on chromatin fibers. Methods Mol Biol. 2010;659:99–115. doi: 10.1007/978-1-60761-789-1_7. [DOI] [PubMed] [Google Scholar]
- Topp CN, Zhong CX, Dawe RK. Centromere-encoded RNAs are integral components of the maize kinetochore. Proc Natl Acad Sci U S A. 2004;101:15986–15991. doi: 10.1073/pnas.0407154101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitfield ML, Sherlock G, Saldanha AJ, Murray JI, Ball CA, Alexander KE, Matese JC, Perou CM, Hurt MM, Brown PO, et al. Identification of genes periodically expressed in the human cell cycle and their expression in tumors. Mol Biol Cell. 2002;13:1977–2000. doi: 10.1091/mbc.02-02-0030.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin H, Takahata S, Blanksma M, McCullough L, Stillman DJ, Formosa T. yFACT induces global accessibility of nucleosomal DNA without H2A-H2B displacement. Mol Cell. 2009;35:365–376. doi: 10.1016/j.molcel.2009.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.