

Glioblastoma is the most prevalent and malignant primary brain tumor, containing self-renewing, tumorigenic cancer stem cells (CSCs) that contribute to tumor initiation and therapeutic resistance. In this review, Lathia et al. discuss how the integration of genetics, epigenetics, and metabolism has shaped our understanding of how CSCs function to drive GBM growth.
Keywords: brain tumor, cancer stem cell, glioblastoma, glioma, stem cell, tumor-initiating cell
Abstract
Tissues with defined cellular hierarchies in development and homeostasis give rise to tumors with cellular hierarchies, suggesting that tumors recapitulate specific tissues and mimic their origins. Glioblastoma (GBM) is the most prevalent and malignant primary brain tumor and contains self-renewing, tumorigenic cancer stem cells (CSCs) that contribute to tumor initiation and therapeutic resistance. As normal stem and progenitor cells participate in tissue development and repair, these developmental programs re-emerge in CSCs to support the development and progressive growth of tumors. Elucidation of the molecular mechanisms that govern CSCs has informed the development of novel targeted therapeutics for GBM and other brain cancers. CSCs are not self-autonomous units; rather, they function within an ecological system, both actively remodeling the microenvironment and receiving critical maintenance cues from their niches. To fulfill the future goal of developing novel therapies to collapse CSC dynamics, drawing parallels to other normal and pathological states that are highly interactive with their microenvironments and that use developmental signaling pathways will be beneficial.
Development is a coordinated summation of the individual cellular dynamics that build an organ, and the programs responsible for this construction are generally preserved in stem cells for organ homeostasis and tissue repair. Tumors are complex systems that recapitulate the complexity of organs or tissues with dynamic regulation and constituent cellular populations during tumor initiation, maintenance, and progression (Hanahan and Weinberg 2011). While many scientists have sought to reduce the complexity of cancer to a one-dimensional process—for example, characterizing cancers solely based on genetics—most advanced cancers unfortunately remain nearly as lethal since the declaration of the War on Cancer in 1971. Targeted therapeutics offer a transient benefit for some cancer types with driving mutations, but even these tumors will develop resistance to overcome initially effective therapies that poison driving oncogenes.
Glioblastoma (GBM; World Health Organization grade IV glioma) is the most prevalent and lethal primary intrinsic brain tumor (Stupp et al. 2009). Unlike other solid tumor cell types, GBM widely invades the surrounding brain but rarely metastasizes to other organs. While halting steps to fight GBM are being made using targeted therapies (e.g., bevacizumab) or immunotherapies, GBM therapy remains focused on achieving maximal surgical resection followed by concurrent radiation therapy with temozolomide (TMZ; an orally available methylation chemotherapy) and subsequent additional adjuvant TMZ therapy. Conventional treatment offers patients with GBM additional survival time with generally acceptable quality of life, but a cure is never achieved. GBM represents one of the most comprehensively genomically characterized cancer types, leading to recognition of groups of tumors defined by transcription profiles (proneural, neural, classical, and mesenchymal), genetics (mutations of isocitrate dehydrogenase 1 [IDH1]), and epigenetics (CpG island methylator phenotype [CIMP]) (Weller et al. 2013). Long-term survivors are often, but not exclusively, patients with tumors harboring mutations in IDH1, which likely represent a different disease than most GBMs. Beyond IDH1 mutations and a few other biomarkers (deletion of chromosomes 1p and 19q in oligodendrogliomas, methyl guanine methyltransferase [MGMT] promoter methylation, etc.), the accumulated genetic characterization of GBMs has failed to impact clinical practice, suggesting that other discovery paradigms should also be considered.
The brain, like other organs with clearly defined cellular hierarchies in development and homeostasis (e.g., blood, breast, skin, and colon), gives rise to tumors with defined cellular hierarchies, suggesting that cancer replicates ontogeny (Reya et al. 2001). Atop the apex of cellular hierarchies are stem cells, which have been assumed to be rare, quiescent, self-renewing cells, but several highly proliferative organs (e.g., the intestine and skin) contain at least two pools of stem cells: one quiescent, and the other highly proliferative (Barker et al. 2010). Stem cells generate transient amplifying cells, which in turn create lineage-restricted progeny that are eventually fated to become the terminally differentiated effector cells.
Neural stem and progenitor cell (NSPC) pools vary in location and control during development, suggesting that different cellular hierarchies may be co-opted by brain tumors (Gibson et al. 2010; Lottaz et al. 2010). Informed by techniques used to enrich and characterize NSPCs, several groups in parallel demonstrated that gliomas and other primary brain tumors contain self-renewing, tumorigenic cells (Ignatova et al. 2002; Hemmati et al. 2003; Singh et al. 2003, 2004; Galli et al. 2004). The nomenclature for these cells has been controversial (as discussed below), with the dominant choice being cancer stem cells (CSCs) or tumor-initiating cells (importantly, these terms are not identical, as a CSC designation is more restrictive but also more informative) (Table 1). Unlike normal brain organization—where the generation of differentiated progeny is stage-specific (neurons and then glia during development) and derived from rapidly dividing progenitor cells and quiescent, multipotent stem cells that persist into adulthood and become activated upon differentiation (Rowitch and Kriegstein 2010)—these populations have yet to be delineated in brain tumors. The ability to distinguish between self-renewing cells with stem and progenitor cell cycle properties and transcriptional signatures is likely to provide clarity with respect to nomenclature and the functional interplay between cells at the apex of the hierarchy. The challenges distinguishing CSCs from their progeny are derived, in part, from the limited recognition of points of relative stability (“attractor states”) in the landscape of cellular identity that define the stem cell state and transitions into (dedifferentiation) and out of (differentiation) a stable state (Chang et al. 2008). Much like the Heisenberg uncertainty principle in physics, our ability to observe the state of a cell is limited by our act of measurement. The presence of subatomic particles is confirmed in retrospect; similarly, the functional definition of both normal and neoplastic stem cells requires retrospective confirmation. The ability to prospectively distinguish glioma stem cells, which reside at the apex of tumor hierarchies, from their differentiated progeny remains challenging; however, stem cell biology faces a similar difficulty with normal stem cell identification. Of note, the CSC hypothesis does not claim a stem cell as the cell of origin for cancers, suggesting that CSCs do not need to adhere to all of the observed features of normal stem cells.
Table 1.
Definitions and functional characteristics of CSCs, tumor-initiating cells, and tumor-propagating cells
In the following sections, we provide an update on intrinsic and extrinsic regulators of the CSC state in GBM and discuss how the integration of genetics, epigenetics, and metabolism has shaped our understanding of how CSCs function to drive GBM growth. We also highlight future opportunities to further understand the complexity of CSC regulation through interaction with other cells (including immune cells) and how the translation of CSC-based therapies needs to take into account the cellular dynamics of CSCs, which rely on developmental signaling programs.
The history of the CSCs
The heterogeneity of tumor cells has long been appreciated, but two decades ago, seminal work from Dick's laboratory (Bonnet and Dick 1997) described the isolation of a leukemia-initiating cell, the first purification of cancer stem-like cells, a population of cells that had originally been proposed to exist >150 years earlier (Sell 2004). The first prospective isolation of human NSPCs was performed using CD133 (Uchida et al. 2000) and prompted a search for brain tumor cells that shared the characteristics of NSPCs. A burst of studies soon followed describing brain CSCs in anaplastic astrocytoma (Ignatova et al. 2002), medulloblastoma, pilocytic astrocytoma, ependymoma, ganglioglioma (Hemmati et al. 2003; Singh et al. 2003), and GBM (Ignatova et al. 2002; Hemmati et al. 2003; Galli et al. 2004). Brain CSCs have subsequently been shown to be resistant to standard-of-care chemotherapy (Chen et al. 2012) and radiotherapy (Bao et al. 2006a), underscoring their role in disease progression and recurrence.
What's in a name?
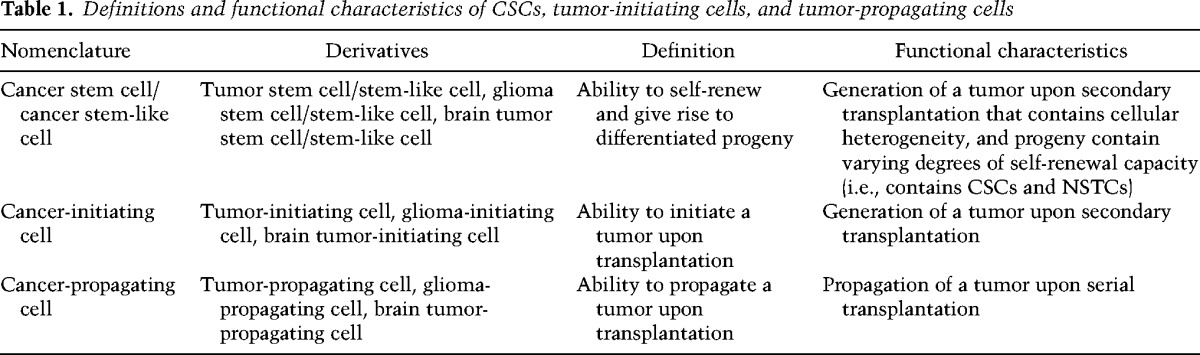
While cellular heterogeneity within CNS tumors is well recognized (Bonavia et al. 2011; Meacham and Morrison 2013), the nomenclature used to describe the self-renewing population of tumor cells with enhanced tumorigenic properties is far from uniform (see Table 1). To date, many terms have been used to describe this population, including cancer/tumor/glioma/brain tumor stem cell, stem-like tumor cell, cancer-/tumor-/glioma-/brain tumor-initiating cell, and cancer-/tumor-/glioma-/brain tumor-propagating cell. This lack of uniformity has generated confusion and controversy by redirecting the focus away from the biology of these cells and their contribution to tumorigenic processes toward identifying markers that the cells express and whether tumor cells can be propagated as free-floating spheres. In addition, while the term “stem cell” is used, it does not necessarily mean these cells have been generated from a transformed stem cell, as there is evidence that multiple cell types—ranging from stem cell to differentiated progeny, depending on the model—are amenable to oncogenic transformation. Therefore, in the current context, it is essential that the strictest functional assays continue to be performed and a singular term be used for studies using models that fulfill this criteria. As the accepted functional definition of a stem cell is the ability to self-renew and generate differentiated progeny, any claims for a CSC population must also demonstrate this capacity (Fig. 1). For brain tumors, this means the ability to generate a tumor upon intracranial transplantation that recapitulates the cellular heterogeneity present in the parental tumor. Unlike the designation of a tumor-initiating cell, CSCs cannot be investigated in isolation due to the required comparison with differentiated progeny. Prospective enrichment and depletion of tumorigenic and nontumorigenic cells demonstrate the presence of a cellular hierarchy. Cells that meet these criteria (tumorigenic and containing a cellular hierarchy) should be referred to as CSCs (or alternatives such as glioma stem cells, glioma CSCs, or brain tumor stem cells in the context of GBM). While the ability to grow as spheres is also evident in CSCs, it is not by default the defining feature of a self-renewing population of cells. In fact, the majority of spheres derived from both normal and neoplastic brain cells come from progenitor cells with limited self-renewal potential, not stem cells (Pastrana et al. 2011). Furthermore, high-passage cell lines, which do not offer the ability to accurately represent tumor complexity in vivo (Lee et al. 2006), should not be replaced with cells grown in long-term passage as spheres but rather with functionally validated CSC models, as this offers the best opportunity to more deeply model the complexity of brain tumors. Thus, although culture of glioma cells as neurospheres may not be required to maintain stemness (Pollard et al. 2009; Cheng et al. 2012), the microenvironment, including medium composition and culture conditions, does necessarily affect the characteristics of CSCs (Pastrana et al. 2011).
Figure 1.
Functional criteria of CSCs. CSCs are defined by functional characteristics that include sustained self-renewal, persistent proliferation, and tumor initiation upon secondary transplantation, which is the definitive functional CSC assay. CSCs also share features with somatic stem cells, including frequency within a tissue (or tumor), stem cell marker expression (examples relevant to GBM and the brain are provided), and the ability to generate progeny of multiple lineages.
CSC enrichment
Great energy and passion have been devoted to the discovery, validation, and use of CSC enrichment methods. Demonstration of a cellular hierarchy demands methods to separate populations that can be functionally studied. Ideally, an enrichment method would be based on a property that defines an essential CSC feature (self-renewal, tumor initiation, etc.) that is immediately lost upon differentiation (i.e., a digital readout) and is usable with live cells. Currently, no such system exists for any cell type (normal or neoplastic) because biologic systems rarely exhibit “all or none” phenomena. Critics of the CSC hypothesis have held this limitation up as proof against CSCs; while the same limitations exist for even the best-characterized normal stem cell (hematopoietic stem cell), no scientists deny the existence of hematopoietic stem cells. Leukemia stem cells are considered a definitive tumor population, yet no marker signature for these cells is definitive (Eppert et al. 2011). A more sophisticated and nuanced use of enrichment systems that is informed by recognition of the diversity of GBMs can lead to context-specific methods to produce matched tumorigenic and nontumorigenic populations.
Most glioma CSC markers have been appropriated from normal stem cells, but the linkage between glioma CSCs and normal stem cells remains controversial. Many of the transcription factors or structural proteins essential for normal NSPC function also mark glioma CSCs, including SOX2 (Hemmati et al. 2003), NANOG (Ben-Porath et al. 2008; Suva et al. 2014), OLIG2 (Ligon et al. 2007), MYC (Kim et al. 2010), MUSASHI1 (Hemmati et al. 2003), BMI1 (Hemmati et al. 2003), NESTIN (Tunici et al. 2004), and inhibitor of differentiation protein 1 (ID1) (Anido et al. 2010). However, because of the limited utility of intracellular proteins for enriching CSCs from nonstem tumor cells (NSTCs) using traditional methods such as flow cytometry, a multitude of potential cell surface markers have been suggested, including CD133 (Hemmati et al. 2003), CD15 (also called Lewis x and SSEA-1 [stage-specific embryonic antigen 1]) (Son et al. 2009), integrin α6 (Lathia et al. 2010), CD44 (Liu et al. 2006), L1CAM (Bao et al. 2008), and A2B5 (Ogden et al. 2008). These types of cell surface markers mediate interactions between cells and the microenvironment, but dissociation of cells from their surroundings rapidly degrades the informational content of markers, requiring rapid utilization.
The first proposed marker, CD133 (Prominin-1), a cell surface glycoprotein expressed on neural stem cells, enriches for cells with higher rates of self-renewal and proliferation and increased differentiation ability (Singh et al. 2003). However, CD133 expression, rather than the AC133 surface epitope, should be used with care to enrich for any cells: Surface CD133 marks stem cells and decreases with differentiation, but the expression of Prominin-1 mRNA is not regulated with stemness (Kemper et al. 2010), suggesting that only the glycosylated surface protein CD133 is CSC-specific. The AC133 antigen marks the glycosylated molecule localized in lipid rafts that signals through PI3K and other key pathways to mediate interactions between a cell and its microenvironment (Wei et al. 2013). Most studies fail to recognize this role and use CD133 as a marker in cells that have been extensively cultured out of their microenvironment. Furthermore, the information contained in CD133 is context-dependent. CD133 mRNA, protein lysates, immunofluorescence, and FACS analysis for the AC133 glycoprotein have very different relationships to cell biology. Unfortunately, the complexity of these biomarkers has led to a reductionist view that has challenged the field due to the lack of consistency in methodology and models. It is nearly certain that CD133 is not universally informative in all tumors and has a false-negative rate for identifying CSCs (CD133-negative cells can be tumor-propagating in some tumors) (Beier et al. 2007). Additionally, the use of CD133 as a stem marker is complicated by the observation that expression of CD133 can be regulated at the level of the cell cycle, with potentially slow-cycling NSPCs lacking CD133 expression during G0/G1 cell cycle phase but still maintaining multipotency (Sun et al. 2009).
Although CD133 continues to be the most commonly used cell surface marker, other markers, such as integrin α6, have been proposed to segregate CSCs and NSTCs (Lathia et al. 2010). CD15/SSEA-1 and CD44 have also been proposed as possible markers, potentially with an association with specific subgroups of GBM (Bhat et al. 2013). These markers have utility but must be approached with caution. Each can mark a large percentage of cells, consistent with a high false-positive rate. Due to the current limitations in the functional assays defining CSCs, false-positive markers are sometimes claimed to be superior to functional identification, but markers lack significant utility in discovery studies, which benefit from greater specificity. Additionally, it is likely that no marker will ever be uniformly informative for CSCs because most tissue types contain multiple populations of stem cells expressing different markers and due to the inherent adaptability of cancer cells.
Several methods other than marker expression have been used to enrich for glioma CSCs, such as the abilities to grow as neurospheres in serum-free medium or efflux fluorescent dyes (Goodell et al. 1996; Kondo et al. 2004). Many investigators have used neurosphere culture to select for progenitor cells in the normal and neoplastic brain cells, but there are challenges with this approach. Neurosphere culture selects for a small fraction of the original tumor cells with bias toward progenitor features and expression of epidermal growth factor receptor (EGFR) and FGFR based on growth factors (EGF and FGF) added to the medium (Pastrana et al. 2011). This selection process eliminates the ability to prospectively enrich and deplete stem-like cells, preventing the delineation of a cellular hierarchy required to prove the presence of CSCs. Neurosphere culture selects for cells that can grow in stem cell medium; however, the selection of CSCs simply based on culture methods fails to recapitulate the heterogeneity of the original tumor in vivo as assessed by histological morphology, differentiated cell lineage, and gene expression (Lee et al. 2006; Lathia et al. 2011; Venere et al. 2011), a characteristic that CSCs acutely isolated using marker expression maintain (Singh et al. 2004). An alternative approach to CSC enrichment is the use of flow cytometry to isolate a side population containing CSCs, which is based on the hypothesis that stem cells contain drug efflux transporters (Yu et al. 2008). While this approach has identified a population of self-renewing cells in a mouse glioma model (Bleau et al. 2009), it has not been used successfully to enrich for self-renewing cells in human GBM (Broadley et al. 2011; Golebiewska et al. 2013), highlighting the model- and species-specific challenges of enrichment methods.
Functional validation—when is a stem cell not a stem cell?
CSC markers, although useful to enrich populations of stem cells from nonstem cells, are not sufficient to define either population due to the lack of definitive markers. Functional validation—the observation of differences in stem cell characteristics of CSCs and NSTCs—is essential to ensure that the enriched cells truly exhibit the functional characteristics of stem cells (Fig. 1). Various methods, both in vitro and in vivo, are employed to assess stem cell characteristics (self-renewal, proliferation, and ability to reproduce the complexity of the original tumor) of enriched cells. In vitro neurosphere formation assays test for both proliferation and self-renewal but fail to address cellular hierarchy and do not recapitulate the tumor microenvironment. Sphere formation is a surrogate of self-renewal capability and—when performed in a limiting dilution format—stem cell frequency, but in vivo tumor formation assays are essential to claim the presence of CSCs.
The gold standard for CSC determination remains the ability of a limiting dilution of cells to recapitulate the complexity of the original patient tumor when transplanted orthotopically. The ability to derive heterogeneity is essential because populations of transit-amplifying cells may form a tumor but will only give rise to cells from their specific lineage. Heterotypic transplantation of cells—for example, into the flank of the animal—may also be informative, but this technique lacks the proper microenvironmental cues of orthotopic implantation.
CSC regulation
Glioma CSCs are regulated by six main mechanisms, which include intrinsic factors such as genetics, epigenetics, and metabolism as well as extrinsic qualities of niche factors, cellular microenvironment, and the host immune system (Fig. 2). The following sections describe the key features of each of these factors and highlight new advances in the topics of epigenetics mapping, single-cell heterogeneity, metabolism, and immunotherapy.
Figure 2.

Regulation of CSCs. Cell-autonomous (intrinsic) and external (extrinsic) forces regulate the CSC state. Key intrinsic regulators include genetic, epigenetic, and metabolic regulation, while extrinsic regulators include interaction with the microenvironment, including niche factors and the immune system.
Intrinsic CSC regulatory mechanisms
Genetics and epigenetics
Through advances in genomic technologies, we now have a comprehensive picture of the genetic mutations and structural variants present in GBM (Atlas 2008; Brennan et al. 2013). Some of the most recurrent alterations include EGFR, IDH1, PDGFRA, HDM2, PIK3CA, and TERT promoter and PI3KR1 gain-of-function mutations or amplifications and mutations or deletions of the tumor suppressors PTEN, TP53, CDKN2A, NF1, ATRX, and RB1. While many of these mutations are prevalent in several other cancer genomes, several mutations are highly enriched in GBM, such as IDH1 mutations, which lead to a CIMP (G-CIMP) (Noushmehr et al. 2010). These studies highlight the significant degree of intertumoral heterogeneity present in GBM, which is further captured at both the transcriptional and epigenetic levels (Phillips et al. 2006; Verhaak et al. 2010), and also underscore the complexity of the clonal evolution and clonal diversity that occur during the genesis of GBM and their bearing on the shape and structure of the CSC hierarchy. While both genetic and epigenetic landscapes define functionally distinct clones during tumor evolution, epigenetic differences likely account for the functional differences between cells within the hierarchy.
Epigenetic maintenance of the CSC state is regulated largely at the level of transcriptional and chromatin regulation. CSC regulation converges on MYC, which occurs in the presence of MYC-mediated cancer cell survival and proliferation programs (Wang et al. 2008; Zheng et al. 2008; Wurdak et al. 2010; Chan et al. 2012; Fang et al. 2014). Additional transcription factors have been identified as important for CSC identity, including STAT3 (Sherry et al. 2009), SOX2 (Gangemi et al. 2009), FOXM1 (Joshi et al. 2013), FOXG1 (Verginelli et al. 2013), GLI1 (Clement et al. 2007), ASCL1 (Rheinbay et al. 2013), ZFX (Fang et al. 2014), NANOG (Zbinden et al. 2010), and ZFHX4 (Chudnovsky et al. 2014), which recruit necessary chromatin remodeling factors to promote maintenance of the glioma CSC state. By using epigenome-wide mapping of cellular chromatin state, Suva et al. (2014) identified a core set of four transcription factors in proneural GBM able to reprogram differentiated tumor cells into glioma CSCs. These investigators showed that POU3F2, SOX2, SALL2, and OLIG2 are master transcription factors required to maintain the tumor-forming capability of these cells, suggesting that mediators of stem cell programs could capture the oncogenic capacity of CSCs. In addition to transcription factors, regulators of nucleosome structure have also been reported to maintain the CSC state. The mixed lineage leukemia 1 (MLL1) protein has been shown to maintain the CSC phenotype through activation of HOXA10, which subsequently regulates a network of homeobox genes that is required for tumor maintenance (Heddleston et al. 2012; Gallo et al. 2013). Similarly, the H3K27 methylase EZH2 has been shown to be important for CSC maintenance through its function as a regulator of both Polycomb-repressive domains and STAT3 signaling (Kim et al. 2013). The BMI1 Polycomb ring finger oncogene regulates both normal neural stem cells and GBM cells (Bruggeman et al. 2007).
These studies highlight the importance of understanding the dynamics of core transcription factors in maintaining stem cell state and the effect that these factors have on shaping the chromatin landscape of cells within the tumor hierarchy.
Single-cell RNA sequencing (RNA-seq) interrogation of cellular heterogeneity within GBMs identified novel genes predominantly present in GBM CSCs compared with differentiated cells and provocatively detected cells of multiple GBM subtypes within single tumors, drawing into question the utility of subtyping tumors and targeting specific subtypes (Patel et al. 2014). Furthermore, these investigators described an inverse correlation between stem signature and cell cycle gene expression, suggesting that the cells that form neurospheres in culture cycle more slowly compared with differentiated and differentiating tumor cells. A parallel, single-cell functional analysis of GBMs confirmed a strong variation of genomics and response to therapy (Meyer et al. 2015). Additional detailed analysis of heterogeneity of this type will greatly expand our understanding of the differences between tumor cells both within and among GBM patients and improve the characterization of glioma CSCs.
Metabolism
GBM CSCs reside in varied tumor microenvironments that limit nutrients, such as glucose and oxygen. Under such conditions, cancer cells, including glioma CSCs, exhibit the “Warburg” effect, a metabolic shift toward aerobic glycolysis and the accumulation of lactate in exchange for sustained ATP production and metabolite generation for macromolecule synthesis. Glioma CSCs demonstrate plasticity in the metabolic pathways used in response to metabolic restrictions and may shift toward the use of the pentose phosphate shunt (Vlashi et al. 2011; Kathagen et al. 2013). This inherent persistence of CSCs under hypoxic and acidic conditions as well as the preferential utilization of HIF-2α signaling compared with NSTCs and normal progenitors promote the maintenance of self-renewal, proliferation, and survival (Li et al. 2009b). Similarly, in conditions of nutrient deprivation such as low glucose, glioma CSCs outcompete neighboring NSTCs for glucose uptake through preferential up-regulation of the high-affinity GLUT3 transporter (Flavahan et al. 2013). A consequence of altered metabolic state is the production of reactive oxygen species. Glioma CSCs not only are dependent on NOS2 activity for promoting tumor growth but also synthesize nitric oxide through the specific up-regulation of NOS2 protein (Eyler et al. 2011). Importantly, in GBM, cellular metabolic characteristics are often genetically hardwired, such as recurrent IDH1 mutations, which are commonly observed in proneural GBM. Mutant IDH1 leads to a gain-of-function enzymatic activity, causing accumulation of 2-hydroxyglutarate, an oncometabolite that inhibits the TET1 and TET2 demethylases to cause aberrant hypermethylation of DNA and histones. While the function of IDH1 mutations in the context of CSCs is not directly defined, IDH1 mutations induce a loss of differentiation, preventing the terminal differentiation of lineage-specific progenitors (Lu et al. 2012). Moving forward, integrated metabolomic and epigenomic profiling may reveal other examples of intricate relationships between metabolism and epigenetic programs and their influence on the glioma CSC state.
Extrinsic CSC regulatory factors
Niche factors
Brain development is orchestrated by a series of regulatory pathways with spatially and temporally controlled activity. Notch and NF-κB (nuclear factor κB) signaling instructs the fate of NSPCs, with the guidance and lineage commitment of progeny dictated by pathways that include the ephrins and bone morphogenetic proteins (BMPs). In a manner that mimics aberrant differentiation, CSCs co-opt developmental programs to maintain an undifferentiated state, increasing their survival and maintenance. Common pathways activated in CSCs include Notch, BMP, NF-κB, and Wnt signaling (Li et al. 2009a; Day et al. 2013; Rheinbay et al. 2013; Lubanska et al. 2014; Yan et al. 2014). Collectively, niche factors represent an overriding theme in CSC biology, where stem and progenitor cell features provide selective advantages to maintain tumor growth (Fig. 2). These pathways may be activated through a combination of genetic and epigenetic alterations in addition to microenvironmental and metabolic factors.
The Notch pathway plays a role during neural development, functioning to inhibit neuronal differentiation and sustain NSPC populations. This pathway is co-opted in GBM, where aberrant NOTCH activation stimulates astrocytes to assume a stem-like state accompanied by increased proliferation (Jeon et al. 2008). The importance of Notch signaling in glioma CSC biology is highlighted by the convergence on this pathway from other pathways and exogenous factors, such as hypoxia, eNOS signaling, and response to radiation (Charles et al. 2010; Wang et al. 2010; Qiang et al. 2012). The dependence of glioma CSCs on Notch signaling is further supported by experiments demonstrating depletion of CSCs by treatment with γ-secretase inhibitors (Fan et al. 2006, 2010).
As BMPs direct NSPC fate toward an astroglial lineage, these signals have been proposed as a possible differentiation therapy for GBM (Piccirillo et al. 2006). Despite the presence of BMP expression in primary GBM tissue, glioma CSCs are highly resistant to the differentiation effects of BMPs in a process that occurs through at least two distinct cell-autonomous mechanisms: the shift to a fetal BMP receptor expression in glioma CSCs through recruitment of the transcriptional repressor EZH2 (Lee et al. 2008) and the secretion of BMP antagonists, specifically Gremlin1, by CSCs to protect against endogenous BMP-mediated differentiation (Yan et al. 2014). In this manner, CSCs generate differentiated progeny that provide supportive cues to the parental cells (e.g., Notch ligands, interleukin-6 [IL-6], and extracellular matrix) while resisting differentiation signals.
The NF-κB pathway has emerged as an important regulator of GBM cell survival and identity through an endogenous cell stress response transcriptional program (Bhat et al. 2013). The A20 protein (TNFAIP3), a mediator of cell survival and the NF-κB pathway, is overexpressed in CSCs compared with NSTCs (Hjelmeland et al. 2010). Supporting these findings, Sema3C and its receptors, PlexinA2 and PlexinD1, are also coordinately expressed in CSCs and activate Rac1 and NF-κB in an autocrine/paracrine loop to promote CSC survival (Man et al. 2014).
GBM CSCs have also been shown to be highly dependent on Ephrin receptor signaling for survival and the maintenance of stem cell properties. Specifically, Ephrin A molecules and the EPHA2 and EPHA3 receptors are highly expressed in glioma CSCs and potentially function through the negative regulation of mitogen-activated protein kinase (MAPK) signaling (Binda et al. 2012; Day et al. 2013).
Wnt signaling is highly active in CSCs and is critical for the maintenance of the stem cell phenotype. An integrated genomic and biological analysis identified PLAGL2 as highly amplified in gliomas with functional suppression of CSC differentiation through modulation of Wnt/β-catenin signaling (Zheng et al. 2010). Comprehensive mapping of chromatin modifications in CSCs and their NSTC counterparts revealed widespread activation of Wnt pathway genes through loss of Polycomb-mediated repression. The CSC chromatin landscape is thought to be dependent on achaete scute family basic helix–loop–helix (bHLH) transcription factor 1 (ASCL1), which activates Wnt signaling through negative regulation of dickkopf WNT signaling pathway inhibitor 1 (DKK1) (Rheinbay et al. 2013). Hedgehog signaling in the CNS is mediated in part by NSPC communication with the cerebrovascular fluid through primary cilia. Gliomas contain primary cilia, and the resulting CSCs are dependent on hedgehog signaling (Bar et al. 2007; Clement et al. 2007; Ehtesham et al. 2007).
Given the role of growth factors in normal brain development, it is not unexpected that numerous canonical growth factor signaling pathways have been shown to contribute to GBM maintenance and function. PDGFRβ signaling promotes CSC survival, self-renewal, and invasion and tumor growth through downstream STAT3 activation (Kim et al. 2012). Similarly, glioma CSCs preferentially express the IL-6 receptor, which also promotes convergent signaling upon STAT3 activation (Wang et al. 2009).
EGFR signaling has also been reported to contribute to CSC maintenance through the activation of AKT, the recruitment of SMAD5, and the induction of ID3, IL-6, and IL-8. This suggests a potential hypothesis in which the EGFR and PDGFRβ pathways are linked by IL-6 signaling. A potential alternate hypothesis is the presence of distinct CSC populations dependent on different growth factor receptor signaling pathways. Supporting this latter notion, EGFR inhibition promotes expansion of a cMET growth factor receptor-positive population of CSCs (Jun et al. 2014). Furthermore, elevated cMET expression is important for CSC maintenance, tumorigenicity, and resistance to radiation (Joo et al. 2012).
Aligned with its role in stress responses, transforming growth factor β (TGF-β) stimulates CSC self-renewal. Autocrine TGF-β signaling permits retention of stemness through positive regulation of SOX2 and SOX4 expression (Ikushima et al. 2009). A distinct subset of TGF-β-dependent CSCs expresses CD44 and ID1 (Anido et al. 2010), which are markers of functionally distinct CSCs. A crucial mediator of the TGF-β response in CSCs is the BMI1 protein, which connects stem cell programs and ER stress pathways through the transcriptional repressor ATF3 (Gargiulo et al. 2013).
Immune system
Immune suppression is a hallmark of cancer (Hanahan and Weinberg 2011); while the brain possesses a unique series of immune surveillance mechanisms that become active during pathogenic states (Ransohoff and Engelhardt 2012), brain tumors have been characterized as immunosuppressive (Platten et al. 2001; Fecci et al. 2006). There is increasing enthusiasm for immunotherapy strategies based on the limited success of signaling pathway inhibitors and anti-angiogenic agents in brain tumors and the success of immunotherapy in melanoma. Immunotherapies for brain tumors include cellular (adoptive T-cell transfer and chimeric antigen receptor engineered T cells), vaccination, and immunomodulatory therapies targeting immune checkpoints (including anti-programmed death 1 [PD1], PD ligand 1 [PD-L1], and cytotoxic T lymphocyte-associated protein 4 [CTLA4] antibodies) (Reardon et al. 2014). Reversing tumor-induced immune suppression by increasing cytotoxic cell function and reducing suppressor cell function may unleash the endogenous immune response. Immunologic therapies may offer an additional benefit, as most strategies do not require intracranial delivery, a major restriction point for many oncologic treatments. While CSCs are key drivers of tumor growth, CSC interactions with the immune system and potential exploitation in immunotherapy are under active investigation (Fig. 3). These studies will require innovative approaches, as the majority of CSC studies involve xenograft models that lack major immune cell components, and many mouse models have reduced cellular heterogeneity. However, the information obtained from mouse model approaches is likely to be informative for the human immune response, as genetically engineered mouse models can recapitulate key aspects of brain tumor immunosuppression (Kong et al. 2010).
Figure 3.
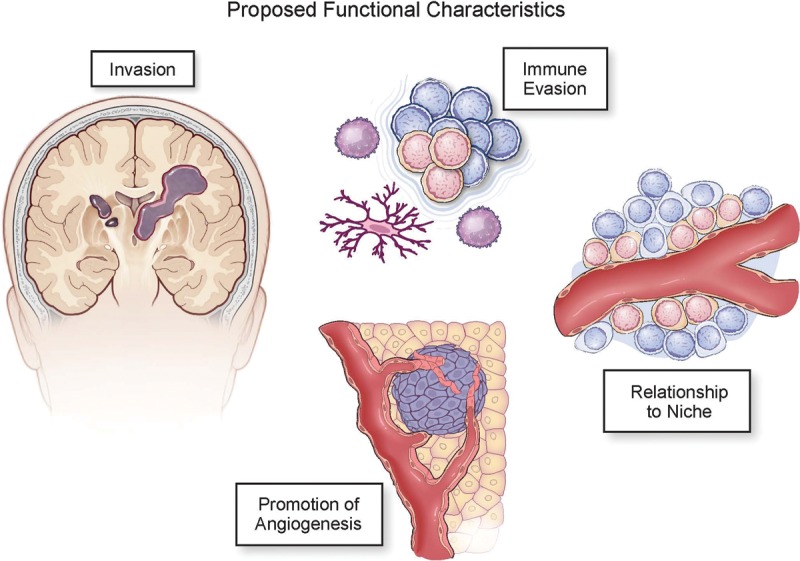
Proposed features of CSCs. Non-cell-autonomous aspects of CSCs may drive tumor growth but also serve as points of fragility. These include the increased ability to invade through the brain parenchyma, immune evasion, relationship with a niche, and promotion of angiogenesis.
Despite these challenges, there is building evidence that CSCs directly modulate the immune system. In coculture studies, CSCs induced regulatory T cells while inhibiting proliferation and cytotoxic T-cell activation with a concomitant induction of cytotoxic T-cell apoptosis, mediated via PD1 and soluble galectin-3 (Di Tomaso et al. 2010; Wei et al. 2010). Other CSC-secreted factors include IL-10 and TGF-β, which also suppresses tumor-associated microglia/macrophage function and generates a more immunosuppressive (M2) phenotype (Wu et al. 2010). Another immunotherapy approach that may benefit from CSC targeting is the development of anti-tumor vaccines. Current vaccine efforts have focused on tumor-specific antigens (such as EGFRvIII) or whole tumor cell lysates, and there is evidence from preclinical models that CSC lysates are more effective in generating dendritic cell (DC) vaccines than differentiated cells (Pellegatta et al. 2006; Xu et al. 2009). CSCs modulate T-cell and tumor-associated microglia/macrophage function through secreted factors (Zhou et al. 2015), which may be exploited in the development of vaccine strategies or in combination with other drugs (Sarkar et al. 2014). These data provide a rationale for future studies investigating how the interaction between CSCs and other immune cell populations may drive immune suppression and in vivo interrogations into how CSC targeting may alter the immune activation status. Evaluating changes in CSC populations as a result of immunotherapy will also be essential, as will be evaluating combinatorial targeting strategies using immunotherapies and anti-CSC approaches.
Microenvironment
Most conventional anti-neoplastic therapies target proliferating cells, but the malignancy of advanced cancers also derives from effects on the immune system, vasculature, and invasion/metastasis (Fig. 3). GBMs infiltrate the surrounding brain, precluding curative surgical resection. Infiltrative tumors must adapt to new environments, including the formation of new vessels to obtain nutrients. GBMs express proangiogenic growth factors (Batchelor et al. 2007), with CSCs driving neoangiogenesis with high levels of VEGF (Bao et al. 2006b). The humanized monoclonal antibody bevacizumab was developed to target VEGF to inhibit angiogenesis and has been used to treat recurrent GBM (Cohen et al. 2009). Bevacizumab attenuates tumor size, but the surviving tumor may display increased invasion in human and mouse models (de Groot et al. 2010), potentially due to a release of c-MET inhibition (Lu et al. 2012). Cancer cells often activate redundant angiogenic pathways in response to VEGF pathway inhibition (Atlas 2008). CSCs located at the perivascular niche are in close contact with the endothelial cells (Calabrese et al. 2007), permitting engagement of endothelial cell Notch ligands with glioma CSC Notch receptors to activate Notch signaling, which supports self-renewal of glioma CSCs (Zhu et al. 2011). CSCs also contribute to vascular structure through transdifferentiation into pericytes to promote tumor growth (Cheng et al. 2013). Inhibition of CSC-derived pericytes disrupts angiogenesis and inhibits tumor growth, directing attention toward nonendothelial cell targeting strategies. Anti-angiogenic drugs in current use have failed to provide a significant survival benefit to GBM patients (Gilbert et al. 2014), suggesting that a benefit may exist to investigating the mechanisms by which tumor cells regulate angiogenesis and that contribute to tumor growth and maintenance to efficiently target the GBM vasculature.
Therapeutic resistance
The mainstay treatment of GBM involves surgery, concurrent radiation with chemotherapy, and adjuvant chemotherapy with TMZ (Stupp et al. 2009). Despite advances in the field, the overall survival rate remains only 15–19 mo (Stupp et al. 2009). The high degree of tumor heterogeneity in GBM contributes to treatment failure, to which functional and molecular heterogeneity and aberrant receptor tyrosine kinase (RTK) activity all contribute. CSCs located at the top of the hierarchy initiate and maintain the tumor after treatment (Chen et al. 2012). Glioma CSCs have also been shown to contribute to radiation resistance by increasing the DNA damage response machinery (Bao et al. 2006a). In terms of molecular heterogeneity, different subtypes of GBM with distinct molecular profiles coexist within the same tumor and likely exhibit differential therapeutic responses (Sottoriva et al. 2013). For example, several RTKs, including PDGFR in the proneural and EGFR in the classical subtype, are altered in GBM (Verhaak et al. 2010). The abnormal activation of RTKs involves many pathways that are redundant and can initiate and maintain downstream signaling, making tumors refractory to treatment (Stommel et al. 2007). A recent single-cell analysis of primary GBM patients showed that cells from the same tumor have differential expression of genes involved in oncogenic signaling, proliferation, immune response, and hypoxia (Patel et al. 2014). Furthermore, an increase in tumor heterogeneity was associated with a decrease in patient survival. The addition of TMZ to radiation has increased median survival by several months (Stupp et al. 2009), but lineage tracing studies in mouse models demonstrate that CSCs repopulate brain tumors after TMZ treatment (Chen et al. 2012). A number of molecular mechanisms have been identified that mediate the therapeutic resistance of CSCs to cytotoxic therapies, including the DNA damage checkpoint, Notch, NF-κB, EZH2, and PARP (Bao et al. 2006a; Wang et al. 2010; Bhat et al. 2013; Venere et al. 2014; Kim et al. 2015), which suggests that CSCs develop multiple mechanisms of resistance that may require combinations of targeted agents. Moving forward, these studies demonstrate the importance of understanding the molecular alterations that are present in recurrent tumors and how these influence the structure of cells within the tumor hierarchy. In addition, it is necessary to consider that therapeutic resistance mechanisms may not be solely innate but may evolve from exposure to microenvironmental factors such as hypoxia and acidic and metabolic stress (Heddleston et al. 2009; Li et al. 2009b; Hjelmeland et al. 2011; Flavahan et al. 2013; Xie et al. 2015).
Therapeutic targeting
Conventional treatment for GBM promotes a transient elimination of the tumor and is almost always followed by tumor recurrence, possibly with an increase in the percentage of CSCs (Auffinger et al. 2014), as CSCs are involved in tumor recurrence and therapeutic resistance (Bao et al. 2006a; Chen et al. 2012). To effectively eliminate CSCs, it is critical to target their essential functions and their interactions with the microenvironment. Treatment with TMZ may kill CSCs that contain higher expression of the DNA repair protein MGMT; however, TMZ cannot prevent self-renewal of CSCs that contain MGMT (Beier et al. 2008). Another feature of CSCs is their ability to evade apoptosis. A potential therapeutic strategy would be the use of PARP inhibitors to enhance apoptosis under genotoxic damage. When the PARP inhibitor ABT-888 was used in combination with TMZ and radiation in GBM cell lines, apoptosis increased, and cells were sensitized to therapy (Barazzuol et al. 2013). GBMs thrive in harsh microenvironments characterized by hypoxia and limited nutrient availability. The HIF family of transcription factors is involved in promoting angiogenesis and cell migration in hypoxic regions (Kaur et al. 2005), and several drugs have been developed to target this gene family, with a few undergoing clinical trials. For example, as described previously, glioma CSCs reprogram their metabolic machinery and preferentially take up glucose to survive in environments with limited nutrients by expressing the high-affinity glucose transporter GLUT3 (Flavahan et al. 2013). GLUT3 therefore represents a promising therapeutic target for potential selective inhibition of CSCs. Epigenetic modifications are manifest in tumor recurrence (Nagarajan and Costello 2009). Histone acetylation and methylation are reversible and can be targeted by drugs; the histone deacetylase (HDAC) inhibitor vorinostat is currently in clinical trials (Bezecny 2014). Immunotherapy is an additional emerging therapeutic approach for GBM. The development of vaccines based on heat-shock proteins, EGFRvIII (Del Vecchio et al. 2012), and DCs (Terasaki et al. 2011) has shown promising results in clinical trials. ICT-107, a patient-derived DC vaccine developed against six antigens highly expressed in glioma CSCs (Phuphanich et al. 2013), is currently under clinical evaluation for use in patients.
Some of the challenges of developing therapeutic targeting agents are derived from the lack of universally informative markers to identify CSCs and the common molecular pathways shared by CSCs and NSPCs. The understanding of the biology of the CSCs and how these cells interact with their microenvironment in combination with the genetic and epigenetic landscape in GBM will be essential to develop more effective therapies.
Reducing complexity through mathematical modeling
As biological observations have revealed increasing levels of complexity, mathematical modeling approaches have provided a framework to understand the dynamic complexity of stem cell self-renewal and differentiation. By use of proliferation data and lineage tracing analysis, quantitative models have been generated for tissue-specific stem cells that have provided insight into the kinetics of cell fate choice and tissue development (Blanpain and Simons 2013). Similar approaches have been taken to reduce the complexity of CSCs. A network-based model has suggested that CSCs can transition between plastic (proliferative, symmetrically dividing, and less invasive) and rigid (quiescent, asymmetrically dividing, and more invasive) networks that can be modulated by extrinsic stressors, such as hypoxia, inflammation, and therapies (Csermely et al. 2015). Testing this model with biological data is likely to provide additional insights into the complexity of CSCs and identify points of fragility for additional therapeutic development. Mathematical approaches have also been used to evaluate the dynamics of GBM growth. Proliferation and invasion are phenotypes that have been modeled (Harpold et al. 2007). By use of a model that takes into account rates of proliferation and invasion in combination with imaging data, it has been proposed that IDH1 mutant tumors are actually less proliferative and more invasive (Baldock et al. 2014). Clinically relevant parameters, such as identifying optimal radiation schedules, have also been modeled using genetically engineered mice (Leder et al. 2014). Additionally, quantitative approaches have been developed to model the events leading to intertumoral and intratumoral heterogeneity in both human patient specimens (Sottoriva et al. 2013) and mouse models (Cheng et al. 2012). Integrating mathematical approaches into future CSC studies will provide an opportunity to identify key pathways essential for self-renewal and will predict responses to therapeutic perturbations.
Future directions
GBM provides an excellent system to investigate the importance of CSCs. While there is a standard set of assays used to enrich for and identify CSCs, it remains unclear whether multiple CSC populations exist in different niches (perivascular and hypoxic) and possess different characteristics (slow vs. rapid cycling) as well as how key developmental signaling pathways are used by each of these populations. In addition, while a hierarchy is in place for GBM, the current view of CSCs and NSTCs is mutually exclusive and lacks a progenitor cell population that serves as an intermediate for differentiated progeny generation from somatic stem cells. Mouse studies have revealed that multiple stem and progenitor cell populations have the capacity to give rise to tumors upon oncogenic transformation, but it remains unclear whether there is a single cell of origin for the human disease or, more likely, whether multiple cells of origin exist and how this may be linked to genetic diversity. Making inroads into these unresolved questions will refine the experimental foundation upon which translational studies aiming to develop novel anti-CSC therapies are built and provide key signaling pathways responsible for CSC maintenance that are amenable for targeting.
The extensive molecular characterization of gliomas of all grades has permitted the recognition that the continuum of tumor grade has hidden a set of genetically distinct diseases. IDH1 mutations produce an oncometabolite, 2-hydroxyglutarate, that reprograms cellular chromatin to assume a stem-like state (Lu et al. 2012). Thus, IDH1 mutant gliomas may have a relatively flat hierarchy, with most tumor cells acquiring stem-like features early in tumor initiation. In contrast, primary GBMs accumulate a greater diversity of genetic and epigenetic alterations, which is associated with a more vertical cellular hierarchy. This duality of tumor biology resembles that of the two forms of head and neck cancers. Human papilloma virus-induced head and neck cancers are morphologically uniform and, like IDH1 mutant gliomas, are more responsive to therapies. Alcohol- and tobacco-associated head and neck cancers harbor more mutations and display a worse outcome with a reliable cellular hierarchy. Large-scale genomic sequencing has informed commonalities among cancer types based on driving genetic lesions. It is possible that similar patterns will be appreciated with cancer types based on epigenetic and cellular hierarchies, creating broader opportunities to improve diagnostics and therapeutics. In fact, expanding the organizational structures is likely to be a useful approach to increase our understanding of complex disease states. Many diseases display heterogeneous aspects that are governed by both cell-autonomous and microenvironmental forces. With the success of immunotherapy approaches to activate the immune system via immune checkpoint inhibition in cancers such as melanoma, understanding how GBM and, in particular, CSCs interface with the immune system is an immediate priority. An alternative view of heterogeneity and therapeutic response may also be informative for future studies. For example, bacterial infections contain distinct populations of cells that have different proliferative potential and responses to therapy. Viable but nonculturable bacteria and latent infections, including tuberculosis, may be found in particular niches associated with inflammation, hypoxia, acidic and nitrosative stress, and nutrient restriction (Oliver 2010). Most antibiotics, like anti-neoplastic agents, are directed against the proliferative population, leaving a resistant population behind. Novel methods are being used to screen for new agents that target resistant bacteria, such as latent tuberculosis (Bryk et al. 2008). Nathan (2004) suggested that “essentiality is conditional, and the conditions defining essentiality are multiple” in the context of latent infections. An identical view can instruct CSC targeting efforts as we grow in our understanding of the governing stimuli both internal and external to CSCs.
One infrequently discussed point is a re-equilibration of a cellular hierarchy in tumors generated from CSCs. If cell-autonomous advantages were the sole determinant of the differentiation state of tumor cells, CSCs would represent the majority of tumor cells, as the evolutionary drive toward increased fitness would provide a selective advantage to CSCs. At steady state (in distinction from homeostasis), tissues balance competing requirements through multiple levels of interaction among stem cells, progenitor cells, and differentiated progeny. Collectively, the individual cellular dynamics in cancer permits tumors to respond to exogenous insults (cytotoxic therapies, immunologic attack, etc.) to maintain the aberrant organ system. These dynamics are also at play within the cellular hierarchy in which CSCs give rise to NSTCs, and, when necessary, NSTCs give rise to CSCs to maintain the cellular equilibrium necessary for optimal tumor growth. CSCs should not be considered a model to simplify the modeling of GBMs and other cancers, but rather the CSC hypothesis constitutes an additional level of complexity that contributes to the malignancy of cancers. As CSCs reside in multiple niches governed by different molecular programs, there will not be single anti-CSC targeted therapeutics with broad activity; instead, CSCs will demand multitargeted approaches. Patients with GBMs are in desperate need of improved therapies. The real validation of CSCs will come with better treatments due to the integration of CSCs into drug development.
Acknowledgments
We sincerely apologize to those investigators whose work we were unable to cite due to space limitations. We thank Amanda Mendelsohn (Center of Medical Art and Photography, Cleveland Clinic) for assistance with figure preparation. We also thank our funding sources: The National Institutes of Health (grants CA154130, CA171652, CA169117, NS087913, and NS089272 to J.N.R., and CA157948, CA191263, and NS083629 to J.D.L.); Sontag Foundation (J.D.L.); Research Programs Committees of Cleveland Clinic (J.N.R); and James S. McDonnell Foundation (J.N.R). S.C.M. is supported by a Canadian Institutes of Health Research Banting Fellowship. Work in the Lathia laboratory is also supported by the Lerner Research Institute, Case Comprehensive Cancer Center, Voices Against Brain Cancer, Blast GBM, the Ohio Cancer Research Associates, Research Scholar Award from the American Cancer Society, V Scholar Award from the V Foundation for Cancer Research, and grant IRG-91-022-18 to the Case Comprehensive Cancer Center from the American Cancer Society.
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.261982.115.
References
- Anido J, Saez-Borderias A, Gonzalez-Junca A, Rodon L, Folch G, Carmona MA, Prieto-Sanchez RM, Barba I, Martinez-Saez E, Prudkin L, et al. 2010. TGF-β receptor inhibitors target the CD44(high)/Id1(high) glioma-initiating cell population in human glioblastoma. Cancer Cell 18: 655–668. [DOI] [PubMed] [Google Scholar]
- Atlas TCG. 2008. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455: 1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auffinger B, Tobias AL, Han Y, Lee G, Guo D, Dey M, Lesniak MS, Ahmed AU. 2014. Conversion of differentiated cancer cells into cancer stem-like cells in a glioblastoma model after primary chemotherapy. Cell Death Differ 21: 1119–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldock AL, Yagle K, Born DE, Ahn S, Trister AD, Neal M, Johnston SK, Bridge CA, Basanta D, Scott J, et al. 2014. Invasion and proliferation kinetics in enhancing gliomas predict IDH1 mutation status. Neuro Oncol 16: 779–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN. 2006a. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444: 756–760. [DOI] [PubMed] [Google Scholar]
- Bao S, Wu Q, Sathornsumetee S, Hao Y, Li Z, Hjelmeland AB, Shi Q, McLendon RE, Bigner DD, Rich JN. 2006b. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res 66: 7843–7848. [DOI] [PubMed] [Google Scholar]
- Bao S, Wu Q, Li Z, Sathornsumetee S, Wang H, McLendon RE, Hjelmeland AB, Rich JN. 2008. Targeting cancer stem cells through L1CAM suppresses glioma growth. Cancer Res 68: 6043–6048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar EE, Chaudhry A, Lin A, Fan X, Schreck K, Matsui W, Piccirillo S, Vescovi AL, DiMeco F, Olivi A, et al. 2007. Cyclopamine-mediated hedgehog pathway inhibition depletes stem-like cancer cells in glioblastoma. Stem Cells 10: 2524–2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barazzuol L, Jena R, Burnet NG, Meira LB, Jeynes JC, Kirkby KJ, Kirkby NF. 2013. Evaluation of poly (ADP-ribose) polymerase inhibitor ABT-888 combined with radiotherapy and temozolomide in glioblastoma. Radiat Oncol 8: 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker N, Bartfeld S, Clevers H. 2010. Tissue-resident adult stem cell populations of rapidly self-renewing organs. Cell Stem Cell 7: 656–670. [DOI] [PubMed] [Google Scholar]
- Batchelor TT, Sorensen AG, di Tomaso E, Zhang WT, Duda DG, Cohen KS, Kozak KR, Cahill DP, Chen PJ, Zhu M, et al. 2007. AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell 11: 83–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beier D, Hau P, Proescholdt M, Lohmeier A, Wischhusen J, Oefner PJ, Aigner L, Brawanski A, Bogdahn U, Beier CP. 2007. CD133+ and CD133− glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res 67: 4010–4015. [DOI] [PubMed] [Google Scholar]
- Beier D, Rohrl S, Pillai DR, Schwarz S, Kunz-Schughart LA, Leukel P, Proescholdt M, Brawanski A, Bogdahn U, Trampe-Kieslich A, et al. 2008. Temozolomide preferentially depletes cancer stem cells in glioblastoma. Cancer Res 68: 5706–5715. [DOI] [PubMed] [Google Scholar]
- Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A, Weinberg RA. 2008. An embryonic stem cell–like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet 40: 499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezecny P. 2014. Histone deacetylase inhibitors in glioblastoma: pre-clinical and clinical experience. Med Oncol 31: 985. [DOI] [PubMed] [Google Scholar]
- Bhat KP, Balasubramaniyan V, Vaillant B, Ezhilarasan R, Hummelink K, Hollingsworth F, Wani K, Heathcock L, James JD, Goodman LD, et al. 2013. Mesenchymal differentiation mediated by NF-κB promotes radiation resistance in glioblastoma. Cancer Cell 24: 331–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binda E, Visioli A, Giani F, Lamorte G, Copetti M, Pitter KL, Huse JT, Cajola L, Zanetti N, DiMeco F, et al. 2012. The EphA2 receptor drives self-renewal and tumorigenicity in stem-like tumor-propagating cells from human glioblastomas. Cancer Cell 22: 765–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanpain C, Simons BD. 2013. Unravelling stem cell dynamics by lineage tracing. Nat Rev Mol Cell Biol 14: 489–502. [DOI] [PubMed] [Google Scholar]
- Bleau AM, Hambardzumyan D, Ozawa T, Fomchenko EI, Huse JT, Brennan CW, Holland EC. 2009. PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell Stem Cell 4: 226–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonavia R, Inda MM, Cavenee WK, Furnari FB. 2011. Heterogeneity maintenance in glioblastoma: a social network. Cancer Res 71: 4055–4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet D, Dick JE. 1997. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 3: 730–737. [DOI] [PubMed] [Google Scholar]
- Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, et al. 2013. The somatic genomic landscape of glioblastoma. Cell 155: 462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broadley KW, Hunn MK, Farrand KJ, Price KM, Grasso C, Miller RJ, Hermans IF, McConnell MJ. 2011. Side population is not necessary or sufficient for a cancer stem cell phenotype in glioblastoma multiforme. Stem Cells 29: 452–461. [DOI] [PubMed] [Google Scholar]
- Bruggeman SW, Hulsman D, Tanger E, Buckle T, Blom M, Zevenhoven J, van Tellingen O, van Lohuizen M. 2007. Bmi1 controls tumor development in an Ink4a/Arf-independent manner in a mouse model for glioma. Cancer Cell 12: 328–341. [DOI] [PubMed] [Google Scholar]
- Bryk R, Gold B, Venugopal A, Singh J, Samy R, Pupek K, Cao H, Popescu C, Gurney M, Hotha S, et al. 2008. Selective killing of nonreplicating mycobacteria. Cell Host Microbe 3: 137–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, Oh EY, Gaber MW, Finklestein D, Allen M, et al. 2007. A perivascular niche for brain tumor stem cells. Cancer Cell 11: 69–82. [DOI] [PubMed] [Google Scholar]
- Chan XH, Nama S, Gopal F, Rizk P, Ramasamy S, Sundaram G, Ow GS, Ivshina AV, Tanavde V, Haybaeck J, et al. 2012. Targeting glioma stem cells by functional inhibition of a prosurvival oncomiR-138 in malignant gliomas. Cell Rep 2: 591–602. [DOI] [PubMed] [Google Scholar]
- Chang HH, Hemberg M, Barahona M, Ingber DE, Huang S. 2008. Transcriptome-wide noise controls lineage choice in mammalian progenitor cells. Nature 453: 544–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles N, Ozawa T, Squatrito M, Bleau AM, Brennan CW, Hambardzumyan D, Holland EC. 2010. Perivascular nitric oxide activates notch signaling and promotes stem-like character in PDGF-induced glioma cells. Cell Stem Cell 6: 141–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Li Y, Yu TS, McKay RM, Burns DK, Kernie SG, Parada LF. 2012. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 488: 522–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng YK, Beroukhim R, Levine RL, Mellinghoff IK, Holland EC, Michor F. 2012. A mathematical methodology for determining the temporal order of pathway alterations arising during gliomagenesis. PLoS Comput Biol 8: e1002337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng L, Huang Z, Zhou W, Wu Q, Donnola S, Liu JK, Fang X, Sloan AE, Mao Y, Lathia JD, et al. 2013. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 153: 139–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chudnovsky Y, Kim D, Zheng S, Whyte WA, Bansal M, Bray MA, Gopal S, Theisen MA, Bilodeau S, Thiru P, et al. 2014. ZFHX4 interacts with the NuRD core member CHD4 and regulates the glioblastoma tumor-initiating cell state. Cell Rep 6: 313–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement V, Sanchez P, de Tribolet N, Radovanovic I, Ruiz i Altaba A. 2007. HEDGEHOG–GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr Biol 17: 165–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen MH, Shen YL, Keegan P, Pazdur R. 2009. FDA drug approval summary: bevacizumab (Avastin) as treatment of recurrent glioblastoma multiforme. Oncologist 14: 1131–1138. [DOI] [PubMed] [Google Scholar]
- Csermely P, Hodsagi J, Korcsmaros T, Modos D, Perez-Lopez AR, Szalay K, Veres DV, Lenti K, Wu LY, Zhang XS. 2015. Cancer stem cells display extremely large evolvability: alternating plastic and rigid networks as a potential Mechanism: network models, novel therapeutic target strategies, and the contributions of hypoxia, inflammation and cellular senescence. Semin Cancer Biol 30: 42–51. [DOI] [PubMed] [Google Scholar]
- Day BW, Stringer BW, Al-Ejeh F, Ting MJ, Wilson J, Ensbey KS, Jamieson PR, Bruce ZC, Lim YC, Offenhauser C, et al. 2013. EphA3 maintains tumorigenicity and is a therapeutic target in glioblastoma multiforme. Cancer Cell 23: 238–248. [DOI] [PubMed] [Google Scholar]
- de Groot JF, Fuller G, Kumar AJ, Piao Y, Eterovic K, Ji Y, Conrad CA. 2010. Tumor invasion after treatment of glioblastoma with bevacizumab: radiographic and pathologic correlation in humans and mice. Neuro Oncol 12: 233–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Vecchio CA, Li G, Wong AJ. 2012. Targeting EGF receptor variant III: tumor-specific peptide vaccination for malignant gliomas. Expert Rev Vaccines 11: 133–144. [DOI] [PubMed] [Google Scholar]
- Di Tomaso T, Mazzoleni S, Wang E, Sovena G, Clavenna D, Franzin A, Mortini P, Ferrone S, Doglioni C, Marincola FM, et al. 2010. Immunobiological characterization of cancer stem cells isolated from glioblastoma patients. Clin Cancer Res 16: 800–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehtesham M, Sarangi A, Valadez JG, Chanthaphaychith S, Becher MW, Abel TW, Thompson RC, Cooper MK. 2007. Ligand-dependent activation of the hedgehog pathway in glioma progenitor cells. Oncogene 26: 5752–5761. [DOI] [PubMed] [Google Scholar]
- Eppert K, Takenaka K, Lechman ER, Waldron L, Nilsson B, van Galen P, Metzeler KH, Poeppl A, Ling V, Beyene J, et al. 2011. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat Med 17: 1086–1093. [DOI] [PubMed] [Google Scholar]
- Eyler CE, Wu Q, Yan K, MacSwords JM, Chandler-Militello D, Misuraca KL, Lathia JD, Forrester MT, Lee J, Stamler JS, et al. 2011. Glioma stem cell proliferation and tumor growth are promoted by nitric oxide synthase-2. Cell 146: 53–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X, Matsui W, Khaki L, Stearns D, Chun J, Li YM, Eberhart CG. 2006. Notch pathway inhibition depletes stem-like cells and blocks engraftment in embryonal brain tumors. Cancer Res 66: 7445–7452. [DOI] [PubMed] [Google Scholar]
- Fan X, Khaki L, Zhu TS, Soules ME, Talsma CE, Gul N, Koh C, Zhang J, Li YM, Maciaczyk J, et al. 2010. NOTCH pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells 28: 5–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X, Huang Z, Zhou W, Wu Q, Sloan AE, Ouyang G, McLendon RE, Yu JS, Rich JN, Bao S. 2014. The zinc finger transcription factor ZFX is required for maintaining the tumorigenic potential of glioblastoma stem cells. Stem Cells 32: 2033–2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fecci PE, Mitchell DA, Whitesides JF, Xie W, Friedman AH, Archer GE, Herndon JE II, Bigner DD, Dranoff G, Sampson JH. 2006. Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer Res 66: 3294–3302. [DOI] [PubMed] [Google Scholar]
- Flavahan WA, Wu Q, Hitomi M, Rahim N, Kim Y, Sloan AE, Weil RJ, Nakano I, Sarkaria JN, Stringer BW, et al. 2013. Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nat Neurosci 16: 1373–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S, Fiocco R, Foroni C, Dimeco F, Vescovi A. 2004. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res 64: 7011–7021. [DOI] [PubMed] [Google Scholar]
- Gallo M, Ho J, Coutinho FJ, Vanner R, Lee L, Head R, Ling EK, Clarke ID, Dirks PB. 2013. A tumorigenic MLL-homeobox network in human glioblastoma stem cells. Cancer Res 73: 417–427. [DOI] [PubMed] [Google Scholar]
- Gangemi RM, Griffero F, Marubbi D, Perera M, Capra MC, Malatesta P, Ravetti GL, Zona GL, Daga A, Corte G. 2009. SOX2 silencing in glioblastoma tumor-initiating cells causes stop of proliferation and loss of tumorigenicity. Stem Cells 27: 40–48. [DOI] [PubMed] [Google Scholar]
- Gargiulo G, Cesaroni M, Serresi M, de Vries N, Hulsman D, Bruggeman SW, Lancini C, van Lohuizen M. 2013. In vivo RNAi screen for BMI1 targets identifies TGF-β/BMP-ER stress pathways as key regulators of neural- and malignant glioma-stem cell homeostasis. Cancer Cell 23: 660–676. [DOI] [PubMed] [Google Scholar]
- Gibson P, Tong Y, Robinson G, Thompson MC, Currle DS, Eden C, Kranenburg TA, Hogg T, Poppleton H, Martin J, et al. 2010. Subtypes of medulloblastoma have distinct developmental origins. Nature 468: 1095–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA, Colman H, Chakravarti A, Pugh S, Won M, et al. 2014. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med 370: 699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golebiewska A, Bougnaud S, Stieber D, Brons NH, Vallar L, Hertel F, Klink B, Schrock E, Bjerkvig R, Niclou SP. 2013. Side population in human glioblastoma is non-tumorigenic and characterizes brain endothelial cells. Brain 136: 1462–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodell MA, Brose K, Paradis G, Conner AS, Mulligan RC. 1996. Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. J Exp Med 183: 1797–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. 2011. Hallmarks of cancer: the next generation. Cell 144: 646–674. [DOI] [PubMed] [Google Scholar]
- Harpold HL, Alvord EC Jr, Swanson KR. 2007. The evolution of mathematical modeling of glioma proliferation and invasion. J Neuropathol Exp Neurol 66: 1–9. [DOI] [PubMed] [Google Scholar]
- Heddleston JM, Li Z, McLendon RE, Hjelmeland AB, Rich JN. 2009. The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle 8: 3274–3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heddleston JM, Wu Q, Rivera M, Minhas S, Lathia JD, Sloan AE, Iliopoulos O, Hjelmeland AB, Rich JN. 2012. Hypoxia-induced mixed-lineage leukemia 1 regulates glioma stem cell tumorigenic potential. Cell Death Differ 19: 428–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmati HD, Nakano I, Lazareff JA, Masterman-Smith M, Geschwind DH, Bronner-Fraser M, Kornblum HI. 2003. Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci 100: 15178–15183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjelmeland AB, Wu Q, Wickman S, Eyler C, Heddleston J, Shi Q, Lathia JD, Macswords J, Lee J, McLendon RE, et al. 2010. Targeting A20 decreases glioma stem cell survival and tumor growth. PLoS Biol 8: e1000319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjelmeland AB, Wu Q, Heddleston JM, Choudhary GS, MacSwords J, Lathia JD, McLendon R, Lindner D, Sloan A, Rich JN. 2011. Acidic stress promotes a glioma stem cell phenotype. Cell Death Differ 18: 829–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignatova TN, Kukekov VG, Laywell ED, Suslov ON, Vrionis FD, Steindler DA. 2002. Human cortical glial tumors contain neural stem-like cells expressing astroglial and neuronal markers in vitro. Glia 39: 193–206. [DOI] [PubMed] [Google Scholar]
- Ikushima H, Todo T, Ino Y, Takahashi M, Miyazawa K, Miyazono K. 2009. Autocrine TGF-β signaling maintains tumorigenicity of glioma-initiating cells through Sry-related HMG-box factors. Cell Stem Cell 5: 504–514. [DOI] [PubMed] [Google Scholar]
- Jeon HM, Jin X, Lee JS, Oh SY, Sohn YW, Park HJ, Joo KM, Park WY, Nam DH, DePinho RA, et al. 2008. Inhibitor of differentiation 4 drives brain tumor-initiating cell genesis through cyclin E and notch signaling. Genes Dev 22: 2028–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo KM, Jin J, Kim E, Ho Kim K, Kim Y, Gu Kang B, Kang YJ, Lathia JD, Cheong KH, Song PH, et al. 2012. MET signaling regulates glioblastoma stem cells. Cancer Res 72: 3828–3838. [DOI] [PubMed] [Google Scholar]
- Joshi K, Banasavadi-Siddegowda Y, Mo X, Kim SH, Mao P, Kig C, Nardini D, Sobol RW, Chow LM, Kornblum HI, et al. 2013. MELK-dependent FOXM1 phosphorylation is essential for proliferation of glioma stem cells. Stem Cells 31: 1051–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun HJ, Bronson RT, Charest A. 2014. Inhibition of EGFR induces a c-MET-driven stem cell population in glioblastoma. Stem Cells 32: 338–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kathagen A, Schulte A, Balcke G, Phillips HS, Martens T, Matschke J, Günther HS, Soriano R, Modrusan Z, Sandmann T, et al. 2013. Hypoxia and oxygenation induce a metabolic switch between pentose phosphate pathway and glycolysis in glioma stem-like cells. Acta Neuropathol 126: 763–780. [DOI] [PubMed] [Google Scholar]
- Kaur B, Khwaja FW, Severson EA, Matheny SL, Brat DJ, Van Meir EG. 2005. Hypoxia and the hypoxia-inducible-factor pathway in glioma growth and angiogenesis. Neuro Oncol 7: 134–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemper K, Sprick MR, de Bree M, Scopelliti A, Vermeulen L, Hoek M, Zeilstra J, Pals ST, Mehmet H, Stassi G, et al. 2010. The AC133 epitope, but not the CD133 protein, is lost upon cancer stem cell differentiation. Cancer Res 70: 719–729. [DOI] [PubMed] [Google Scholar]
- Kim J, Woo AJ, Chu J, Snow JW, Fujiwara Y, Kim CG, Cantor AB, Orkin SH. 2010. A Myc network accounts for similarities between embryonic stem and cancer cell transcription programs. Cell 143: 313–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Kim E, Wu Q, Guryanova O, Hitomi M, Lathia JD, Serwanski D, Sloan AE, Weil RJ, Lee J, et al. 2012. Platelet-derived growth factor receptors differentially inform intertumoral and intratumoral heterogeneity. Genes Dev 26: 1247–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Kim M, Woo DH, Shin Y, Shin J, Chang N, Oh YT, Kim H, Rheey J, Nakano I, et al. 2013. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell 23: 839–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Joshi K, Ezhilarasan R, Myers TR, Siu J, Gu C, Nakano-Okuno M, Taylor D, Minata M, Sulman EP, et al. 2015. EZH2 protects glioma stem cells from radiation-induced cell death in a MELK/FOXM1-dependent manner. Stem Cell Rep 4: 226–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo T, Setoguchi T, Taga T. 2004. Persistence of a small subpopulation of cancer stem-like cells in the C6 glioma cell line. Proc Natl Acad Sci 101: 781–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong LY, Wu AS, Doucette T, Wei J, Priebe W, Fuller GN, Qiao W, Sawaya R, Rao G, Heimberger AB. 2010. Intratumoral mediated immunosuppression is prognostic in genetically engineered murine models of glioma and correlates to immunotherapeutic responses. Clin Cancer Res 16: 5722–5733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lathia JD, Gallagher J, Heddleston JM, Wang J, Eyler CE, Macswords J, Wu Q, Vasanji A, McLendon RE, Hjelmeland AB, et al. 2010. Integrin α6 regulates glioblastoma stem cells. Cell Stem Cell 6: 421–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lathia JD, Venere M, Rao MS, Rich JN. 2011. Seeing is believing: are cancer stem cells the Loch Ness monster of tumor biology? Stem Cell Rev 7: 227–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leder K, Pitter K, Laplant Q, Hambardzumyan D, Ross BD, Chan TA, Holland EC, Michor F. 2014. Mathematical modeling of PDGF-driven glioblastoma reveals optimized radiation dosing schedules. Cell 156: 603–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM, Pastorino S, Purow BW, Christopher N, Zhang W, et al. 2006. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 9: 391–403. [DOI] [PubMed] [Google Scholar]
- Lee J, Son MJ, Woolard K, Donin NM, Li A, Cheng CH, Kotliarova S, Kotliarov Y, Walling J, Ahn S, et al. 2008. Epigenetic-mediated dysfunction of the bone morphogenetic protein pathway inhibits differentiation of glioblastoma-initiating cells. Cancer Cell 13: 69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Wang H, Eyler CE, Hjelmeland AB, Rich JN. 2009a. Turning cancer stem cells inside out: an exploration of glioma stem cell signaling pathways. J Biol Chem 284: 16705–16709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Bao S, Wu Q, Wang H, Eyler C, Sathornsumetee S, Shi Q, Cao Y, Lathia J, McLendon RE, et al. 2009b. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 15: 501–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligon KL, Huillard E, Mehta S, Kesari S, Liu H, Alberta JA, Bachoo RM, Kane M, Louis DN, Depinho RA, et al. 2007. Olig2-regulated lineage-restricted pathway controls replication competence in neural stem cells and malignant glioma. Neuron 53: 503–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Yuan X, Zeng Z, Tunici P, Ng H, Abdulkadir IR, Lu L, Irvin D, Black KL, Yu JS. 2006. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer 5: 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lottaz C, Beier D, Meyer K, Kumar P, Hermann A, Schwarz J, Junker M, Oefner PJ, Bogdahn U, Wischhusen J, et al. 2010. Transcriptional profiles of CD133+ and CD133− glioblastoma-derived cancer stem cell lines suggest different cells of origin. Cancer Res 70: 2030–2040. [DOI] [PubMed] [Google Scholar]
- Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A, et al. 2012. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483: 474–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubanska D, Market-Velker BA, deCarvalho AC, Mikkelsen T, Fidalgo da Silva E, Porter LA. 2014. The cyclin-like protein Spy1 regulates growth and division characteristics of the CD133+ population in human glioma. Cancer Cell 25: 64–76. [DOI] [PubMed] [Google Scholar]
- Man J, Shoemake J, Zhou W, Fang X, Wu Q, Rizzo A, Prayson R, Bao S, Rich JN, Yu JS. 2014. Sema3C promotes the survival and tumorigenicity of glioma stem cells through Rac1 activation. Cell Rep 9: 1812–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meacham CE, Morrison SJ. 2013. Tumour heterogeneity and cancer cell plasticity. Nature 501: 328–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer M, Reimand J, Lan X, Head R, Zhu X, Kushida M, Bayani J, Pressey JC, Lionel AC, Clarke ID, et al. 2015. Single cell-derived clonal analysis of human glioblastoma links functional and genomic heterogeneity. Proc Natl Acad Sci 112: 851–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan RP, Costello JF. 2009. Epigenetic mechanisms in glioblastoma multiforme. Semin Cancer Biol 19: 188–197. [DOI] [PubMed] [Google Scholar]
- Nathan C. 2004. Antibiotics at the crossroads. Nature 431: 899–902. [DOI] [PubMed] [Google Scholar]
- Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, et al. 2010. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 17: 510–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogden AT, Waziri AE, Lochhead RA, Fusco D, Lopez K, Ellis JA, Kang J, Assanah M, McKhann GM, Sisti MB, et al. 2008. Identification of A2B5+CD133− tumor-initiating cells in adult human gliomas. Neurosurgery 62: 505–514. [DOI] [PubMed] [Google Scholar]
- Oliver JD. 2010. Recent findings on the viable but nonculturable state in pathogenic bacteria. FEMS Microbiol Rev 34: 415–425. [DOI] [PubMed] [Google Scholar]
- Pastrana E, Silva-Vargas V, Doetsch F. 2011. Eyes wide open: a critical review of sphere-formation as an assay for stem cells. Cell Stem Cell 8: 486–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, et al. 2014. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344: 1396–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegatta S, Poliani PL, Corno D, Menghi F, Ghielmetti F, Suarez-Merino B, Caldera V, Nava S, Ravanini M, Facchetti F, et al. 2006. Neurospheres enriched in cancer stem-like cells are highly effective in eliciting a dendritic cell-mediated immune response against malignant gliomas. Cancer Res 66: 10247–10252. [DOI] [PubMed] [Google Scholar]
- Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, Misra A, Nigro JM, Colman H, Soroceanu L, et al. 2006. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 9: 157–173. [DOI] [PubMed] [Google Scholar]
- Phuphanich S, Wheeler CJ, Rudnick JD, Mazer M, Wang H, Nuno MA, Richardson JE, Fan X, Ji J, Chu RM, et al. 2013. Phase I trial of a multi-epitope-pulsed dendritic cell vaccine for patients with newly diagnosed glioblastoma. Cancer Immunol Immunother 62: 125–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccirillo SG, Reynolds BA, Zanetti N, Lamorte G, Binda E, Broggi G, Brem H, Olivi A, Dimeco F, Vescovi AL. 2006. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature 444: 761–765. [DOI] [PubMed] [Google Scholar]
- Platten M, Wick W, Weller M. 2001. Malignant glioma biology: role for TGF-β in growth, motility, angiogenesis, and immune escape. Microsc Res Tech 52: 401–410. [DOI] [PubMed] [Google Scholar]
- Pollard SM, Yoshikawa K, Clarke ID, Danovi D, Stricker S, Russell R, Bayani J, Head R, Lee M, Bernstein M, et al. 2009. Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell 4: 568–580. [DOI] [PubMed] [Google Scholar]
- Qiang L, Wu T, Zhang HW, Lu N, Hu R, Wang YJ, Zhao L, Chen FH, Wang XT, You QD, et al. 2012. HIF-1α is critical for hypoxia-mediated maintenance of glioblastoma stem cells by activating Notch signaling pathway. Cell Death Differ 19: 284–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff RM, Engelhardt B. 2012. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat Rev Immunol 12: 623–635. [DOI] [PubMed] [Google Scholar]
- Reardon DA, Freeman G, Wu C, Chiocca EA, Wucherpfennig KW, Wen PY, Fritsch EF, Curry WT Jr, Sampson JH, Dranoff G. 2014. Immunotherapy advances for glioblastoma. Neuro Oncol 16: 1441–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reya T, Morrison SJ, Clarke MF, Weissman IL. 2001. Stem cells, cancer, and cancer stem cells. Nature 414: 105–111. [DOI] [PubMed] [Google Scholar]
- Rheinbay E, Suva ML, Gillespie SM, Wakimoto H, Patel AP, Shahid M, Oksuz O, Rabkin SD, Martuza RL, Rivera MN, et al. 2013. An aberrant transcription factor network essential for Wnt signaling and stem cell maintenance in glioblastoma. Cell Rep 3: 1567–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowitch DH, Kriegstein AR. 2010. Developmental genetics of vertebrate glial-cell specification. Nature 468: 214–222. [DOI] [PubMed] [Google Scholar]
- Sarkar S, Döring A, Zemp FJ, Silva C, Lun X, Wang X, Kelly J, Hader W, Hamilton M, Mercier P, et al. 2014. Therapeutic activation of macrophages and microglia to suppress brain tumor-initiating cells. Nat Neurosci 17: 46–55. [DOI] [PubMed] [Google Scholar]
- Sell S. 2004. Stem cell origin of cancer and differentiation therapy. Crit Rev Oncol Hematol 51: 1–28. [DOI] [PubMed] [Google Scholar]
- Sherry MM1, Reeves A, Wu JK, Cochran BH. 2009. STAT3 is required for proliferation and maintenance of multipotency in glioblastoma stem cells. Stem Cells 10: 2383–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB. 2003. Identification of a cancer stem cell in human brain tumors. Cancer Res 63: 5821–5828. [PubMed] [Google Scholar]
- Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. 2004. Identification of human brain tumour initiating cells. Nature 432: 396–401. [DOI] [PubMed] [Google Scholar]
- Son MJ, Woolard K, Nam DH, Lee J, Fine HA. 2009. SSEA-1 is an enrichment marker for tumor-initiating cells in human glioblastoma. Cell Stem Cell 4: 440–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sottoriva A, Spiteri I, Piccirillo SG, Touloumis A, Collins VP, Marioni JC, Curtis C, Watts C, Tavare S. 2013. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc Natl Acad Sci 110: 4009–4014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stommel JM, Kimmelman AC, Ying H, Nabioullin R, Ponugoti AH, Wiedemeyer R, Stegh AH, Bradner JE, Ligon KL, Brennan C, et al. 2007. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science 318: 287–290. [DOI] [PubMed] [Google Scholar]
- Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K, et al. 2009. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol 10: 459–466. [DOI] [PubMed] [Google Scholar]
- Sun Y, Kong W, Falk A, Hu J, Zhou L, Pollard S, Smith A. 2009. CD133 (Prominin) negative human neural stem cells are clonogenic and tripotent. PLoS One 4: e5498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suva ML, Rheinbay E, Gillespie SM, Patel AP, Wakimoto H, Rabkin SD, Riggi N, Chi AS, Cahill DP, Nahed BV, et al. 2014. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell 157: 580–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terasaki M, Shibui S, Narita Y, Fujimaki T, Aoki T, Kajiwara K, Sawamura Y, Kurisu K, Mineta T, Yamada A, et al. 2011. Phase I trial of a personalized peptide vaccine for patients positive for human leukocyte antigen–A24 with recurrent or progressive glioblastoma multiforme. J Clin Oncol 29: 337–344. [DOI] [PubMed] [Google Scholar]
- Tunici P, Bissola L, Lualdi E, Pollo B, Cajola L, Broggi G, Sozzi G, Finocchiaro G. 2004. Genetic alterations and in vivo tumorigenicity of neurospheres derived from an adult glioblastoma. Mol Cancer 3: 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida N, Buck DW, He D, Reitsma MJ, Masek M, Phan TV, Tsukamoto AS, Gage FH, Weissman IL. 2000. Direct isolation of human central nervous system stem cells. Proc Natl Acad Sci 97: 14720–14725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venere M, Fine HA, Dirks PB, Rich JN. 2011. Cancer stem cells in gliomas: identifying and understanding the apex cell in cancer's hierarchy. Glia 59: 1148–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venere M, Hamerlik P, Wu Q, Rasmussen RD, Song LA, Vasanji A, Tenley N, Flavahan WA, Hjelmeland AB, Bartek J, et al. 2014. Therapeutic targeting of constitutive PARP activation compromises stem cell phenotype and survival of glioblastoma-initiating cells. Cell Death Differ 21: 258–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verginelli F, Perin A, Dali R, Fung KH, Lo R, Longatti P, Guiot MC, Del Maestro RF, Rossi S, di Porzio U, et al. 2013. Transcription factors FOXG1 and Groucho/TLE promote glioblastoma growth. Nat Commun 4: 2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, et al. 2010. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17: 98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlashi E, Lagadec C, Vergnes L, Matsutani T, Masui K, Poulou M, Popescu R, Della Donna L, Evers P, Dekmezian C, et al. 2011. Metabolic state of glioma stem cells and nontumorigenic cells. Proc Natl Acad Sci 108: 16062–16067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Wang H, Li Z, Wu Q, Lathia JD, McLendon RE, Hjelmeland AB, Rich JN. 2008. c-Myc is required for maintenance of glioma cancer stem cells. PLoS One 3: e3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Lathia JD, Wu Q, Wang J, Li Z, Heddleston JM, Eyler CE, Elderbroom J, Gallagher J, Schuschu J, et al. 2009. Targeting interleukin 6 signaling suppresses glioma stem cell survival and tumor growth. Stem Cells 27: 2393–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Wakeman TP, Lathia JD, Hjelmeland AB, Wang XF, White RR, Rich JN, Sullenger BA. 2010. Notch promotes radioresistance of glioma stem cells. Stem Cells 28: 17–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Barr J, Kong LY, Wang Y, Wu A, Sharma AK, Gumin J, Henry V, Colman H, Sawaya R, et al. 2010. Glioma-associated cancer-initiating cells induce immunosuppression. Clin Cancer Res 16: 461–473. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Wei Y, Jiang Y, Zou F, Liu Y, Wang S, Xu N, Xu W, Cui C, Xing Y, Liu Y, et al. 2013. Activation of PI3K/Akt pathway by CD133–p85 interaction promotes tumorigenic capacity of glioma stem cells. Proc Natl Acad Sci 110: 6829–6834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weller M, Pfister SM, Wick W, Hegi ME, Reifenberger G, Stupp R. 2013. Molecular neuro-oncology in clinical practice: a new horizon. Lancet Oncol 14: e370–e379. [DOI] [PubMed] [Google Scholar]
- Wu A, Wei J, Kong LY, Wang Y, Priebe W, Qiao W, Sawaya R, Heimberger AB. 2010. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol 12: 1113–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurdak H, Zhu S, Romero A, Lorger M, Watson J, Chiang CY, Zhang J, Natu VS, Lairson LL, Walker JR, et al. 2010. An RNAi screen identifies TRRAP as a regulator of brain tumor-initiating cell differentiation. Cell Stem Cell 6: 37–47. [DOI] [PubMed] [Google Scholar]
- Xie Q, Wu Q, Horbinski CM, Flavahan WA, Yang K, Zhou W, Dombrowski SM, Huang Z, Fang X, Shi Y, et al. 2015. Mitochondrial control by DRP1 in brain tumor initiating cells. Nat Neurosci 18: 501–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Liu G, Yuan X, Xu M, Wang H, Ji J, Konda B, Black KL, Yu JS. 2009. Antigen-specific T-cell response from dendritic cell vaccination using cancer stem-like cell-associated antigens. Stem Cells 27: 1734–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan K, Wu Q, Yan DH, Lee CH, Rahim N, Tritschler I, DeVecchio J, Kalady MF, Hjelmeland AB, Rich JN. 2014. Glioma cancer stem cells secrete Gremlin1 to promote their maintenance within the tumor hierarchy. Genes Dev 28: 1085–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu SC, Ping YF, Yi L, Zhou ZH, Chen JH, Yao XH, Gao L, Wang JM, Bian XW. 2008. Isolation and characterization of cancer stem cells from a human glioblastoma cell line U87. Cancer Lett 265: 124–134. [DOI] [PubMed] [Google Scholar]
- Zbinden M, Duquet A, Lorente-Trigos A, Ngwabyt SN, Borges I, Ruiz i Altaba A. 2010. NANOG regulates glioma stem cells and is essential in vivo acting in a cross-functional network with GLI1 and p53. EMBO J 29: 2659–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Ying H, Yan H, Kimmelman AC, Hiller DJ, Chen AJ, Perry SR, Tonon G, Chu GC, Ding Z, et al. 2008. p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature 455: 1129–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Ying H, Wiedemeyer R, Yan H, Quayle SN, Ivanova EV, Paik JH, Zhang H, Xiao Y, Perry SR, et al. 2010. PLAGL2 regulates Wnt signaling to impede differentiation in neural stem cells and gliomas. Cancer Cell 17: 497–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Ke SQ, Huang Z, Flavahan W, Fang X, Paul J, Wu L, Sloan AE, McLendon RE, Li X, et al. 2015. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat Cell Biol 17: 170–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu TS, Costello MA, Talsma CE, Flack CG, Crowley JG, Hamm LL, He X, Hervey-Jumper SL, Heth JA, Muraszko KM, et al. 2011. Endothelial cells create a stem cell niche in glioblastoma by providing NOTCH ligands that nurture self-renewal of cancer stem-like cells. Cancer Res 71: 6061–6072. [DOI] [PMC free article] [PubMed] [Google Scholar]