

In this study, Chen et al. investigate the physiological importance of YAP phosphorylation in regulating endogenous YAP activity. Using mutant YapS112A knock-in mice, they found that the mice show a compensatory decrease in YAP protein levels due to increased phosphorylation at a mammalian-specific phosphodegron site on YAP. The results provide novel insight into a homeostatic mechanism that maintains physiological levels of YAP activity, which could work to prevent oncogenic activation of YAP.
Keywords: 14-3-3, Hippo signaling, YAP oncoprotein, phosphorylation, tumorigenesis
Abstract
The Hippo signaling pathway converges on YAP to regulate growth, differentiation, and regeneration. Previous studies with overexpressed proteins have shown that YAP is phosphorylated by its upstream kinase, Lats1/2, on multiple sites, including an evolutionarily conserved 14-3-3-binding site whose phosphorylation is believed to inhibit YAP by excluding it from the nucleus. Indeed, nuclear localization of YAP or decreased YAP phosphorylation at this site (S168 in Drosophila, S127 in humans, and S112 in mice) is widely used in current literature as a surrogate of YAP activation even though the physiological importance of this phosphorylation event in regulating endogenous YAP activity has not been defined. Here we address this question by introducing a YapS112A knock-in mutation in the endogenous Yap locus. The YapS112A mice are surprisingly normal despite nuclear localization of the mutant YAP protein in vivo and profound defects in cytoplasmic translocation in vitro. Interestingly, the mutant YapS112A mice show a compensatory decrease in YAP protein levels due to increased phosphorylation at a mammalian-specific phosphodegron site on YAP. These findings reveal a robust homeostatic mechanism that maintains physiological levels of YAP activity and caution against the assumptive use of YAP localization alone as a surrogate of YAP activity.
The Hippo tumor suppressor pathway regulates organ size in Drosophila and mammals, and its dysregulation contributes to human cancers (Zeng and Hong 2008; Pan 2010; Zhao et al. 2010a; Halder and Johnson 2011; Barry and Camargo 2013; Piccolo et al. 2013). Central to the Hippo pathway is a kinase cascade involving sequential activation of the Ste20-like kinase Hpo (Mst1/2 in mammals) and the nuclear Dbf2-related (NDR) family kinase Wts (Lats1/2 in mammals), which in turn phosphorylates and inactivates the transcriptional coactivator Yki (YAP/TAZ in mammals). Supporting the physiological importance of Hippo signaling in growth control, inactivation of Hippo pathway tumor suppressors such as Hpo/Mst or overexpression of the Yki/YAP oncogene leads to massive tissue overgrowth in both Drosophila and mammals (Harvey et al. 2003; Wu et al. 2003; Huang et al. 2005; Camargo et al. 2007; Dong et al. 2007; Zhou et al. 2009, 2011; Lee et al. 2010; Lu et al. 2010).
Since phosphorylation of Yki/YAP represents the key signaling output of the Hippo pathway, there has been significant interest in understanding the mechanism of Yki/YAP inactivation by Hippo signaling. Previous studies with overexpressed proteins have shown that Yki/YAP can be phosphorylated by its upstream kinase, Wts/Lats, on multiple HxRxxS motifs (Fig. 1A). Among the three HxRxxS motifs in Yki and five HxRxxS motifs in YAP, one motif (YkiS168 in Drosophila, YAPS127 in humans, and YAPS112 in mice) serves as a 14-3-3-binding site that is required for phosphorylation-induced cytoplasmic translocation (Dong et al. 2007; Zhao et al. 2007). Besides the evolutionarily conserved 14-3-3-binding site, YAP is phosphorylated by Lats1/2 at another HxRxxS motif (YAPS381 in humans and YAPS366 in mice) (Zhao et al. 2010b). Phosphorylation of YAP at this site primes YAP for subsequent phosphorylation by casein kinase 1δ/ε (CK1δ/ε), which results in recruitment of the E3 ubiquitin ligase SCFβ-TRCP and YAP degradation by the ubiquitin–proteasome system. Interestingly, NIH3T3 cells could be transformed only by YAP protein carrying mutations in both the 14-3-3 site and the phosphodegron sites (Zhao et al. 2010b), suggesting that both mechanisms contribute to YAP regulation. However, unlike the 14-3-3-binding site, the phosphodegron site is not conserved in Drosophila Yki, suggesting that evolutionarily divergent mechanisms likely have evolved to regulate Yki/YAP in different animals.
Figure 1.
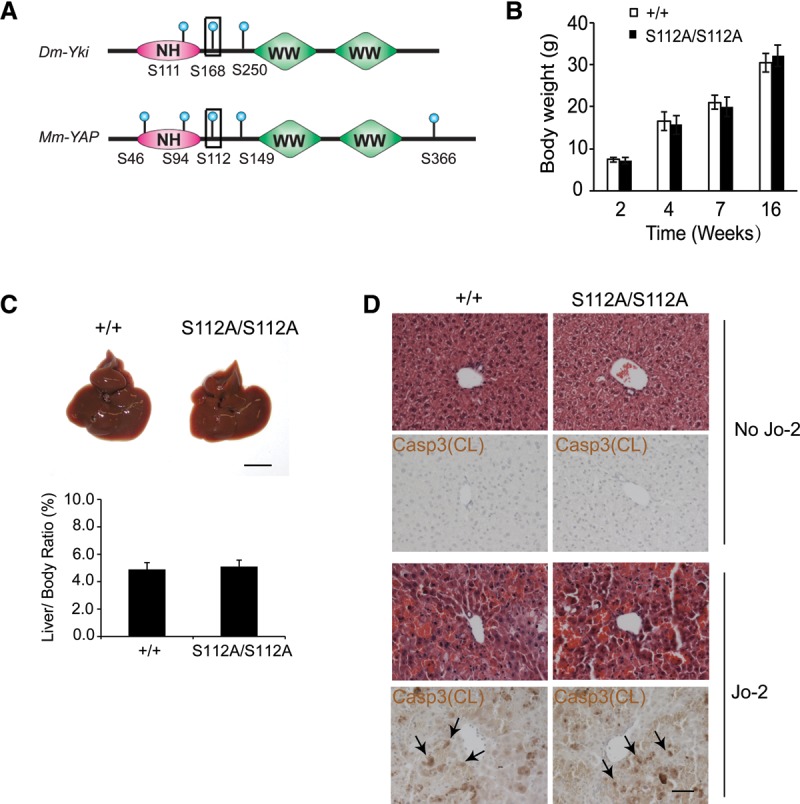
YAPS112 phosphorylation is dispensable for normal mouse development. (A) Schematic comparison of Drosophila Yki and murine YAP proteins showing the multiple HxRxxS phosphorylation sites, the WW domains, and the N-terminal homology (NH) domain required for Sd/TEAD binding. The conserved 14-3-3 site is also marked (boxed site). (B) Body weight of wild-type and YapS112A/S112A littermates. (C) Gross appearance of livers and quantification of the liver/body weight ratio from wild-type and YapS112A/S112A littermates at 2 mo of age. Data are mean ± SD from five animals of each genotype. Bar, 1 cm. (D) H&E and cleaved Caspase-3 staining of liver sections from 3-mo-old wild-type and YapS112A/S112A littermates before and 3 h after Jo-2 injection. Bar, 50 µm.
How the remaining HxRxxS motifs besides the 14-3-3-binding site and the phosphodegron site contribute to YAP inactivation has not been reported to date.
An important caveat with these studies is that they were almost exclusively based on the analysis of exogenously expressed constructs. One exception is in Drosophila, in which Halder and colleagues (Zhao et al. 2007) had isolated three gain-of-function alleles of endogenous yki surrounding the S168 site (H163Y, H163L, and P170S). These alleles are homozygous lethal, and the heterozygotes show tissue overgrowth due to gain-of-function Yki activity (Zhao et al. 2007). When these mutations were introduced into YAP, they were found to decrease (but not eliminate) YAP phosphorylation and/or 14-3-3 binding (Zhao et al. 2007). Furthermore, a transgene that expresses YkiS168A, but not wild-type Yki, close to endogenous Yki levels (using the tubulin promoter) results in tissue overgrowth and fly lethality (Dong et al. 2007). While these findings support the importance of the 14-3-3-binding site in Drosophila, the importance of this motif in endogenous mammalian YAP has not been determined. This is particularly relevant given the increasing appreciation of the evolutionary divergence of Hippo pathway regulation between Drosophila and mammals (Bossuyt et al. 2014). Indeed, due to the presence of both 14-3-3 binding and phosphodegron sites in mammalian YAP, one cannot predict a priori which phosphorylation site is essential.
Another unanswered issue in the Hippo research field concerns the exact contribution of the S127/S112 14-3-3-binding site to YAP subcellular localization. Initial studies of Hippo signaling in cultured mammalian cells revealed density-dependent localization of YAP, whereby YAP is nuclear in sparsely cultured cells and localized more to the cytoplasm upon confluence (Zhao et al. 2007). This nuclear-to-cytoplasmic YAP translocation was later extended to other conditions, such as disruption of actin cytoskeleton (Zhao et al. 2012) or treatment with certain GPCR (G-protein-coupled receptor) ligands (Yu et al. 2012). The literature, however, was ambiguous about whether such translocation is mediated exclusively through the S127/S112 site. While the YAPS127A mutant was shown to abolish 14-3-3 binding (Zhao et al. 2007) and should in principle eliminate cytoplasmic translocation induced by confluency or cytoskeleton disruption, many studies actually resorted to a YAP mutant lacking all HxRxxS motifs (YAP5SA) (Zhao et al. 2007, 2012). This is further complicated by reports suggesting that YAP can be translocated to the cytoplasm independently of Lats phosphorylation under certain culture conditions (Dupont et al. 2011). Irrespective of whether cytoplasmic translocation is mediated exclusively through S127/S112 phosphorylation, since previous studies were based on exogenously expressed constructs, the extent to which S127/S112 phosphorylation regulates endogenous YAP localization needs to be defined. From a broader perspective, how relevant is contact-induced cytoplasmic YAP translocation in cultured cells to the developmental regulation of organ size in vivo?
Here we investigated the physiological role of YAPS112 phosphorylation by introducing a YapS112A mutation in the endogenous Yap locus. Consistent with an essential role for 14-3-3 binding in cytoplasmic localization of YAP, the knock-in animals show nuclear localization of the mutant YAP protein in intact tissues, and cultured cells derived from the animals are resistant to YAP cytoplasmic translocation induced by multiple signals. Surprisingly, the YapS112A mice are phenotypically normal but show a compensatory decrease in YAP protein levels due to increased phosphorylation at YAP's phosphodegron site. These findings suggest the existence of a robust homeostatic mechanism that maintains physiological levels of YAP activity. Our studies suggest that YAP localization alone may not always be a reliable surrogate of YAP activity and offer an unprecedented example in which profound defects in YAP subcellular localization have no visible developmental consequences in YAP-mediated growth regulation in vivo.
Results
YAPS112 phosphorylation is dispensable for normal mouse development
We used homologous recombination to introduce a S112A mutation into the second exon and a loxP-STOP-loxP cassette in the first intron of Yap (Supplemental Fig. S1A). This strategy was initially designed to enable Cre-mediated excision of the STOP cassette, thus allowing conditional expression of the endogenous S112A allele. However, after germline transmission of the conditional S112A allele, we found that constitutive removal of the STOP cassette had no visible effect on animal development. We therefore used the constitutive S112A allele for all subsequent studies. Correct targeting of the YapS112A mutation was confirmed by PCR (Supplemental Fig. S1B,C) and DNA sequencing (Supplemental Fig. S1D) as well as phospho-specific antibody against the S112 phosphorylation site (for examples, see Fig. 5, below).
Figure 5.

Compensatory reduction of YAP protein levels in the YapS112A mice. (A) Decreased YAP protein levels in the YapS112A livers. Liver lysates of wild-type, YapS112A/+, and YapS112A/S112A mice (4 mo old) were probed with the indicated antibodies. The graphs show quantification of YAP protein and mRNA levels. Note the absence of S112 phosphorylation and the decrease in YAP protein but not mRNA levels. Values are means ± SD. n = 3. (*) P < 0.05, t-test. (B) Decreased YAP protein levels and increased YAPS366 phosphorylation in multiple organs of the YapS112A mice (2 mo old). Note that although the p-YAPS366 level was comparable between mutant and wild-type brains, the ratio of p-YAPS366 relative to total YAP was higher in the mutant due to the dramatic decrease of YAP protein level (see quantification in C).Values are means ± SD. n = 3. (C) Quantification of relative YAP protein level (left graph) and P-YAP (S366)/YAP ratio (right graph) in different organs of wild-type and YapS112A mice. Data are mean ± SD from three animals of each genotype. (*) P < 0.05, t-test. (D) Western blot analysis of cell lysates from the wild-type and YapS112A MEFs.
Intercrossing of YapS112A/+ animals revealed that wild-type (Yap+/+), heterozygote (YapS112A/+), and homozygote (YapS112A/S112A) littermates were born with the expected Mendelian ratio. Careful examination of the YapS112A/S112A homozygotes did not reveal any change in body weight or liver size in mice of all ages (Fig. 1B,C). Examination of the distal colon, another organ that is known to be sensitive to hyperactive YAP (Cai et al. 2010; Zhou et al. 2011), also did not reveal any abnormalities in crypt morphology or cell proliferation (Supplemental Fig. S1E,F). The YapS112A/S112A homozygotes also show normal fertility and have been maintained as healthy colonies in our laboratory for >7 years. For simplicity, we refer to the YapS112A/S112A homozygotes as YapS112A mice here unless otherwise specified.
Since activation of YAP is known to confer resistance to Fas-mediated apoptosis (Dong et al. 2007), we subjected the YapS112A mice to this experimental paradigm. Administration of the Fas agonist antibody (Jo-2) induced similar liver hemorrhage and hepatocyte death in the YapS112A and wild-type littermates (Fig. 1D). These findings further suggest that the YapS112A animals have a grossly normal level of YAP activity.
The YapS112A livers show subtle changes of YAP target gene expression and are more prone to carcinogen-induced hepatocellular carcinoma (HCC)
To investigate the possibility that the YapS112A mutation may confer a weak gain-of-function activity, we examined the expression of several commonly studied YAP target genes in the liver. Real-time PCR analysis revealed a modest increase in the mRNA levels of Birc5/Survivin, Cyr61, and Ctgf in the YapS112A livers compared with livers from age-matched control littermates (Fig. 2A). These findings suggest that the YapS112A mice may have a weak gain of function of YAP activity, although it was too weak to cause visible morphological and/or developmental defects under normal conditions.
Figure 2.

Mild gain of function of YAP in the YapS112A mice as revealed by YAP target expression and susceptibility to hepatocellular carcinogenesis. (A) Real-time PCR analysis of Birc5, Cyr61, Ctgf, Yap, and Taz mRNA levels in liver tissues of wild-type and YapS112A mice. Data are mean ± SD. n = 3 for each genotype. (*) P < 0.05, t-test. (B) Gross appearance of livers and quantification of liver/body weight ratio of wild-type and YapS112A mice 6.5 mo after diethylnitrosoamine (DEN) treatment. Data are mean ± SD from five animals of each genotype. (*) P < 0.05, t-test. Bar, 1 cm. (C) H&E staining of liver sections from wild-type and YapS112A mice 6.5 mo after DEN treatment. A large tumor in the YapS112A liver is indicated by “T.” Bar, 100 μm. (D) Wild-type, YapS112A/S112A, and Lats1/2 knockout mouse embryonic fibroblasts (MEFs) were subjected to wound healing assay. Shown are representative images of wound healing from 0 to 24 h after wound scratch. The white dashed lines mark the edges of the wound. Bar, 500 μm. Cell migration into the wound scratch was quantified as the percent wound closure relative to the open wound and compared with that of wild-type cells at each time point. All values are the means of triplicate experiments ± SD. (*) P < 0.05, t-test.
We reasoned that the weak gain of function of YAP in the YapS112A animals may be amplified under certain oncogenic conditions. To test this possibility, we subjected the animals to the DNA alkylating agent diethylnitrosoamine (DEN), a commonly used carcinogen for inducing HCC (Pitot et al. 1978). Interestingly, we found that the YapS112A mice were more prone to DEN-induced HCC formation. At 6.5 mo after DEN treatment, all of the YapS112A mice developed massive HCC (n = 10), whereas none of the matched wild-type mice developed macroscopically visible liver cancers (n = 10) (Fig. 2B,C). These findings further support the gain-of-function nature of the YapS112A mutation. This conclusion is also supported by the analysis of mouse embryonic fibroblasts (MEFs) derived from the YapS112A animals, which showed increased expression of YAP target genes (Supplemental Fig. S2A) and increased migration in a wound healing assay (Fig. 2D), a cellular property that is known to be enhanced by YAP activity (Sorrentino et al. 2014).
Lats1 and Lats2 are required for liver homeostasis by restricting YAP activity
The absence of visible developmental defects in the YapS112A animals is unexpected given that mutations affecting residues surrounding the analogous site in Drosophila Yki confer obvious growth advantage in vivo and are homozygous lethal (Zhao et al. 2007). Although Lats1/2 have been previously reported to directly phosphorylate YAP based on in vitro studies (Zhao et al. 2007; Hao et al. 2008; Zhang et al. 2008), it has also been suggested that Lats1/2 may not be the kinase responsible for YAPS112 phosphorylation in the liver (Zhou et al. 2009). To test this formal possibility, we examined the consequence of removing Lats1/2 function in the liver. First, we generated a floxed allele of Lats2 (Lats2flox) by homologous recombination (Supplemental Fig. S3A,B). In this allele, exon 5 and exon 6 of Lats2 were flanked by two LoxP sites. The Lats2flox allele was crossed to Lats1−/− mice (Yabuta et al. 2013) to obtain Lats1−/−; Lats2flox/flox animals. Adenovirus expressing the Cre recombinase was injected into the Lats1−/−; Lats2flox/flox animals to induce loss of Lats1/2 function in the liver. The Ad-Cre-injected Lats1−/−; Lats2flox/flox mice developed massive hepatomegaly, with liver size reaching ∼30% of the body weight 8 wk after Ad-Cre delivery (Fig. 3A,B). Microscopically, the Ad-Cre-injected Lats1−/−; Lats2flox/flox liver showed massive proliferation of the cytokeratin (CK)-positive biliary epithelial cells (BECs) (Fig. 3C,D). The overproliferating BECs first appeared as clusters of cells in the portal triads, which eventually overtook the hepatocytes to be the predominant cell type in the liver (Fig. 3C,D). Immunostaining of the Ad-Cre-injected Lats1−/−; Lats2flox/flox liver showed diminished YAP-S112 phosphorylation accompanied by an increase of overall YAP protein levels (Fig. 3D). These phenotypes resemble those described for liver-specific knockouts of other Hippo pathway tumor suppressors, such as NF2 and Mst1/2 (Zhou et al. 2009; Benhamouche et al. 2010; Lee et al. 2010; Lu et al. 2010; Zhang et al. 2010). Importantly, introducing loss of YAP and TAZ into the Lats1/2 mutant background (Ad-Cre; Lats1−/−; Lats2flox/flox; Yapflox/flox; Tazflox/flox) nearly completely abolished hepatomegaly (Fig. 3E) and BEC overproliferation (Supplemental Fig. S3C) of the Lats1/2 mutant liver. The rare BEC clusters remaining in these animals were due to escaper cells that failed to delete Yap and/or Taz, as revealed by positive staining with an antibody that recognizes both YAP and TAZ in these clusters (Supplemental Fig. S3C). Taken together, these findings demonstrate that Lats1/2 are indeed required for restricting YAP activity in vivo.
Figure 3.
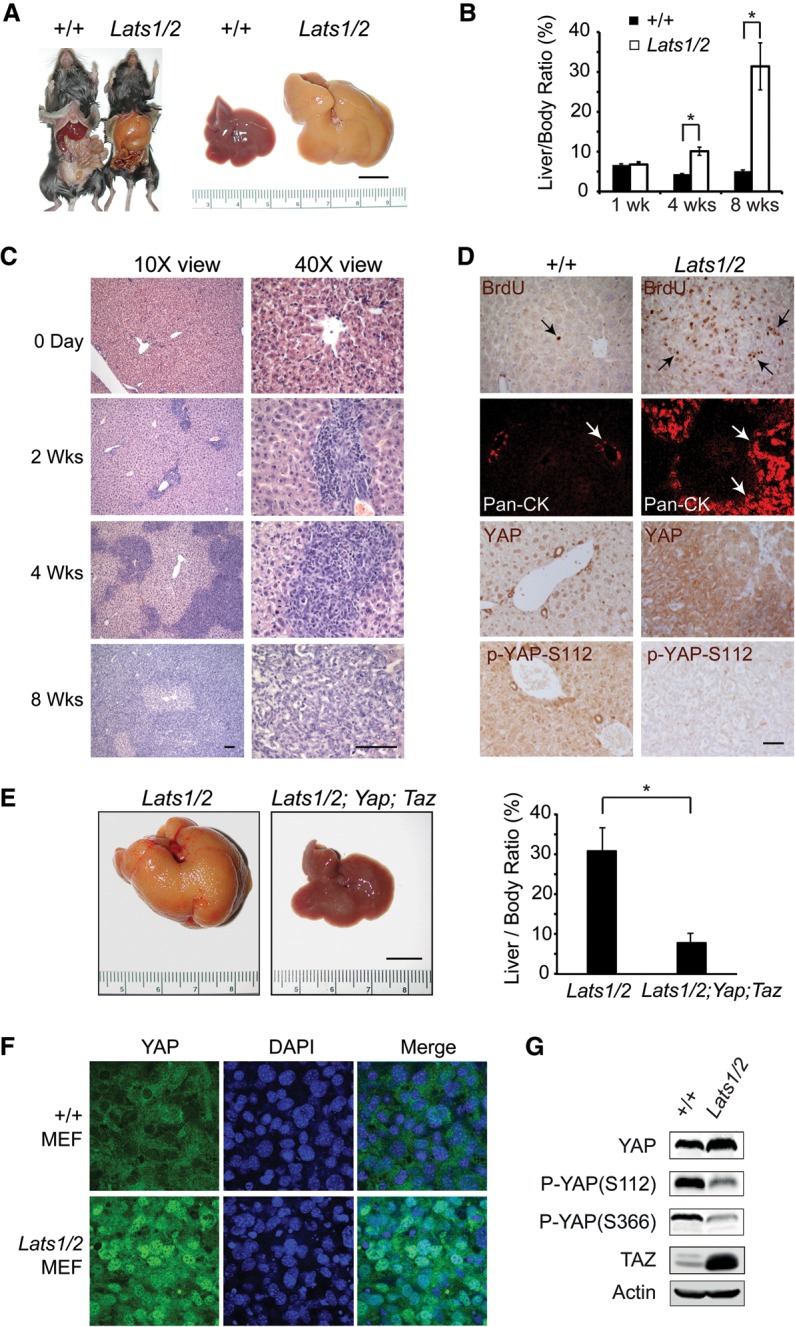
Lats1 and Lats2 are required for liver homeostasis by restricting YAP activity. (A) Gross appearance of control and Lats1−/−; Lats2flox/flox mouse livers 8 wk after Ad-Cre injection. Bar, 1 cm. (B) Quantification of liver/body ratio in wild-type and Lats1−/−; Lats2flox/flox littermates after Ad-Cre injection. Data are mean ± SD from five animals of each genotype. (*) P < 0.05, t-test. (C) H&E staining of liver sections from Lats1−/−; Lats2flox/flox animals before and after Ad-Cre injection. Bar, 50 μm. (D) BrdU, wide-spectrum CK (pan-CK), YAP, and phospho-YAP (S112) staining of liver sections from wild-type and Lats1−/−; Lats2flox/flox littermates 4 wk after Ad-Cre injection. Bar, 50 μm. (E) Gross appearance of livers and quantification of liver/body weight ratio of Lats1−/−; Lats2flox/flox and Lats1−/−; Lats2flox/flox; Yapflox/flox; Tazflox/flox mice 8 wk after Ad-Cre injection. Data are mean ± SD from five animals of each genotype. (*) P < 0.05, t-test. Bar, 1 cm. (F) Loss of cell contact-induced YAP translocation in the Lats1/2 mutant MEFs. Wild-type and Lats1/2 mutant MEFs grown at high density were immunostained for endogenous YAP (green) and nuclear dye DAPI (blue). Note the prominent nuclear staining of YAP in the Lats1/2 mutant MEFs compared with the wild-type MEFs. (G) Western blot analysis of cell lysates from the wild-type and Lats1/2 mutant MEFs.
To further examine the regulatory relationship between Lats1/2, YAP, and TAZ, we generated MEFs from the Lats1/2 mutant and wild-type littermates. Like the YapS112A MEFs, the Lats1/2 mutant MEFs also showed increased migration in a wound healing assay (Fig. 2D). Consistent with our analysis in the liver, YAP phosphorylation at both S112 and the phosphodegron site S366 was significantly decreased in the Lats1/2 MEFs (Fig. 3G). We also observed a dramatic increase in TAZ protein levels (Fig. 3G), consistent with YAP and TAZ being regulated by a similar phosphodegron mechanism by Hippo signaling (Liu et al. 2010; Zhao et al. 2010b). It is worth noting that although YAPS112 and YAPS366 phosphorylation was greatly diminished in the Lats1/2 mutant MEFs, it was not completely blocked (Fig. 3G). This is consistent with recent studies that have implicated the existence of additional YAP kinases besides Lats1/2 (Zhang et al. 2015).
S112 phosphorylation is required for 14-3-3 binding and cytoplasmic translocation of endogenous YAP induced by multiple signals
Previous studies that have implicated S127/S112 phosphorylation in 14-3-3-dependent cytoplasmic translocation were based on exogenously expressed YAP proteins. Thus, a formal possibility is that S127/S112 may not be as essential for 14-3-3 binding of endogenous YAP. Such a scenario could, in principle, explain the absence of developmental defects in the YapS112A animals. We tested this possibility by examining MEFs derived from the YapS112A or wild-type littermates. The physical interactions between endogenous YAP and 14-3-3 were completely abolished in the YapS112A MEFs (Fig. 4A). Thus, as for exogenous YAP, S112 phosphorylation is required for 14-3-3 binding of endogenous YAP.
Figure 4.

S112 phosphorylation is required for 14-3-3 binding and cytoplasmic translocation of endogenous YAP induced by contact inhibition in cell culture or developmental Hippo signaling in intact tissues. (A) Coimmunoprecipitation assay. Cell lysates of wild-type and YapS112A/S112A MEFs were immunoprecipitated with α-14-3-3 antibody and immunoblotted with α-YAP antibody. (B) Loss of cell contact-induced YAP translocation in the YapS112A/S112A MEFs. Wild-type and YapS112A/S112A MEFs grown at low or high density were immunostained for endogenous YAP (green) and nuclear dye DAPI (blue). Endogenous YAP shows nuclear-to-cytoplasmic translocation at high cell density in the wild-type cells but not in the YapS112A/S112A cells. Bar, 50 μm. (C) Immunostaining of YAP in liver sections from wild-type and YapS112A mice. Note the more prominent nuclear localization of YAP in the YapS112A liver compared with the wild-type liver (arrows). Also note the overall decrease of YAP staining intensity in the YapS112A liver. Tissue sections were processed in parallel and stained under identical conditions. Bar, 50 μm. (D) Immunostaining of YAP in colon sections from wild-type and YapS112A mice. Note the decrease of overall YAP staining and the more prominent nuclear localization in the colonic epithelial cells in YapS112A mice compared with the wild-type mice (arrows). Tissue sections were processed and stained under identical conditions. Bar, 50 μm.
In cultured mammalian cells, cell–cell contact induces cytoplasmic translocation of YAP (Zhao et al. 2007). Based on studies of exogenously expressed YAP constructs, the 14-3-3-binding site is believed to play a key role in this process (Zhao et al. 2007). Even though S127/S112 phosphorylation is required for 14-3-3 binding of endogenous YAP, it remains possible that such binding may not be as essential for controlling the subcellular localization of endogenous YAP. To investigate whether S112 phosphorylation is required for cell contact-induced translocation of endogenous YAP, we examined YAP protein localization in MEFs derived from the YapS112A or wild-type littermates. In sparse cultures, the YapS112A and wild-type MEFs showed similar nuclear localization of endogenous YAP (Fig. 4B). In confluent cultures, while wild-type MEFs showed a clear nuclear-to-cytoplasmic translocation of endogenous YAP, YAP remained nuclear in the YapS112A MEFs (Fig. 4B). Consistent with Lats1/2 being the major kinases for S112 phosphorylation, Lats1/2 mutant MEFs also showed a defect in nuclear-to-cytoplasmic translocation in confluent cultures (Fig. 3F). Thus, S112 phosphorylation is required for cell contact-induced cytoplasmic translocation of endogenous YAP.
Besides cell–cell contact, we also tested a number of other conditions that have been reported to induce cytoplasmic translocation of YAP. These include actin polymerization inhibitor latrunculin B (LatB) (Zhao et al. 2012), GPCR ligand epinephrine (Yu et al. 2012), adenylyl cyclase activator forskolin (Yu et al. 2013), and ROCK (rho-associated protein kinase) inhibitor Y-27632 (Dupont et al. 2011; Wada et al. 2011). While treatment with any of these agents induced a clear nuclear-to-cytoplasmic translocation of endogenous YAP in wild-type MEFs, the mutant YAPS112A protein remained nuclear in the YapS112A MEFs under the same treatment conditions (Supplemental Figs. S4–S7). Taken together, these results indicate that S112 phosphorylation is required for not only 14-3-3 binding but also cytoplasmic translocation of endogenous YAP induced by a wide spectrum of signals ranging from contact inhibition to disruption of actin cytoskeleton. Since YAP cytoplasmic translocation in cultured cells is widely used as a convenient assay for Hippo signaling, we caution that this cell culture assay per se does not necessarily predict Hippo-mediated growth regulation in vivo.
The YapS112A mice show abnormal subcellular localization of endogenous YAP in intact tissues despite the absence of visible developmental defects
The profound defects in nuclear-to-cytoplasmic translocation of YAP in the YapS112A MEFs prompted us to examine the subcellular localization of the mutant YAPS112A protein in intact animals. For this purpose, we analyzed YAP immunostaining in the liver and colon, two tissues in which the role of Hippo signaling in growth control has been well characterized (Camargo et al. 2007; Dong et al. 2007; Zhou et al. 2009; Cai et al. 2010; Lu et al. 2010; Song et al. 2010; Zhou et al. 2011). In the wild-type liver, YAP is diffusely distributed throughout the hepatocytes without discernible nuclear accumulation (Fig. 4C). In contrast, the mutant YAPS112A protein in the YapS112A hepatocytes stains the nuclei more prominently than the cytoplasm (Fig. 4C). A similar pattern was observed in the colonic epithelial cells, in which the mutant YAPS112A protein shows more discernible nuclear staining compared with the wild-type YAP (Fig. 4D). Given the normal development of the liver (Fig. 1C,D) and the colon (Supplemental Fig. S1E,F), the YapS112A mice provide an unprecedented example in which abnormal subcellular localization of YAP has no visible developmental consequences (albeit with a very modest effect on gene expression) (see Fig. 2A). This is in striking contrast to other genetic models, such as inactivation of Sav1 and Mst1/2, in which nuclear localization of YAP correlates with robust activation of its growth-promoting activity (Zhou et al. 2009, 2011; Cai et al. 2010; Song et al. 2010).
Compensatory reduction of YAP protein levels in the YapS112A mice
The results described above present a conundrum in which nuclear localization of the YAP transcriptional activator in the YapS112A animals is not translated into robust activation of YAP activity. The first clue to resolving this puzzle came from analysis of YAP immunostaining in the YapS112A animals. In our analysis of YAP immunostaining in the liver and colon, we noted that besides the more discernible YAP nuclear localization in the mutant tissues, the overall intensity of YAP staining in the YapS112A tissue appears lower than that in the corresponding wild-type tissue (Fig. 4C,D). This is very different from Sav1 or Mst1/2 mutant mice, which display not only nuclear localization of YAP but also an increase in overall YAP intensity (Zhou et al. 2009, 2011; Cai et al. 2010; Song et al. 2010). An attractive possibility is that there may be a compensatory reduction of YAP protein levels in the YapS112A mice; despite its nuclear localization and thus enhanced intrinsic transcriptional activity, a reduction in YAPS112A protein levels may render the overall YAP activity in the cell close to the normal levels.
To examine this possibility more quantitatively, we analyzed liver lysates by Western blotting. Consistent with our immunostaining results, YAP protein levels were significantly lower in mouse livers of the YapS112A animals compared with those from the wild-type littermates (Fig. 5A). A similar compensatory reduction of YAPS112A protein levels was also observed in other organs in the YapS112A mice (such as the brain, lung, heart, and spleen) (Fig. 5B,C) as well as the YapS112A MEFs (Fig. 5D). These results suggest that the compensatory reduction of YAPS112A protein levels is a general phenomenon across multiple tissue and cell types.
To understand the molecular mechanism underlying this compensation, we first examined Yap mRNA levels. The Yap mRNA levels were unchanged in the YapS112A livers compared with the wild-type littermates (Fig. 5A), suggesting that the reduction in YAPS112A protein levels is mediated by a post-transcriptional mechanism. To investigate this mechanism further, we treated the YapS112A and wild-type MEFs with the protein synthesis inhibitor cycloheximide (CHX) and compared the half-lives of endogenous YAP protein in the two different cells. This analysis revealed that the YAP protein was degraded much faster in the YapS112A MEFs than in the wild-type MEFs (Fig. 6A). These data suggest that the YAP protein is intrinsically less stable in the YapS112A cells.
Figure 6.

Compensatory decrease of YAP stability and activation of the Hippo kinase cascade in the YapS112A mice. (A) Faster turnover of endogenous YAPS112A protein compared with endogenous wild-type (WT) YAP protein. Confluent cultures of wild-type and YapS112A MEFs were treated with 50 µg/mL CHX for different periods of time and probed with the indicated antibodies. A representative blot is shown. YAP protein levels were quantified by the LI-COR Odyssey imaging system from three parallel experiments, normalized to actin, and arbitrarily set as 1 at time 0 in the graph shown at the right. Note that twice as much YapS112A cell lysates were loaded in each lane to adjust for the lower levels of YAP protein in the YapS112A cells (*). (B) Western blot analysis of liver protein lysates from wild-type and YapS112A mice. The graphs show quantification of YAP, TAZ, NF2, Mst1, Mst2, Lats1, Lats2, p-Lats, TEAD1-4, and CTGF levels in mutant livers relative to wild-type livers. Data are mean ± SD from three animals for each genotype. (*) P < 0.05, t-test.
YAPS112A induces a compensatory activation of the Hippo kinase cascade in the YapS112A mice
Since the best-characterized mechanism of destabilizing YAP involves its Hippo signaling-responsive phosphodegron motif (Zhao et al. 2010b), we hypothesized that the decreased stability of the endogenous YAPS112A protein may be due to increased Hippo signaling in the YapS112A cells, which should promote phosphorylation of its phosphodegron motif and thus induce YAPS112A degradation. In agreement with this hypothesis, phosphorylation of the phosphodegron site YAPS366 (corresponding to human YAPS381) was significantly increased in mouse livers from the YapS112A animals compared with those from the wild-type animals (Fig. 5B,C). This compensatory increase in YAPS366 phosphorylation was observed in not only the livers but also other organs in the YapS112A animals (such as the brain, lung, heart, and spleen) (Fig. 5B,C) as well as the YapS112A MEFs (Fig. 5D). Thus, the YapS112A animals are characterized by decreased YAP protein levels and a corresponding increase in YAPS366 phosphorylation across multiple tissue and cell types.
Consistent with enhanced Hippo signaling in the YapS112A animals, we found that the protein abundance of TAZ, a YAP paralog that is destabilized by Hippo signaling through a similar phosphodegron-dependent mechanism (Liu et al. 2010), was dramatically decreased in the YapS112A livers compared with the wild-type livers (Fig. 6B) even though Taz mRNA levels were normal in the YapS112A livers (Fig. 2A). A similar decrease of TAZ protein levels was also observed in the YapS112A MEFs (Fig. 5D). These data further support the notion that the increased YAPS366 phosphorylation and YAP degradation in the YapS112A animals is due to a compensatory increase in overall Hippo signaling activity rather than any intrinsic property of the YAP protein that autonomously biased the YAPS112A protein for phosphorylation on alternative sites when the S112 site is blocked.
Feedback activation of Hippo signaling protects against developmental defects in the YapS112A animals
The compensatory activation of Hippo signaling in the YapS112A animals is reminiscent of negative feedback previously described in Drosophila, in which increased Yki activity is accompanied by increased expression of upstream Hippo pathway tumor suppressors (Hamaratoglu et al. 2006). Consistent with a similar negative feedback being engaged in the YapS112A animals, the protein and mRNA abundance of NF2, Mst1, and Lats2 was increased in the YapS112A livers compared with wild-type littermates (Fig. 6B; Supplemental Fig. S2B). In contrast, the expression of Lats1, Mst2, and TEAD1–4 were similar between the YapS112A and wild-type livers (Fig. 6B). The protein abundance of CTGF is modestly increased in the YapS112A livers (Fig. 6B), consistent with the mild increase of Ctgf mRNA expression in the YapS112A livers (Fig. 2A).
To investigate the functional significance of this negative feedback, we engineered YapS112A mice with acute inactivation of NF2 in the adult liver induced by Ad-Cre injection. It had been reported previously that loss of NF2 in the adult liver results in very mild phenotypes manifested as focal periportal BEC hyperplasia even 8 mo after Ad-Cre injection (Benhamouche et al. 2010). Interestingly, although the YapS112A mutation does not display any visible developmental defects by itself, it greatly enhanced the Nf2-deficient phenotypes in adult livers. Two months after Ad-Cre injection, all of the Ad-Cre; Nf2flox/flox; YapS112A mice developed massive bile duct hamartomas (n = 5) with greatly enlarged liver size, whereas none of the matched Ad-Cre; Nf2flox/flox mice developed macroscopically visible hamartomas (Fig. 7A). Histological analysis revealed massive islands of proliferating CK-positive BECs in the Ad-Cre; Nf2flox/flox; YapS112A livers, in contrast to the mild focal proliferation of BECs in the matched Ad-Cre; Nf2flox/flox livers (Fig. 7B). Interestingly, although the YAP protein levels were lower in the YapS112A animals compared with wild-type animals, the difference in YAP protein levels between the two genotypes was greatly diminished after Ad-Cre-induced Nf2 gene deletion (Fig. 7C), supporting our model that activation of Hippo signaling contributes to the compensatory decrease of YAP protein abundance in the YapS112A mice. Thus, although the YapS112A mice do not show any visible developmental defects due to feedback activation of Hippo signaling, removing a critical Hippo signaling component can compromise this negative feedback and thus genetically “expose” the oncogenic potential of the YAPS112A protein.
Figure 7.
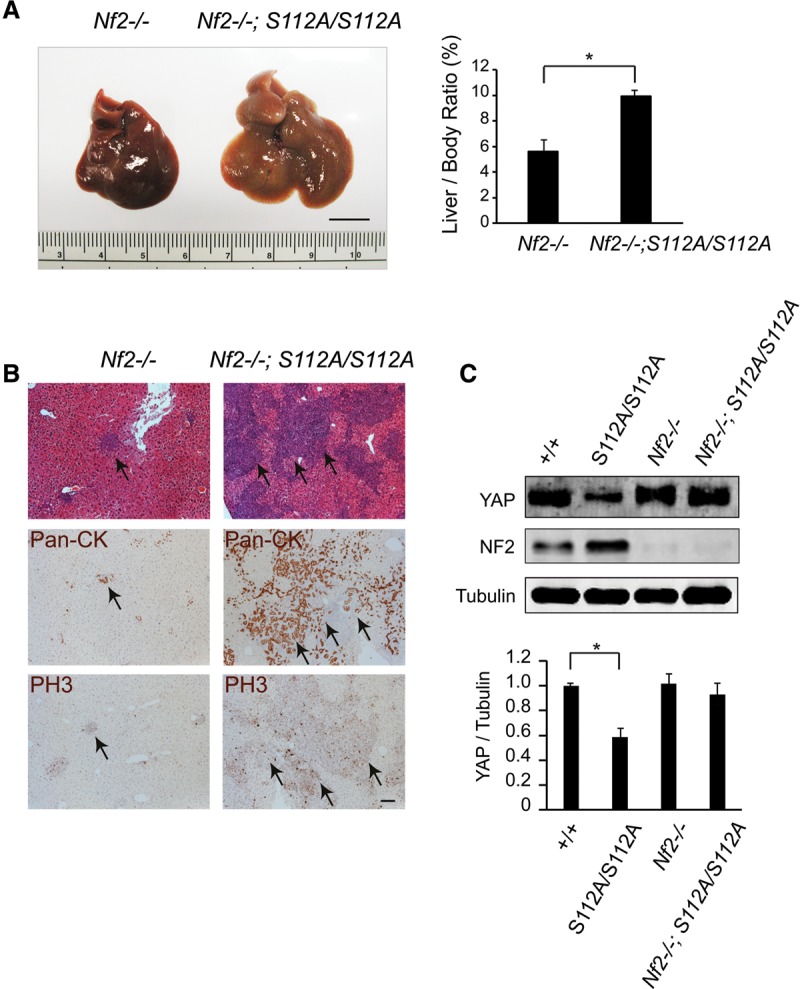
Feedback activation of Hippo signaling protects against developmental defects in the YapS112A animals. (A) Gross appearance of livers and quantification of liver/body weight ratio of Nf2flox/flox and Nf2flox/flox; YapS112A/S112A mice 8 wk after Ad-Cre injection. Data are mean ± SD from five animals of each genotype. (*) P < 0.05, t-test. Bar, 1 cm. (B) H&E, pan-CK, and phospho-Histone H3 (PH3) staining of liver sections from Nf2flox/flox and Nf2flox/flox; YapS112A/S112A littermates 8 wk after Ad-Cre injection. Note the increased BEC proliferation in Nf2flox/flox; YapS112A/S112A mutant livers compared with those of Nf2flox/flox mutant livers. Bar, 100 µm. (C) Western blot analysis of liver protein lysates from wild-type, YapS112A/S112A, Nf2flox/flox and Nf2flox/flox; YapS112A/S112A mice 2 mo after Ad-Cre injection. The graph shows quantification of YAP relative to tubulin from the four genotypes. Data are mean ± SD from three animals for each genotype. (*) P < 0.05, t-test.
Discussion
Recent mouse genetic studies using conditional loss-of-function and gain-of-function approaches have implicated the Hippo signaling pathway as a potent regulator of tissue growth, regeneration, and stem cell biology in mammals (Zeng and Hong 2008; Pan 2010; Zhao et al. 2010a; Halder and Johnson 2011; Barry and Camargo 2013; Piccolo et al. 2013). Thus, it has been puzzling why loss-of-function mutations in tumor suppressor components of the Hippo pathway or gain-of-function mutations in the oncoprotein YAP are relatively rare from comprehensive surveys of human cancer genomes. Our analysis of an endogenously activating mutation in YAP, which abolishes YAP-14-3-3 interactions and cyoplasmic translocation of YAP under multiple cell culture conditions and developmental contexts, provides at least one possible explanation. We suggest that increased YAP activity leads to compensatory activation of Hippo kinase cascade, which in turn results in decreased YAP protein levels due to phosphorylation of a mammalian-specific phosphodegron site. We suggest that such a feedback provides an intrinsic mechanism to maintain homeostatic levels of YAP activity in a cell and may therefore mask the phenotypic consequences of DNA mutations that would otherwise result in increased YAP activity.
Negative feedback regulation of upstream pathway components is a common feature of many signaling pathways that is generally believed to contribute to the steadiness and robustness of cell signaling in vivo (Stelling et al. 2004). The homeostatic mechanism that we uncovered in mammalian Hippo signaling is conceptually similar to that previously described in Drosophila, in which increased Yki activity is accompanied by increased transcription of upstream Hippo pathway tumor suppressors such as Kibra, Expanded, Crumbs, and Four-jointed (Cho et al. 2006; Hamaratoglu et al. 2006; Genevet et al. 2009, 2010). Indeed, we previously observed increased expression of multiple Hippo pathway components in a transgenic mouse model with liver-specific YAP overexpression (Dong et al. 2007). Although negative feedback appears to be a common feature of the Hippo signaling network in diverse animals, our study highlights a critical difference in the extent to which such negative feedback contributes to the homeostatic regulation of Yki/YAP activity in Drosophila versus mammals. While the YapS112A mice are phenotypically normal under physiological conditions (albeit with a modest effect on YAP target gene expression), mutations surrounding the analogous site in endogenous yki results in obvious gain-of-function phenotypes (overgrowth and homozygous lethality) in Drosophila (Zhao et al. 2007), and a transgene expressing the analogous YkiS168A mutant of Yki (but not wild-type Yki) close to its physiological levels results in tissue overgrowth and fly lethality (Dong et al. 2007). Thus, it appears that the negative feedback triggered by the YapS112A mutation is robust enough to protect the mice from developmental abnormalities, whereas negative feedback triggered by similar mutations in Yki cannot do so in Drosophila. These findings further add to the evolutionary divergence of pathway regulation between Drosophila and mammals despite the evolutionary conservation of many core components of the Hippo pathway (Bossuyt et al. 2014).
Nuclear accumulation of YAP is widely used as a surrogate of YAP activation in both the characterization of signaling events related to YAP regulation and the analysis of cancer samples. As we showed in this study, however, nuclear localization per se in either cultured cells or intact tissues is insufficient to predict increased YAP activity at a cellular level. Therefore, cellular YAP activity must be gauged by a combination of its nuclear localization and protein levels within a cell, perhaps ideally by assays for YAP transcriptional activity. We suggest that overexpression of YAP, instead of nuclear localization of YAP alone, may be more relevant to tumorigenesis. This is consistent with the identification of YAP gene amplification and the widespread YAP overexpression in human cancers (Overholtzer et al. 2006; Zender et al. 2006; Steinhardt et al. 2008). Understanding the genetic and epigenetic control of YAP gene expression in normal and pathological conditions as well as the mechanisms underlying the homeostatic control of YAP activity may shed light on how cancer cells manage to overcome this intrinsic homeostatic control of YAP activity in the course of tumorigenesis.
Materials and methods
Mouse genetics
A targeting vector containing the first two exons of the Yap gene was generated by recombineering as described previously (Liu et al. 2003). The TCC codon (encoding Ser112) located on exon 2 was mutated into the GCC codon (encoding Ala) by site-directed mutagenesis. A Lox-Stop-Lox cassette containing the NLS-β-geo coding region under the CMV enhancer/chicken β-actin promoter (provided by Dr. Jeremy Nathans, Johns Hopkins University School of Medicine) was inserted between exon 1 and exon 2. Transformed embryonic stem cell colonies were screened by long-template PCR with the following primer sets: PKI5F (5′-GATCCAGTTATCATAGCAAGTGTGTTCTCAATTTAAAGGC-3′) and PKI5R (5′-CTAGTCAATAATCAATGTCGACagtAAGCTtGCGGAACCC -3′) to generate a 4.5-kb band for positive clones and PKI3F (5′-TCTTATCATGTCTGGATCCACTAGTTCTAGCTAGTCTAGGTCGAC-3′) and PKI3R (5′-GCACCAAAAGCTGTTGTCTCCTACTCAGTCAGGAAGATGTTAACA-3′) to generate a 4.4-kb band for positive clones. Successfully targeted embryonic stem cell clones (confirmed by both 5′ PCR and 3′ PCR) were microinjected into C57BL/6 blastocysts. Germline transmission from generated chimeric offspring was confirmed by long-template PCR. Mice carrying the targeted allele were bred to CMV-Cre transgenic mice (provided by Dr. Jeremy Nathans, Johns Hopkins University School of Medicine) to remove the Lox-Stop-Lox cassette and generate the Yap S112A mice. Genomic DNA extracted from tail biopsies were genotyped with a PCR primer set (PKI1, 5′-GAACTTGCTTTAGGCTAAAG-3′; and PKI2, 5′-GAGTTTATTTAGCCGAGCAG-3′) that generated a 258-base-pair (bp) band from the wild-type allele and a 336-bp band from the S112A allele.
Lats1 mutant and Tazflox mice have been described (Xin et al. 2013; Yabuta et al. 2013). Nf2floxand Yapflox mice were described previously (Zhang et al. 2010). To generate the Lats2flox allele, a targeting vector containing exons 5 and 6 of Lats2 was generated by recombineering as described previously (Liu et al. 2003). Gene targeting in embryonic stem cells was screened by long-template PCR with the following primer sets: PL5F (5′-AGGCTTCCCGTGGTAAACGAGGACATTGTAAGGGCTTCCCGAGGG-3′) and P5LR (5′-TTAAGGGTTATTGAATATGATCGGAATTGGGCTGCAGGAA-3′) to generate a 4.4-kb band for positive clones and P3LF (5′-GCTCTATGGCTTCTGAGGCGGAAAGAACCAGCTGGGGCTCGAC-3′) and P3R (5′-GGTCCTCAGAGGGCTAATTTGAAATCATGTTGACTCTTCCTGTTG-3′) to generate a 4.5-kb band for positive clones (Supplemental Fig. S3A,B). Successfully targeted embryonic stem cell clones were microinjected into C57BL/6 blastocysts. Germline transmission from the chimeric founders was confirmed by PCR analysis. Mice carrying the targeted allele were bred to Flp recombinase transgenic mice to remove the FRT-flanked Neo cassette and generate the Lats2flox allele. Genomic DNA extracted from tail biopsies were genotyped with a PCR primer set (PL1, 5′-GAACTACTGAAATACTAACTAGAT-3′; PL2, 5′-GTCTTTGGCTAGAGAACTTTCTG-3′; or PL3, 5′-CCAGGTTGGACTAGGTCTAAAGACATGAG-3′) that generated a 252-bp band from the wild-type allele, a 356-bp band from the floxed allele, and a 550-bp band from the knockout allele.
Ad-Cre was injected into 3-wk-old wild-type, Lats1−/−; Lats2flox/flox, or Lats1−/−; Lats2flox/flox; Yapflox/flox; Tazflox/flox mice at 2 × 109 pfu per milliliter in 0.15 mL of sterile saline via retro-orbital sinus using a 30-gauge needle and analyzed at the indicated time points afterward. A similar amount of Ad-Cre was injected into 8-wk-old Nf2flox/flox and Nf2flox/flox; YapS112A littermates. For DEN treatment, 2-wk-old mice received a one-time intraperitoneal injection of DEN at a dose of 25 μg per gram of body weight. The animals were analyzed 6.5 mo after DEN treatment.
Immunofluorescence staining
MEFs were isolated from embryonic day 12.5 (E12.5) wild-type and YapS112A/S112A embryos and grown in DMEM (Invitrogen) supplemented with 10% fetal bovine serum and 50 µg/mL penicillin/streptomycin. Cells were maintained in a 37°C incubator at 5% CO2. The YapS112A and wild-type MEFs were cultured in six-well plates to confluence and trypsinized. Equal numbers of the YapS112A, Lats1/2 mutant and wild-type MEFs (5 × 104 for low density and 5 × 105 for high density) were plated into each well of a four-well Lab-Tek II chamber slide (Thermo Scientific) and incubated for 24 h at 37°C. Cells were treated with different chemicals (epinephrine, forskolin, LatB, or Y-27632) at the indicated doses and times. Cells were fixed with 4% paraformaldehyde for 15 min and then permeabilized with 0.3% Triton X-100. After blocking in 5% goat serum for 1 h, slides were incubated with anti-YAP antibody (1:300 dilution; Novus) diluted in 5% goat serum overnight at 4°C. After washing with PBS, slides were incubated with Alexa fluor 488-conjugated secondary antibodies (1:1000 dilution) for 1 h. The slides were incubated with DAPI (1:1000 dilution) for 5 min, washed, and mounted.
Quantitative real-time PCR (qPCR)
Total cellular or liver RNA was extracted using TRIzol reagent (Invitrogen). RNA was reverse-transcribed using iScript cDNA synthesis kit (Bio-Rad). qPCR was performed using the iQ SYBR Green supermix (Bio-Rad) on a iQ5 multicolor real-time PCR detection system (Applied Biosystems). qPCR was done in triplicate using Gapdh as a housekeeping control. Relative differences in the expression of the candidate genes in different experimental mouse livers were determined using 2−ΔΔCt method (Livak and Schmittgen 2001). The primer sequences used are available on request.
Histological analysis and immunostaining
Mouse liver and intestinal tissues were fixed overnight in 10% neutral buffered formalin solution (Sigma), embedded in paraffin, and sectioned at 5 µm. Sections were stained with hematoxylin–eosin for histologic examination or with Sirius Red to visualize fibrosis. For BrdU labeling, mice were injected with BrdU at 30 μg per gram of body weight 2 h prior to dissection. Primary antibodies used for immunofluorescence were rabbit anti-pan-CK (1:500; DAKO, Z0622) and rabbit anti-YAP (1:100; Novus Biologicals). Cy3-conjugated goat anti-mouse (1:250) and Alexa488-conjugated goat anti-rabbit (1:250; Molecular Probes) secondary antibodies were used for immunofluorescence. Immunohistochemistry staining was performed on paraffin sections using a Vectastain ABC kit according to manufacturer's instructions (Vector Laboratories). Primary antibodies used for immunohistochemistry were mouse anti-BrdU (1:100; Developmental Studies Hybridoma Bank), rabbit anti-YAP (1:100; Cell Signaling), rabbit anti-P-YAP (1:400; Cell Signaling), anti-Ki67 (1:1000; Novocastra), anti-phospho-Histone H3 (1:400; Millipore), and anti-cleaved Caspase-3 (1:100; Cell Signaling).
Mouse colonic crypts were isolated as described (Cai et al. 2010). In brief, colons at ∼1 cm away from the anus were cut longitudinally and incubated in PBS with 5 mM EDTA for 15 min at 37°C. Crypts were released by vigorous shaking. The width of crypts was measured in AxioVision release 4.8.
Protein lysate, Western blot analysis, and coimmunoprecipitation
Cells or tissues were lysed in RIPA buffer (150 mM sodium chloride, 50 mM Tris-HCl at pH 7.4, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM PMSF) with protease inhibitors (Roche). The proteins were separated on SDS–polyacrylamide gels and transferred onto PVDF membranes (Millipore). The blots were probed with antibodies against YAP (Cell Signaling, #4912), phospho-YAP (Ser112; Cell Signaling, #4911), phospho-YAP (Ser366; Cell Signaling, #13619), Lats1 (Cell Signaling, #3477), Lats2 (Cell Signaling, #13646), Mst1 (Cell Signaling, #ab51134), NF2 (Sigma, HPA003097), Mst2 (Epitomics, 1943-1), CTGF (Santa Cruz Biotechnology, sc-14939), α-tubulin (Developmental Studies Hybridoma Bank, 12G10), PAN-TEAD (Cell Signaling, #13295), MOB1 (Cell Signaling, #3863), phospho-MOB1 (Cell Signaling, #8699), YAP, and TAZ (Cell Signaling, #8418) and normalized by Actin (Millipore, MAB1501). Signals were detected and quantified by a LI-COR infrared imaging system. The antibodies used for coimmunoprecipitation were 14-3-3 (Santa Cruz Biotechnology, sc-732) and YAP (Novus Biologicals, NB110-58358).
CHX treatment and cell migration assay
Cells were plated at 3 × 106 per well in six-well plates and cultured for 24 h. CHX (50 μg/mL) was added to block new protein synthesis. Cell lysates were collected in 1× sample buffer at the indicated time points and analyzed by immunoblotting with antibodies against YAP, P-YAP-S112, and actin. Signal intensities were measured by the LI-COR Odyssey imaging system.
Wild-type, YapS112A/S112A, and Lats1/2 mutant MEFs were plated onto six-well plates and allowed to form a confluent monolayer. The cell monolayer was then scratched in a straight line to make a “scratch wound” with a 0.2-mL pipette tip. Pictures of the closure of the scratch were captured at 0, 10, and 24 h.
Supplementary Material
Acknowledgments
We thank Dr. Jeremy Nathans for providing CMV-Cre mice, and Dr. Eric Olson for Tazflox mice. We thank Haibo Bai, Yuchen Duan, Nathan Law, Suresh Nayar, Vanessa Moore, and Christopher Maltbie for technical assistance. This study was supported in part by grants from the National Institutes of Health (EY015708). D.P. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.264234.115.
References
- Barry ER, Camargo FD. 2013. The Hippo superhighway: signaling crossroads converging on the Hippo/Yap pathway in stem cells and development. Curr Opin Cell Biol 25: 247–253. [DOI] [PubMed] [Google Scholar]
- Benhamouche S, Curto M, Saotome I, Gladden AB, Liu CH, Giovannini M, McClatchey AI. 2010. Nf2/Merlin controls progenitor homeostasis and tumorigenesis in the liver. Genes Dev 24: 1718–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossuyt W, Chen CL, Chen Q, Sudol M, McNeill H, Pan D, Kopp A, Halder G. 2014. An evolutionary shift in the regulation of the Hippo pathway between mice and flies. Oncogene 33: 1218–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai J, Zhang N, Zheng Y, de Wilde RF, Maitra A, Pan D. 2010. The Hippo signaling pathway restricts the oncogenic potential of an intestinal regeneration program. Genes Dev 24: 2383–2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camargo FD, Gokhale S, Johnnidis JB, Fu D, Bell GW, Jaenisch R, Brummelkamp TR. 2007. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr Biol 17: 2054–2060. [DOI] [PubMed] [Google Scholar]
- Cho E, Feng Y, Rauskolb C, Maitra S, Fehon R, Irvine KD. 2006. Delineation of a Fat tumor suppressor pathway. Nat Genet 38: 1142–1150. [DOI] [PubMed] [Google Scholar]
- Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, Gayyed MF, Anders RA, Maitra A, Pan D. 2007. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 130: 1120–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le DJ, Forcato M, Bicciato S, et al. 2011. Role of YAP/TAZ in mechanotransduction. Nature 474: 179–183. [DOI] [PubMed] [Google Scholar]
- Genevet A, Polesello C, Blight K, Robertson F, Collinson LM, Pichaud F, Tapon N. 2009. The Hippo pathway regulates apical-domain size independently of its growth-control function. J Cell Sci 122: 2360–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genevet A, Wehr MC, Brain R, Thompson BJ, Tapon N. 2010. Kibra is a regulator of the salvador/warts/hippo signaling network. Dev Cell 18: 300–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halder G, Johnson RL. 2011. Hippo signaling: growth control and beyond. Development 138: 9–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamaratoglu F, Willecke M, Kango-Singh M, Nolo R, Hyun E, Tao C, Jafar-Nejad H, Halder G. 2006. The tumour-suppressor genes NF2/Merlin and Expanded act through Hippo signalling to regulate cell proliferation and apoptosis. Nat Cell Biol 8: 27–36. [DOI] [PubMed] [Google Scholar]
- Hao Y, Chun A, Cheung K, Rashidi B, Yang X. 2008. Tumor suppressor LATS1 is a negative regulator of oncogene YAP. J Biol Chem 283: 5496–5509. [DOI] [PubMed] [Google Scholar]
- Harvey KF, Pfleger CM, Hariharan IK. 2003. The Drosophila Mst ortholog, hippo, restricts growth and cell proliferation and promotes apoptosis. Cell 114: 457–467. [DOI] [PubMed] [Google Scholar]
- Huang J, Wu S, Barrera J, Matthews K, Pan D. 2005. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila homolog of YAP. Cell 122: 421–434. [DOI] [PubMed] [Google Scholar]
- Lee KP, Lee JH, Kim TS, Kim TH, Park HD, Byun JS, Kim MC, Jeong WI, Calvisi DF, Kim JM, et al. 2010. The Hippo–Salvador pathway restrains hepatic oval cell proliferation, liver size, and liver tumorigenesis. Proc Natl Acad Sci 107: 8248–8253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Jenkins NA, Copeland NG. 2003. A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res 13: 476–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C-Y, Zha Z-Y, Zhou X, Zhang H, Huang W, Zhao D, Li T, Chan SW, Lim CJ, Hong W. 2010. The hippo tumor pathway promotes TAZ degradation by phosphorylating a phosphodegron and recruiting the SCFβ-TrCP E3 ligase. J Biol Chem 285: 37159–37169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔC(T) method. Methods 25: 402–408. [DOI] [PubMed] [Google Scholar]
- Lu L, Li Y, Kim SM, Bossuyt W, Liu P, Qiu Q, Wang Y, Halder G, Finegold MJ, Lee JS, et al. 2010. Hippo signaling is a potent in vivo growth and tumor suppressor pathway in the mammalian liver. Proc Natl Acad Sci 107: 1437–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overholtzer M, Zhang J, Smolen GA, Muir B, Li W, Sgroi DC, Deng CX, Brugge JS, Haber DA. 2006. Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proc Natl Acad Sci 103: 12405–12410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan D. 2010. The hippo signaling pathway in development and cancer. Dev Cell 19: 491–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccolo S, Cordenonsi M, Dupont S. 2013. Molecular pathways: YAP and TAZ take center stage in organ growth and tumorigenesis. Clin Cancer Res 19: 4925–4930. [DOI] [PubMed] [Google Scholar]
- Pitot HC, Barsness L, Goldsworthy T, Kitagawa T. 1978. Biochemical characterisation of stages of hepatocarcinogenesis after a single dose of diethylnitrosamine. Nature 271: 456–458. [DOI] [PubMed] [Google Scholar]
- Song H, Mak KK, Topol L, Yun K, Hu J, Garrett L, Chen Y, Park O, Chang J, Simpson RM, et al. 2010. Mammalian Mst1 and Mst2 kinases play essential roles in organ size control and tumor suppression. Proc Natl Acad Sci 107: 1431–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorrentino G, Ruggeri N, Specchia V, Cordenonsi M, Mano M, Dupont S, Manfrin A, Ingallina E, Sommaggio R, Piazza S. 2014. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat Cell Biol 16: 357–366. [DOI] [PubMed] [Google Scholar]
- Steinhardt AA, Gayyed MF, Klein AP, Dong J, Maitra A, Pan D, Montgomery EA, Anders RA. 2008. Expression of Yes-associated protein in common solid tumors. Hum Pathol 39: 1582–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelling J, Sauer U, Szallasi Z, Doyle FJ III, Doyle J. 2004. Robustness of cellular functions. Cell 118: 675–685. [DOI] [PubMed] [Google Scholar]
- Wada K, Itoga K, Okano T, Yonemura S, Sasaki H. 2011. Hippo pathway regulation by cell morphology and stress fibers. Development 138: 3907–3914. [DOI] [PubMed] [Google Scholar]
- Wu S, Huang J, Dong J, Pan D. 2003. hippo encodes a Ste-20 family protein kinase that restricts cell proliferation and promotes apoptosis in conjunction with salvador and warts. Cell 114: 445–456. [DOI] [PubMed] [Google Scholar]
- Xin M, Kim Y, Sutherland LB, Murakami M, Qi X, McAnally J, Porrello ER, Mahmoud AI, Tan W, Shelton JM, et al. 2013. Hippo pathway effector Yap promotes cardiac regeneration. Proc Natl Acad Sci 110: 13839–13844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yabuta N, Mukai S, Okamoto A, Okuzaki D, Suzuki H, Torigata K, Yoshida K, Okada N, Miura D, Ito A, et al. 2013. N-terminal truncation of Lats1 causes abnormal cell growth control and chromosomal instability. J Cell Sci 126: 508–520. [DOI] [PubMed] [Google Scholar]
- Yu FX, Zhao B, Panupinthu N, Jewell JL, Lian I, Wang LH, Zhao J, Yuan H, Tumaneng K, Li H, et al. 2012. Regulation of the Hippo–YAP pathway by G-protein-coupled receptor signaling. Cell 150: 780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu FX, Zhang Y, Park HW, Jewell JL, Chen Q, Deng Y, Pan D, Taylor SS, Lai ZC, Guan KL. 2013. Protein kinase A activates the Hippo pathway to modulate cell proliferation and differentiation. Genes Dev 27: 1223–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zender L, Spector MS, Xue W, Flemming P, Cordon-Cardo C, Silke J, Fan ST, Luk JM, Wigler M, Hannon GJ, et al. 2006. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell 125: 1253–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Q, Hong W. 2008. The emerging role of the hippo pathway in cell contact inhibition, organ size control, and cancer development in mammals. Cancer Cell 13: 188–192. [DOI] [PubMed] [Google Scholar]
- Zhang J, Smolen GA, Haber DA. 2008. Negative regulation of YAP by LATS1 underscores evolutionary conservation of the Drosophila Hippo pathway. Cancer Res 68: 2789–2794. [DOI] [PubMed] [Google Scholar]
- Zhang N, Bai H, David KK, Dong J, Zheng Y, Cai J, Giovannini M, Liu P, Anders RA, Pan D. 2010. The Merlin/NF2 tumor suppressor functions through the YAP oncoprotein to regulate tissue homeostasis in mammals. Dev Cell 19: 27–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Tang F, Terracciano L, Hynx D, Kohler R, Bichet S, Hess D, Cron P, Hemmings BA, Hergovich A, et al. 2015. NDR functions as a physiological YAP1 kinase in the intestinal epithelium. Curr Biol 25: 296–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, Xie J, Ikenoue T, Yu J, Li L, et al. 2007. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev 21: 2747–2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Li L, Lei Q, Guan KL. 2010a. The Hippo–YAP pathway in organ size control and tumorigenesis: an updated version. Genes Dev 24: 862–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Li L, Tumaneng K, Wang CY, Guan KL. 2010b. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCFβ-TRCP. Genes Dev 24: 72–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Li L, Wang L, Wang CY, Yu J, Guan KL. 2012. Cell detachment activates the Hippo pathway via cytoskeleton reorganization to induce anoikis. Genes Dev 26: 54–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D, Conrad C, Xia F, Park JS, Payer B, Yin Y, Lauwers GY, Thasler W, Lee JT, Avruch J, et al. 2009. Mst1 and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer Cell 16: 425–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D, Zhang Y, Wu H, Barry E, Yin Y, Lawrence E, Dawson D, Willis JE, Markowitz SD, Camargo FD, et al. 2011. Mst1 and Mst2 protein kinases restrain intestinal stem cell proliferation and colonic tumorigenesis by inhibition of Yes-associated protein (Yap) overabundance. Proc Natl Acad Sci 108: E1312–E1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.