

In this study, Patel and colleagues determined the crystal structure of the conserved C terminus of the hSpt2C histone chaperone bound to an H3/H4 tetramer. The results suggest that Spt2 interacts with the periphery of the H3/H4 tetramer and promotes its recycling.
Keywords: H3/H4 tetramer, epigenetics, histone chaperone, histone maintenance, Spt2, spurious transcription, transcription elongation
Abstract
Cells use specific mechanisms such as histone chaperones to abrogate the inherent barrier that the nucleosome poses to transcribing polymerases. The current model postulates that nucleosomes can be transiently disrupted to accommodate passage of RNA polymerases and that histones H3 and H4 possess their own chaperones dedicated to the recovery of nucleosomes. Here, we determined the crystal structure of the conserved C terminus of human Suppressors of Ty insertions 2 (hSpt2C) chaperone bound to an H3/H4 tetramer. The structural studies demonstrate that hSpt2C is bound to the periphery of the H3/H4 tetramer, mimicking the trajectory of nucleosomal-bound DNA. These structural studies have been complemented with in vitro binding and in vivo functional studies on mutants that disrupt key intermolecular contacts involving two acidic patches and hydrophobic residues on Spt2C. We show that contacts between both human and yeast Spt2C with the H3/H4 tetramer are required for the suppression of H3/H4 exchange as measured by H3K56ac and new H3 deposition. These interactions are also crucial for the inhibition of spurious transcription from within coding regions. Together, our data indicate that Spt2 interacts with the periphery of the H3/H4 tetramer and promotes its recycling in the wake of RNA polymerase.
In the context of transcription elongation, earlier studies suggested that nucleosomes are disassembled in front of RNA polymerase II and recovered immediately after its passage (Kireeva et al. 2002; Belotserkovskaya et al. 2003). The appropriate redeposition of histones upon RNA polymerase II passage depends on histone chaperones and histone marks (Smolle and Workman 2013). A defect in this function leads to inaccurate transcription from within coding regions, a significant change in the transcription profile, and the production of thousands of cryptic transcripts (Rufiange et al. 2007; Smolle and Workman 2013). Interestingly, in transcribed regions, H2A/H2B exchange rates are substantially higher (at least 20-fold) than H3/H4 exchange rates (Kimura and Cook 2001; Thiriet and Hayes 2005), suggesting that nucleosomes could be transiently disrupted to accommodate the passage of RNA polymerases and that histones H3 and H4 have their own recycling mechanisms during transcription elongation (Dion et al. 2007). Recently, several reports confirmed that the majority of H3/H4 tetramers are maintained, rarely split, and recycled back during transcription (Xu et al. 2010; Katan-Khaykovich and Struhl 2011). Importantly, several classes of histone chaperones are involved in histone H3/H4 dynamics on transcribed genes, including Asf1, HIRA, FACT (facilitates chromatin transcription complex), Spt6 (encoded by Suppressors of Ty insertions gene family 6), and Spt2 (Orphanides et al. 1999; Kaplan et al. 2003; Nourani et al. 2006; Jamai et al. 2009; Avvakumov et al. 2011; Osakabe et al. 2013). Most of these conserved factors are essential to the maintenance of nucleosomal structure and the redeposition of H3/H4 within coding regions of genes (Avvakumov et al. 2011; Thebault et al. 2011; Smolle and Workman 2013). However, the mechanism of H3/H4 recycling by these factors during transcription elongation remains unclear. The histone chaperones Asf1 (English et al. 2006; Natsume et al. 2007) and Daxx (Elsasser et al. 2012; Liu et al. 2012) have been cocrystalized with H3/H4, with their structures confirming earlier biochemical studies indicating that these histone chaperones interact with, exchange, and deposit H3/H4 dimers (English et al. 2006; Natsume et al. 2007; Elsasser et al. 2012; Liu et al. 2012). Here we focused on the newly discovered histone chaperone hSpt2 (Osakabe et al. 2013) and its complex with histone H3/H4 from a combined structural and functional perspective. Spt2 protein functions in the remodeling and/or maintenance of chromatin structure during transcription through its ability to bind chromatin in vivo (Nourani et al. 2006). We solved the crystal structure of the human histone chaperone hSpt2 C-terminal domain (hSpt2C) bound to the Xenopus histone H3/H4 tetramer. Our structural results identify a novel mode of chaperone histone recognition and intermolecular contacts between hSpt2C and the H3/H4 tetramer that are distinct from those observed in previous structural studies of H3/H4 dimers bound by chaperones Asf1 (English et al. 2006; Natsume et al. 2007) and DAXX (Elsasser et al. 2012; Liu et al. 2012). This finding expands on previous nonstructural observations suggesting that histone chaperones Nap1 and Vps75 can also bind to H3/H4 in a tetrameric conformation (Bowman et al. 2011). Interestingly, we identified two acidic patches and several hydrophobic residues in hSpt2C that play key roles in H3/H4 tetramer recognition, with in vivo functional studies highlighting that these conserved hSpt2C residues are essential for recovery of the old H3/H4 in the wake of RNA polymerase II readthrough during transcription in yeast.
Results
Generation of a stable hSpt2C–H3/H4 complex
Previous studies established that full-length hSpt2 bound to both H2A/H2B and H3/H4 under low-salt (0.15 M NaCl) conditions as monitored by gel shift assays (Osakabe et al. 2013). Furthermore, The C terminus (571–685) of hSpt2, but not its N-terminal counterpart (1–570), bound His-tagged H3/H4 under low-salt conditions as monitored by pull-down assays (Osakabe et al. 2013). We independently confirmed that the C terminus (571–685) of GST-tagged hSpt2 bound H3/H4 under low-salt conditions as monitored by pull-down assays (data not shown).
Thus, our structural efforts focused on the generation and crystallization of hSpt2C bound to H3/H4, and, in order to facilitate formation of a stable complex, studies were undertaken under high-salt (2.0 M NaCl) conditions, which also favor H3/H4 tetramer formation. Size exclusion chromatography (SEC) established that hSpt2C (571–685) forms a stable complex with Xenopus histone H3/H4, allowing for generation and purification of the complex for crystallization trials (Supplemental Fig. S1A,B). Xenopus and human histone H3 differ at only two residue positions, whereas histone H4 is identical between the two species (Supplemental Fig. S2A,B). GST pull-down assays verify that hSpt2C binds similarly to Xenopus and human H3/H4 (Supplemental Fig. S2C).
Crystallization and structure determination of the hSpt2C–H3/H4 tetramer complex
Crystallization trials were undertaken on various hSpt2C and H3/H4 constructs as listed in Supplemental Table S1, with the best-diffracting crystals obtained for hSpt2(571–685) and H3(27–135)/H4(1–102). The structure of this complex was determined at 3.3 Å using the H3/H4 tetramer in the nucleosome (Protein Data Bank [PDB] code 1AOI) (Luger et al. 1997) as a search model during molecular replacement carried out using the program Phaser (McCoy et al. 2007) as implemented in the Phenix suite (Adams et al. 2010). The model building was carried out manually by using the program Coot (Emsley et al. 2010), and the structure was refined using Refmac (Murshudov et al. 1997). Given the modest 3.3 Å resolution and the fact that phases were obtained via molecular replacement method, 4.6 Å Se-Met data (details of data processing outlined in the Materials and Methods) were used to validate the registry of chain tracing of bound hSpt2C in the complex. The X-ray data statistics for native and Se-Met data are listed in Supplemental Table S2.
Two views of the complex containing hSpt2C in a ribbon representation and histone H3/H4 in ribbon and surface representations are shown in Figure 1, B and C, respectively. We were able to trace 308 of the 379 side chains (81.3%) in the complex. A key feature of this complex is that hSpt2C binds to the H3/H4 tetramer. In the structure, one hSpt2C molecule adopts a two-α-helical (labeled αC1 and αC2) fold (positions 607–675), while only αC1 (positions 604–626) could be traced in the second hSpt2C molecule of the complex (Fig. 1B,C). This implies that αC2 of the second hSpt2C molecule is most likely disordered in the structure of the complex. Notably, no density was observed connecting the long and short hSpt2C helices between adjacent complexes in the crystal lattice (Supplemental Fig. S3A,B). We are able to trace α1, α2, and α3 of both H3 (positions 60–135) and H4 (positions 26–95) but not the αN of H3 (positions 45–56), which was disordered in the complex (Fig. 1B,C).
Figure 1.

Crystal structure of human Spt2 histone-binding domain 571–685 (hSpt2C) bound to the histone H3/H4 tetramer in the hSpt2C–H3/H4 tetramer complex. (A) Schematic of α-helical distribution within the histone-binding domain of chaperone hSpt2 (magenta) and histones H3 (blue) and H4 (green). (B,C) Ribbon and surface views of the H3/H4 tetramer in the crystal structure of hSpt2C–H3/H4 tetramer complex. The complex contains one hSpt2C molecule where the density can be traced for αC1–linker–αC2 and a second molecule where the density can be traced solely for αC1. (D,E) Ribbon (D) and surface (E) views of the interaction of αC2 and the tip of the H3/H3 four-helix bundle in the crystal structure of the hSpt2C–H3/H4 tetramer complex. (F) Superposition of the structures of the hSpt2C–H3/H4 tetramer complex and mononucleosomes (PDB code 1AOI) (only DNA shown in silver) highlighting the clash between αC2 of hSpt2C and nucleosomal DNA. (G) Interaction between αC1 of hSpt2C and H3/H4 in the complex. (H) The same interface following superposition of the structures of the hSpt2C–H3/H4 tetramer complex (in color) and mononucleosomes (PDB code: 1AOI) (in silver) highlighting the clash between αC1 of hSpt2C and H2A/H4.
There is room for accommodation of only one αC2 helix of hSpt2C within the H3/H3 four-helix bundle interface of the H3/H4 tetramer as shown in Figure 1D,E. Thus, it is likely that the αC1 of the second molecule of hSpt2C that appears to be involved in the stabilization of the complex could reflect a contribution from crystal-packing interactions. We monitored the contribution of αC1 and αC2 helical segments within hSpt2C to binding with the H3/H4 tetramer by isothermal titration calorimetry (ITC). hSpt2C (637–685) composed of only αC2 binds the H3/H4 tetramer with a Kd of 2.3 μM and ΔH of −38.9 Kcal/mol (N = 1), while hSpt2C (610–644) composed of only αC1 binds the H3/H4 tetramer with a reduced Kd of 55.6 μM and a much lower ΔH of −9.2 Kcal/mol (Supplemental Figs. S6, S7; Supplemental Table S3), thereby highlighting the major contribution of αC2 of hSpt2C, which interacts with the tip of the H3/H3 four-helix bundle made up of the α2 and α3 helices of H3 (Fig. 1B).
In order to establish the stoichiometry of the complex in solution, we used gel filtration experiments coupled with multiple-angle light scattering (SEC-MALS) to measure the molecular mass of the complex under high-salt (2 M NaCl) crystallization conditions. The molecular mass of H3–H4 was measured at 55.8 kDa (error 4%, expected histone H3/H4 tetramer mass was 53.4 kDa), that of hSpt2C(571–685) was measured at 14.2 kDa (error 10%, expected monomer mass was 14.3 kDa), and that of the complex of hSpt2C(571–685)–H3/H4 was measured at 65.9 kDa (error 2%, expected complex of one hSpt2C bound to one histone H3/H4 tetramer mass was 67.7 kDa) (Fig. 2A,C). The same stoichiometry of one chaperone bound per H3/H4 tetramer was also observed for binding of SUMO-tagged hSpt2C(571–685) to the H3/H4 tetramer (Fig. 2B,C).
Figure 2.

SEC-MALS analysis of the hSpt2C–histone H3/H4 complex. (A) Analysis of histone H3/H4 (blue), hSpt2(571–685) (green), and H3/H4–hSpt2(571–685) (red) under 2 M NaCl conditions. Reyleigh ratio in continuous lines as a function of the elution volume. The calculated molecular mass is shown as a dashed line in the corresponding color with the secondary scale at the right. (B) Analysis of histone H3/H4 (blue), SUMO-tagged hSpt2(571–685) (green), H3–H4–hSpt2(571–685)SUMO (red) under 2 M NaCl conditions. Reyleigh ratio in continuous lines as a function of the elution volume. The calculated molecular mass is shown as a dashed line in the corresponding color with the secondary scale at the right. (C) Tables of mass measurements by SEC-MALS.
Intermolecular contacts in the hSptC–H3/H4 tetramer complex
We observed extensive intermolecular contacts between αC2, adjacent linker (Fig. 3A; electron density in Supplemental Fig. S8A), and αC1 (Fig. 3B) of bound hSpt2C and segments spanning the periphery of the H3/H4 tetramer in the structure of the hSpt2C–H3/H4 tetramer complex. Intermolecular hydrophobic contacts were observed involving M641 within the linker of hSpt2C, with hydrophobic side chains of H4 (V43 and methylene side chains of R35 and R39) in the complex (Fig. 3C; Supplemental Fig. S8C). In addition, adjacent E642 is involved in a hydrogen-bonding interaction with the tyrosine hydroxyl of Y51 of H4 (Fig. 3D; Supplemental Fig. S8D). Intermolecular hydrogen bonds are also formed by a pair of acidic patches on hSpt2C involving residues E651 and E652 toward the N terminus of αC2 (Fig. 3E,F; Supplemental Fig S8E,F) and E662 and D663 toward the center of αC2 (Fig. 3G,H; Supplemental Fig. S8G,H) with basic and polar side chains of the H3/H4 tetramer in the complex. The importance of these intermolecular contacts has been probed by ITC binding studies of hSpt2C(637–685) amino acid mutants potentially impacting on the recognition process. ITC data established that the M641A mutation involved in hydrophobic interactions as well as dual mutations of hSpt2C acidic patch residues E651A/E652A (designated patch 1) and E662A/D663A (patch 2) involved in hydrogen-bonding interactions and L658A/G659N resulted in complete loss in binding affinity (Fig. 3I). In contrast, non-histone-interacting Spt2C residue mutations K650A and E671A had minimal impact on hSpt2C–H3/H4 tetramer complex formation (Supplemental Fig. S9).
Figure 3.

Intermolecular contacts between the segment spanning αC1–linker–αC2 of hSpt2C and the histone H3/H4 tetramer in the structure of the hSpt2C–H3/H4 tetramer complex. (A,B) Overview of intermolecular contacts involving linker–αC2 segment (A) and αC1–linker segment (B) in the hSpt2C–H3/H4 tetramer complex. (C–H) Expanded views labeled C–H associated with A are outlined below. Intermolecular contacts between residue M641 (C) and E642 (D) of hSpt2C and residues within H4, between residues E651 (E) and E652 (F) of hSpt2C and residues within H3, and between E662 (G) and D663 (H) of hSpt2C and residues within H3 in the complex. Hydrogen-bonding interactions are indicated by blue dashed lines together with bond distance. (I) ITC binding curves for wild type and mutants of human hSpt2C (637–685; linker-αC2) to histone H3/H4 tetramer in 0.5 M NaCl solution. (J,K) Expanded views labeled J and K associated with B are outlined below. Intermolecular contacts between residues Y625 (J) and E633 (K) of hSpt2C and residues within H4.
Pull-down assays using full-length wild-type and mutant hSpt2 demonstrate observable but weaker H3/H4 binding of mutants E651A/E652A, L685A/G659N, and E662A/E663A, with further reduced binding for the M641A mutant (Supplemental Fig. S10). This result implies that weak interactions are likely to exist between full-length hSpt2 and H3/H4 that are outside the hSpt2C 637–685 segment. We also monitored the ability of full-length wild-type and mutant hSpt2 in the presence of all four histones to convert open circular (OC) to supercoiled (SC) φX174 RF DNA. We observed a significant decrease in supercoiling for mutants E651A/E652A and E662A/D663A and an even more pronounced decrease for mutant M641A (Supplemental Fig. S11), paralleling our observations in pull-down experiments mentioned above for the same full-length hSpt2 mutants.
There are other critical contacts in the hSpt2C–H3/H4 tetramer complex between the αC1–linker segment of bound hSpt2C and the H3/H4 tetramer as shown in Figure 3B. Thus, Y625 of hSpt2C participates in hydrophobic interactions (Fig. 3J; Supplemental Fig. S8J), while E633 of hSpt2C participates in hydrogen-bonding interactions (Fig. 3K; Supplemental Fig. S8K). Notably, αC1 of hSpt2C is anchored in a groove between α2 of H3 and α3 of H4 (Fig. 3B).
Comparison of the Spt2–H3/H4 tetramer complex with the nucleosome
The H3/H4 tetramers exhibit a root mean square deviation (RMSD) of 1.67 Å following superposition of the structures of the hSpt2C–H3/H4 tetramer complex with that of the Xenopus mononucleosome (PDB code 1AOI) (stereo pair in Supplemental Fig. S12A; Luger et al. 1997). Nevertheless, and keeping in mind the limitations in interpretation associated with the resolution (3.3 Å) of the Spt2 complex, differences were observed in the alignment of helices α3 (Supplemental Fig. S12B) and α2 (Supplemental Fig. S12C) of H4 following comparison between structures. It should be noted that hSpt2C bound along the periphery of the H3/H4 tetramer would result in a steric clash between its αC2 helix and nucleosomal DNA bound around the histone octamer core (Fig. 1F). This implies competition between DNA and the αC2 helix of hSpt2C for the H3/H3 four-helix bundle. To address this question, we used a gel shift assay that monitors binding of DNA to H3/H4 to form a tetrasome. The experimental data in Supplemental Figure S13 support DNA-mediated tetrasome formation, which cannot be disrupted with increasing concentrations of hSpt2C. Our data imply that hSpt2 can bind the H3/H4 tetramer only when it is off the DNA. It is conceivable that Spt2 is able to/could trap any H3/H4 tetramer that comes off the DNA during transcription. This implies that Spt2 is not in competition with DNA but instead suggests that Spt2 could absorb or stabilize the tetramers as they come off the DNA during transcription.
The alignment of αC1 of hSpt2C relative to the H3/H4 tetramer in the complex is shown in Figure 1G. Indeed, αC1 occupies the same position as a pair of adjacently aligned β sheets formed by C-terminal segments of H2A and H4 in the structure of the nucleosome (Fig. 1H), and hence αC1 of bound hSpt2C in the complex would clash with the H2A/H4 interaction in the nucleosome.
hSpt2C forms a complex with the H3/H4 tetramer under physiological salt conditions
The hSpt2C–H3/H4 tetramer complex for structure determination was assembled under high-salt conditions to facilitate stable complex formation (Supplemental Fig. S1), as were crystallization conditions (see the Materials and Methods). It is known that the H3/H4 dimer–tetramer equilibrium shifts toward the tetramer under high-salt conditions (Banks and Gloss 2003), and hence we needed to independently establish that the hSpt2C–H3/H4 tetramer complex also formed under physiological salt (0.15 M NaCl) conditions. To address this issue, we undertook disuccinimidyl suberate (DSS)-mediated cross-linking experiments on both the αC1–αC2-containing hSpt2C(571–685)–H3/H4 complex (Fig. 4A) and the αC2-containing hSpt2C(637–685)–H3/H4 complex (Fig. 4B). In Figure 4, A and B, control lanes 1–6 and complex formation lanes 8–10 are in 0.15 M NaCl, while lane 7, which is a control marker for H3/H4 tetramer formation, is in 0.5 M NaCl. Our DSS-mediated cross-linking data establish that hSpt2C(571–685) containing αC1–αC2 forms a complex with the H3/H4 tetramer (Fig. 4A, lanes 8,9), as does hSpt2C(637–685) containing only αC2, which also forms a complex with the H3/H4 tetramer (Fig. 4B, lanes 8–10) under physiological salt conditions.
Figure 4.
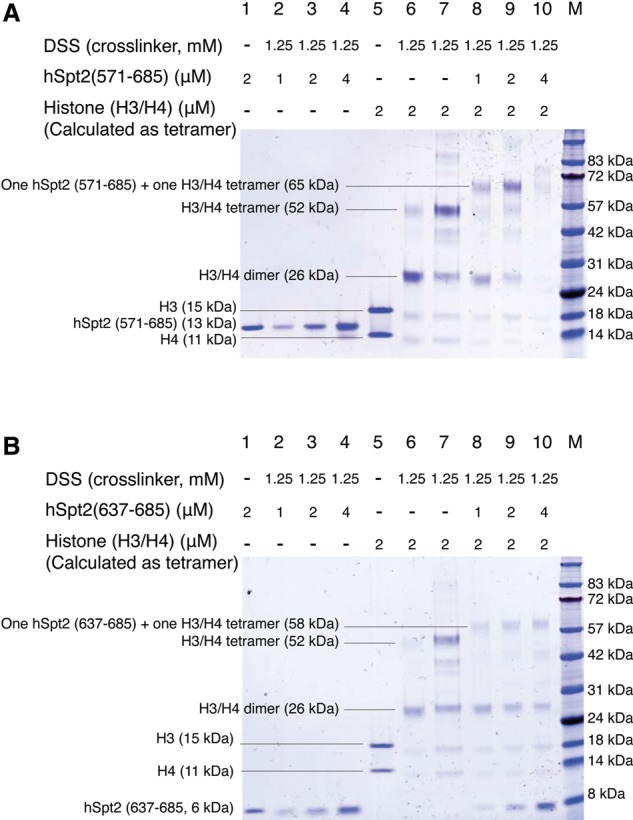
DSS-mediated cross-linking assays of hSpt2C(571–685) and hSpt2C(637–683) to histone H3/H4 under physiological salt (0.15 M NaCl) condition. (A) DSS cross-linking assays of hSpt2C(571–685) to histone H3/H4. (Lane 1) Input of hSpt2C(571–685) protein. (Lanes 2–4) Cross-linking of hSpt2C(571–685) alone with the indicated concentrations. (Lane 5) Input of histone H3/H4. (Lane 6) Cross-linking of histone H3/H4 alone under 0.15 M NaCl salt condition. (Lane 7) Cross-linking of histone H3/H4 alone under 0.5 M NaCl salt condition. (Lanes 8–10) Cross-linking of the indicated concentrations of hSpt2C(571–685) to histone H3/H4 under 0.15 M NaCl salt condition. (Lane M) Protein marker. (B) DSS cross-linking assays of hSpt2C(637–685) to histone H3/H4. (Lane 1) Input of hSpt2C(637–685) protein. (Lanes 2–4) Cross-linking of hSpt2C(637–685) alone with the indicated concentrations. (Lane 5) Input of histone H3/H4. (Lane 6) Cross-linking of histone H3/H4 alone under 0.15 M NaCl salt condition. (Lane 7) Cross-linking of histone H3/H4 alone under 0.5 M NaCl salt condition. (Lanes 8–10) Cross-linking of the indicated concentrations hSpt2C(637–685) to histone H3/H4 under 0.15 M NaCl salt condition. (Lane M) protein marker.
In contrast, hSpt2C(610–644) containing only αC1 does not facilitate histone H3/H4 tetramer formation (Supplemental Fig. S14A) under physiological salt conditions. We also investigated complex formation between hSpt2(571–685) containing αC1–αC2 and H3(L126E/I130E)/H4, a double H3 mutant that disrupts H3/H4 tetramer formation. We observed very weak bands corresponding to the formation of a complex of hSpt2(571–685) with the H3(L126E/I130E)/H4 dimer under 0.15 M NaCl conditions (Supplemental Fig. S14B).
Collectively, these data establish that hSpt2C constructs containing αC1–αC2 and αC2 alone, but not αC1 alone, facilitate the transition of H3/H4 from a dimer to a tetramer, thereby generating hSpt2C–H3/H4 tetramer complex formation.
Yeast Spt2C interacts with H3/H4 tetramer in vitro and in vivo
The αC1–αC2 segment of hSpt2C(571–685) shares sequence homology with the Saccharomyces cerevisiae Spt2 protein (ySpt2/Sin1) (Fig. 5A; Sternberg et al. 1987; Osakabe et al. 2013). Most of the hSpt2C residues involved in intermolecular recognition of the complex with H3/H4 are conserved in S. cerevisiae (ySpt2C) (designated by an asterisk in Fig. 5A). In contrast to its hSpt2C counterpart, ySpt2 function has been extensively studied (Nourani et al. 2006; Thebault et al. 2011). It promotes recycling of H3/H4 in transcribed regions, inhibits replication-independent H3/H4 exchange, stabilizes nucleosomes, and thereby contributes to the repression of spurious transcription (Nourani et al. 2006; Thebault et al. 2011). These functions could be associated with a possible interaction with the H3/H4 tetramer. To investigate the role of the ySpt2–H3/H4 interaction domain, we prepared stable H3/H4 complexes with ySpt2C(259–333) containing the αC1–αC2 domains (Supplemental Fig. S15), known to favor H3/H4 tetramer formation, and performed ITC studies (Fig. 5B; Supplemental Fig. S7). While wild-type ySpt2C(259–333) binds the H3/H4 tetramer with a Kd of 1.5 μM and ΔH of −19.2 Kcal/mol (Fig. 5B), the corresponding acidic patch residue dual mutants E299A/E300A (patch 1) and E310A/D311A (patch 2) as well as linker residues mutants M289A/E290A completely lose binding affinity (Fig. 5B). These results indicate that it is very likely that hSpt2C and ySpt2C chaperones target the H3/H4 tetramer in a similar manner. We next wanted to confirm these in vitro data and asked whether yeast Spt2 interacts with histone H3 in vivo. For that, we conducted coimmunoprecipitation experiments on nuclear extracts expressing wild-type Spt2 tagged with the myc epitope. To avoid interactions through DNA, these extracts were first digested with benzonase nuclease and subsequently incubated with anti-myc antibodies. In the absence of DNA, as indicated in Supplemental Figure S16, we observed an interaction with histone H3 (Fig. 5C). Interestingly, this binding is reduced by mutations of H3/H4 interaction residues, as shown for the acidic patch E299/E300 mutants as well as linker residue M289A/E290A mutants (Fig. 5C). This is consistent with the in vitro data and indicates that ySpt2 interacts with histone H3 in vivo, and the residues identified by the structural data play a major role in this interaction.
Figure 5.

The yeast Spt2 protein interacts in vitro and in vivo with H3/H4. (A) Sequence alignment highlighting conserved domains of human hSpt2C(571–685) and yeast ySpt2C(259–333). Key conserved Spt2C amino acids are shown in a red background. Mutated residues in hSpt2C are indicated by asterisks. (B) ITC binding curves for wild-type and mutants of yeast ySpt2C(259–333) to histone H3/H4 tetramer in 0.5 M NaCl solution. The Kd values, when measurable, are shown in the figure, while nonmeasurable values are indicated by ND. (C) Mutations in the ySpt2C H3/H4 interaction interface affect the in vivo binding of ySpt2 to histone H3. Coimmunoprecipitation experiment using nuclear extracts from yeast cells expressing different versions of Spt2 tagged with Myc epitope. To avoid interaction through binding to DNA, we digested the nuclear extracts with benzonase nuclease. As shown in Supplemental Figure S10, after benzonase treatment, DNA was undetected in the extracts used for coimmunoprecipitation. The immunoprecipitations were performed with anti-Myc antibodies cross-linked to protein-A sepharose beads. The proteins were analyzed with anti-myc and anti-H3 Western blots. Spt2–H3 coimmunoprecipitation was affected by mutations in Spt2 residues that are important for the interaction with H3/H4 in vitro.
The interaction of ySpt2 with H3/H4 is required for regulation of the H3K56ac level and replication-independent H3/H4 exchange in coding regions
We next analyzed, in vivo, the contribution of the ySpt2–H3/H4 tetramer interaction interface to the function of ySpt2 in the regulation of H3/H4 exchange. The replication-independent H3/H4 turnover is tightly associated with the H3K56ac level (Rufiange et al. 2007). Thus, we assessed by chromatin immunoprecipitation (ChIP) the level of H3K56ac in G1-arrested cells expressing wild type or the ySpt2 mutants tested above. The ySpt2 mutated in residues important for the interaction with the H3/H4 tetramer are generally associated with higher specific incorporation of new H3K56ac specifically in the coding regions of transcribed genes (Fig. 6A; Supplemental Fig. S17). In contrast, no such effect was observed in the nontranscribed gene AZR1 (Fig. 6A) or the intergenic region of chromosome V (NoORF) (Supplemental Fig. S17). Consistent with the ITC assays (Fig. 5B), the M306A/A307N mutation in yeast (equivalent to L658A/G659N in humans) does not have a striking effect on H3K56ac. Finally, it should be mentioned that mutations in Spt2 do not change the chaperone's levels in G1-arrested cells used in these ChIP assays, as indicated in Supplemental Figure S18.
Figure 6.

The Spt2–H3/H4 tetramer interaction interface is required for the regulation of replication-independent H3/H4 exchange and spurious transcription in coding regions. (A) ChIPs assessing the H3K56ac level in G1-arrested cells expressing different versions of Spt2. The values shown represent H3K56ac levels relative to histone H3 occupancy. The coding regions of different genes were tested, and Rpb1 association (shown in the left panel) indicates the transcription level of each gene. H3K56ac level was used as a surrogate of H3/H4 exchange. ChIP experiments were done at least in triplicates. (*) P-value < 0.05; (**) P-value < 0.01; (***) P-value < 0.001. (B) Growth tests on the indicated media of cells expressing different versions of Spt2 and containing the FLO8-HIS3 spurious transcription reporter or the Lys2-128δ SPT reporter. Growth of these cells on −histidine (galactose) or −lysine medium indicates a loss of Spt2 function in the refolding of chromatin structure at transcribed regions. (C) ChIPs assessing the H3K56ac level in G1-arrested cells expressing wild-type Spt2 and mutants (Spt2E298A and Spt2L318A) of αC2 residues that are not in contact with H3/H4 histones. As for A, the values shown represent H3K56ac levels relative histone H3 occupancy in the coding regions of different genes. The H3K56ac level was used as a surrogate of H3/H4 exchange. ChIP experiments were done at least in triplicates. (D) Growth tests on the indicated media of cells expressing different versions of Spt2, including mutants in αC2 residues (Spt2-E298A and Spt2-L318A) that are not in contact with H3/H4 histones. These strains contain the FLO8-HIS3 spurious transcription reporter or the Lys2-128δ SPT reporter. Growth of cells on −histidine (galactose) or −lysine medium indicate a loss of Spt2 function in the refolding of chromatin structure at transcribed regions.
We also tested directly the H3 exchange (Supplemental Fig. S19) by measuring the level of newly synthesized histone H3 incorporation at various locations as described previously in G1-arrested cells (Rufiange et al. 2007). Similar to H3K56ac, mutations of residues important for H3/H4 tetramer interaction resulted in higher incorporation of new Flag-H3 into coding regions of active genes. Together, these data indicate that the Spt2–H3/H4 tetramer interaction interface is crucial for the recycling of old H3/H4 tetramers in transcribed regions of active genes. Importantly, as shown in Supplemental Figure S20 by Western blots and ChIP assays, mutating Spt2–tetramer interaction residues has no effect on the production of ySpt2 or its recruitment to the sites of its function but instead hinders its ability to chaperone the H3/H4 tetramer.
The interaction of ySpt2 with H3/H4 is required for the regulation of spurious transcription from within coding regions
The deletion of the SPT2 gene derepresses spurious transcription from within coding regions and suppresses the effect of Ty and δ insertion mutations (Spt− phenotype) in the HIS4 or LYS2 promoters (Simchen et al. 1984; Winston et al. 1984; Nourani et al. 2006). We examined the consequence of mutating ySpt2 residues that are crucial to ySpt2–tetramer interaction on the repression of internal spurious transcription from the FLO8 cryptic promoter that is fused to a HIS3 reporter (Fig. 6B, left panel) and in the suppression of Ty insertion at LYS2 (Fig. 6B, right panel). The expression from the FLO8 cryptic promoter, as measured by growth in the absence of histidine, indicates spurious transcription, reflecting a defect in chromatin refolding during transcription (Cheung et al. 2008). Importantly, most of the mutants have phenotypes (Spt− and FLO8 spurious transcripts) indicating a loss of Spt2 chromatin function. Consistent with the other assays, the M306A/A307N mutant retains some of the Spt2 function (Fig. 6B). The H3K36 methyltransferase Set2 and the histone chaperone Asf1 play a crucial role in the regulation of histone H3 exchange in yeast. Deletion of SET2 and ASF1 in strains expressing ySpt2 mutants did not change the Spt− or the cryptic transcription phenotypes associated with spt2 alleles (Supplemental Fig. S21). In addition, spt2 mutant phenotypes are associated with an equal production of Spt2 protein, as shown in Supplemental Figure S18.
Our data strongly suggest that Spt2 residues that contact histone H3/H4 play a crucial role in the maintenance of the H3/H4 tetramer in coding regions. However, it is still possible that Spt2 αC2 is a sensitive structure, and any change of residues in this part of the chaperone could lead to a loss of function. To address this important issue, we mutated other residues of αC2, E298, and L318 (corresponding to residues K650 and E671 in human Spt2 protein via sequence alignment, as shown in Fig. 5A), which are not predicted to contact the H3/H4 tetramer. As described earlier, mutating the human K650 and E671 had no effect on the in vitro binding of hSpt2C to the H3/H4 tetramer (Supplemental Fig. S9). For the yeast mutants, we first tested H3/H4 turnover as measured by the H3K56ac level. As shown in Figure 6C, in the absence of yeast Spt2, the level of H3K56ac is increased at coding regions of active genes ACT1, FSK1, and PMA1. Interestingly, similarly to yeast Spt2 wild type, expressing Spt2-E298A and Spt2-L318A suppresses specifically H3K56ac incorporation at these loci. This indicates that mutating two different residues in αC2 that are not predicted to contact histone H3/H4 does not affect Spt2 function in rescuing old H3/H4 tetramers and inhibiting the incorporation of newly synthesized histone H3/H4. We next asked whether Spt2-E298A and Spt2-L318A mutants are capable of inhibiting spurious transcription from the FLO8 cryptic promoter. We expressed these two proteins (Spt2-E298A and Spt2-L318A) in a strain bearing the FLO8-HIS3 reporter and compared the growth with strains expressing wild type and histone H3/H4 contact mutants. Interestingly, while cells expressing Spt2 histone contact mutants grow very well in the absence of histidine, this was not the case for strains producing wild-type Spt2, Spt2-E298A, and Spt2-L318A. This indicates clearly that Spt2-E298A and Spt2-L318A mutants repress spurious transcription, as does the wild-type protein. We also found that Spt2-E298A and Spt2-L318A suppress the Spt− phenotype of the spt2Δ mutant in contrast to Spt2 histone contact mutants. Altogether, our data rule out the possibility that the phenotypes of Spt2 histone interaction mutants are associated with a sensitivity of the αC2 structure to any change of residues. Thus, we conclude that the Spt2–H3/H4 interaction interface in αC2 is required for the normal refolding of chromatin structure in the coding regions of transcribed genes.
Human Spt2C suppresses the incorporation of newly synthesized histone H3 in the coding regions of active genes and inhibits spurious transcription in yeast
We next analyzed the human hSpt2C chaperone domain in the functional assays described earlier. For that, we replaced the ySpt2C chaperone domain (257–333) with the hSpt2 chaperone domain (571–685) and tested the level of histone H3K56ac as described in Figure 6A. Interestingly, the human domain suppresses new H3 incorporation, as measured by H3K56ac levels, at the transcribed genes (Fig. 7A; Supplemental Fig. S22A). Interestingly, this is associated with the restoration of Spt2 chromatin function shown by the suppression of growth on histidine-lacking medium of strains containing the FLO8-HIS3 reporter (Fig. 7B, left panel, fourth lane; Supplemental Fig. S22B). Thus, addition of the human Spt2 chaperone domain to the yeast protein lacking the same domain restores the inhibition of spurious transcription from the FLO8 cryptic promoter. We also tested the suppression of the phenotype associated with Ty insertion at LYS2 (Fig. 7B, right panel, fourth lane; Supplemental Fig. S22C). Similar to cryptic transcription, we found that the human Spt2 chaperone domain inhibits growth on the lysine-lacking medium, indicating a restoration of Spt2 function in yeast. This clearly indicates that the human Spt2 histone chaperone is functionally related to the yeast protein and suppresses in vivo the loss of its chaperone domain.
Figure 7.

Expression of human Spt2C in yeast suppresses ySpt2C mutation. (A) ChIPs assessing the H3K56ac level in G1-arrested cells expressing different versions of Spt2 deleted or not in the H3/H4 interaction C-terminal domain (258–333). This domain was replaced by the human Spt2C(571–685) that interacts with H3/H4 (fourth lane) and by different hSpt2C mutants tested in vitro in Figure 3I. ChIP experiments were done at least in triplicates. (*) P-value < 0.05; (**) P-value < 0.01; (***) P-value < 0.001. (B) Growth tests on the indicated media of cells expressing different versions of Spt2 fused or not to wild-type and mutants of hSpt2-C(571–685) and containing the FLO8-HIS3 spurious transcription reporter or the Lys2-128δ SPT reporter.
Next, we mutated human Spt2C residues assayed earlier by ITC (Fig. 3I) and tested the mutants similarly to wild-type hSpt2C for H3K56ac levels. Importantly, we found that while the fusion of the human chaperone domain to yeast Spt2 decreases the incorporation of H3K56ac in coding regions of transcribed genes, mutating any of the residues that are important to the interaction with H3/H4 tetramer abolishes this suppression and leads to a higher deposition of newly synthesized histone H3 (Fig. 7A). This indicates that the human Spt2 histone H3/H4 interaction interface is crucial to the functional complementation and the recycling of old tetramers by the fusion protein. We next asked whether the human H3/H4 interaction residues identified in our structure are required for the suppression of H3 exchange, spurious transcription, and the spt− phenotype (Fig. 7A,B). Importantly, we found that mutations of all human Spt2C residues that are crucial in the interaction with the H3/H4 tetramer led to increased H3K56ac; growth on histidine- or lysine-lacking medium, indicating a failure in the repression of H3 exchange; the inhibition of spurious transcription; and a loss of insertion suppression. Therefore, the human Spt2 residues that contact the histone H3/H4 tetramer are required for the in vivo function of hSpt2C in the suppression of H3 turnover and Ty insertion and the repression of spurious transcription. Altogether, our in vivo data show that hSpt2 and ySpt2 histone H3/H4 tetramer interaction determinants are important for the maintenance of low H3/H4 exchange in coding regions, for H3/H4 tetramer integrity, and for repression of spurious transcription.
Discussion
Conclusions from a structural perspective
Our results highlight the ability of chaperone hSpt2C to bind selectively to the H3/H4 tetramer. This conclusion holds for X-ray studies on the complex generated under high-salt conditions (Fig. 1) as well as from cross-linking studies undertaken under physiological salt conditions (Fig. 4). The αC2 helix of hSpt2 is positioned over a four-helix bundle associated with the H3/H3 interface, which can be generated only on H3/H4 tetramer formation (Fig. 1D,E). This four-helix bundle interface can accommodate only one αC2 helix of hSpt2, and hence the stoichiometry of the complex was one hSpt2C bound per H3/H4 tetramer (Fig. 4). Our studies also establish that αC2 makes a greater contribution than αC1 of hSpt2C to recognition of the H3/H4 tetramer (Supplemental Fig. S6).
The hSpt2C–H3/H4 tetramer complex is stabilized by both electrostatic and hydrophobic intermolecular interactions (Fig. 3). Indeed, mutation of residues lining a pair of acidic patches on Spt2C results in impaired binding to the H3/H4 tetramer, a feature common for both human (Fig. 3I) and yeast (Fig. 5B) chaperones.
Our demonstration of selective binding of the hSpt2C chaperone to the H3/H4 tetramer contrasts with earlier examples of selective binding of Asf1 (English et al. 2006; Natsume et al. 2007) and Daxx (Elsasser et al. 2012; Liu et al. 2012), chaperones to the H3/H4 dimer. Thus, while hSpt2C binds to the periphery of the H3/H4 tetramer (Fig. 1B,C) in part by overlapping with the trajectory of nucleosomal DNA (Fig. 1F), Asf1 targets the H3/H4 dimer by binding to the H3/H4 tetramerization interface (English et al. 2006; Natsume et al. 2007), and DAXX generates an all-α-helical scaffold to envelop the H3.3/H4 dimer (Elsasser et al. 2012; Liu et al. 2012). This Spt2 tetramer mode of interaction provides new insights into how the H3/H4 tetramer could be recycled in cells.
Spt2 interaction with H3/H4 contributes to the rescue of the H3/H4 tetramer upon RNA polymerase II passage
The H3/H4 tetramer contains most of the functionally important histone epigenetic marks, and the question of how tetramers are formed and maintained is a primary issue. It has long been known that nucleosome assembly on DNA during replication and transcription involves the redeposition of old H3/H4 tetramers without splitting them into two dimers (Annunziato 2005; Xu et al. 2010; Katan-Khaykovich and Struhl 2011). However, these observations have been difficult to reconcile with the currently characterized mechanisms of histone chaperone systems involving interactions with dimers that are consistent with splitting of tetramers (English et al. 2006; Natsume et al. 2007; Elsasser et al. 2012; Liu et al. 2012). One possible way of reconciling the persistence of H3/H4 tetramers during transcription and replication is the existence of alternate modes of interaction with different classes of histone chaperones. Interestingly, previous elegant nonstructural studies using pulsed electron double resonance (PELDOR) measurements and protein cross-linking indicated that histone chaperones Nap1 and Vps75 can bind to H3/H4 in a tetrameric conformation (Bowman et al. 2011). These important observations suggested the existence of alternate mode of histone interaction that could contribute to the maintenance of histone tetramer integrity during cycles of nucleosome reassembly. However, to date, a precise mechanism explaining how histone chaperones could interact with the H3/H4 tetramer and rescue this important oligomer in vivo has never been reported.
In the present study, we provide evidence that the new class of conserved histone chaperone Spt2 binds directly to the H3/H4 tetramer. In vivo analyses indicate that this interaction is required for the recycling of H3/H4 in the wake of transcription and therefore suggests a new mechanism of maintaining the H3/H4 tetramer during transcription. Interestingly, two conservative acidic patches involving E651/E652 and E662/D663 positioned within αC2 in hSpt2C and conserved in ySpt2 directly bind to SIN (SWI/SNF-independent) mutational residues R116 and T118 located at the tip of the four-helix bundle formed by H3/H3′. Spt2 is also called Sin1, and the gene encoding for this protein was found in the same genetic screen that identified SIN mutations in histones genes (Sternberg et al. 1987). When mutated in yeast, R116 and T118 led to similar spt2/sin1 loss-of-function mutation phenotypes (Kruger et al. 1995; Luger et al. 1997). Additionally, the conserved M641 in hSpt2 (M289 in ySpt2) interacts with V43 of H4, which is also a SIN mutation residue, indicating that H4 stabilization is also important for H3/H4 tetramer recognition by Spt2C. Thus, our structural data give important new insights into the understanding of the Spt2–H3/H4 functional link that was discovered almost three decades ago.
Spt2 interacts functionally and physically with the histone chaperone Spt6 (Nourani et al. 2006; Thebault et al. 2011; Bhat et al. 2013). Evidence linking these two proteins led to the proposition that Spt2 may simply assist, with its DNA-binding activity, Spt6 during the reaction of histone redeposition in the wake of transcription (Nourani et al. 2006; Bhat et al. 2013). Our new observations advance further the understanding of Spt2's role during transcription elongation and show that Spt2 interacts directly with the H3/H4 tetramer. Importantly, this interaction is required for the control of H3/H4 recycling. Thus, without excluding other possibilities, we propose that Spt2 travels with RNA polymerase II, interacts with the H3/H4 tetramers that are displaced, and deposits them behind the transcription machinery. Alternatively, Spt2 could simply stabilize the H3/H4 tetramer on DNA after a wave of transcription. In the absence of this protein, the H3/H4 tetramer would come off the DNA, split into H3/H4 dimers, join the free pool of histones, and mix with new H3/H4 dimers. In any case, with its H3/H4 tetramer interaction, Spt2 inhibits H3/H4 turnover and maintains the old oligomers during transcription. In the absence of Spt2, this function is impaired, leading to the modification of chromatin structure with important consequences on spurious transcription.
Finally, our identification of Spt2 as a histone chaperone that targets the H3/H4 tetramer expands on earlier studies of chaperones Asf1 (English et al. 2006; Natsume et al. 2007) and Daxx (Elsasser et al. 2012; Liu et al. 2012) binding to H3/H4 dimers. Thus, H3/H4-binding chaperones use different folding topologies to target different surfaces and oligomerization states of H3/H4, thereby highlighting their functional diversity.
Materials and methods
Protein expression and purification
The gene of hSpt2 was PCR-amplified from a human brain pool and has a sequence identical to that of Homo sapiens SPT2 cDNA NM194285. The C-terminal domains of hSpt2 protein hSpt2C (residues 571–685, 585–685, 610–644, 637–685, and 610–685) were expressed as fusion proteins with an N-terminal hexahistidine tag plus a yeast Sumo tag in Escherichia coli strain BL21 (DE3) RIL (Stratagene). The cells were cultured at 37°C until OD600 reached 0.8, after which the medium was cooled to 20°C, and IPTG was added to a final concentration of 0.4 mM to induce protein expression overnight at 20°C. The cells were harvested by centrifugation at 4°C and disrupted by sonication in buffer W (500 mM NaCl, 20 mM imidazole, 50 mM Tris-HCl at pH 8.0) supplemented with 1 mM phenylmethylsulfonyl fluoride (PMSF) protease inhibitor and 2 mM β-mercaptoethanol. After centrifugation, the supernatant was loaded onto a HisTrap FF column (GE Healthcare). After extensive washing by buffer W, the target protein was eluted with buffer W supplemented with 300 mM imidazole (pH 8.0). The hexahistidine-Sumo tag was cleaved by Ulp1 protease and removed by further elution through a HisTrap FF column. Individual pooled target proteins were further purified by passage through a Hiload 16/60 Superdex 200 column (GE Healthcare) with buffer E (200 mM NaCl, 20 mM Tris-HCl at pH 8.0, 5 mM DTT).
GST-tagged proteins were overexpressed with pGEX-6p-1 vector (GE Healthcare) in E. coli strain BL21 (DE3) cells in LB medium. The cells were induced by 0.4 mM IPTG when OD600 reached a value of 0.8 and were cultured overnight at 20°C in a shaker. The cell extract was prepared using Emulsiflex C3 homogenizer (Avestin). The proteins were first purified from the soluble fraction by glutathione sepharose 4 Fast Flow (GE Healthcare) in 1× PBS buffer (137 mM NaCl, 10 mM phosphate, 2.7 mM KCl at pH 7.4) followed by further purification using HiTrap Heparin HP columns (GE Healthcare) with buffer containing 20 mM Tris-HCl (pH 7.5), 1 mM dithiotheritol, and 200 mM NaCl. All of the mutants were constructed using a QuikChange mutagenesis kit (Stratagene) and purified with the same protocol as the wild-type protein.
Reconstitution of the hSpt2C–histone complex
We used previously published methods with modifications for reconstitution of hSpt2C (571–685) with H3/H4 (Dyer et al. 2004). Briefly, Xenopus histones H3/H4 were produced and purified by gel filtration and HPLC/ion exchange chromatography under denaturing conditions. The purified H3/H4 histones were dialyzed against water, freeze-dried, and stored at −80°C. For generation of the hSpt2C–H3/H4 tetramer complex, purified histone H3 (residues 27–135), histone H4 (full-length), and hSpt2C proteins were dissolved at 1:1:1 stoichiometry under denaturing conditions containing 7 M guanidine-HCl, 20 mM Tris-HCl (pH 7.5), and 10 mM DTT. The mixtures were then dialyzed for 8 h against 2 M NaCl buffer containing 10 mM Tris-HCl (pH 7.5), 1 mM EDTA, and 5 mM β-mercaptoethanol at least three times. The reconstituted complexes were fractionated by Hiload 16/60 Superdex 200 (GE Healthcare) gel filtration chromatography under 2 M NaCl buffer containing 10 mM Tris-HCl (pH 7.5), 1 mM EDTA, and 1 mM DTT and analyzed with NuPAGE Novex 4%–12% Bis-Tris Gel (Invitrogen). For generation of the Se-Met-labeled complex, hSpt2C(571–685, Se-Met, I615M mutant) was reconstituted with native H3 (residues 27–135) and H4 (full-length) histones at 1:1:1 stoichiometry under similar refolding conditions.
Crystallization and data collection
The reconstituted hSpt2C–H3/H4 tetramer complex was collected by pooling Hiload 16/60 Superdex 200 (GE Healthcare) gel filtration chromatographic peak fractions to a concentration of 5 mg/mL for crystallization trials. Crystals of the hSpt2C–H3/H4 tetramer complex were grown from a solution containing 0.02 M NaCl (Fluke), 0.2 M HEPES 7.5 (Sigma-Aldrich), and 1.5 M ammonium sulfate (Sigma-Aldrich) at 20°C using the hanging drop vapor diffusion method. For data collection, crystals were flash-frozen (100 K) in the above reservoir solution supplemented with 2 M Li-sulfate (Sigma-Aldrich). A diffraction data set was collected to 3.3 Å resolution at wavelength 0.9798 Å on NE-CAT beam line 24ID-E at the Advanced Photon Source, Argonne National Laboratory. Crystals of the hSpt2C(Se-Met, I615M)–H3/H4 tetramer complex were grown under conditions similar to that of the native complex from a solution containing 0.02 M NaCl (Fluke), 0.2 M HEPES 7.5 (Sigma-Aldrich), and 1.6 M ammonium sulfate (Sigma-Aldrich) at 20°C using the hanging drop vapor diffusion method. For data collection, crystals were flash-frozen (100 K) in the above reservoir solution supplemented with 20% glycerol (Sigma-Aldrich). A diffraction data set was collected to 4.6 Å resolution at wavelength 0.9792 Å on NE-CAT beam line 24ID-E at the Advanced Photon Source, Argonne National Laboratory.
Structure determination and refinement
The X-ray data set on the hSpt2C–H3/H4 tetramer complex was processed using HKL2000 (Otwinowski and Minor 1997), and the structure was solved using the structure of the H3/H4 tetramer in the nucleosome (PDB code 1AOI) as a search model during molecular replacement carried out using the program Phaser (McCoy et al. 2007) as implemented in the Phenix program suite (Adams et al. 2010). The model building was carried out manually by using the program Coot (Emsley et al. 2010). Of 379 residues (81.3%), 308 could be placed with side chains in the complex structure. The validation of the hSpt2C structure was performed with the Se-Met method following the introduction of the I615M mutation.
The X-ray data set on the hSpt2C(Se-Met, I615M)–H3/H4 tetramer complex was indexed and integrated using the program HKL2000 (Otwinowski and Minor 1997). The data extended to 4.6 Å with an average multiplicity of 6, counting the Bijvoet pairs separately. Overall completeness was 99%, with 89% of the reflections measured at the highest-resolution shell (4.68–4.6 Å). Analysis of χ2 values indicated the presence of anomalous signal up to a resolution of ∼5 Å. Four selenium sites were located using the program SHELXD (Sheldrick 2010). Phases after 100 cycles of density modification with SHELXE clearly displayed the overall architecture of the molecule, with tubular density for helices. We were able to place the molecule (H3, H4, and Spt2C) in the electron density map deduced from SHELXE (Sheldrick 2010) using the program MOLREP (Vagin and Teplyakov 2010). Next, we used the selenium sites and the MOLREP-derived model in Phaser (McCoy et al. 2007) for experimental phasing with phase combination with the known model. Phaser was able to locate one additional Se-Met site. The anomalous difference Fourier map calculated from these phases showed five peaks above the 5σ cutoff. These account for the five expected Se-Met sites (Se-Met615, Se-Met641, Se-Met660, Se-Met667, and Se-Met674) for one molecule of Spt2C and unequivocally fix the orientation of Spt2C. The statistics of the diffraction data and refinement are summarized in Supplemental Table S2.
Pull-down assays
GST-tagged hSpt2C(571–685) was expressed as fusion proteins with an N-terminal GST tag in E. coli strain BL21 (DE3) RIL (Stratagene). The cells were cultured at 37°C until OD600 reached 0.8, after which the medium was cooled to 20°C, and IPTG was added to a final concentration of 0.4 mM to induce protein expression overnight at 20°C. Glutathione resins (GenScript) were used for pull-downs with purified proteins. Eight micrograms of purified GST-tagged hSpt2 full-length protein, 20 μg of GST-tagged hSpt2C, or 20 μg of GST protein (control) was mixed with 3 μg of H3/H4 tetramer in binding buffer (20 mM Tris-HCl at pH 7.6, 700 mM NaCl, 1 mM dithiotheritol). The glutathione resins were added to the reaction mixture, and the samples were then incubated for 1 h at 4°C. After incubation, glutathione resin mixtures were pelleted and washed three times with wash buffer (1% Triton-X100, 20 mM Tris-HCl at pH 7.6, 700 mM NaCl, 1 mM dithiotheritol). The proteins captured by glutathione resins were analyzed by NuPAGE Novex 4%–12% Bis-Tris gel (Invitrogen). The bands were visualized by Coomassie Brilliant blue staining.
ITC measurements
All of the binding experiments were performed on a Microcal ITC 200 calorimeter at 20°C. First, hSpt2(610–685) protein, hSpt2(610–644), hSpt2(637–685) samples, ySpt2 (259–333), ySpt2 (282–333), and mutants were dialyzed for 3 h against buffer A (0.5 M NaCl, 2 mM β-mercaptoethanol, 20 mM HEPES at pH 7.5) at room temperature, and the histone H3/H4 tetramer samples were diluted in the same buffer. The exothermic heat of the reaction was measured by 17 sequential 2.2-μL injections of 600 μM Spt2 protein into 200 μL of 35 μM histone H3/H4 solution (calculated as H3/H4 tetramer), spaced at intervals of 250 sec. The heat of dilution was obtained by injecting assay buffer into the histone H3/H4 solution and was subtracted from the heat of reaction before the fitting process. Binding curves were analyzed by nonlinear least squares fitting of the data using Microcal Origin software.
Supercoiling assays for nucleosome assembly activity
The supercoiling assay (topological assay) was performed according to the method described previously (Osakabe et al. 2013). H2A/H2B (10 ng/μL) and 10 ng/μL H3.1/H4 were preincubated with the indicated amounts of full-length hSpt2 for 15 min at 37°C. To initiate the reaction, 10 ng/μL relaxed φX174 RF1 DNA (Thermo Scientific), prepared by an incubation with 0.8 U of wheat germ topoisomerase I (Promega) per 100 ng of DNA for 150 min at 37°C, was added to the reaction mixture, and the reactions were continued in 20 μL of 20 mM Tris-HCl buffer (pH 8.0) containing 140 mM NaCl, 2 mM MgCl2, and 5 mM DTT for 60 min at 37°C. The samples were then incubated for 60 min at 42°C, and the proteins were removed by incubation with 60 μL of a proteinase K solution (20 mM Tris-HCl at pH 8.0, 20 mM EDTA, 0.5% SDS, 0.5 mg/mL proteinase K) for 15 min at 37°C followed by phenol-chloroform extraction. During DNA extraction, the reaction mixture was diluted with 280 μL of H2O, and extract was diluted with an equal volume of phenol:CHCl3:isoamyl alcohol (25:24:1). Next, the samples were spun for 15 min at room temperature followed by addition of 0.5 μg of 20 mg/mL glycogen, 40 μL of 3 M sodium acetate (pH 7.0), and 800 μL of ethanol into 340 μL of supernatant and left for 15 min on ice. The samples were then collected via spinning for 15 min at 4°C. The pellets were dried briefly and dissolved with 15 μL of H2O. The samples were then analyzed by 1% agarose gel electrophoresis in 1× TAE buffer (40 mM Tris acetate, 1 mM EDTA) at 4.4 V/cm for 4 h and visualized by ethidium bromide (Bio-Rad) staining.
DSS-mediated cross-linking assays
hSpt2C [including hSpt2(571–685), hSpt2(637–685), and hSpt2(610–644)] proteins were purified, and the histone (H3/H4)2 tetramer and histone H3(L126E/I130E)/H4 dimer were refolded as previously described, except the Tris-HCl buffer was replaced with HEPES-Na (pH 7.5) buffer. Cross-linker DSS was dissolved in DMSO at 12.5 mM concentration. First, histones and hSpt2C [including hSpt2(571–685), hSpt2(637–685), and hSpt2(610–644)] proteins were mixed with the indicated final concentration at 0.15 M NaCl and 10 mM HEPES-Na (pH 7.5) buffer for 15 min at room temperature. Next, DSS were added into protein mixtures with a 1.25 mM final concentration, and the reaction mixture was incubated at room temperature for 20 min. Adding 1 M Tris 7.5 to a final concentration of 50 mM quenched the reactions. Samples were analyzed by NuPAGE Novex 4%–12% Bis-Tris Gel (Invitrogen). The bands were visualized by Coomassie Brilliant blue staining. It should be noted that histone tetramer cross-linking samples (Fig. 3, all lanes 7; Supplemental Fig. S8) were mixed and reacted under 0.5 M NaCl and 10 mM HEPES-Na (pH 7.5) buffer.
SEC-MALS
For molar mass determination, purified proteins or protein complexes were analyzed using an ÄKTA-MALS system. Note that 200 μg of protein solution was injected on Superdex 200 10/300 GL (GE Healthcare) equilibrated in 10 mM Tris 7.5, 0.5 mM DTT, 0.5 mM EDTA (pH 8.0), and 2 M NaCl at a flow rate of 0.3 mL per minute. Separation and ultraviolet detection were performed using an ÄKTA system (GE Healthcare), light scattering was monitored using a miniDAWN TREOS system (Wyatt Technology), and concentration was measured by an Optilab T-rEX differential refractometer (Wyatt Technology). Molar masses of proteins were calculated using the Astra 6.1 program (Wyatt Technology) with a dn/dc value of 0.185 mL/g.
S. cerevisiae media, strains, and plasmids
Strains were constructed using standard genetic methods, are isogenic to S288C (Winston et al. 1995), and are listed in Supplemental Table S4. The plasmid pRS415 containing the wild-type SPT2, SPT2-Myc, SPT2-Flag, or the fusion ySpt2–hSpt2C gene was described before (Bhat et al. 2013). Mutagenesis was done on these constructs with the QuikChange mutagenesis kit from Stratagene and the instructions therein. The yeast plasmids used in this study were all checked by sequencing. Yeast cells expressing different spt2 alleles were constructed by transformation of SPT2-deleted strains with pRS415 plasmid containing the indicated alleles. In the coimmunoprecipitation experiments, the Spt2-Myc isoforms were expressed from a p426-GAL1 overexpression plasmid. In the phenotypic assays (Fig. 6B,D), growth of different strains was monitored by spot tests on the indicated medium. In Figure 6D, the Gal-Histidine medium contained 1 mM 3-AT.
Coimmunoprecipitation experiments
Coimmunoprecipitations were conducted on yeast nuclear extracts treated with benzonase (Millipore, 71205-25KUN) at 200 U/mL during 15 min. The extracts were clarified by centrifugation and incubated overnight with 10 μL of anti-Flag M2 beads (Sigma, A2220) or 30 μL of anti-Myc (Covance, MMS-150R) protein-A sepharose beads (GE, 17-0780-01). The immunoprecipitated proteins were subsequently analyzed by anti-Flag, anti-myc, or anti-H3 Western blots.
ChIP
ChIP experiments were performed as described previously (Rufiange et al. 2007). Efficient G1 arrest (at least 95%) of yeast cells was achieved by adding 500 ng/mL α factor for 2–3 h. Immunoprecipitation of Rpb1 was performed using 2 μL of the 8WG16 anti-CTD antibody per immunoprecipitation (Covance). The immunoprecipitations of H3 and H3K56ac were done with 0.5 μL of rabbit anti-H3 antibody per immunoprecipitation (Abcam, 1791) and 0.2 μL of anti-H3K56 antibody per immunoprecipitation (Millipore, 07-677-I), respectively. Finally, the Flag ChIPs were done using 10 μL of anti-Flag agarose beads per immunoprecipitation (Sigma, A2220), and Myc ChIPs were done using 1 μL of anti-Myc 9E10 per immunoprecipitation (Covance monoclonal antibodies, MMS-150R). All ChIP experiments were done at least in triplicates; P-value < 0.05 (*), P-value < 0.01 (**), and P-value < 0.001 (***).
Accession numbers
The coordinates and structure factors for the hSpt2(571-685)–H3/H4 tetramer and the SeMet- hSpt2(571-685)–H3/H4 tetramer complexes have been deposited in the PDB under accession numbers 5BS7 and 5BSA, respectively.
Supplementary Material
Acknowledgments
We thank Dhirendra Simanshu for assistance with processing of the X-ray data sets, and Qi Qiao in the laboratory of Hao Wu (Harvard Medical School) for assistance on SEC-MALS data collection and analysis. We also thank Martin Simard and Jacques Côté for their critical reading of the manuscript. This work was supported by a Leukemia and Lymphoma Program Project grant (LLS-SCOR 7132-08) and STARR Foundation grant (I5-A554) to D.J.P. and a Canadian Institutes of Health Research MOP81245 grant to A.N. A.N. holds a CRC Tier 2 chair. This work is based on research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P41 GM103403). The Pilatus 6M detector on the 24-ID-C beamline is funded by a National Institutes of Health-Office of Research Infrastructure Programs HEI grant (S10 RR029205). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science user facility operated for the DOE Office of Science by Argonne National Laboratory under contract number DE-AC02-06CH11357. The structural and in vitro binding research was undertaken by S.C. with the assistance of H.H. under the supervision of D.J.P., while the in vivo functional studies were undertaken by A.R. under the supervision of A.N. K.R.R. assisted with the analysis of the native and Se-Met X-ray data sets of the complexes.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.261115.115.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. 2010. Phenix: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66: 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annunziato AT. 2005. Split decision: what happens to nucleosomes during DNA replication? J Biol Chem 280: 12065–12068. [DOI] [PubMed] [Google Scholar]
- Avvakumov N, Nourani A, Cote J. 2011. Histone chaperones: modulators of chromatin marks. Mol Cell 41: 502–514. [DOI] [PubMed] [Google Scholar]
- Banks DD, Gloss LM. 2003. Equilibrium folding of the core histones: the H3–H4 tetramer is less stable than the H2A–H2B dimer. Biochemistry 42: 6827–6839. [DOI] [PubMed] [Google Scholar]
- Belotserkovskaya R, Oh S, Bondarenko VA, Orphanides G, Studitsky VM, Reinberg D. 2003. FACT facilitates transcription-dependent nucleosome alteration. Science 301: 1090–1093. [DOI] [PubMed] [Google Scholar]
- Bhat W, Boutin G, Rufiange A, Nourani A. 2013. Casein kinase 2 associates with the yeast chromatin reassembly factor Spt2/Sin1 to regulate its function in the repression of spurious transcription. Mol Cell Biol 33: 4198–4211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman A, Ward R, Wiechens N, Singh V, El-Mkami H, Norman DG, Owen-Hughes T. 2011. The histone chaperones Nap1 and Vps75 bind histones H3 and H4 in a tetrameric conformation. Mol Cell 41: 398–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung V, Chua G, Batada NN, Landry CR, Michnick SW, Hughes TR, Winston F. 2008. Chromatin- and transcription-related factors repress transcription from within coding regions throughout the Saccharomyces cerevisiae genome. PLoS Biol 6: e277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dion MF, Kaplan T, Kim M, Buratowski S, Friedman N, Rando OJ. 2007. Dynamics of replication-independent histone turnover in budding yeast. Science 315: 1405–1408. [DOI] [PubMed] [Google Scholar]
- Dyer PN, Edayathumangalam RS, White CL, Bao Y, Chakravarthy S, Muthurajan UM, Luger K. 2004. Reconstitution of nucleosome core particles from recombinant histones and DNA. Methods Enzymol 375: 23–44. [DOI] [PubMed] [Google Scholar]
- Elsasser SJ, Huang H, Lewis PW, Chin JW, Allis CD, Patel DJ. 2012. DAXX envelops a histone H3.3–H4 dimer for H3.3-specific recognition. Nature 491: 560–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66: 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English CM, Adkins MW, Carson JJ, Churchill ME, Tyler JK. 2006. Structural basis for the histone chaperone activity of Asf1. Cell 127: 495–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamai A, Puglisi A, Strubin M. 2009. Histone chaperone spt16 promotes redeposition of the original h3-h4 histones evicted by elongating RNA polymerase. Mol Cell 35: 377–383. [DOI] [PubMed] [Google Scholar]
- Kaplan CD, Laprade L, Winston F. 2003. Transcription elongation factors repress transcription initiation from cryptic sites. Science 301: 1096–1099. [DOI] [PubMed] [Google Scholar]
- Katan-Khaykovich Y, Struhl K. 2011. Splitting of H3–H4 tetramers at transcriptionally active genes undergoing dynamic histone exchange. Proc Natl Acad Sci 108: 1296–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura H, Cook PR. 2001. Kinetics of core histones in living human cells: little exchange of H3 and H4 and some rapid exchange of H2B. J Cell Biol 153: 1341–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kireeva ML, Walter W, Tchernajenko V, Bondarenko V, Kashlev M, Studitsky VM. 2002. Nucleosome remodeling induced by RNA polymerase II: loss of the H2A/H2B dimer during transcription. Mol Cell 9: 541–552. [DOI] [PubMed] [Google Scholar]
- Kruger W, Peterson CL, Sil A, Coburn C, Arents G, Moudrianakis EN, Herskowitz I. 1995. Amino acid substitutions in the structured domains of histones H3 and H4 partially relieve the requirement of the yeast SWI/SNF complex for transcription. Genes Dev 9: 2770–2779. [DOI] [PubMed] [Google Scholar]
- Liu CP, Xiong C, Wang M, Yu Z, Yang N, Chen P, Zhang Z, Li G, Xu RM. 2012. Structure of the variant histone H3.3-H4 heterodimer in complex with its chaperone DAXX. Nat Struct Mol Biol 19: 1287–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. 1997. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389: 251–260. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. 2007. Phaser crystallographic software. J Appl Crystallogr 40: 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr 53: 240–255. [DOI] [PubMed] [Google Scholar]
- Natsume R, Eitoku M, Akai Y, Sano N, Horikoshi M, Senda T. 2007. Structure and function of the histone chaperone CIA/ASF1 complexed with histones H3 and H4. Nature 446: 338–341. [DOI] [PubMed] [Google Scholar]
- Nourani A, Robert F, Winston F. 2006. Evidence that Spt2/Sin1, an HMG-like factor, plays roles in transcription elongation, chromatin structure, and genome stability in Saccharomyces cerevisiae. Mol Cell Biol 26: 1496–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orphanides G, Wu WH, Lane WS, Hampsey M, Reinberg D. 1999. The chromatin-specific transcription elongation factor FACT comprises human SPT16 and SSRP1 proteins. Nature 400: 284–288. [DOI] [PubMed] [Google Scholar]
- Osakabe A, Tachiwana H, Takaku M, Hori T, Obuse C, Kimura H, Fukagawa T, Kurumizaka H. 2013. Vertebrate Spt2 is a novel nucleolar histone chaperone that assists in ribosomal DNA transcription. J Cell Sci 126: 1323–1332. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol, 276: 307–326. [DOI] [PubMed] [Google Scholar]
- Rufiange A, Jacques PE, Bhat W, Robert F, Nourani A. 2007. Genome-wide replication-independent histone H3 exchange occurs predominantly at promoters and implicates H3 K56 acetylation and Asf1. Mol Cell 27: 393–405. [DOI] [PubMed] [Google Scholar]
- Sheldrick GM. 2010. Experimental phasing with SHELXC/D/E: combining chain tracing with density modification. Acta Crystallogr D Biol Crystallogr 66: 479–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simchen G, Winston F, Styles CA, Fink GR. 1984. Ty-mediated gene expression of the LYS2 and HIS4 genes of Saccharomyces cerevisiae is controlled by the same SPT genes. Proc Natl Acad Sci 81: 2431–2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolle M, Workman JL. 2013. Transcription-associated histone modifications and cryptic transcription. Biochim Biophys Acta 1829: 84–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternberg PW, Stern MJ, Clark I, Herskowitz I. 1987. Activation of the yeast HO gene by release from multiple negative controls. Cell 48: 567–577. [DOI] [PubMed] [Google Scholar]
- Thebault P, Boutin G, Bhat W, Rufiange A, Martens J, Nourani A. 2011. Transcription regulation by the noncoding RNA SRG1 requires Spt2-dependent chromatin deposition in the wake of RNA polymerase II. Mol Cell Biol 31: 1288–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiriet C, Hayes JJ. 2005. Replication-independent core histone dynamics at transcriptionally active loci in vivo. Genes Dev 19: 677–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin A, Teplyakov A. 2010. Molecular replacement with MOLREP. Acta Crystallogr D Biol Crystallogr 66: 22–25. [DOI] [PubMed] [Google Scholar]
- Winston F, Chaleff DT, Valent B, Fink GR. 1984. Mutation affecting TY-mediated expression of the HIS4 gene of Saccharomyces cerevisiae. Genetics 107: 179–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winston F, Dollard C, Ricupero-Hovasse SL. 1995. Construction of a set of convenient Saccharomyces cerevisiae strains that are isogenic to S288C. Yeast 11: 53–55. [DOI] [PubMed] [Google Scholar]
- Xu M, Long C, Chen X, Huang C, Chen S, Zhu B. 2010. Partitioning of histone H3-H4 tetramers during DNA replication-dependent chromatin assembly. Science 328: 94–98. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.