

Abstract
Purpose of Review
In this review we discuss how the Genetic Hypercalciuric Stone-Forming (GHS) rats, which closely model idiopathic hypercalciuria and stone formation in humans, provide insights into the pathophysiology and consequences of clinical hypercalciuria.
Recent Findings
Hypercalciuria in the GHS rats is due to a systemic dysregulation of calcium transport, as manifest by increased intestinal calcium absorption, increased bone resorption and decreased renal tubule calcium reabsorption. Increased levels of vitamin D receptor in intestine, bone and kidney appear to mediate these changes. The excess receptors are biologically active and increase tissue sensitivity to exogenous vitamin D. Bones of GHS rats have decreased bone mineral density (BMD) as compared with Sprague Dawley rats, and exogenous 1,25(OH)2D3 exacerbates the loss of BMD. Thiazide diuretics improve the BMD in GHS rats.
Summary
Studying GHS rats allows direct investigation of the effects of alterations in diet and utilization of pharmacologic therapy on hypercalciuria, urine supersaturation, stone formation and bone quality in ways that are not possible in humans.
Keywords: nephrolithiasis, calcium, bone mineral density, bone resorption
Introduction
The majority of patients who form calcium (Ca)-containing kidney stones are hypercalciuric as compared to non-stone formers(1-5). At least a third of these patients have no demonstrable metabolic cause for the increased urinary (u) Ca excretion and are classified as having idiopathic hypercalciuria (IH). Patients with idiopathic hypercalciuria (IH), defined as excessive uCa without a demonstrable metabolic cause, generally have normal serum (s) Ca, normal or elevated s1,25(OH)2D3 (1,25D), normal or elevated s parathyroid hormone (sPTH), normal or low s phosphate (sP). These patients are often in negative Ca balance, that is they excrete more calcium in their urine than they absorb from their diet, indicating a net loss of total body calcium(1, 3, 6-14) and they often have a reduction in bone mass(3, 6, 15-28) and an increase in the rate of fracture(20, 27). IH exhibits a polygenic mode of inheritance(2, 29, 30).
To study IH in ways that are impossible in man, we have established a novel rat strain, the Genetic Hypercalciuric Stone-Forming (GHS) rat, by inbreeding the most hypercalciuric progeny of successive generations of Sprague-Dawley (SD) rats (31-56). When fed a standard, ample Ca diet, each GHS rat now consistently excretes ~10 fold more uCa than SD controls (31-56). Like patients with IH, GHS rats have normal sCa(32), increased intestinal Ca absorption(38) and enhanced bone resorption(37), decreased renal tubule Ca reabsorption(39), and normal s1,25D levels(36, 38, 45, 46, 53), in addition to decreased bone mineral density(35, 40). Hypercalciuria is a polygenic trait in GHS rats(43) as it is in humans(2, 29, 30). When fed a standard, ample Ca diet all GHS rats develop kidney stones(33, 52, 54, 55) which are composed of calcium phosphate (CaP) (33, 49, 50, 54). Addition of the amino acid hydroxyproline to the diet of GHS rats results in calcium oxalate (CaOx) stone formation (31, 34, 47). The GHS rats have been maintained with continuous selection for hypercalciuria for over 100 generations (Fig. 1). We have used the GHS rats to address fundamental questions in the pathophysiology of hypercalciuria and Ca stone formation and resultant effects on bone.
Figure 1. Urine calcium excretion over successive generations of Genetic Hypercalciuric Stone-Forming rats.
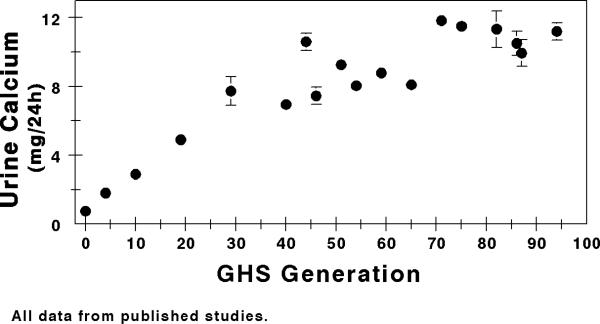
The GHS rats have been maintained with continuous selection for hypercalciuria for over 100 generations. All data from published studies (31-36, 39, 40, 42, 44, 47, 49-52, 54-60*).
What is the Origin of the Increased Urinary Calcium in GHS Rats?
All uCa must originate from intestinal absorption of dietary Ca and bone Ca is, by far, the largest repository of Ca in the body (26). An increase in uCa may result from increased intestinal Ca absorption, increased bone resorption or a failure of renal tubular Ca reabsorption which would then drive increased Ca absorption and/or resorption to maintain serum Ca.
To determine whether the GHS rats have increased intestinal Ca absorption we measured Ca transport in isolated duodenum using an Ussing chamber (32). We found that the mucosal to serosal Ca flux (absorption) in GHS was higher than that in SD and there was no difference in the serosal to mucosal flux (secretion) indicating greater net Ca absorption in the GHS compared to SD rats.
That there is an increase in intestinal Ca absorption in the GHS rats does not preclude that enhanced bone resorption or a defect in renal tubular Ca reabsorption contributes to the hypercalciuria. Using a low Ca diet (LCD) to effectively eliminate dietary Ca absorption, uCa was decreased in both GHS and SD rats(36). However, even on LCD, uCa from GHS rats was higher than from SD and higher than the total dietary Ca, indicating that bone is an important contributor to increased uCa in GHS rats. However from this study it is not clear if the contribution from bone is due to a primary increase in bone resorption and/or a decrease in renal tubular Ca reabsorption leading to enhanced bone resorption.
To determine the contribution of bone to the hypercalcuria we cultured calvariae from the GHS rats and found greater Ca resorption from bone in response to graded doses of 1,25D than calvariae from SD rats(37), consistent with a primary increase in bone resorption in the GHS rats. The increased levels of VDR found in GHS bone cells (37, 53, 61) were apparently inducing increased bone resorption. To further assess the contribution of bone resorption to hypercalciuria, GHS or SD rats were fed LCD and injected with the bisphosphonate alendronate to inhibit bone resorption(52). While uCa from GHS was greater than that from SD with LCD alone, alendronate eliminated the difference and restored a positive Ca balance.
However increased intestinal Ca absorption and bone resorption does not exclude a contribution to the hypercalciuria from decreased renal tubular Ca reabsorption. Using parathyroidectomized GHS and SD rats we found that at a similar filtered Ca load there was far less renal tubular reabsorption in GHS rats compared to controls(39). Diuretic studies strongly suggested that there was decreased tubular Ca reabsorption in the thick ascending limb of Henles’ loop; however, that study could not exclude other sites of disordered Ca reabsorption(39). Thus the GHS rats appear to have a systemic dysregulation of Ca transport in the principal Ca transporting sites in the body, the intestine, the kidney and the bone.
Contribution of Increased Levels of VDR to Hypercalciuria
GHS rats have increased intestinal Ca absorption(62), increased bone Ca resorption(37) and reduced renal tubular Ca reabsorption(39) compared to their parental strain, SD rats. In each of these tissues, GHS rats have increased numbers of vitamin D receptors (VDR), while circulating levels of 1,25D are generally normal(36, 38, 45, 46, 53). Increased VDR have been found in one study of human IH (61) but not in others (63, 64). The increased number of VDR coupled to a normal level of 1,25D suggest that in GHS rats, the VDR is undersaturated with 1,25D. Indeed, when GHS rats were fed a low Ca diet their intestinal Ca absorption increased to a greater extent than SD, even though their s1,25D did not increase as much (32). Thus, the increased VDR in GHS led to a greater increase in intestinal Ca absorption even with a smaller increase in circulating 1,25D compared to SD.
With more VDR in GHS, addition of exogenous 1,25D should increase hypercalciuria in GHS to a greater extent than in SD rats. To test this hypothesis, GHS and SD rats were fed a diet containing ample Ca and injected with either 1,25D or vehicle for 16 days (57). With 1,25D, uCa in SD increased by 22.4 ± 1.5 mg/24h while in GHS uCa increased by 29.8 ± 1.8 (p=0.003, Fig. 2). These data demonstrate that the increased number of VDR in GHS as compared to SD leads to a greater physiological effect (increase in uCa), indicating that the additional VDR in GHS rats are biologically active. We would expect 1,25D-induced increased intestinal Ca absorption(32) and 1,25D-induced increased bone resorption(37) each to contribute to the increased uCa observed in GHS with 1,25D.
Figure 2. Urine calcium excretion in SD and GHS, without or with 1,25D.
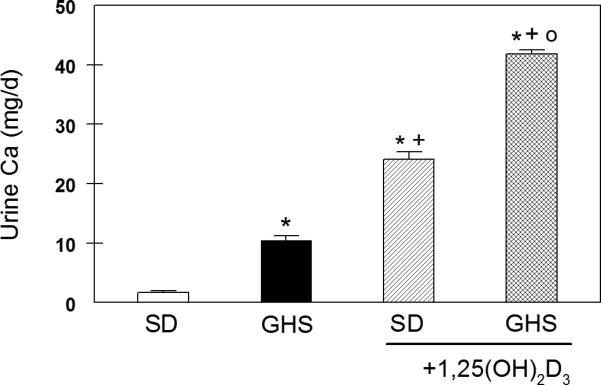
UCa was measured in 24 hour collected urine and expressed as mg/d, mean±SEM. *, p<0.05 compared to SD; +, p<0.05 compared to GHS; o, p<0.05 compared to SD+1,25D. From (57)
In IH patients(65) and GHS rats(36), decreased dietary Ca leads to decreased uCa. To study the effects of 1,25D on kidney and bone Ca handling we utilized a low Ca diet to effectively eliminate the contribution of increased intestinal absorption on the 1,25D-induced increase in uCa in GHS. GHS and SD rats were fed a low-Ca (0.02%) diet (LCD) and injected with 1,25D(58). Any increase in uCa would arise from increased bone resorption and/or decreased renal tubular reabsorption. With 1,25D, uCa in SD increased by 8.1±1.0 mg/24h and increased to an even greater extent in GHS by 16.8±0.8 mg/24h (P < 0.001). In GHS rats on LCD with or without 1,25D, uCa far exceeded daily Ca intake (no more than 2.6 mg/day), indicating that the biological activity of the increased VDR in bone cells and/or renal tubules must be a major contributor to the increased uCa in GHS rats. Thus on a very low Ca diet the decrease in net intestinal Ca absorption (in spite of an increase in percent Ca absorption) is greater than the decrease in uCa leading to an exacerbation of the net negative Ca balance.
The increased biological activity of the VDR in GHS intestine and bone leads to a greater influx of Ca into the systemic circulation. Basal renal tubular Ca reabsorption in GHS rats is decreased relative to SD(39), but the effects of 1,25D on renal tubular Ca reabsorption are not clear. To determine if 1,25D alters RNA expression of components of renal tubular Ca reabsorption (reviewed in(66-68)) associated with hypercalciuria while rats were fed a normal Ca diet, we measured kidney RNA expression of transient receptor potential vanilloid (TRPV)5 and calbindin D28K which were increased similarly in SD+1,25D and GHS+1,25D. The Na+/Ca2+ exchanger (NCX1) was increased in GHS+1,25D. Klotho, which activates TRPV5, was decreased in SD+1,25D and GHS+1,25D. TRPV6 was increased in SD+1,25D and increased further in GHS+1,25D. Claudin 14, 16, and 19, Na/K/2Cl transporter (NKCC2), and secretory K channel (ROMK) did not differ between SD+1,25D and GHS+1,25D. While the changes in RNA abundance are more consistent with increased Ca reabsorption, the observed increase in uCa, which must come from increased intestinal absorption and/or increased bone resorption, must exceed any effect of 1,25D on TRPV6 or NCX1-mediated renal Ca reabsorption. We next determined if there was also a 1,25D-mediated decrease in RNA expression of the components of renal Ca transport while rats were fed LCD. 1,25D administration resulted in a suppression of klotho, an activator of the renal Ca reabsorption channel TRPV5, in both SD and GHS rats. This fall in klotho would be expected to decrease tubular reabsorption of the Ca released from bone by 1,25D-induced resorption. Further studies of actual transport activity are necessary to directly address the effects of 1,25D on tubular Ca transport in GHS rats.
To eliminate the component of bone resorption in 1,25D-stimulated hypercalciuria in GHS rats, we utilized the bisphosphonate alendronate (alen)(59*). SD and GHS rats were fed LCD and half were injected daily with 1,25D. After 8 days all were also injected with alen until euthanasia at day 16. At 8 days, 1,25D increased uCa in SD and to an even greater extent in GHS rats (Fig. 3). At 16 days, alen eliminated the 1,25D-induced increase in uCa in SD. However, in GHS rats, alen decreased, but did not eliminate, the 1,25D-induced hypercalciuria, suggesting that even a large dose of alen cannot completely prevent the 1,25D-induced bone resorption in GHS. This partial resistance to alen may be due to the increased VDR in GHS tissues. While decreased renal tubular Ca reabsorption would contribute to the hypercalciuria in GHS rats fed LCD and given alen, there were no consistent differences in mRNA expression of renal transcellular or paracellular Ca transporters between GHS and SD(59*). If these results can be confirmed in humans with IH, the use of bisphosphonates, such as alen, may not completely prevent the decreased bone density observed in these patients (3, 6, 15-28).
Figure 3. Urine calcium excretion in SD and GHS rats fed low calcium diet (LCD), without or with exogenous 1,25D, before and during alendronate (alen).
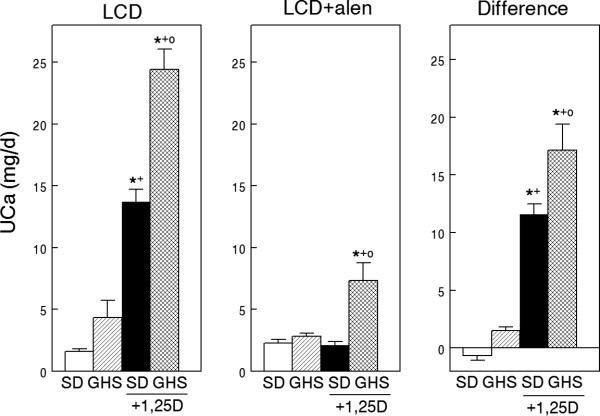
Left panel, LCD diet. Middle panel, LCD diet with alen injection. Right panel, difference (right panel) calculated as (uCa before alen) – (uCa after alen). Data expressed as mean±SEM. Significance was measured within groups. *, p<0.05 compared to SD; +, p<0.05 compared to GHS; o, p<0.05 compared to SD+1,25D. From (59*)
How does the Hypercalciuria of GHS Rats Affect Bone Quality?
In humans, kidney stone patients often have decreased BMD(15, 18, 69, 70) and our studies indicate that bone is an important contributor to the hypercalciuria in GHS rats. The hypercalciuria-induced loss of Ca from existing bone would be expected to decrease bone quality in GHS rats. GHS and control SD rats were fed a constant amount of either a low Ca (LCD) or a high Ca (HCD) diet for 6 wks(35). Urine Ca was greater in the GHS rats on either diet. GHS rats fed HCD had reduced cortical (humerus) and trabecular (L(1)-L(5) vertebrae) BMD as compared to SD (Fig. 4). GHS rats fed HCD had a decrease in trabecular volume and thickness, whereas LCD led to an approximately 20-fold increase in both osteoid surface and volume. GHS rats fed HCD had no change in vertebral strength (failure stress), ductibility (failure strain), stiffness (modulus), or toughness, whereas in the humerus, there was reduced ductibility and toughness and an increase in modulus, indicating that the defect in mechanical properties is mainly manifested in cortical, rather than trabecular, bone. GHS rat cortical bone is more mineralized than trabecular bone and LCD led to a decrease in the mineralization profile. Thus, the GHS rats, fed an ample Ca diet, have reduced BMD with reduced trabecular volume, mineralized volume, and thickness, and their bones are more brittle and fracture prone, indicating that GHS rats have an intrinsic disorder of bone that is not secondary to any reduction in dietary Ca.
Figure 4. GHS BMD.
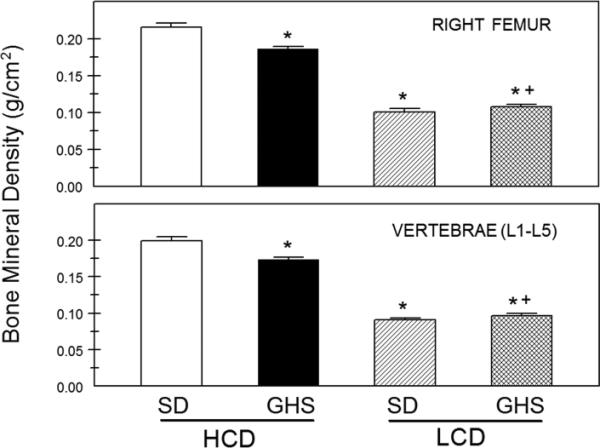
BMD of the whole right femurs, representing cortical bone, and the lumbar vertebrae (L1–L5) representing trabecular bone (mean ± SE). BMD was measured using DXA at the conclusion of the study. *Different from Ctl eating HCD, p < 0.05; +different from GHS eating HCD, p < 0.05. From (35)
As we have shown that 1,25D induces a greater increase in uCa in GHS than SD rats, we hypothesized that 1,25D would exacerbate the bone defects seen in GHS rats. We fed both GHS and SD rats an ample Ca diet and injected either low or high dose 1,25D (LD and HD, respectively or vehicle (veh) daily for 16 days(60*). Femoral areal bone mineral density (aBMD, by DEXA) was decreased in GHS+LD and GHS+HD relative to GHS+veh, while there was no effect on SD. Vertebral aBMD was lower in GHS compared to SD and further decreased in GHS+HD. Both femoral and L6 vertebral volumetric BMD (by micro-computerized tomography) were lower in GHS and further reduced by HD. Histomorphometry indicated a decreased osteoclast number in GHS+HD compared to GHS+veh or SD+HD. In tibiae, GHS+HD trabecular thickness and number increased, with a 12-fold increase in osteoid volume but only a threefold increase in bone volume (Fig. 5). Bone formation rate was decreased in GHS+HD relative to GHS+veh, confirming the mineralization defect. The loss of BMD and the mineralization defect in GHS rats contribute to increased hypercalciuria; if these effects persist, they would result in decreased bone strength, making the bones from GHS rats more fracture-prone. The deleterious effects of exogenous 1,25D on bone quality in GHS rats indicates that the increased amount of VDR in these animals plays an important role in determining their bone health.
Figure 5. Trabecular bone structure.
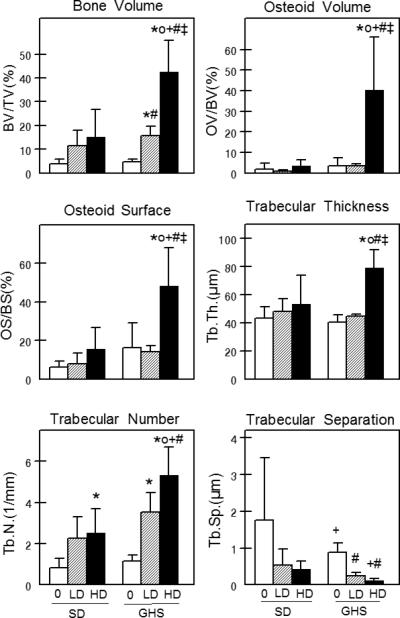
Static histomorphometry was performed on proximal left tibiae from SD and GHS rats. % bone volume, % osteoid volume, % osteoid surface, trabecular thickness, trabecular number and trabecular separation were determined. All data are presented as mean ± standard deviation. * p<0.05 compared to SD+veh; o p<0.05 compared to SD+LD; + p<0.05 compared to SD+HD; # p<0.05 compared to GHS+veh; ‡ p< 0.05 compared to GHS+LD. From (60*)
To examine the mechanism of 1,25D activity on bone in GHS rats, we examined VDR effects on bone morphogenetic protein 2 (BMP2)(71). Both 1,25D and BMP2 are critical for normal maintenance of bone metabolism and bone formation, respectively. The complex nature of bone cell regulation suggests a potential interaction of these two important regulators in GHS rats. BMP2 expression is suppressed by the VDR-1,25D complex in Bone Marrow Stromal Cells (BMSCs) from GHS and SD rats and in the UMR-106 rat osteosarcoma cell line (Fig. 6). Chromatin immunoprecipitation (ChIP) assays identified VDR binding only to one of several potential binding sites within the BMP2 promoter regions, an inhibitory region that also mediates suppressor reporter gene activity. Genes can be epigenetically regulated by modification of DNA and/or histones, producing marks that modulate protein interactions(72). The down-regulation of BMP2 by 1,25D was studied in vitro in BMSCs and UMR-106 cells using the DNA methyltransferase inhibitor 5-aza-2′-deoxycytidine (DAC) and the histone deacetylase inhibitor trichostatin A (TSA). Both DAC and TSA activate BMP2 expression in combination with 1,25D. Bisulfite DNA pyrosequencing reveals that 1,25D completely hypermethylates a single CpG site in the same BMP2 promoter region as identified by the ChIP and reporter gene assays. ChIP assays also show that 1,25D can increase the repressive histone H3K9me2 mark and reduce the acetylation of histone H3 at the same BMP2 promoter region. Taken together, our results indicate that 1,25D binding to VDR down-regulates BMP2 gene expression in BMSCs and osteoblast-like UMR-106 cells by binding to the BMP2 promoter region via DNA methylation and histone modification. These results suggest that exogenously supplied BMP2 might enhance bone quality in GHS rats and perhaps in IH patients.
Figure 6. VDR levels and BMP2 expression in GHS and SD rats.
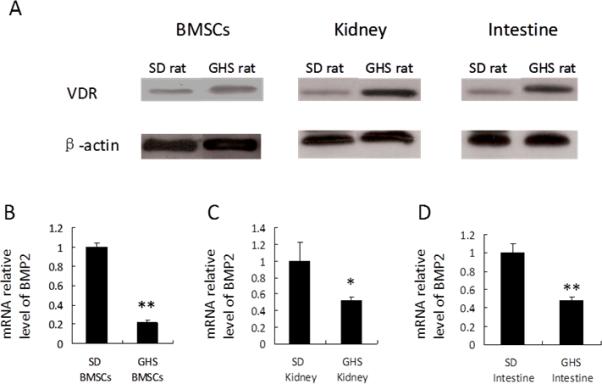
Nuclear proteins were isolated from BMSCs and tissues of SD and GHS rats and were subjected to Western blot using anti- VDR. Representative data are shown in (A). Expression of BMP2 in BMSCs, kidney and intestine tissues from GHS rats decrease relative to SD rats (B, C, D). (*, p<0.05; **, p<0.01). From (71)
Do Strategies Directed Toward Reducing Recurrent Stone Formation Ameliorate the Decreased Bone Quality in GHS rats?
Clinically, thiazide diuretics reduce uCa and prevent recurrent stone formation in human stone-forming patients(73-76); however, whether they also lead to an improvement in bone quality is not clear. We used GHS rats to test the hypothesis that the thiazide diuretic chlorthalidone (CTD) would have a favorable effect on bone density and quality(40). GHS rats were fed a 1.2% Ca diet, and half were also fed CTD. As anticipated, rats fed CTD had a marked reduction in uCa. An increase in trabecular mineralization was observed with CTD compared with controls. CTD also improved the architecture of trabecular bone. Using micro-computed tomography (μCT), trabecular bone volume (BV/TV), trabecular thickness, and trabecular number were increased with CTD. A significant increase in trabecular thickness with CTD was confirmed by static histomorphometry. CTD also improved the connectivity of trabecular bone. Significant improvements in vertebral strength and stiffness were measured by vertebral compression. Conversely, a slight loss of bending strength was detected in the femoral diaphysis with CTD. Thus results obtained in GHS rats suggest that CTD can favorably influence vertebral fracture risk. CTD did not alter formation parameters, suggesting that the improved vertebral bone strength was due to decreased bone resorption and retention of bone structure, rather than enhanced bone formation.
Summary
In GHS rats the hypercalciuria results from a systemic dysregulation of Ca transport affecting the major Ca transporting organs: intestine(38), bone(37) and kidney(39). In the GHS rats an increased level of VDR has been found in each of these organs and we have found that 1,25D leads to greater alterations in Ca transport at each of these sites than in SD. Thus in GHS rats the hypercalciuria appears to be directly related to excessive VDR activity. A similar role for VDR activity appears important in human hypercalciuria. Lemann fed normal men vitamin D and replicated hypercalciuria and negative Ca balance(77). If human IH can also be shown to be due to increased VDR leading to an increased effect of circulating 1,25D, then strategies can be devised to reduce the hypercalciuria and improve bone quality and perhaps ultimately eliminate recurrent Ca stone disease.
Summary Points.
Genetic Hypercalciuric Stone-forming (GHS) rats provide a useful model of human idiopathic hypercalciuria (IH).
Similar to many stone-forming patients, GHS rats display decreased bone mineral density (BMD).
This reduction in BMD can be ameliorated in GHS by treatment of the hypercalciuria with thiazide diuretics.
The hypercalciuria in GHS seems to be a consequence of increased vitamin D receptor levels in tissues important in calcium transport (intestine, kidney, bone).
Acknowlegements
none
Financial support and sponsorship
This work was supported by Grant RO1 DK075462 from the National Institutes of Health
Footnotes
Conflicts of Interest
none
Reference List
- 1.Monk RD, Bushinsky DA. Nephrolithiasis and nephrocalcinosis. In: Frehally J, Floege J, Johnson RJ, editors. Comprehensive Clinical Nephrology. Elsevier; St. Louis: 2010. pp. 687–701. [Google Scholar]
- 2.Bushinsky DA, Coe FL, Moe OW. Nephrolithiasis. In: Brenner BM, editor. The Kidney. W.B. Saunders; Philadelphia: 2012. pp. 1455–507. [Google Scholar]
- 3.Monk RD, Bushinsky DA. Kidney stones. In: Kronenberg HM, Melmed S, Polonsky KS, Larsen PR, editors. Williams Textbook of Endocrinology. W.B.Saunders; Philadelphia: 2011. pp. 1350–67. [Google Scholar]
- 4.Bushinsky DA. Calcium Nephrolithiasis. In: Rosen CJ, editor. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. American Society of Bone and Mineral Research; Washington, D.C.: 2008. pp. 460–4. [Google Scholar]
- 5.Worcester EM, Coe FL. Calcium Kidney Stones. New England Journal of Medicine. 2010;363(10):954–63. doi: 10.1056/NEJMcp1001011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bushinsky DA. Recurrent hypercalciuric nephrolithiasis - does diet help? N Eng J Med. 2002;346:124–5. doi: 10.1056/NEJM200201103460210. [DOI] [PubMed] [Google Scholar]
- 7.Bushinsky DA. Renal Lithiasis. In: Humes HD, editor. Kelly's Textbook of Medicine. Lippincott Williams & Wilkens; New York: 2000. pp. 1243–8. [Google Scholar]
- 8.Bushinsky DA, Parker WR, Alexander KM, Krieger NS. Metabolic, but not respiratory, acidosis increases bone PGE2 levels and calcium release. Am J Physiol (Renal Fluid Electrolyte Physiol) 2001;281:F1058–F66. doi: 10.1152/ajprenal.0355.2000. [DOI] [PubMed] [Google Scholar]
- 9.Bushinsky DA. Nephrolithiasis. Journal of the American Society of Nephrology. 1998;9:917–24. doi: 10.1681/ASN.V95917. [DOI] [PubMed] [Google Scholar]
- 10.Consensus C. Prevention and treatment of kidney stones. JAMA. 1988;260:977–81. [PubMed] [Google Scholar]
- 11.Pak CYC. Pathophysiology of calcium nephrolithiasis. In: Seldin DW, Giebisch G, editors. The Kidney: Physiology and Pathophysiology. Raven Press, Ltd.; New York: 1992. pp. 2461–80. [Google Scholar]
- 12.Coe FL, Parks JH, Asplin JR. The pathogenesis and treatment of kidney stones. New England Journal of Medicine. 1992;327:1141–52. doi: 10.1056/NEJM199210153271607. [DOI] [PubMed] [Google Scholar]
- 13.Coe FL, Favus MJ, Asplin JR. Nephrolithiasis. In: Brenner BM, Rector FC Jr., editors. The Kidney. W.B. Saunders Company; Philadelphia: 2004. pp. 1819–66. [Google Scholar]
- 14.Coe FL, Bushinsky DA. Pathophysiology of hypercalciuria. Am J Physiol (Renal Fluid Electrolyte Physiol) 1984;247:F1–F13. doi: 10.1152/ajprenal.1984.247.1.F1. [DOI] [PubMed] [Google Scholar]
- 15.Vezzoli G, Soldati L, Ardila M, et al. Urinary calcium is a determinant of bone mineral density in elderly men participating in the InCHIANTI study. Kid Int. 2005;67:2006–14. doi: 10.1111/j.1523-1755.2005.00302.x. [DOI] [PubMed] [Google Scholar]
- 16.Giannini S, Nobile M, Sartori L, Calo L, Tasca A, Dalle Carbonare L, et al. Bone density and skeletal metabolism are altered in idiopathic hypercalciuria. Clin Nephrol. 1998;50:94–100. [PubMed] [Google Scholar]
- 17.Tasca A, Cacciola A, Ferrarese P, Ioverno E, Visona E, Bernardi C, et al. Bone alterations in patients with idiopathic hypercalciuria and calcium nephrolithiasis. Urology. 2002;59:865–9. doi: 10.1016/s0090-4295(02)01626-6. [DOI] [PubMed] [Google Scholar]
- 18.Asplin JR, Bauer KA, Kinder J, Muller G, Coe BJ, Parks JH, et al. Bone mineral density and urine calcium excretion among subjects with and without nephrolithiasis. Kidney Int. 2003;63:662–9. doi: 10.1046/j.1523-1755.2003.00763.x. [DOI] [PubMed] [Google Scholar]
- 19.Sakhaee K. Nephrolithiasis as a systemic disorder. CurrOpinNephrol Hypertens. 2008;17(3):304–9. doi: 10.1097/MNH.0b013e3282f8b34d. [DOI] [PubMed] [Google Scholar]
- 20.Melton LJ, III, Crowson CS, Khosla S, Wilson DM, Fallon WM. Fracture risk among patients with urolithiasis: a population based cohort study. Kidney International. 1998;53:459–64. doi: 10.1046/j.1523-1755.1998.00779.x. [DOI] [PubMed] [Google Scholar]
- 21.Pietschmann F, Breslau NA, Pak CYC. Reduced vertebral bone density in hypercalciuric nephrolithiasis. J Bone Miner Res. 1992;7:1383–8. doi: 10.1002/jbmr.5650071205. [DOI] [PubMed] [Google Scholar]
- 22.Jaeger P, Lippuner K, Casez JP, Hess B, Ackerman D, Hug C. Low bone mass in idiopathic renal stone formers: magnitude and significance. Journal of Bone and Mineral Research. 1994;9:1525–32. doi: 10.1002/jbmr.5650091004. [DOI] [PubMed] [Google Scholar]
- 23.Misael da Silva AM, dos Reis LM, Pereira RC, Futata E, Branco-Martins CT, Noronha IL, et al. Bone involvement in idiopathic hypercalciuria. Clin Nephrol. 2002;57:183–91. doi: 10.5414/cnp57183. [DOI] [PubMed] [Google Scholar]
- 24.Heilberg IP, Martini LA, Teixeira SH, Szejnfeld VL, Carvalho AB, Lobao R, et al. Effect of etidronate treatment on bone mass of male nephrolithiasis patients with idiopathic hypercalciuria and osteopenia. Nephron. 1998;79:430–7. doi: 10.1159/000045089. [DOI] [PubMed] [Google Scholar]
- 25.Bataille P, Achard JM, Fournier A, Boudailliez B, Westell PF, Esper NE, et al. Diet, vitamin D and vertebral mineral density in hypercalciuric calcium stone formers. Kidney International. 1991;39:1193–205. doi: 10.1038/ki.1991.151. [DOI] [PubMed] [Google Scholar]
- 26.Heilberg IP, Weisinger JR. Bone disease in idiopathic hypercalciuria. Curr Opin Nephrol Hypertens. 2006;15:394–402. doi: 10.1097/01.mnh.0000232880.58340.0c. [DOI] [PubMed] [Google Scholar]
- 27.Lauderdale DS, Thisted RA, Wen M, Favus M. Bone mineral density and fracture among prevalent kidney stone cases in the Third National Health and Nutrition Examination Survey. J Bone Miner. 2001;16:1893–8. doi: 10.1359/jbmr.2001.16.10.1893. [DOI] [PubMed] [Google Scholar]
- 28.Cauley JA, Fullman RL, Stone KL, et al. Factors associated with the lumbar spine and proximal femur bone mineral density in older men. Osteoporos Int. 2005;16:1525–37. doi: 10.1007/s00198-005-1866-8. [DOI] [PubMed] [Google Scholar]
- 29.Moe OW, Bonny O. Genetic hypercalciuria. Journal of the American Society of Nephrology. 2005;16(3):729–45. doi: 10.1681/ASN.2004100888. [DOI] [PubMed] [Google Scholar]
- 30.Stechman MJ, Loh NY, Thakker RV. Genetics of hypercalciuric nephrolithiasis: renal stone disease. Acad Sci. 2007;1116:461–84. doi: 10.1196/annals.1402.030. [DOI] [PubMed] [Google Scholar]
- 31.Bushinsky DA, Asplin JR, Grynpas MD, Evan AP, Parker WR, Alexander KM, et al. Calcium oxalate stone formation in genetic hypercalciuric stone-forming rats. Kidney Int. 2002;61:975–87. doi: 10.1046/j.1523-1755.2002.00190.x. [DOI] [PubMed] [Google Scholar]
- 32.Bushinsky DA, Favus MJ. Mechanism of hypercalciuria in genetic hypercalciuric rats: inherited defect in intestinal calcium transport. J Clin Invest. 1988;82:1585–91. doi: 10.1172/JCI113770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bushinsky DA, Grynpas MD, Nilsson EL, Nakagawa Y, Coe FL. Stone formation in genetic hypercalciuric rats. Kidney Int. 1995;48:1705–13. doi: 10.1038/ki.1995.468. [DOI] [PubMed] [Google Scholar]
- 34.Evan AP, Bledsoe SB, Smith SB, Bushinsky DA. Calcium oxalate crystal localization and osteopontin immunostaining in genetic hypercalciuric stone-forming rats. Kidney Int. 2004;65:154–61. doi: 10.1111/j.1523-1755.2004.00396.x. [DOI] [PubMed] [Google Scholar]
- 35.Grynpas M, Waldman S, Holmyard D, Bushinsky DA. Genetic hypercalciuric stone-forming rats have a primary decrease in BMD and strength. J Bone Miner Res. 2009;24(8):1420–6. doi: 10.1359/JBMR.090223. Epub 2009/03/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim M, Sessler NE, Tembe V, Favus MJ, Bushinsky DA. Response of genetic hypercalciuric rats to a low calcium diet. Kidney Int. 1993;43(1):189–96. doi: 10.1038/ki.1993.31. [DOI] [PubMed] [Google Scholar]
- 37.Krieger NS, Stathopoulos VM, Bushinsky DA. Increased sensitivity to 1,25(OH)2D3 in bone from genetic hypercalciuric rats. Am J Physiol (Cell Physiol) 1996;271:C130–C5. doi: 10.1152/ajpcell.1996.271.1.C130. [DOI] [PubMed] [Google Scholar]
- 38.Li XQ, Tembe V, Horwitz GM, Bushinsky DA, Favus MJ. Increased intestinal vitamin D receptor in genetic hypercalciuric rats: a cause of intestinal calcium hyperabsorption. J Clin Invest. 1993;91:661–7. doi: 10.1172/JCI116246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tsuruoka S, Bushinsky DA, Schwartz GJ. Defective renal calcium reabsorption in genetic hypercalciuric rats. Kidney Int. 1997;51:1540–7. doi: 10.1038/ki.1997.212. [DOI] [PubMed] [Google Scholar]
- 40.Bushinsky DA, Willett T, Asplin JR, Culbertson C, Che SPY, Grynpas M. Chlorthalidone improves vertebral bone quality in genetic hypercalciuric stone-forming rats. Journal of Bone and Mineral Research. 2011;26:1904–12. doi: 10.1002/jbmr.374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bai S, Wang H, Shen J, Zhou R, Bushinsky DA, Favus MJ. Elevated vitamin D receptor levels in genetic hypercalciuric stone-forming rats are associated with downregulation of Snail. Journal of Bone and Mineral Research. 2010;25:830–40. doi: 10.1359/jbmr.091010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Asplin JR, Donahue SE, Lindeman C, Michalenka A, Strutz KL, Bushinsky DA. Thiosulfate reduces calcium phosphate nephrolithiasis. Journal of the American Society of Nephrology : JASN. 2009;20(6):1246–53. doi: 10.1681/ASN.2008070754. Epub 2009/04/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoopes RR, Jr., Middleton FA, Sen S, Hueber PA, Reid R, Bushinsky DA, et al. Isolation and confirmation of a calcium excretion quantitative trait locus on chromosome 1 in genetic hypercalciuric stone-forming congenic rats. Journal of the American Society of Nephrology. 2006;17(5):1292–304. doi: 10.1681/ASN.2005080828. [DOI] [PubMed] [Google Scholar]
- 44.Bushinsky DA, LaPlante K, Asplin JR. Effect of cinacalcet on urine calcium excretion and supersaturation in genetic hypercalciuric stone-forming rats. Kidney Int. 2006;69:1586–92. doi: 10.1038/sj.ki.5000324. [DOI] [PubMed] [Google Scholar]
- 45.Yao J, Karnauskas AJ, Bushinsky DA, Favus MJ. Regulation of renal calcium-sensing receptor gene expression in response to 1,25(OH)2D3 in genetic hypercalciuric stone forming rats. Journal of the American Society of Nephrology. 2005;16:1300–8. doi: 10.1681/ASN.2004110991. [DOI] [PubMed] [Google Scholar]
- 46.Karnauskas AJ, van Leeuwen JP, van den Bemd GJ, Kathpalia PP, DeLuca HF, Bushinsky DA, et al. Mechanism and function of high vitamin D receptor levels in genetic hypercalciuric stone-forming rats. Journal of Bone and Mineral Research. 2005;20:447–54. doi: 10.1359/JBMR.041120. [DOI] [PubMed] [Google Scholar]
- 47.Bushinsky DA, Asplin JR. Thiazides reduce brushite, but not calcium oxalate, supersaturation and stone formation in genetic hypercalciuric stone-forming rats. Journal of the American Society of Nephrology. 2005;16:417–24. doi: 10.1681/ASN.2004070543. [DOI] [PubMed] [Google Scholar]
- 48.Hoopes RR, Reid R, Sen S, Szpirer C, Dixon P, Pannet A, et al. Quantitative trait loci for hypercalciuria in a rat model of kidney stone disease. Journal of the American Society of Nephrology. 2003;14:1844–50. doi: 10.1097/01.asn.0000073920.43848.a3. [DOI] [PubMed] [Google Scholar]
- 49.Bushinsky DA, Grynpas MD, Asplin JR. Effect of acidosis on urine supersaturation and stone formation in genetic hypercalciuric stone forming rats. Kidney Int. 2001;59(4):1415–23. doi: 10.1046/j.1523-1755.2001.0590041415.x. [DOI] [PubMed] [Google Scholar]
- 50.Bushinsky DA, Parker WR, Asplin JR. Calcium phosphate supersaturation regulates stone formation in genetic hypercalciuric stone-forming rats. Kidney Int. 2000;57:550–60. doi: 10.1046/j.1523-1755.2000.00875.x. [DOI] [PubMed] [Google Scholar]
- 51.Bushinsky DA, Bashir MA, Riordon DR, Nakagawa Y, Coe FL, Grynpas MD. Increased dietary oxalate does not increase urinary calcium oxalate saturation in hypercalciuric rats. Kidney Int. 1999;55:602–12. doi: 10.1046/j.1523-1755.1999.00281.x. [DOI] [PubMed] [Google Scholar]
- 52.Bushinsky DA, Neumann KJ, Asplin J, Krieger NS. Alendronate decreases urine calcium and supersaturation in genetic hypercalciuric rats. Kidney Int. 1999;55:234–43. doi: 10.1046/j.1523-1755.1999.00247.x. [DOI] [PubMed] [Google Scholar]
- 53.Yao J, Kathpalia P, Bushinsky DA, Favus MJ. Hyperresponsiveness of vitamin D receptor gene expression to 1,25-dihydroxyvitamin D3: A new characteristic of genetic hypercalciuric stone-forming rats. J Clin Invest. 1998;101:2223–32. doi: 10.1172/JCI1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Asplin JR, Bushinsky DA, Singharetnam W, Riordon D, Parks JH, Coe FL. Relationship between supersaturation and crystal inhibition in hypercalciuric rats. Kidney Int. 1997;51:640–5. doi: 10.1038/ki.1997.93. [DOI] [PubMed] [Google Scholar]
- 55.Bushinsky DA, Kim M, Sessler NE, Nakagawa Y, Coe FL. Increased urinary saturation and kidney calcium content in genetic hypercalciuric rats. Kidney Int. 1994;45:58–65. doi: 10.1038/ki.1994.7. [DOI] [PubMed] [Google Scholar]
- 56.Krieger N, Asplin JR, Frick KK, Granja I, Culbertson CD, Ng A, Grynpas MD, Bushinsky DA. Effect of potassium citrate on calcium phosphate stones in genetic hypercalciuric stone-forming rats. Journal of American Society of Nephrology. 2015 doi: 10.1681/ASN.2014121223. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Frick KK, Asplin JR, Favus MJ, Culbertson C, Krieger NS, Bushinsky DA. Increased biological response to 1,25(OH)2D3 in genetic hypercalciuric stone-forming rats. Am J Physiol Renal Physiol. 2013;304(6):F718–26. doi: 10.1152/ajprenal.00645.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Frick KK, Asplin JR, Krieger NS, Culbertson CD, Asplin DM, Bushinsky DA. Enhanced hypercalciuria in genetic hypercalciuric stone-forming rats fed a low calcium diet. Am J Physiol Renal Physiol. 2013;305:F1132–F8. doi: 10.1152/ajprenal.00296.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59*.Frick KK, Asplin JR, Culbertson CD, Granja I, Krieger NS, Bushinsky DA. Persistence of 1,25(OH)2D3-induced hypercalciuria in alendronate treated genetic hypercalciuric stone-forming rats fed a low calcium diet. American Journal of Physiology. 2014;306:F1081–7. doi: 10.1152/ajprenal.00680.2013. [Provides strong support for the role of bone in GHS hypercalciuria.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60*.Ng AH, Frick KK, Krieger NS, Asplin JR, Cohen-McFarlane M, Culbertson CD, Kyker-Snowman K, Grynpas MD, Bushinsky DA. 1,25(OH)2D3-enhanced hypercalciuria in genetic hypercalciuric stone-forming rats. Calc Tiss Int. 2014;94:531–43. doi: 10.1007/s00223-014-9838-7. [Describes intrinsic defect of bone mineralization in GHS.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Favus MJ, Karnauskas AJ, Parks JH, Coe FL. Peripheral blood monocyte vitamin D receptor levels are elevated in patients with idiopathic hypercalciuria. J Clin Endocrinol Metab. 2004;89:4937–43. doi: 10.1210/jc.2004-0412. [DOI] [PubMed] [Google Scholar]
- 62.Li XQ, Tembe V, Horwitz GM, Bushinsky DA, Favus MJ. Increased intestinal vitamin D receptor in genetic hypercalciuric rats. A cause of intestinal calcium hyperabsorption. J Clin Invest. 1993;91(2):661–7. doi: 10.1172/JCI116246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zerwekh JE, Hughes MR, Reed BY, Breslau NA, Heller HJ, Lemke M, et al. Evidence for normal vitamin D receptor messenger ribonucleic acid and genotype in absorptive hypercalciuria. J Clin Endocrinol Metab. 1995;80:2960–5. doi: 10.1210/jcem.80.10.7559881. [DOI] [PubMed] [Google Scholar]
- 64.Zerwekh JE, Reed BY, Heller HJ, Gonzalez GB, Haussler MR, Pak CY. Normal vitamin D receptor concentration and responsiveness to 1,25-dihydroxyvitamin D3 in skin fibroblasts from patients with absorptive hypercalciuria. Miner Electrolyte Metab. 1998;24:307–13. doi: 10.1159/000057388. [DOI] [PubMed] [Google Scholar]
- 65.Liberman UA, Sperling O, Atsmon A, Frank M, Modan M, deVries A. Metabolic and calcium kinetic studies in idiopathic hypercalciuria. J Clin Invest. 1968;47:2580–90. doi: 10.1172/JCI105940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Frick KK, Bushinsky DA. Molecular mechanisms of primary hypercalciuria. J Am Soc Neph. 2003;14:1082–95. doi: 10.1097/01.asn.0000062960.26868.17. [DOI] [PubMed] [Google Scholar]
- 67.Hoenderop JGJ, Nilius B, Bindels RJM. Calcium absorption across epithelia. Physiological Reviews. 2005;85(1):373–422. doi: 10.1152/physrev.00003.2004. [DOI] [PubMed] [Google Scholar]
- 68.Kumar R, Vallon V. Reduced Renal Calcium Excretion in the Absence of Sclerostin Expression: Evidence for a Novel Calcium-Regulating Bone Kidney Axis. Journal of the American Society of Nephrology. 2014;25(10):2159–68. doi: 10.1681/ASN.2014020166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Giannini S, Nobile M, Dalle Carbonare L, et al. Hypercalciuria is a common and important finding in postmenopausal women with osteoporosis. Eur J Endocrinol. 2003;149:209–13. doi: 10.1530/eje.0.1490209. [DOI] [PubMed] [Google Scholar]
- 70.Sakhaee K, Maalouf NM, Kumar R, Pasch A, Moe OW. Nephrolithiasis-associated bone disease: pathogenesis and treatment options. Kidney Int. 2011;79(4):393–403. doi: 10.1038/ki.2010.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fu B, Wang H, Wang J, Barouhas I, Liu W, Shuboy A, et al. Epigenetic regulation of BMP2 by 1,25-dihydroxyvitamin D3 through DNA methylation and histone modification. PLoS One. 2013;8:e61423. doi: 10.1371/journal.pone.0061423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chopra M, Bohlander SK. Disturbing the histone code in leukemia: translocations and mutations affecting histone methyl transferases. Cancer Genetics. 2014 doi: 10.1016/j.cancergen.2014.10.005. Epub doi:10.1016/j.cancergen.2014.10.005. [DOI] [PubMed] [Google Scholar]
- 73.Pearle MS, Roehrborn CG, Pak CYC. Meta-analysis of randomized trials for medical prevention of calcium oxalate nephrolithiasis. Journal of Endurology. 1999;13:679–85. doi: 10.1089/end.1999.13.679. [DOI] [PubMed] [Google Scholar]
- 74.Escribano J, Balaguer A, Pagone F, Feliu A, Roqué I, Figuls M. Pharmacological interventions for preventing complications in idiopathic hypercalciuria. Cochrane Database Syst Rev. 2009:CD004754. doi: 10.1002/14651858.CD004754.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kairaitis L. (CARI) CfAwRI. The CARI guidelines. Kidney stones: prevention of recurrent calcium nephrolithiasis. Nephrology (Carlton) 2007;(Suppl 1):S11–20. doi: 10.1111/j.1440-1797.2006.00723.x. [DOI] [PubMed] [Google Scholar]
- 76.Fink HA, Wilt TJ, Eidman KE, Garimella PS, MacDonald R, Rutks IR, et al. Medical Management to Prevent Recurrent Nephrolithiasis in Adults: A Systematic Review for an American College of Physicians Clinical Guideline. Annals of Internal Medicine. 2013;158(7):535–43. doi: 10.7326/0003-4819-158-7-201304020-00005. [DOI] [PubMed] [Google Scholar]
- 77.Adams ND, Gray RW, Lemann JJ. Effects of calcitriol administration on calcium metabolism in healthy men. Kid Int. 1982;21:90–7. doi: 10.1038/ki.1982.13. [DOI] [PubMed] [Google Scholar]