

Abstract
The past 15 years have seen enormous advances in our understanding of the receptor and signalling systems that allow dendritic cells (DCs) to respond to pathogens or other danger signals and initiate innate and adaptive immune responses. We are now beginning to appreciate that many of these pathways not only stimulate changes in the expression of genes that control DC immune functions, but also affect metabolic pathways, thereby integrating the cellular requirements of the activation process. In this Review, we focus on this relatively new area of research and attempt to describe an integrated view of DC immunometabolism.
Dendritic cells (DCs) are a diverse group of related, haematopoietic cell types that are specialized for recognizing pathogens1 (BOX 1). They express various pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs), nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs) and C-type lectins, which are able to bind molecular motifs that are characteristic of particular pathogens or that are associated with cellular damage2–4. Ligation of PRRs initiates signalling pathways that lead to cellular activation and marked changes in gene expression and cellular biology3. DCs activated via PRRs have central roles in both innate and adaptive immunity, in which they drive the activation of antigen-specific T cells. As such, DCs have a central role in the immune system.
Box 1. Dendritic cell subsets.
Dendritic cells (DCs) are defined by their uniquely efficient ability to activate naive T cells. Although originally defined as an apparently homogeneous population of adherent stellate cells in the spleen110, they are now known to comprise numerous subsets and to be present, during the steady state, within all lymphoid organs and the majority of peripheral tissues (reviewed recently in REF. 1). DCs are resting cells that have the characteristic ability to respond to pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs), and/or to cytokines, and become activated. In general, DCs express CD11c and MHC class II, but once activated, they increase their expression levels of surface MHC–peptide complexes and of co-stimulatory molecules, and this allows them to effectively activate T cells. There are four major subsets of DCs: conventional DCs (cDCs), Langerhans cells, monocyte-derived DCs and plasmacytoid DCs (pDCs). These cells are related to each other as they have a common myeloid progenitor. cDCs within lymphoid organs are comprised of two major subpopulations, which are distinguished by the expression of CD8α or CD4. There are tissue-resident cells that are equivalent to, and related by lineage to, these populations and marked by the expression of CD103 and CD11b, respectively. These cells will migrate to lymph nodes in the steady state, and to a notably greater extent following peripheral infection, immunization, or other disturbances that lead to DC activation. Cells equivalent to CD8α+ DCs, CD4+ DCs and pDCs can be grown from bone marrow in vitro by stimulation with FMS-like tyrosine kinase 3 ligand (FLT3L). Langerhans cells are skin-resident cells that are similar to macrophages in many ways, but which can assume cDC-like properties when they migrate to lymphoid organs. Monocytes can develop into TNF and iNOS-producing (TIP)-DCs at inflammatory sites, but the extent to which these cells represent true DCs is questioned. CD11c+MHC class IIhi DCs can be grown from bone marrow cultured with granulocyte–macrophage colony-stimulating factor (GM-CSF); these bone marrow-derived DCs have been proposed to be equivalent to in vivo monocyte-derived DCs, but it is debatable how closely these cells are related to any in vivo population. pDCs are a distinct lineage of DCs that are more specialized for cytokine production, particularly type I interferon production, rather than antigen presentation. In this context, however, it should be stressed that upon activation, all DCs begin to secrete a variety of cytokines that markedly influence the cells that they are interacting with, and it seems reasonable to consider this facet of DC behaviour as integral to their biology. It is likely that additional subsets of DCs with specialized functions remain to be identified.
It is becoming increasingly clear that different stages of immune cell activation coincide with, and are underpinned by, different types of cellular metabolism that are tailored towards the bioenergetic and biosynthetic needs of these cells. The relevance of this to lymphocytes and macrophages has been extensively covered in a number of recent reviews5–7. The metabolic requirements of an activated DC are also distinct from those of a quiescent DC and, as such, changes in metabolism must be integral to the successful activation of these cells. This realization has led to interest in the cellular metabolism of DCs, not least because it is possible that manipulation of the metabolic state of DCs could be used to modify inflammatory and immune responses for therapeutic purposes8. Interest in this area has increased with the realization that cells can respond to metabolites themselves, thereby sensing metabolic changes within themselves or in other cells that may presage danger9. In this Review, we discuss the current understanding of DC metabolism, highlighting areas that we feel hold particular promise for future research in this fast-moving area.
Metabolism in developing and resting DCs
Conventional DCs (cDCs) and plasmacytoid DCs (pDCs) originate from committed DC progenitors1 (CDPs) (BOX 1). DCs can also develop from monocytes, and CDPs and monocytes may have a common origin in a population of macrophage and DC progenitors (MDPs). At this time, little is known about the metabolism of CDPs (or that of MDPs). However, the differentiation of human monocytes into DCs in response to granulocyte–macrophage colony-stimulating factor (GM-CSF) and interleukin-4 (IL-4) is accompanied by increased expression of peroxisome proliferator-activated receptor-γ (PPARγ)10,11, which is a key transcription factor controlling lipid metabolism, and PPARγ co-activator 1α (PGC1α), which is a master regulator of mitochondrial biogenesis. The upregulation of PPARγ and PGC1α is followed by increased mitochondrial biogenesis12. Moreover, inhibition of mitochondrial respiration in monocytes by the electron transport chain (ETC) inhibitor rotenone is able to block DC differentiation12,13. In these studies, there was a close association between increased citrate synthase activity and DC differentiation. Citrate gives rise to isocitrate and subsequently α-ketoglutarate (αKG) in the tricarboxylic acid cycle (TCA cycle), but it is also a precursor for fatty acid synthesis (BOX 2). In this role, citrate is exported from mitochondria and converted into cytosolic acetyl-CoA, which is an important intermediate in several pathways, including fatty acid synthesis. The differentiation of monocytes into DCs in vitro and the development of DCs in lymphoid organs and peripheral tissues in vivo have been shown to depend on fatty acid synthesis14, so it is reasonable to conclude that the differentiation processes that give rise to DCs are dependent on the metabolic pathways that integrate mitochondrial function with the synthesis of fatty acids (FIG. 1).
Box 2. Glucose metabolism.
The glycolysis pathway allows the import of glucose and its conversion into pyruvate in the cytosol. There are offshoots of this pathway, including the pentose phosphate pathway (PPP), which allows the production of NADPH, a cofactor that is important for the synthesis of nucleotides and for fatty acid synthesis. Pyruvate has two possible main fates. The first is conversion into lactic acid and then conversion into lactate, which produces NAD+ that can then be reused for the production of ATP by glycolysis (see the figure; indicated by the dashed arrow). Alternatively, pyruvate enters mitochondria via mitochondrial pyruvate carrier 1 (MPC1), where it is converted into acetyl-CoA. The importance of this decision point in pyruvate metabolism pathways is indicated by the fact that it is regulated at many levels. Key enzymes in this regard are encoded by splice variants of transcripts of the Pkm gene (which encodes pyruvate kinase), which catalyse the production of pyruvate from its precursor phosphoenolpyruvic acid. Oxidation of pyruvate in the mitochondria is promoted by PKM1, but PKM2 promotes the expression of hypoxia-inducible factor 1α (HIF1α), which drives the conversion of pyruvate into lactate by inducing the expression of lactate dehydrogenase A (LDHA)30,111,112. Regulation of the balance of PKM1 and PKM2 activity is therefore an area of considerable interest (see REF. 113), but there are currently no studies on the biology of these enzymes in dendritic cells (DCs). In mitochondria, acetyl-CoA from pyruvate enters the tricarboxylic acid (TCA) cycle as shown in the figure. Reactions in the cycle lead to the production of NADH and FADH, which serve as substrates for the electron transport chain (ETC), and thereby support oxidative phosphorylation (OXPHOS) and the production of ATP. Fatty acids and glutamine can also fuel the TCA cycle as indicated. Citrate — which can be made from glucose, fatty acids or glutamine — can be exported from mitochondria to fuel the production of acetyl-CoA in the cytoplasm, which is a substrate for fatty acid synthesis, a process which is crucial for Toll-like receptor (TLR)-induced activation of DCs33, and an acetate donor for protein acetylation. αKGDH, α-ketoglutarate dehydrogenase; F6P, fructose 6−phosphate; G6P, glucose 6−phosphate; HK, hexokinase; IDH, isocitrate dehydrogenase; MDH, malate dehydrogenase; OAA, oxaloacetate; PDH, pyruvate dehydrogenase; ROS, reactive oxygen species.
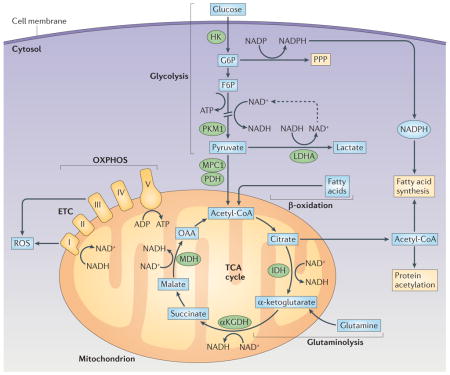
Figure 1. Changes in dendritic cell metabolism through development, quiescence and activation.
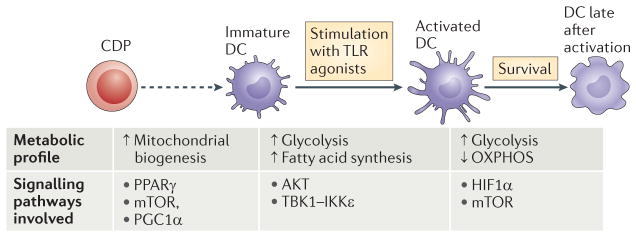
The development of dendritic cells (DCs) from progenitor cells is associated with mitochondrial biogenesis, which is driven by peroxisome proliferator-activated receptor-γ (PPARγ) co-activator 1α (PGC1α) and promoted by PPARγ, mammalian target of rapamycin (mTOR) and MYC. Differentiated DCs populate their niches as immature DCs. Immature DCs use fatty acid oxidation as a core metabolic process. Activation of DCs by Toll-like receptor (TLR) agonists leads to a rapid increase in flux through glycolysis and the associated pentose phosphate pathway, with an accompanying increase in spare respiratory capacity and fatty acid synthesis. These metabolic changes are initiated by a pathway downstream of TLRs that involves AKT, TANK-binding kinase 1 (TBK1), inhibitor of nuclear factor-κB kinase subunit-ε (IKKε) and hexokinase 2 (HK2), and they are crucial for DC activation. After being activated, DCs remain glycolytic. This process is essential for continued DC survival and is controlled by mTOR and hypoxia-inducible factor 1α (HIF1α). CDP, committed DC progenitor; OXPHOS, oxidative phosphorylation.
Regulation by mammalian target of rapamycin
Anabolic processes are centrally regulated in cells by the serine/threonine kinase mammalian target of rapamycin (mTOR), which comprises two complexes — mTORC1 and mTORC2 — that together have key roles in the regulation of cellular metabolism15. These complexes are downstream of signalling pathways that sense growth factors, nutrient levels and energy status, and they are generally involved in the control of anabolic metabolism. Consistent with coordinated roles for mTORC1 and fatty acid synthesis in DC differentiation, inhibition of mTORC1 with rapamycin prevents the FMS-like tyrosine kinase 3 (FLT3) ligand (FLT3L)-driven outgrowth of mouse pDCs and cDCs from bone marrow cultures in vitro and reduces DC numbers in vivo16,17. Additionally, the differentiation and survival of human monocyte-derived DCs is impaired by rapamycin18,19.
Consistent with a role for mTOR signalling in DC development, the FLT3L-driven development of pDCs and cDCs from bone marrow is enhanced in the absence of phosphatase and tensin homologue (PTEN), which is a negative regulator of mTOR, and populations of CD8+ cDCs and CD103+ cDCs are expanded in vivo in mice in which Pten has been conditionally deleted in DCs using the Cd11c–Cre system17. These effects can be inhibited by rapamycin17. Moreover, the Cd11c–Cre-mediated deletion of Raptor, which encodes a key component of mTORC1, alters steady-state DC populations, increasing the numbers of splenic cDCs20 but reducing the numbers of epidermal Langerhans cells21. Generally consistent with these findings, the tamoxifen-induced oestrogen receptor–Cre-mediated deletion of floxed Tsc1 (which encodes tuberous sclerosis 1, another negative regulator of mTORC1), results in enhanced outgrowth of DCs from GM-CSF-stimulated bone marrow cultures22. However, other studies have shown that releasing cell-intrinsic inhibition of mTOR signalling by deleting Tsc1 (again using the oestrogen receptor–Cre system) reduces the number of cDCs and pDCs in vivo, and prevents the outgrowth of cDCs and pDCs from FLT3L-stimulated bone marrow cultures23. This is associated with increased expression of MYC (a molecule that is discussed in more detail below) and downstream dysregulation of mitochondrial respiration, fatty acid synthesis and glycolysis, diminished life expectancy and a reduced ability to activate T cells.
The divergent findings on the effects of deleting two different negative regulators of mTORC1 (PTEN and TSC1) may reflect detailed differences in the mode of action of these two proteins23. Moreover, the different effects of Tsc1 deletion on DC development in GM-CSF bone marrow cultures compared with FLT3L-stimulated cultures could reflect the growth of distinct DC subsets in these two culture systems. Regardless, on balance, the data point to a crucial role for mTORC1 in the regulation of DC development and survival.
Regulation by MYC
mTORC1 induces the expression of MYC, which acts as a transcription factor to promote the expression of genes encoding proteins in the glycolysis pathway24. There are three MYC paralogues: MYC, MYCN and MYCL. During DC development, MYC expression is downregulated as MYCL expression is turned on in cDC progenitors25. CD103+ cDCs fail to develop normally in the absence of MYCL and, although CD8α+ cDCs do develop in MYCL-deficient mice, they are less capable of activating T cells during infections with Listeria monocytogenes or vesicular stomatitis virus, suggesting that they are unable to mature into efficient antigen-presenting cells25. Although the extent to which this reflects an important role of MYCL in DC metabolism is unclear at this point, it is intriguing that the ability of GM-CSF — which acts as a growth factor and signals through phosphoinositide 3-kinase (PI3K)–AKT and presumably mTORC1 — to promote CD8α+ cDC survival ex vivo is substantially diminished in the absence of Mycl25. Moreover, key metabolism genes, including complex I (NADH oxidase) of the ETC, are expressed to a lesser extent in CD8α+ cDCs in the absence of Mycl, indicating that this gene has a role in the regulation of metabolic processes; this finding may reflect a broader role of MYC in mitochondrial biogenesis26. MYC is also crucial in the regulation of glutaminolysis27, the process by which glutamine is converted to αKG for use in the TCA cycle (BOX 2). However, the role of glutamine in DC biology has yet to be thoroughly addressed.
Metabolism of resting DCs
In resting cells that face relatively few anabolic demands, the catabolism of complex molecules can provide substrates for the TCA cycle within mitochondria. For instance, the catabolism of proteins and triacylglycerols provides amino acids and fatty acids, respectively, and this fuels ATP production by oxidative phosphorylation (OXPHOS) (BOX 2; FIG. 2). Resting GM-CSF-induced bone marrow-derived DCs (BMDCs) — which differ from activated DCs as they are relatively sessile, less secretory, and less able to interact with and activate T cells — were shown to use fatty acid oxidation to fuel OXPHOS28. It is currently unclear whether resting cDCs or pDCs similarly fuel OXPHOS with fatty acid oxidation. Resting BMDCs also consume glucose, but whether this is primarily used to fuel OXPHOS or is used as a carbon source for other anabolic pathways is also unclear28.
Figure 2. Toll-like receptor signalling integrates endoplasmic reticulum stress and changes in metabolism to support activation.

In resting dendritic cells (DCs), fatty acid oxidation is engaged and the endoplasmic reticulum (ER) is relatively constrained. Following exposure to agonists of pattern recognition receptors (PRRs), signalling pathways are activated that lead to the expression of a broad array of nuclear factor-κB (NF-κB)- and interferon-regulatory factor (IRF)-responsive genes. This may lead to ER stress and the activation of the unfolded protein response (UPR) as the cells attempt to accommodate the production of a large set of proteins destined for secretion. A downstream effector of the UPR is X-box-binding protein 1 (XBP1), which transcriptionally activates the genes encoding enzymes for fatty acid synthesis; the UPR seems to be constitutively active in DCs. For Toll-like receptors (TLRs), and potentially other PRRs, this is coupled with activation of AKT, TANK-binding kinase 1 (TBK1), inhibitor of NF-κB kinase subunit-ε (IKKε) and hexokinase 2 (HK2), which promotes increased flux through the glycolysis pathway with resultant increases in citrate export for fatty acid synthesis. This is supported by the coincident increase in activity of the pentose phosphate pathway (PPP), which facilitates the production of NADPH, a crucial cofactor for fatty acid synthesis. Synthesis of new fatty acids allows expansion of the ER, which is likely to relieve ER stress and lead to the production and secretion of effector molecules that are central to DC activation. Thick arrow indicates that fatty acid oxidation is the primary metabolic signature of resting DCs. TCA, tricarboxylic acid.
It is feasible that DCs use glucose to synthesize fatty acids that they subsequently oxidize. Although this cycle may seem futile, it has recently been shown to be essential for the development of memory CD8+ T cells29. It has been argued that the process may be beneficial in quiescent T cells as it enables the simultaneous maintenance of mitochondrial health and the glycolysis and fatty acid synthesis machinery that allows the T cells to respond strongly following restimulation by antigen29. The same could be true for DCs, which, as is addressed in the next section, have to be ready to respond rapidly when their PRRs are engaged.
Metabolism of activated DCs
In order to be used as a substrate for ATP synthesis, glucose has to be processed by the cytosolic glycolysis pathway to generate pyruvate (BOX 2). Pyruvate can enter mitochondria via mitochondrial pyruvate carrier 1 (MPC1) and be converted into acetyl-CoA, which can enter the TCA cycle. However, pyruvate can have an alternative fate whereby it is converted to lactic acid, which is secreted into the extracellular environment as lactate. The conversion of pyruvate to lactate allows the coupled regeneration of NAD+ and the production of ATP via the glycolysis pathway (BOX 2). This allows cells to produce ATP when oxygen is limiting and places glucose, rather than fatty acids or amino acids, in the unique position of being essential for cells to survive under hypoxic conditions.
In some cells — notably, tumour cells — this pathway is used even when oxygen is not limiting, a process known as aerobic glycolysis or, famously, Warburg metabolism30. The TLR-driven activation of macrophages leads to the adoption of Warburg metabolism6. Similarly, BMDCs show an increase in their consumption of glucose and production of lactate following stimulation with TLR agonists28,31. It is now clear that these observations reflect two metabolic changes linked to activation: an early phase of increased flux through the glycolysis pathway that is common to monocyte-derived DCs and cDCs, followed by a fundamental change in cellular metabolism, whereby BMDCs commit to Warburg metabolism (FIG. 1). A detailed understanding of these issues was made possible, in part, by the development of techniques for measuring real-time changes in extracellular acidification and oxygen consumption rates32. When this approach was used to examine DC metabolism, it was found that increased glycolysis occurs in DCs within minutes of activation by a broad array of TLR agonists33. This rapid increase in glycolysis also occurs in activated CD8α+ and CD11b+ cDCs, suggesting that it is a fundamental feature of DC activation33. Blocking glycolysis using 2-deoxyglucose (an inhibitor of hexokinase (HK), the first enzyme in the glycolysis pathway; BOX 2) results in marked inhibition of DC activation, showing the importance of glucose as a substrate to support the response of DCs to TLR agonists28,33.
Function of glycolysis in DC activation
The early increase in glycolysis that occurs in activated BMDCs is associated with an increase in lactate production, but does not reflect the mere engagement of Warburg metabolism, as OXPHOS is sufficient to supply all of the ATP needed by the cells33, and the transport of pyruvate (the end-product of glycolysis) into mitochondria via MPC1 is crucial for early DC activation33. Thus, a major function of increased glycolysis seems to be to increase the production of pyruvate to fuel the TCA cycle. This facilitates a transient increase in spare respiratory capacity34,35, an indication that the cells are more metabolically capable than resting BMDCs, which is consistent with the fact that there are more demands placed on them for cell–cell interactions, mediator secretion and migration.
It seems likely that increased spare respiratory capacity indicates that cells have the ability to use TCA cycle intermediates for purposes other than continuing to run the cycle. Consistent with this, a major consequence of the entry of glucose-derived carbons into the TCA cycle is the increased production of fatty acids downstream of citrate export into the cytosol33 (BOX 2). As discussed above, fatty acid synthesis is implicated in the differentiation of DCs from progenitor cells, and it is intriguing that it becomes important again during the transition of DCs from quiescence to activation following stimulation with TLR agonists (FIG. 1). Although it remains unclear why fatty acid synthesis is required during DC differentiation, the role of this pathway in activated DCs seems to be linked to the necessity to increase the mass of both endoplasmic reticulum (ER) and Golgi to support the increased demand for protein synthesis33 (FIG. 2). This is consistent with the fact that the major effects of pharmacologically inhibiting glycolysis or fatty acid synthesis during the initial stages of activation of BMDCs are post-transcriptional, indicating that both pathways are mainly required not for gene expression downstream of TLRs, but rather for the synthesis of proteins from transcripts made in response to activation33.
The promotion of fatty acid synthesis in response to TLR stimulation also leads to increased lipid storage in lipid droplets33,36. In other cells, lipid stores of this type can be accessed by regulated pathways of lipolysis to provide free fatty acids for both catabolic (for example, fatty acid oxidation) or anabolic (for example, membrane synthesis) purposes37. Free fatty acids also serve as second messengers for signal transduction pathways initiated by nuclear hormone receptors38 (see below). In the liver, lipid content in DCs is positively correlated with immunogenicity and is dependent on de novo fatty acid synthesis39, whereas in the context of tumours, high lipid content in DCs is associated with impaired immune priming40,41. If and how neutral lipid stores are contributing to DC biology in either of these cases remains to be determined.
A link between ER stress and metabolic changes in activated DCs
As highly secretory cells, such as activated DCs, attempt to coordinate increases in gene expression with protein output they can experience ER stress, which is marked by the build-up of unfolded proteins in the ER lumen42. Unchecked, this can lead to cell death; however, the unfolded protein response (UPR) has evolved as a means for cells to mitigate this outcome42. The UPR enables the coordinated expansion of the ER through increased synthesis of fatty acids for ER membranes and of proteins that constitute the folding machinery. As one of the three sensors of ER stress, inositol-requiring protein 1α (IRE1α) is particularly important in DC biology. It targets mRNAs encoding X-box-binding protein 1 (XBP1), generating a cleaved product that is a transcription factor that controls ER biogenesis43, thereby alleviating ER stress and promoting cellular survival42 (FIG. 2). Importantly, IRE1α and its target XBP1 have been found to be constitutively active in resting cDCs44. Moreover, XBP1 expression in early progenitor cells is essential for cDC and pDC development45, and its deletion at later times in DC development results in defects in CD8α+ cDCs (but not CD11b+ cDCs), including a disorganized ER, diminished expression of CD8α and CD11c, and the inability to cross-present dead-cell-associated antigens on MHC class I molecules to CD8+ T cells44.
Taken together, these findings indicate that the coordination of ER events has a particularly crucial role in DC biology. This is of note, given the recent interest in the link between innate immunity and the ER stress response46,47. For example, stimuli that promote ER stress increase TLR-driven macrophage activation, and both TLR2 and TLR4 activate IRE1α and XBP1, with XBP1 being essential for maximal cytokine production in response to TLR agonists in these cells46.
Changes in metabolism at times late after activation
BMDCs that have been activated by TLR agonists for more than 12 hours exclusively use Warburg metabolism to meet their bioenergetic needs. In these cells, there is very little (if any) measureable mitochondrial oxygen consumption, and OXPHOS is essentially switched off28,48 (FIG. 1). It is now clear that commitment of BMDCs to Warburg metabolism following activation is a direct response to the cellular expression of inducible nitric oxide synthase (iNOS), and the inhibition of the ETC by nitric oxide (NO). In the absence of functional OXPHOS, due to inhibition of the ETC by NO, BMDCs activated by TLR agonists are dependent on glycolysis for the synthesis of ATP for survival48. The ability of NO, made during inflammation, to inhibit OXPHOS is of considerable interest and shows how this effector gas is likely to influence the metabolism of pathogenic microorganisms that are themselves dependent on OXPHOS.
It is also clear that the metabolic status of bystander cells that are incapable of making NO themselves can be affected by exogenous NO49. Therefore, NO-producing inflammatory DCs and macrophages may exert metabolic control over cells with which they are interacting. This could be particularly pertinent to T cell activation by NO-producing DCs, as T cells translocate their mitochondria towards the immunological synapse50, making them particularly vulnerable to NO-mediated intoxication of their mitochondria51. Interestingly, neither mouse cDCs nor human DCs in general express iNOS; consistent with this, these cells do not commit to Warburg metabolism following activation in vitro48. Nevertheless, cDCs activated by TLR agonists in vivo do exhibit diminished mitochondrial activity and enhanced glycolysis over the long term, which they rely on for survival, similar to iNOS-expressing DCs. However, these changes are reported to be iNOS independent and driven by autocrine type I interferon (IFN) signalling through hypoxia-inducible factor 1α (HIF1α)52.
Metabolic regulators of DC activation
As discussed above, mTOR and its upstream activators PI3K and AKT are central regulators of cellular activation and proliferation due to their ability to control glycolysis and anabolic metabolism. Consistent with a role for mTOR in regulating DC activation, in the absence of TSC1, cDCs display an increased expression of maturation markers, such as CD40, CD80 and CD86, at steady state23. Furthermore, in mouse and human pDCs, there is a clear dependence on mTOR for IFNα production, and in mouse BMDCs and/or human monocyte-derived DCs, rapamycin selectively inhibits certain aspects of TLR-driven DC activation, including the expression of IL-6 and IL-10, and possibly tumour necrosis factor (TNF) 19,53–56.
Several studies have documented a positive role for the mTOR target HIF1α in the TLR-driven activation of BMDCs and cDCs31,52,57. HIF1 comprises α- and β-subunits, and is classically considered to be activated by hypoxia. It has been implicated in promoting glycolysis in several systems by inducing the expression of many of the enzymes in the glycolysis pathway30 (BOX 2). However, recent evidence suggests that the early TLR-driven induction of glycolysis that occurs during DC activation does not require mTOR- or HIF1α-mediated signalling33,58, but instead depends on AKT35,59. Interestingly, activation of AKT downstream of TLRs is dependent on TANK-binding kinase 1 (TBK1) and inhibitor of nuclear factor-κB kinase subunit-ε(IKKε) rather than PI3K33, which is more usually implicated in this process60. AKT is crucial because it directly phosphorylates and thereby activates HK2 to associate with the mitochondrial surface, where it is thought to be able to more effectively catalyse one of the rate-limiting steps of glycolysis, the phosphorylation of glucose to glucose 6-phosphate61,59 (BOX 2). Targeted inhibition of TBK1, IKKε or AKT, or blocking mitochondrial association of HK2 with mitochondria, markedly diminishes the ability of BMDCs to respond to TLR agonists and become activated33 (FIG. 2).
Notably, as TBK1 and IKKε are also activated downstream of RLR signalling, it is possible that rapid induction of glycolysis is a common response to any innate sensing of pathogens by DCs, enabling a rapid metabolic response to these danger signals (FIG. 3). This suggests that the mTOR–HIF1α signalling axis modulates DC activation independently from regulation of glycolytic metabolism62,63. However, this signalling axis is important for the long-term commitment of DCs to glycolysis following activation by TLR agonists28,32,54,55 (FIG. 1). Interestingly, rapamycin-treated BMDCs survive longer following TLR activation than their untreated counterparts. Furthermore, they exhibit prolonged and increased expression of co-stimulatory molecules and are more effective at inducing T cell responses that promote antitumour immune responses54. These findings seem at odds with the well-known reduced immunogenicity of rapamycin-conditioned human monocyte-derived DCs19. This discrepancy might be explained by the fact that in mouse BMDCs, mTOR inhibition reduces iNOS expression, with the subsequent maintenance of mitochondrial function and prolonged lifespan of these cells54,64. By contrast, in human DCs, the immunosuppressive effects of rapamycin may dominate in the absence of iNOS expression.
Figure 3. Mitochondria are foci for the integration of metabolism and innate responses.

The mitochondria-associated membranes (MAMs) are areas of close interaction between mitochondria and the endoplasmic reticulum (ER). The MAMs are the sites at which mitochondrial antiviral signalling protein (MAVS) and the NOD-, LRR- and pyrin domain-containing 3 (NLRP3) inflammasome localize and they serve as a Ca2+ store to maintain mitochondrial calcium concentrations. MAVS interacts with retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs) that have sensed viral RNA. The MAVS–RLR complex is then able to initiate signalling to induce expression of cytokines and theoretically promote increased glycolytic flux to support dendritic cell (DC) activation and type I interferon (IFN) responses (indicated by the dashed arrows). MAVS–RLR signalling is dependent on active mitochondria with a high mitochondrial membrane potential (Δψm) and reciprocally serves to promote mitochondrial expansion. NLRP3 is sensitive to reactive oxygen species (ROS) that are produced by the electron transport chain (ETC) and also senses ATP-driven decreases in intracellular potassium concentrations. Once activated, NLRP3 activates caspase 1, which is able to cleave and thereby activate the pro-forms of interleukin-1β (IL-1β) and IL-18. Increased ROS production can be promoted by Toll-like receptor 1 (TLR1), TLR2 and TLR4 signalling through the activation of tumour necrosis factor receptor-associated factor 6 (TRAF6), which relocates to the mitochondria. There, in conjunction with evolutionarily conserved signalling intermediate in Toll pathway (ECSIT), TRAF6 promotes ROS production by the ETC. IKKε, inhibitor of nuclear factor-κB kinase subunit-ε; IRF, IFN-regulatory factor; NF-κB, nuclear factor-κB; P2X7, P2X purinoceptor 7; TBK1, TANK-binding kinase 1.
Recent reports have revealed a crucial role for the histidine solute carrier SLC15A4 in the activation of pDCs by agonists of TLR7 and TLR9, which are TLRs that are located in endosomes65. Based on recent work in B cells, SLC15A4 is necessary for endolysosomal acidification, and in the absence of this, TLR-initiated, mTOR-dependent, IFN-regulatory factor 7 (IRF7)-mediated type I IFN production is inhibited66. Moreover, autocrine signalling by type I IFNs through the IFNα/β receptor — which leads to further increased type I IFN production and the activation of additional genes, and is mTOR dependent — is also inhibited in Slc15a4-deficient cells66. These data are consistent with the fact that mTORC1 localizes to lysosomes where it acts as sensor of lysosomal amino acid concentrations, which are an indicator of metabolic status67. The data from the Slc15a4 studies imply that mTORC responsiveness to lysosomal amino acid levels, or perhaps acidity, is a crucial component in cellular responsiveness to agonists of endolysosomal TLRs.
A key signalling pathway that opposes mTOR-controlled anabolic metabolism is regulated by the metabolic sensor AMP kinase (AMPK). AMPK is known to antagonize biosynthetic pathways, such as fatty acid synthesis, and has instead been shown to promote catabolic processes via several pathways, including the activation of PGC1α, which promotes mitochondrial biogenesis to increase OXPHOS6,68. In DCs, knockdown of AMPK potentiates TLR-induced DC activation, whereas pharmacological activation of AMPK suppresses TLR-induced glucose consumption and concomitant activation of DCs28,69. This indicates that AMPK signalling has a prominent role in the metabolic control of DC activation. Consistent with a role for PGC1α in regulating DC activation, treatment with resveratrol — a drug that, in addition to its many other effects, is thought to favour catabolic metabolism through activation of the histone deacetylase (HDAC) sirtuin 1, leading to suppressed HIF1α expression but enhanced PGC1α expression70–72 — reduced TLR-driven DC activation and rendered these cells more tolerogenic. Conversely, DCs deficient for nuclear factor erythroid 2-related factor 2 (NRF2) or PPARγ, which are downstream targets of PGC1α, display increased maturation and T cell-priming capacity73–75. Hence, these studies suggest an important role for the AMPK–PGC1α axis in antagonizing metabolic pathways that promote activation and immunogenicity of DCs, and may point to the intriguing possibility that the immunogenicity or tolerogenicity of DCs is determined by the balance between anabolic versus catabolic metabolic pathways in these cells.
Signalling by intracellular metabolites in DCs
Recent findings have led to the realization that cellular metabolism is not only a source of building blocks and ATP to support proper DC differentiation and function, but also that products of metabolic pathways act as signalling molecules to trigger cellular responses (TABLE 1). One example of this is provided by the important recent finding that in M1 macrophages, HIF1α is activated by succinate — an intermediate in the TCA cycle that is produced from glutamate in cells that are committed to Warburg metabolism (BOX 2) — and has a crucial role in the production of IL-1β63. Interestingly, extracellular succinate is also able to promote DC activation by binding to the G protein-coupled receptor (GPCR) succinate receptor 1 (REF. 76). Another example concerns reactive oxygen species (ROS), which are a normal byproduct of the function of the ETC. Cellular concentrations of ROS are increased when the ETC is inhibited — for example, with rotenone or antimycin77 — and this activates the NOD-, LRR- and pyrin domain-containing 3 (NLRP3) inflammasome77, leading to the activation of caspase 1 and the production of IL-1β and IL-18 (REF. 9) (FIG. 3). This pathway can be induced by agonists of TLR1, TLR2 or TLR4, which promote the activation of TNF receptor-associated factor 6 (TRAF6) and its relocation to mitochondria, where it interacts with evolutionarily conserved signalling intermediate in Toll pathway (ECSIT) leading to increased ROS production78 (FIG. 3). In DCs, inflammasome activation is important for cellular activation and for the ability of the cells to induce adaptive antitumour immunity in certain settings79. A third example is provided by the NAD+/NADH ratio, which has a crucial role in the regulation of protein acetylation and has far-reaching consequences for cellular activation and metabolism (BOX 3; TABLE 1).
Table 1.
A selection of key metabolites to which dendritic cells can respond*
| Source | Metabolite | Receptor or sensor | Molecular effect | DC response |
|---|---|---|---|---|
| Exogenous | Lactate | GPR81? | HIF1α stabilization |
|
| ATP |
|
ND |
|
|
| Butyrate |
|
Deacetylase inhibition |
|
|
| Propionate |
|
Deacetylase inhibition | Reduced rate of DC differentiation | |
| Endogenous | Succinate | Prolyl hydroxylases |
|
Induction of IL-1β expression |
| Reactive oxygen species | NLRP3 | Caspase 1 activation | Production of active IL-1β and IL-18 | |
| NAD+ | Sirtuins | PGC1α deacetylation | Altered mitochondrial metabolism | |
| Endogenous and exogenous | All trans retinoic acid | RAR | Suppression of pro-inflammatory genes | Induction of tolerogenic DCs |
| Oxysterols | LXR | Expression of pro-inflammatory genes | Enhanced DC immunogenicity | |
| Polyunsaturated or oxidized fatty acids |
|
Inhibition of NF-κB signalling |
|
|
| Vitamin D | VDR | Suppression of pro-inflammatory genes | Induction of tolerogenic DCs |
Note that this list is not exhaustive. A2A, adenosine receptor A2A; DC, dendritic cell; GPR, G protein-coupled receptor; HIF1α, hypoxia-inducible factor 1α; IL, interleukin; LXR, liver X receptor; ND, not defined; NF-κB, nuclear factor-κB; NLRP3, NOD-, LRR- and pyrin domain-containing 3; PGC1α, PPARγ co-activator 1α; PPAR, peroxisome proliferator-activated receptor; P2XR, P2X purinoceptor; P2YR, P2Y purinergic receptor; RAR, retinoic acid receptor; SLC, solute carrier; VDR, vitamin D receptor.
Box 3. Acetylation, methylation and glycosylation in dendritic cell biology.
Post-translational modification of proteins by acetylation, methylation and/or glycosylation has a crucial role in modulating protein function108. Acetyl-CoA converted from citrate is a major acetyl donor for the acetylation pathway. The importance of histone acetylation in the epigenetic regulation of gene expression is now well recognized, but many other proteins — including the majority of enzymes in glycolysis and fatty acid metabolism — are also acetylated, and acetylation of key participants, such as pyruvate kinase M2 (PKM2) and peroxisome proliferator-activated receptor-γ (PPARγ) co-activator 1α (PGC1α) modulates function in these pathways114,115. Among deacetylases, certain sirtuins are regulated by NAD+/NADH ratios, such that they become activated when NAD+ levels are high. In muscle cells, activation of the AMP/ATP sensor AMP kinase (AMPK) results in increased NAD+/NADH ratios and sirtuin 1 (SIRT1) activation116, resulting in deacetylation of PGC1α and promotion of metabolic pathways that restore NAD+/NADH ratios. In dendritic cells (DCs), SIRT1 mediates the deacetylation of interferon (IFN)-regulatory factor 1 (IRF1) and, in doing so, inhibits the ability of cells to produce interleukin-27 (IL-27) and IFNβ117. SIRT1 has also been reported to regulate the balance of IL-12p70 versus IL-23 production118 and to repress PPARγ activity in DCs119. In other cell types, SIRT6 has been shown to repress the ability of hypoxia-inducible factor 1α (HIF1α) to promote glycolysis and of MYC to induce glutaminolysis, by acting to deacetylate histone H3 (REFS 120,121). Reversible methylation of histones H3 and H4 at different sites can also repress or promote transcription, and inducible inflammatory genes can be regulated in this fashion in DCs122. Histone methyltransferases catalyse the transfer of methyl groups to key lysines or arginines in histones, using S-adenosyl methionine, an intermediate of sulphur metabolism, as the methyl donor123; S-adenosyl methionine is made from methionine plus ATP. Glycosylation events, which have crucial roles in the folding and secretion of proteins destined for export, and in regulating intracellular protein function, are dependent on the sugar donor uridine diphosphate N-acetylglucosamine (UDP-GlcNAc), which is the major product of the hexosamine biosynthetic pathway106, a metabolic pathway that has received very little attention in DCs. Indeed, relatively little is known of the role of regulated acetylation, methylation or glycosylation in the biology of DCs and these areas are ripe for investigation.
Signalling by exogenous metabolites in DCs
Lipid sensing by GPCRs
We have discussed the ability of PRRs to promote changes in metabolism that support parallel changes in gene expression during activation. DCs are also able to respond directly to exogenous metabolites. For example, the short-chain fatty acid butyrate — which is one of three short-chain fatty acids (the other two being propionate and acetate) that are made by commensal bacteria as a byproduct of the fermentation of dietary fibre — has a range of effects on DC biology. Through the GPCR GPR109A; (also known as HCAR2), butyrate promotes the expression of IL-10 and the enzyme aldehyde dehydrogenase, which catalyses the conversion of retinal to retinoic acid80. Retinoic acid and IL-10 mediate many of the anti-inflammatory and tolerogenic effects exerted by DCs in the context of the normal interaction of the immune system with the intestinal microbiota (TABLE 1). Of considerable interest, GPR109A is also a receptor for niacin (also known as nicotinic acid and vitamin B3), which has effects on DCs that phenocopy those of butyrate80.
In addition to its effects through GPCRs, butyrate that enters cells through the Na+-coupled monocarboxylate transporter SLC5A8 can act as a potent HDAC inhibitor, and consequently regulate gene expression through this pathway. In this way, butyrate prevents the GM-CSF-driven development of DCs in vitro81,82. Of acetate and propionate, which are also taken into DCs by SLC5A8, only propionate is a HDAC inhibitor, and it inhibits DC development in a similar fashion to butyrate81. In BMDCs, butyrate simultaneously promotes IL-23 expression while suppressing IL-12 production in response to TLR agonists82.
As alluded to below, oxysterols can act as ligands for nuclear hormone receptors. However, the oxysterol 7α,25-dihydroxycholesterol (7α,25-OHC), which is made by stromal cells within lymphoid organs, is a ligand for the GPCR GPR183 (also known as EBI2) and, in addition to having important roles in regulating B cell movement, the 7α,25-OHC–EBI2 axis was recently shown to be essential for the differentiation and appropriate localization of CD4+ cDCs in the spleen83.
Lipid sensing by nuclear hormone receptors
DCs can sense lipids and lipid-soluble ligands through nuclear hormone receptors, which act as transcription factors when activated by ligand binding. There has been considerable interest in the role of a subset of these receptors that form heterodimers with the retinoid X receptor (RXR) on DC biology. RXR can form heterodimers with retinoic acid receptors (RARs), liver X receptors (LXRs), PPARs and vitamin D receptors (VDRs), and mediate recognition and responsiveness to a variety of external ligands including retinoic acid (RXR–RAR), oxysterols (RXR–LXR), polyunsaturated and oxidized fatty acids (RXR–PPAR), and 1α,25-dihydroxyvitamin.D3 (for a recent, exhaustive review on this subject, see REF. 38). In brief, ligation of these receptors in DCs is generally associated with reduced responsiveness to TLR agonists and attendant anti-inflammatory effects. Moreover, it is important to realize that, as they become activated, DCs can acquire the ability to make ligands for many of these receptors. For example, TLR2 signalling induces the expression of the enzymes that enable DCs to produce retinoic acid from vitamin A84, and this can have a marked effect on whether regulatory or effector T cell responses are induced, especially in the intestine85–88.
Adenosine and ATP sensing
DCs are also able to respond to extracellular adenosine and ATP. Adenosine is recognized by the A2A GPCR on DCs and it conditions the cells to become anti-inflammatory, limiting DC activation89,90. ATP can also be sensed by P2YRs, which are GPCRs that promote DC migration91, or through ligand-gated ion channels (P2XRs) that provide an activating signal92 and stimulate the caspase 1-dependent cleavage and secretion of IL-1β93 (FIG. 3).
Lactic acid
Recent reports have indicated that lactic acid may also directly affect DC biology. Lactate promotes alternative activation of macrophages, reflected by the expression of genes typically associated with IL-4 stimulation, such as Arg1 (which encodes arginase), Fizz1 (also known as Retnla), Mgl1 (which encodes macrophage galactose-type lectin; also known as CLEC10A) and Mgl2. This effect was found to be largely dependent on HIF1α, which is stabilized by lactate under normoxic conditions. This is interesting because it is likely that DCs will be exposed to increased levels of lactate as they become activated and as they infiltrate tumours or sites of infection. In this context, tumour cell-derived lactic acid was shown to inhibit IL-12 production and antigen presentation by DCs in co-culture assays94. Moreover, high lactate concentrations affect the differentiation of DCs from monocytes, favouring the emergence of less inflammatory DCs that are biased towards producing IL-10 (REF. 95).
At this time, the molecular mechanisms underlying the effects of lactate on DC biology are unknown, but it is interesting to speculate that the lactate receptor GPR81 (also known as HCAR1) may be involved. This receptor, which binds lactate and inhibits lipolysis in adipocytes96, is reported to suppress pro-inflammatory TLR- and inflammasome-mediated responses in macrophages97.
Mitochondrial metabolism and danger sensing
In addition to the TLRs, which sense exogenous signals either outside the cell or within endosomal or lysosomal compartments, cells express cytoplasmic RNA sensors known as the RLRs98, which promote antiviral cDC responses99. RLR agonists induce the interaction of these receptors with mitochondrial antiviral signalling protein (MAVS) — a protein that localizes to the mitochondria100 and to mitochondria-associated membranes (MAMs)101 (FIG. 3) — and this initiates signalling for induction of expression of antiviral genes including those encoding type I IFNs. MAMs are junctional regions between mitochondria and the ER, and are structures of increasing interest in terms of the link between cellular metabolism and innate immune functions102. They are primarily sites for the regulated transfer of Ca2+ and lipids between the ER and mitochondria103. In mitochondria, Ca2+ has a crucial role in the activation of pyruvate dehydrogenase, αKG dehydrogenase and isocitrate dehydrogenase. As such, the regulated supply of Ca2+ to mitochondria is essential for maintaining TCA cycle activity103. Moreover, uptake of substrates such as glutamate into the mitochondrial matrix is Ca2+ dependent104. Enzymes that are important in lipid and glucose metabolism also localize to MAMs, as do ER stress sensors103.
Available data suggest a close functional link between MAMs, mitochondrial activity and antiviral responses78,102,105. For MAVS to function in RLR signalling, it has to be localized to mitochondria100. Moreover, the function of MAVS is enhanced by interactions with mitofusin 1 (MFN1), a protein that is involved in mitochondrial elongation and fusion events106. In addition, consistent with a role for RLR-mediated signalling in modulating mitochondrial function, mitochondria have been found to elongate when RLRs are activated106. Interestingly, it is clear that RLR signalling is dependent on mitochondrial membrane potential107 and that other mitochondrial proteins — such as MFN2, NLR family member X1 and receptor for globular head domain of complement component 1q — negatively regulate MAVS78, suggesting that the reverse may also be true in that mitochondrial metabolism regulates innate sensing by RLRs.
Conclusion
There is a growing appreciation for the role of metabolic changes in the phenotype and function of DCs. Moreover, evidence is accumulating that many fundamental cellular processes in DCs — such as transcription factor activation, gene and protein expression, organelle homeostasis, danger sensing and stress responses — are regulated by metabolic processes or the metabolites that they generate. However, we are only beginning to understand the extent to which metabolism is interlinked with these processes and how this affects the functional properties of DCs. For instance, most studies to date have used well-controlled in vitro experiments with ample nutrient availability and perturbations in only a single gene or metabolite. However, the extent to which this models the metabolic complexity of the microenvironment in which DCs reside in situ — such as in tumours or sites of inflammation where nutrient and/or oxygen availability may be limiting — is still poorly understood. Furthermore, little is known about whether different DC subsets (for example, cDCs and pDCs) have distinct metabolic requirements or whether they rely on common metabolic programmes for their function. Finally, the regulation of generation of metabolites for the post-transcriptional modification of proteins by acetylation, methylation or succinylation, and the effects of these changes on signalling pathways and/or gene expression108,109, remain largely unstudied in DCs and are ripe for investigation (BOX 3). Clearly, it will be important to more fully characterize how metabolism controls the immune priming function of DCs and whether metabolic manipulation of DCs can be used to alter their immune-polarizing properties. This will not only improve our fundamental understanding of the biology of DCs, but will also be key to the development of metabolism-based approaches to improve the efficacy of DC-based immunotherapies.
Acknowledgments
The authors have benefitted greatly from discussions about immune cell metabolism with E. Pearce, S. Huang, E. Amiel, R. Van der Windt, D. O’Sullivan, C.-H. Chang, C. Krawczyk, R. Jones, A. Jha, E. Driggers and M. Artymov. Their research is supported by the US National Institutes of Health (E.J.P.) and the Netherlands Organization for Scientific Research (B.E.).
Glossary
- Electron transport chain (ETC)
The electron transport chain links nutrient oxidation to ATP production by oxidative phosphorylation. It consists of complexes I–V in the mitochondrial inner membrane
- Tricarboxylic acid cycle (TCA cycle; also known as the Krebs cycle or citric acid cycle)
This pathway catalyses the oxidation of acetyl-CoA (from glucose or fatty acids, or indirectly from amino acids) to generate NADH and FADH, which fuel the electron transport chain and thereby oxidative phosphorylation and ATP production. It also serves as a source of precursors for amino acid and lipid synthesis
- Bone marrow-derived DCs (BMDCs)
These monocyte-derived dendritic cells (DCs) are generated from bone marrow cultures supplemented with granulocyte–macrophage colony-stimulating factor and are used to model the behaviour of DCs that develop from monocytes under inflammatory conditions in vivo
- Spare respiratory capacity
The amount of energy generating capacity, beyond that needed for basal biology, that a cell maintains in reserve to be used when called upon
- M1 macrophages
Macrophages that have been activated by a Toll-like receptor agonist, usually in combination with interferon-γ
Footnotes
Competing interests statement
The authors declare no competing interests.
References
- 1.Satpathy AT, Wu X, Albring JC, Murphy KM. Re(de)fining the dendritic cell lineage. Nature Immunol. 2012;13:1145–1154. doi: 10.1038/ni.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hemmi H, Akira S. TLR signalling and the function of dendritic cells. Chem Immunol Allergy. 2005;86:120–135. doi: 10.1159/000086657. [DOI] [PubMed] [Google Scholar]
- 3.Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–650. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 4.Osorio F, Reis e Sousa C. Myeloid C-type lectin receptors in pathogen recognition and host defense. Immunity. 2011;34:651–664. doi: 10.1016/j.immuni.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 5.Pearce EL, Poffenberger MC, Chang CH, Jones RG. Fueling immunity: insights into metabolism and lymphocyte function. Science. 2013;342:1242454. doi: 10.1126/science.1242454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O’Neill LA, Hardie DG. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature. 2013;493:346–355. doi: 10.1038/nature11862. [DOI] [PubMed] [Google Scholar]
- 7.Pollizzi KN, Powell JD. Integrating canonical and metabolic signalling programmes in the regulation of T cell responses. Nature Rev Immunol. 2014;14:435–446. doi: 10.1038/nri3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Everts B, Pearce EJ. Metabolic control of dendritic cell activation and function: recent advances and clinical implications. Frontiers Immunol. 2014;5:203. doi: 10.3389/fimmu.2014.00203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McGettrick AF, O’Neill LA. How metabolism generates signals during innate immunity and inflammation. J Biol Chem. 2013;288:22893–22898. doi: 10.1074/jbc.R113.486464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Le Naour F, et al. Profiling changes in gene expression during differentiation and maturation of monocyte-derived dendritic cells using both oligonucleotide microarrays and proteomics. J Biol Chem. 2001;276:17920–17931. doi: 10.1074/jbc.M100156200. [DOI] [PubMed] [Google Scholar]
- 11.Ishikawa F, et al. The developmental program of human dendritic cells is operated independently of conventional myeloid and lymphoid pathways. Blood. 2007;110:3591–3660. doi: 10.1182/blood-2007-02-071613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zaccagnino P, et al. An active mitochondrial biogenesis occurs during dendritic cell differentiation. Int J Biochem Cell Biol. 2012;44:1962–1969. doi: 10.1016/j.biocel.2012.07.024. [DOI] [PubMed] [Google Scholar]
- 13.Del Prete A, et al. Role of mitochondria and reactive oxygen species in dendritic cell differentiation and functions. Free Radic Biol Med. 2008;44:1443–1451. doi: 10.1016/j.freeradbiomed.2007.12.037. This is an excellent review of some less well-appreciated aspects of mitochondrial biology that relate to DC function. [DOI] [PubMed] [Google Scholar]
- 14.Rehman A, et al. Role of fatty-acid synthesis in dendritic cell generation and function. J Immunol. 2013;190:4640–4649. doi: 10.4049/jimmunol.1202312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hackstein H, et al. Rapamycin inhibits IL-4-induced dendritic cell maturation in vitro and dendritic cell mobilization and function in vivo. Blood. 2003;101:4457–4463. doi: 10.1182/blood-2002-11-3370. [DOI] [PubMed] [Google Scholar]
- 17.Sathaliyawala T, et al. Mammalian target of rapamycin controls dendritic cell development downstream of Flt3 ligand signaling. Immunity. 2010;33:597–606. doi: 10.1016/j.immuni.2010.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Woltman AM, et al. Rapamycin specifically interferes with GM-CSF signaling in human dendritic cells, leading to apoptosis via increased p27KIP1 expression. Blood. 2003;101:1439–1445. doi: 10.1182/blood-2002-06-1688. [DOI] [PubMed] [Google Scholar]
- 19.Haidinger M, et al. A versatile role of mammalian target of rapamycin in human dendritic cell function and differentiation. J Immunol. 2010;185:3919–3931. doi: 10.4049/jimmunol.1000296. [DOI] [PubMed] [Google Scholar]
- 20.Ohtani M, et al. Cutting edge: mTORC1 in intestinal CD11c+ CD11b+ dendritic cells regulates intestinal homeostasis by promoting IL-10 production. J Immunol. 2012;188:4736–4740. doi: 10.4049/jimmunol.1200069. [DOI] [PubMed] [Google Scholar]
- 21.Kellersch B, Brocker T. Langerhans cell homeostasis in mice is dependent on mTORC1 but not mTORC2 function. Blood. 2013;121:298–307. doi: 10.1182/blood-2012-06-439786. [DOI] [PubMed] [Google Scholar]
- 22.Pan H, et al. Critical role of the tumor suppressor tuberous sclerosis complex 1 in dendritic cell activation of CD4 T cells by promoting MHC class II expression via IRF4 and CIITA. J Immunol. 2013;191:699–707. doi: 10.4049/jimmunol.1201443. References 17 and 22 show how the deletion of TSC1 results in metabolic dysregulation with marked effects on DC biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Y, et al. Tuberous sclerosis 1 (Tsc1)-dependent metabolic checkpoint controls development of dendritic cells. Proc Natl Acad Sci USA. 2013;110:E4894–E4903. doi: 10.1073/pnas.1308905110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang R, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. 2011;35:871–882. doi: 10.1016/j.immuni.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.KCW, et al. L-Myc expression by dendritic cells is required for optimal T-cell priming. Nature. 2014;507:243–247. doi: 10.1038/nature12967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li F, et al. Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Mol Cell Biol. 2005;25:6225–6234. doi: 10.1128/MCB.25.14.6225-6234.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wahlstrom T, Henriksson MA. Impact of MYC in regulation of tumor cell metabolism. Biochim Biophys Acta. 2014 doi: 10.1016/j.bbagrm.2014.07.004. http://dx.doi.org/10.1016/j.bbagrm.2014.07.004. [DOI] [PubMed]
- 28.Krawczyk CM, et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood. 2010;115:4742–4749. doi: 10.1182/blood-2009-10-249540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.O’Sullivan D, et al. Memory CD8+ T cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity. 2014;44:75–88. doi: 10.1016/j.immuni.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Palsson-McDermott EM, O’Neill LA. The Warburg effect then and now: from cancer to inflammatory diseases. BioEssays. 2013;35:965–973. doi: 10.1002/bies.201300084. [DOI] [PubMed] [Google Scholar]
- 31.Jantsch J, et al. Hypoxia and hypoxia-inducible factor-1α modulate lipopolysaccharide-induced dendritic cell activation and function. J Immunol. 2008;180:4697–4705. doi: 10.4049/jimmunol.180.7.4697. References 28 and 31 demonstrate the importance of glycolysis for supporting DC activation in response to TLR agonists. [DOI] [PubMed] [Google Scholar]
- 32.Gerencser AA, et al. Quantitative microplate-based respirometry with correction for oxygen diffusion. Anal Chem. 2009;81:6868–6878. doi: 10.1021/ac900881z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Everts B, et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKε supports the anabolic demands of dendritic cell activation. Nature Immunol. 2014;15:323–332. doi: 10.1038/ni.2833. This paper shows that the increased requirement for glucose in activated DCs is linked to a need to synthesize more fatty acids to support the expansion of the ER and Golgi, and to adopt a secretory state. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochem J. 2011;435:297–312. doi: 10.1042/BJ20110162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van der Windt GJ, et al. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity. 2011;36:68–78. doi: 10.1016/j.immuni.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maroof A, English NR, Bedford PA, Gabrilovich DI, Knight SC. Developing dendritic cells become ‘lacy’ cells packed with fat and glycogen. Immunology. 2005;115:473–483. doi: 10.1111/j.1365-2567.2005.02181.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zechner R, et al. FAT SIGNALS—lipases and lipolysis in lipid metabolism and signaling. Cell Metab. 2012;15:279–291. doi: 10.1016/j.cmet.2011.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nagy L, Szanto A, Szatmari I, Szeles L. Nuclear hormone receptors enable macrophages and dendritic cells to sense their lipid environment and shape their immune response. Physiol Rev. 2012;92:739–789. doi: 10.1152/physrev.00004.2011. [DOI] [PubMed] [Google Scholar]
- 39.Ibrahim J, et al. Dendritic cell populations with different concentrations of lipid regulate tolerance and immunity in mouse and human liver. Gastroenterology. 2012;143:1061–1072. doi: 10.1053/j.gastro.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Herber DL, et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nature Med. 2010;16:880–886. doi: 10.1038/nm.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ramakrishnan R, et al. Oxidized lipids block antigen cross-presentation by dendritic cells in cancer. J Immunol. 2014;192:2920–2931. doi: 10.4049/jimmunol.1302801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 43.Reimold AM, et al. Plasma cell differentiation requires the transcription factor XBP-1. Nature. 2001;412:300–307. doi: 10.1038/35085509. [DOI] [PubMed] [Google Scholar]
- 44.Osorio F, et al. The unfolded-protein-response sensor IRE-1α regulates the function of CD8α+ dendritic cells. Nature Immunol. 2014;15:248–257. doi: 10.1038/ni.2808. [DOI] [PubMed] [Google Scholar]
- 45.Iwakoshi NN, Pypaert M, Glimcher LH. The transcription factor XBP-1 is essential for the development and survival of dendritic cells. J Exp Med. 2007;204:2267–2275. doi: 10.1084/jem.20070525. This paper was the first to show that the UPR has a seemingly particularly crucial role in DC biology. It should be read in conjunction with reference 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martinon F, Glimcher LH. Regulation of innate immunity by signaling pathways emerging from the endoplasmic reticulum. Curr Opin Immunol. 2011;23:35–40. doi: 10.1016/j.coi.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Muralidharan S, Mandrekar P. Cellular stress response and innate immune signaling: integrating pathways in host defense and inflammation. J Leukocyte Biol. 2013;94:1167–1184. doi: 10.1189/jlb.0313153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Everts B, et al. Commitment to glycolysis sustains survival of NO-producing inflammatory dendritic cells. Blood. 2012;120:1422–1431. doi: 10.1182/blood-2012-03-419747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Amiel E, et al. Mechanistic target of rapamycin inhibition extends cellular lifespan in dendritic cells by preserving mitochondrial function. J Immunol. 2014;193:2821–2830. doi: 10.4049/jimmunol.1302498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Quintana A, et al. T cell activation requires mitochondrial translocation to the immunological synapse. Proc Natl Acad Sci USA. 2007;104:14418–14423. doi: 10.1073/pnas.0703126104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van der Windt GJ, et al. CD8 memory T cells have a bioenergetic advantage that underlies their rapid recall ability. Proc Natl Acad Sci USA. 2013;110:14336–14341. doi: 10.1073/pnas.1221740110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pantel A, et al. Direct type I IFN but not MDA5/TLR3 activation of dendritic cells is required for maturation and metabolic shift to glycolysis after poly IC stimulation. PLoS Biol. 2014;12:e1001759. doi: 10.1371/journal.pbio.1001759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cao W, et al. Toll-like receptor-mediated induction of type I interferon in plasmacytoid dendritic cells requires the rapamycin-sensitive PI(3)K-mTOR-p70S6K pathway. Nature Immunol. 2008;9:1157–1164. doi: 10.1038/ni.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Amiel E, et al. Inhibition of mechanistic target of rapamycin promotes dendritic cell activation and enhances therapeutic autologous vaccination in mice. J Immunol. 2012;189:2151–2158. doi: 10.4049/jimmunol.1103741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Boor PP, Metselaar HJ, Mancham S, van der Laan LJ, Kwekkeboom J. Rapamycin has suppressive and stimulatory effects on human plasmacytoid dendritic cell functions. Clin Exp Immunol. 2013;174:389–401. doi: 10.1111/cei.12191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hussaarts L, et al. Rapamycin and omega-1: mTOR-dependent and -independent Th2 skewing by human dendritic cells. Immunol Cell Biol. 2013;91:486–489. doi: 10.1038/icb.2013.31. [DOI] [PubMed] [Google Scholar]
- 57.Wobben R, et al. Role of hypoxia inducible factor-1α for interferon synthesis in mouse dendritic cells. Biol Chem. 2013;394:495–505. doi: 10.1515/hsz-2012-0320. [DOI] [PubMed] [Google Scholar]
- 58.Spirig R, et al. Effects of TLR agonists on the hypoxia-regulated transcription factor HIF-1α and dendritic cell maturation under normoxic conditions. PLoS ONE. 2010;5:e0010983. doi: 10.1371/journal.pone.0010983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Miyamoto S, Murphy AN, Brown JH. Akt mediates mitochondrial protection in cardiomyocytes through phosphorylation of mitochondrial hexokinase-II. Cell Death Differ. 2008;15:521–529. doi: 10.1038/sj.cdd.4402285. [DOI] [PubMed] [Google Scholar]
- 60.Ward PS, Thompson CB. Signaling in control of cell growth and metabolism. Cold Spring Harb Perspect Biol. 2012;4:a006783. doi: 10.1101/cshperspect.a006783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.John S, Weiss JN, Ribalet B. Subcellular localization of hexokinases I and II directs the metabolic fate of glucose. PLoS ONE. 2011;6:e17674. doi: 10.1371/journal.pone.0017674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pawlus MR, Hu CJ. Enhanceosomes as integrators of hypoxia inducible factor (HIF) and other transcription factors in the hypoxic transcriptional response. Cell Signall. 2013;25:1895–1903. doi: 10.1016/j.cellsig.2013.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tannahill GM, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. 2013;496:238–242. doi: 10.1038/nature11986. This paper is about macrophages, but it introduces a concept that is likely to be broadly relevant to DC biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lisi L, Navarra P, Feinstein DL, Dello Russo C. The mTOR kinase inhibitor rapamycin decreases iNOS mRNA stability in astrocytes. J Neuroinflamm. 2011;8:1. doi: 10.1186/1742-2094-8-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Blasius AL, et al. Slc15a4, AP-3, and Hermansky-Pudlak syndrome proteins are required for Toll-like receptor signaling in plasmacytoid dendritic cells. Proc Natl Acad Sci USA. 2010;107:19973–19978. doi: 10.1073/pnas.1014051107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kobayashi T, et al. The histidine transporter SLC15A4 coordinates mTOR-dependent inflammatory responses and pathogenic antibody production. Immunity. 2014;41:375–388. doi: 10.1016/j.immuni.2014.08.011. This paper, which focuses on B cell biology, provides mechanistic insight into the important findings on DCs reported in references 53 and 65. [DOI] [PubMed] [Google Scholar]
- 67.Zoncu R, et al. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H+-ATPase. Science. 2011;334:678–683. doi: 10.1126/science.1207056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Waickman AT, Powell JD. mTOR, metabolism, and the regulation of T-cell differentiation and function. Immunol Rev. 2012;249:43–58. doi: 10.1111/j.1600-065X.2012.01152.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Carroll KC, Viollet B, Suttles J. AMPKα1 deficiency amplifies proinflammatory myeloid APC activity and CD40 signaling. J Leukocyte Biol. 2013;94:1113–1121. doi: 10.1189/jlb.0313157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Houtkooper RH, Pirinen E, Auwerx J. Sirtuins as regulators of metabolism and healthspan. Nature Rev Mol Cell Biol. 2012;13:225–238. doi: 10.1038/nrm3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lagouge M, et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1α. Cell. 2006;127:1109–1122. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 72.Svajger U, Obermajer N, Jeras M. Dendritic cells treated with resveratrol during differentiation from monocytes gain substantial tolerogenic properties upon activation. Immunology. 2010;129:525–535. doi: 10.1111/j.1365-2567.2009.03205.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rangasamy T, et al. Nuclear erythroid 2 p45-related factor 2 inhibits the maturation of murine dendritic cells by ragweed extract. Am J Respir Cell Mol Biol. 2010;43:276–285. doi: 10.1165/rcmb.2008-0438OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Aw Yeang HX, et al. Loss of transcription factor nuclear factor-erythroid 2 (NF-E2) p45-related factor-2 (Nrf2) leads to dysregulation of immune functions, redox homeostasis, and intracellular signaling in dendritic cells. J Biol Chem. 2012;287:10556–10564. doi: 10.1074/jbc.M111.322420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Klotz L, et al. Peroxisome proliferator-activated receptor γ control of dendritic cell function contributes to development of CD4+ T cell anergy. J Immunol. 2007;178:2122–2131. doi: 10.4049/jimmunol.178.4.2122. [DOI] [PubMed] [Google Scholar]
- 76.Rubic T, et al. Triggering the succinate receptor GPR91 on dendritic cells enhances immunity. Nature Immunol. 2008;9:1261–1269. doi: 10.1038/ni.1657. [DOI] [PubMed] [Google Scholar]
- 77.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 78.Arnoult D, Soares F, Tattoli I, Girardin SE. Mitochondria in innate immunity. EMBO Rep. 2011;12:901–910. doi: 10.1038/embor.2011.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ghiringhelli F, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1β-dependent adaptive immunity against tumors. Nature Med. 2009;15:1170–1178. doi: 10.1038/nm.2028. [DOI] [PubMed] [Google Scholar]
- 80.Singh N, et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity. 2014;40:128–139. doi: 10.1016/j.immuni.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Singh N, et al. Blockade of dendritic cell development by bacterial fermentation products butyrate and propionate through a transporter (Slc5a8)-dependent inhibition of histone deacetylases. J Biol Chem. 2010;285:27601–27608. doi: 10.1074/jbc.M110.102947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Berndt BE, et al. Butyrate increases IL-23 production by stimulated dendritic cells. Am J Physiol Gastrointest Liver Physiol. 2012;303:G1384–G1392. doi: 10.1152/ajpgi.00540.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gatto D, et al. The chemotactic receptor EBI2 regulates the homeostasis, localization and immunological function of splenic dendritic cells. Nature Immunol. 2013;14:446–453. doi: 10.1038/ni.2555. [DOI] [PubMed] [Google Scholar]
- 84.Manicassamy S, et al. Toll-like receptor 2-dependent induction of vitamin A-metabolizing enzymes in dendritic cells promotes T regulatory responses and inhibits autoimmunity. Nature Med. 2009;15:401–409. doi: 10.1038/nm.1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Coombes JL, et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-β and retinoic acid-dependent mechanism. J Exp Med. 2007;204:1757–1764. doi: 10.1084/jem.20070590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hill JA, et al. Retinoic acid enhances Foxp3 induction indirectly by relieving inhibition from CD4+CD44hi cells. Immunity. 2008;29:758–770. doi: 10.1016/j.immuni.2008.09.018. References 84, 85 and 86 reveal the importance of vitamin A metabolism in DCs in the induction of immune tolerance. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Manicassamy S, Pulendran B. Retinoic acid-dependent regulation of immune responses by dendritic cells and macrophages. Seminars Immunol. 2009;21:22–27. doi: 10.1016/j.smim.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hall JA, Grainger JR, Spencer SP, Belkaid Y. The role of retinoic acid in tolerance and immunity. Immunity. 2011;35:13–22. doi: 10.1016/j.immuni.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hasko G, Pacher P. A2A receptors in inflammation and injury: lessons learned from transgenic animals. J Leukocyte Biol. 2008;83:447–455. doi: 10.1189/jlb.0607359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Li L, et al. Dendritic cells tolerized with adenosine A2AR agonist attenuate acute kidney injury. J Clin Invest. 2012;122:3931–3942. doi: 10.1172/JCI63170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Idzko M, et al. Nucleotides induce chemotaxis and actin polymerization in immature but not mature human dendritic cells via activation of pertussis toxin-sensitive P2y receptors. Blood. 2002;100:925–932. doi: 10.1182/blood.v100.3.925. [DOI] [PubMed] [Google Scholar]
- 92.Granstein RD, et al. Augmentation of cutaneous immune responses by ATPγS: purinergic agonists define a novel class of immunologic adjuvants. J Immunol. 2005;174:7725–7731. doi: 10.4049/jimmunol.174.12.7725. [DOI] [PubMed] [Google Scholar]
- 93.Mariathasan S, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 94.Gottfried E, et al. Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood. 2006;107:2013–2021. doi: 10.1182/blood-2005-05-1795. [DOI] [PubMed] [Google Scholar]
- 95.Nasi A, Rethi B. Disarmed by density: A glycolytic break for immunostimulatory dendritic cells? Oncoimmunology. 2013;2:e26744. doi: 10.4161/onci.26744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Liu C, et al. Lactate inhibits lipolysis in fat cells through activation of an orphan G-protein-coupled receptor, GPR81. J Biol Chem. 2009;284:2811–2822. doi: 10.1074/jbc.M806409200. [DOI] [PubMed] [Google Scholar]
- 97.Hoque R, Farooq A, Ghani A, Gorelick F, Mehal WZ. Lactate reduces liver and pancreatic injury in Toll-like receptor- and inflammasome-mediated inflammation via GPR81-mediated suppression of innate immunity. Gastroenterology. 2014;146:1763–1774. doi: 10.1053/j.gastro.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Goubau D, Deddouche S, Reis e Sousa C. Cytosolic sensing of viruses. Immunity. 2013;38:855–869. doi: 10.1016/j.immuni.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wang X, et al. Differential requirement for the IKKβ/NF-κB signaling module in regulating TLR- versus RLR-induced Type 1 IFN expression in dendritic cells. J Immunol. 2014;193:2538–2545. doi: 10.4049/jimmunol.1400675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF 3. Cell. 2005;122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 101.Horner SM, Liu HM, Park HS, Briley J, Gale M., Jr Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc Natl Acad Sci USA. 2011;108:14590–14595. doi: 10.1073/pnas.1110133108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.van Vliet AR, Verfaillie T, Agostinis P. New functions of mitochondria associated membranes in cellular signaling. Biochim Biophys Acta. 2014;1843:2253–2262. doi: 10.1016/j.bbamcr.2014.03.009. [DOI] [PubMed] [Google Scholar]
- 103.Hayashi T, Rizzuto R, Hajnoczky G, Su TP. MAM: more than just a housekeeper. Trends Cell Biol. 2009;19:81–88. doi: 10.1016/j.tcb.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lasorsa FM, et al. Recombinant expression of the Ca2+-sensitive aspartate/glutamate carrier increases mitochondrial ATP production in agonist-stimulated Chinese hamster ovary cells. J Biol Chem. 2003;278:38686–38692. doi: 10.1074/jbc.M304988200. [DOI] [PubMed] [Google Scholar]
- 105.Koshiba T. Mitochondrial-mediated antiviral immunity. Biochim Biophys Acta. 2013;1833:225–232. doi: 10.1016/j.bbamcr.2012.03.005. [DOI] [PubMed] [Google Scholar]
- 106.Castanier C, Garcin D, Vazquez A, Arnoult D. Mitochondrial dynamics regulate the RIG-I-like receptor antiviral pathway. EMBO Rep. 2010;11:133–138. doi: 10.1038/embor.2009.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Koshiba T, Bashiruddin N, Kawabata S. Mitochondria and antiviral innate immunity. Int J Biochem Mol Biol. 2011;2:257–262. [PMC free article] [PubMed] [Google Scholar]
- 108.Wellen KE, Thompson CB. A two-way street: reciprocal regulation of metabolism and signalling. Nature Rev Mol Cell Biol. 2012;13:270–276. doi: 10.1038/nrm3305. This is an important review for understanding the broader role of metabolism in cellular functions. [DOI] [PubMed] [Google Scholar]
- 109.Mills E, O’Neill LA. Succinate: a metabolic signal in inflammation. Trends Cell Biol. 2014;24:313–320. doi: 10.1016/j.tcb.2013.11.008. [DOI] [PubMed] [Google Scholar]
- 110.Steinman RM, Cohn ZA. Identification of a novel cell type in peripheral lymphoid organs of mice. I Morphology, quantitation, tissue distribution. J Exp Med. 1973;137:1142–1162. doi: 10.1084/jem.137.5.1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Christofk HR, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452:230–233. doi: 10.1038/nature06734. [DOI] [PubMed] [Google Scholar]
- 112.Luo W, Semenza GL. Pyruvate kinase M2 regulates glucose metabolism by functioning as a coactivator for hypoxia-inducible factor 1 in cancer cells. Oncotarget. 2011;2:551–556. doi: 10.18632/oncotarget.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hitosugi T, et al. Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci Signal. 2009;2:ra73. doi: 10.1126/scisignal.2000431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lv L, et al. Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Mol Cell. 2011;42:719–730. doi: 10.1016/j.molcel.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Dominy JE, Jr, Lee Y, Gerhart-Hines Z, Puigserver P. Nutrient-dependent regulation of PGC-1α’s acetylation state and metabolic function through the enzymatic activities of Sirt1/GCN5. Biochim Biophys Acta. 2010;1804:1676–1683. doi: 10.1016/j.bbapap.2009.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Canto C, et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056–1060. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yang H, Lee SM, Gao B, Zhang J, Fang D. Histone deacetylase sirtuin 1 deacetylates IRF1 protein and programs dendritic cells to control Th17 protein differentiation during autoimmune inflammation. J Biol Chem. 2013;288:37256–37266. doi: 10.1074/jbc.M113.527531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Alvarez Y, et al. Sirtuin 1 is a key regulator of the interleukin-12 p70/interleukin-23 balance in human dendritic cells. J Biol Chem. 2012;287:35689–35701. doi: 10.1074/jbc.M112.391839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Legutko A, et al. Sirtuin 1 promotes Th2 responses and airway allergy by repressing peroxisome proliferator-activated receptor-γ activity in dendritic cells. J Immunol. 2011;187:4517–4529. doi: 10.4049/jimmunol.1101493. [DOI] [PubMed] [Google Scholar]
- 120.Sebastian C, et al. The histone deacetylase SIRT6 is a tumor suppressor that controls cancer metabolism. Cell. 2012;151:1185–1199. doi: 10.1016/j.cell.2012.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zhong L, et al. The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1α. Cell. 2010;140:280–293. doi: 10.1016/j.cell.2009.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Fang TC, et al. Histone H3 lysine 9 di-methylation as an epigenetic signature of the interferon response. J Exp Med. 2012;209:661–669. doi: 10.1084/jem.20112343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Igarashi K, Katoh Y. Metabolic aspects of epigenome: coupling of S-adenosylmethionine synthesis and gene regulation on chromatin by SAMIT module. Sub-Cell Biochem. 2013;61:105–118. doi: 10.1007/978-94-007-4525-4_5. [DOI] [PubMed] [Google Scholar]