

Abstract
Signal transduction through Ras translates extracellular signals into biological responses, including cell proliferation, cell survival, growth, and differentiation. For these reasons, dysregulating Ras can have dramatic effects at the cellular and organismal levels. Germline mutations that increase Ras signaling disrupt development, whereas mutational activation of Ras in somatic cells can cause cancer. Thus, identifying additional mechanisms that positively or negatively regulate Ras could have profound implications for treating human diseases. New evidence identifies K-Ras monoubiquitination as a previously unknown means to potentiate Ras signaling.
A single Ras isoform in Drosophila and Caenorhabditis elegans expands into a family of Ras proteins in humans—N-Ras, H-Ras, and differently spliced forms of K-Ras, K-Ras4A, and K-Ras4B (referred to here as K-Ras). Mammalian Ras proteins share almost 100% identity in the first 165 amino acids, and all isoforms demonstrate the ability to transform cells in culture. However, they show less than 15% amino acid identity in the C-terminal 24 amino acids, and they demonstrate key differences including compartmental localization, trafficking, post-translational modifications, and pattern of mutations in human disease (1–18).
Activating germline mutations in K-Ras occur in Noonan syndrome and cardio-faciocutaneous syndrome, and similar mutations in H-Ras give rise to Costello syndrome and in N-Ras, to autoimmune lymphoproliferative syndrome (1, 2). Mutational activation in cancer also results in different tumor spectra. Whereas mutational activation of K-Ras occurs in >90% of all pancreatic cancers (11–13), N-Ras is the most commonly mutated Ras in acute myeloid leukemia (14, 15) and is also frequently mutated in melanoma (16, 17), and mutation of H-Ras is common in bladder cancer (18).
Given the importance of Ras, we would expect cells to use multiple mechanisms to potentiate and attenuate Ras activity. Ras becomes active in the guanosine triphosphate (GTP)–bound state and inactive in the guanosine diphosphate (GDP)–bound state. Guanine nucleotide exchange factors (GEFs) promote exchange of GDP for GTP to activate Ras. In contrast, guanosine triphosphatase (GTPase)–activating proteins (GAPs) promote hydrolysis of GTP on Ras to GDP, which inactivates Ras (5). Various posttranslational modifications—including farnesylation, proteolysis, methylation, palmitoylation, and depalmitoylation—modulate the membrane affinity and localization of Ras to define exposure to downstream effectors. However, the identity of proteins other than GEFs and GAPs that directly target Ras to alter its activity has remained elusive.
Normal protein turnover mechanisms regulate the abundance of all proteins; indeed, proteasomal degradation of polyubiquitinated Ras proteins regulates their stability, a process mediated by the ubiquitin ligase SCF/β-TRCP (β-transducin repeat–containing protein) (19). In addition, Wnt signaling decreases β-TRCP–induced polyubiquitination of Ras (19). Several studies also identified a nonproteasomal fate for ubiquitinated Ras. Mono- and diubiquitination of Drosophila Ras and mammalian H-Ras and N-Ras, but not K-Ras, restrict the ability to signal to ERK (extracellular signal–regulated kinase) (20, 21), which suggests a conserved role for ubiquitination in decreasing the activity of Ras. Experiments with mammalian cells transfected with an H-Ras–ubiquitin fusion or an H-Ras mutant that could not be ubiquitinated demonstrated that ubiquitination promotes endosomal localization or retention (or both) of H-Ras (21), which suggests that the restricted ability to signal resulted from sequestration from certain effectors. Subsequent work revealed that the ubiquitin ligase Rabex-5 promotes ubiquitination of Drosophila Ras (22) and mammalian H-Ras, but not K-Ras (23), in a negative feedback mechanism (20–24).
Whereas previous studies reported that mono- and diubiquitination restrict the activity of H-Ras and of N-Ras, but not that of K-Ras, Sasaki et al. (25) report the monoubiquitination of K-Ras and demonstrate that K-Ras ubiquitination promotes its activity. Sasaki et al. found monoubiquitinated K-Ras predominantly in the GTP-loaded state and unconjugated K-Ras predominantly in the GDP-loaded state and, therefore, propose that ubiquitination may enhance GTP-loading of K-Ras. They identified the major site of K-Ras ubiquitination as Lys147, with a minor site at Lys117. Similarly, they identified H-Ras sites of ubiquitination as Lys117, Lys147, and Lys170. Moreover, they showed that ubiquitinated K-Ras demonstrates increased affinity for Raf, PI3K (phosphatidylinositol 3-kinase), and RalGDS in binding assays and that it activated Raf and PI3K more efficiently than unconjugated Ras in in vitro kinase assays. In contrast, a Lys147→Leu (K147L) substitution in an activated Gly12→Val (G12V) K-Ras mutant showed reduced binding to PI3K but not to Raf. Unlike H-Ras ubiquitination, which promotes endosomal localization (21), K-Ras ubiquitination did not change its apparent subcellular localization (15). Compellingly, Sasaki at al. establish an in vivo role for ubiquitination of Lys147 in promoting the oncogenic functions of K-Ras. They observed decreased tumorigenicity of K147L substitution mutants of oncogenic G12V K-Ras in terms of tumor weight and tumor volume in nude mice. K147L/G12V K-Ras tumors that formed exhibited reduced phospho-S6 and a trend of reduced AKT activation compared with G12V K-Ras tumors. This suggests an exciting model in which ubiquitination of K-Ras at Lys147 normally acts to preferentially increase the strength of signaling through select downstream pathways.
The seemingly divergent means by which conjugation to ubiquitin regulates H- or N-Ras (20–23) and K-Ras (25) raise various exciting questions. First, do distinct ubiquitin ligases (specificity components of the ubiquitin pathway) regulate H- or N-Ras and K-Ras, or does the same ubiquitin ligase target all three isoforms? Previous (20–23) and current findings (25) (Fig. 1) suggest that different ubiquitin ligases target H- or N-Ras and K-Ras. For example, GTP-loading affects Ras conformation. Sasaki et al. (25) demonstrated ubiquitination of a form of K-Ras that is mutated at Thr35 and is locked in a GDP-bound conformation, consistent with a model that ubiquitination may precede GTP-loading, whereas a previous study (21) reported that the G12V form of H-Ras (which is locked in a GTP-bound conformation that is distinct from the GDP-bound conformation) remained a suitable substrate for ubiquitination, consistent with ubiquitination occurring after GTP-loading. Targeting disparate conformations suggests that ubiquitination of K-Ras and H-Ras may occur by distinct mechanisms. Also, although formally possible that K-Ras ubiquitination was below detection limits, Xu et al. showed that Rabex-5 promoted ubiquitination of H-Ras but not K-Ras in a purified system and in various cell lines (23).
Fig. 1.
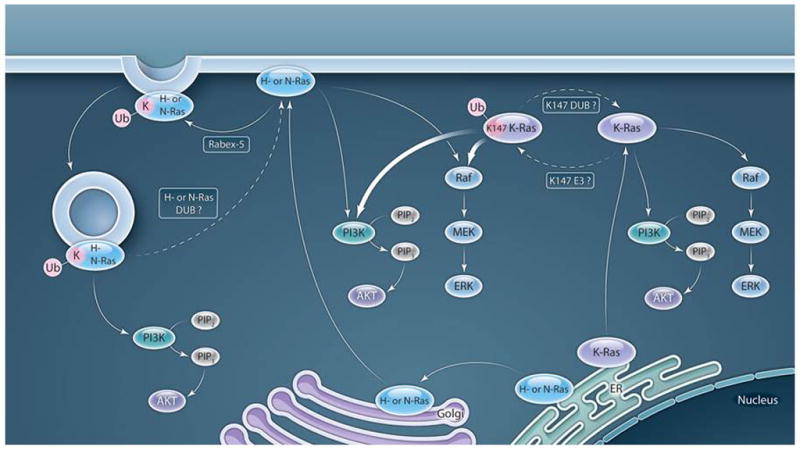
Ubiquitination differentially regulates K-Ras and H- or N-Ras. Previous studies demonstrated that mono- or diubiquitination of H- or N-Ras at an unidentified lysine (K) promotes endosomal localization or retention (depicted here as relocalization), which restricts the ability to signal to ERK, but not to AKT (20–23). Presumably, a DUB could act to remove ubiquitin from H- or N-Ras, and so enable a return to the membrane and restore the ability to signal through ERK. In contrast, Sasaki et al. (25) demonstrated that monoubiquitination of K-Ras at Lys147 (K147) by an unknown ubiquitin E3 ligase increases the ability to signal through PI3K and possibly Raf and does not alter K-Ras trafficking. Removal of the ubiquitin added to Lys147 by a DUB could restore activation of PI3K by K-Ras to normal amounts. Dashed lines indicate putative steps.
Second, does Rabex-5 promote ubiquitination at a lysine residue in H-Ras other than Lys147, or does Lys147 ubiquitination play a different role in H-Ras than in K-Ras? Ubiquitinated H-Ras localizes to the endosomes (21, 23), which compromises signaling through ERK and potentially limits access to other effectors. Sasaki et al. (25) showed that even a low amount of ubiquitination of K-Ras at Lys147 could translate into meaningful outputs through PI3K, which suggests that endosomal H-Ras localization may either not limit access to PI3K, or that increased signaling through PI3K may offset any limited access. Because trafficking of H-Ras and K-Ras exposes each protein to different aspects of the cellular milieu, in terms of downstream effectors and ubiquitin-interacting proteins, modifying the same lysine residue could translate into distinct biological outputs. Alternatively, Sasaki et al. reported modification of both Lys117 and Lys170 in H-Ras (25), and Jura et al. (21) replaced eight lysines to make a form of H-Ras that could not be ubiquitinated, which suggests that modification of another lysine residue could underlie restriction of H-Ras activity.
Third, did Ras ubiquitination arise with a single function in an ancestor with only one Ras? The conserved role of Ras ubiquitination (from Drosophila to mammals) to restrict signaling through ERK indicates that Ras ubiquitination may have originated as an inhibitory mechanism. Subsequent evolutionary expansion with multiple Ras forms could have enabled ubiquitination of distinct Ras proteins to assume different roles depending on tissue type, compartmentalization, and other determinants. The potentiating role of K-Ras ubiquitination reported by Sasaki et al. (25) may reflect a recent adaptation borne of the specialization of each Ras species.
Alternatively, an activating role for Ras ubiquitination has not been evaluated in Drosophila. Previous studies showed that nonspecifically reducing overall ubiquitination promoted increased activation of ERK, but not that of AKT, in whole larvae (20) and that Rabex-5–mediated Ras ubiquitination restricted Drosophila Ras signaling to ERK (22). However, these findings do not preclude a role for Ras ubiquitination to increase signaling through PI3K—for example, in specific tissues or developmental periods not examined in these studies.
Finally, ubiquitination is reversible; deubiquitinating enzymes (DUBs) remove ubiquitin from substrates. Does a DUB attenuate K-Ras signaling specific effector pathways by removing ubiquitin from Lys147? Such a DUB could add a further level of intricacy to Ras regulation and play an important role in fine-tuning signaling outputs through Ras effectors. In light of the in vivo tumor studies performed by Sasaki et al. (25), a DUB acting on K-Ras Lys147 could represent a tumor suppressor that could act on oncogenic K-Ras, a departure from Ras inhibitors such as GAPs.
In identifying that ubiquitination of K-Ras at Lys147 promotes Ras signaling, Sasaki et al. (25) have emphasized that modification of even a small percentage of total Ras can lead to meaningful biological consequences and adds another layer of complexity to our understanding of Ras regulation. Taken together with the current state of the field, these findings implicate various mechanisms by which different cell types and tissues could establish distinct physiological outcomes in specific contexts by expressing different combinations of K-Ras and H- or N-Ras ubiquitin ligases, DUBs, or ubiquitin-interacting trafficking factors. The as-yet-unidentified ubiquitin ligase targeting K-Ras joins the ranks of GEF proteins in its ability to promote K-Ras activity and a K-Ras Lys147 DUB could join GAPs in inhibiting K-Ras.
References
- 1.Tidyman WE, Rauen KA. The RASopathies: Developmental syndromes of Ras/MAPK pathway dysregulation. Curr Opin Genet Dev. 2009;19:230–236. doi: 10.1016/j.gde.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gelb BD, Tartaglia M. Noonan syndrome and related disorders: Dysregulated RAS-mitogen activated protein kinase signal transduction. Hum Mol Genet. 2006;15:R220–R226. doi: 10.1093/hmg/ddl197. [DOI] [PubMed] [Google Scholar]
- 3.Young A, Lyons J, Miller AL, Phan VT, Alarcón IR, McCormick F. Ras signaling and therapies. Adv Cancer Res. 2009;102:1–17. doi: 10.1016/S0065-230X(09)02001-6. [DOI] [PubMed] [Google Scholar]
- 4.Duursma AM, Agami R. Ras interference as cancer therapy. Semin Cancer Biol. 2003;13:267–273. doi: 10.1016/s1044-579x(03)00040-3. [DOI] [PubMed] [Google Scholar]
- 5.Vigil D, Cherfils J, Rossman KL, Der CJ. Ras superfamily GEFs and GAPs: Validated and tractable targets for cancer therapy? Nat Rev Cancer. 2010;10:842–857. doi: 10.1038/nrc2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mor A, Philips MR. Compartmentalized Ras/ MAPK signaling. Annu Rev Immunol. 2006;24:771–800. doi: 10.1146/annurev.immunol.24.021605.090723. [DOI] [PubMed] [Google Scholar]
- 7.Fehrenbacher N, Bar-Sagi D, Philips M. Ras/ MAPK signaling from endomembranes. Mol Oncol. 2009;3:297–307. doi: 10.1016/j.molonc.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Omerovic J, Prior IA. Compartmentalized signalling: Ras proteins and signalling nanoclusters. FEBS J. 2009;276:1817–1825. doi: 10.1111/j.1742-4658.2009.06928.x. [DOI] [PubMed] [Google Scholar]
- 9.Omerovic J, Laude AJ, Prior IA. Ras proteins: Paradigms for compartmentalised and isoform-specific signalling. Cell Mol Life Sci. 2007;64:2575–2589. doi: 10.1007/s00018-007-7133-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Silvius JR. Mechanisms of Ras protein targeting in mammalian cells. J Membr Biol. 2002;190:83–92. doi: 10.1007/s00232-002-1026-4. [DOI] [PubMed] [Google Scholar]
- 11.Smit VTHBM, Boot AJM, Smits AMM, Fleuren GJ, Cornelisse CJ, Bos JL. KRAS codon 12 mutations occur very frequently in pancreatic adenocarcinomas. Nucleic Acids Res. 1988;16:7773–7782. doi: 10.1093/nar/16.16.7773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rozenblum E, Schutte M, Goggins M, Hahn SA, Panzer S, Zahurak M, Goodman SN, Sohn TA, Hruban RH, Yeo CJ, Kern SE. Tumor-suppressive pathways in pancreatic carcinoma. Cancer Res. 1997;57:1731–1734. [PubMed] [Google Scholar]
- 13.Bardeesy N, DePinho RA. Pancreatic cancer biology and genetics. Nat Rev Cancer. 2002;2:897–909. doi: 10.1038/nrc949. [DOI] [PubMed] [Google Scholar]
- 14.Farr CJ, Saiki RK, Erlich HA, McCormick F, Marshall CJ. Analysis of RAS gene mutations in acute myeloid leukemia by polymerase chain reaction and oligonucleotide probes. Proc Natl Acad Sci USA. 1988;85:1629–1633. doi: 10.1073/pnas.85.5.1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vogelstein B, Civin CI, Preisinger AC, Krischer JP, Steuber P, Ravindranath Y, Weinstein H, Elfferich P, Bos J. RAS gene mutations in childhood acute myeloid leukemia: A Pediatric Oncology Group study. Genes Chromosomes Cancer. 1990;2:159–162. doi: 10.1002/gcc.2870020212. [DOI] [PubMed] [Google Scholar]
- 16.Reifenberger J, Knobbe CB, Sterzinger AA, Blaschke B, Schulte KW, Ruzicka T, Reifenberger G. Frequent alterations of Ras signaling pathway genes in sporadic malignant melanomas. Int J Cancer. 2004;109:377–384. doi: 10.1002/ijc.11722. [DOI] [PubMed] [Google Scholar]
- 17.Whitwam T, Vanbrocklin MW, Russo ME, Haak PT, Bilgili D, Resau JH, Koo HM, Holmen SL. Differential oncogenic potential of activated RAS isoforms in melanocytes. Oncogene. 2007;26:4563–4570. doi: 10.1038/sj.onc.1210239. [DOI] [PubMed] [Google Scholar]
- 18.Visvanathan KV, Pocock RD, Summerhayes IC. Preferential and novel activation of H-ras in human bladder carcinomas. Oncogene Res. 1988;3:77–86. [PubMed] [Google Scholar]
- 19.Kim SE, Yoon JY, Jeong WJ, Jeon SH, Park Y, Yoon JB, Park YN, Kim H, Choi KY. H-Ras is degraded by Wnt/beta-catenin signaling via beta-TrCP-mediated polyubiquitylation. J Cell Sci. 2009;122:842–848. doi: 10.1242/jcs.040493. [DOI] [PubMed] [Google Scholar]
- 20.Yan H, Chin ML, Horvath EA, Kane EA, Pfleger CM. Impairment of ubiquitylation by mutation in Drosophila E1 promotes both cell-autonomous and non-cell-autonomous Ras-ERK activation in vivo. J Cell Sci. 2009;122:1461–1470. doi: 10.1242/jcs.042267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jura N, Scotto-Lavino E, Sobczyk A, Bar-Sagi D. Differential modification of Ras proteins by ubiquitination. Mol Cell. 2006;21:679–687. doi: 10.1016/j.molcel.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 22.Yan H, Jahanshahi M, Horvath EA, Liu HY, Pfleger CM. Rabex-5 ubiquitin ligase activity restricts Ras signaling to establish pathway homeostasis in Drosophila. Curr Biol. 2010;20:1378–1382. doi: 10.1016/j.cub.2010.06.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu L, Lubkov V, Taylor LJ, Bar-Sagi D. Feedback regulation of Ras signaling by Rabex-5-mediated ubiquitination. Curr Biol. 2010;20:1372–1377. doi: 10.1016/j.cub.2010.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Colicelli J. Signal transduction: RABGEF1 fingers RAS for ubiquitination. Curr Biol. 2010;20:R630–R632. doi: 10.1016/j.cub.2010.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sasaki AT, Carracedo A, Locasale JW, Anastasiou D, Takeuchi K, Kahoud ER, Haviv S, Asara JM, Pandolfi PP, Cantley LC. Ubiquitination of K-Ras enhances activation and facilitates binding to select downstream effectors. Sci Signal. 2011;4:ra13. doi: 10.1126/scisignal.2001518. [DOI] [PMC free article] [PubMed] [Google Scholar]