

Abstract
Background
Transposable elements are mobile DNA sequences that are widely distributed in prokaryotic and eukaryotic genomes, where they represent a major force in genome evolution. However, transposable elements have rarely been documented in viruses, and their contribution to viral genome evolution remains largely unexplored. Pandoraviruses are recently described DNA viruses with genome sizes that exceed those of some prokaryotes, rivaling parasitic eukaryotes. These large genomes appear to include substantial noncoding intergenic spaces, which provide potential locations for transposable element insertions. However, no mobile genetic elements have yet been reported in pandoravirus genomes.
Results
Here, we report a family of miniature inverted-repeat transposable elements (MITEs) in the Pandoravirus salinus genome, representing the first description of a virus populated with a canonical transposable element family that proliferated by transposition within the viral genome. The MITE family, which we name Submariner, includes 30 copies with all the hallmarks of MITEs: short length, terminal inverted repeats, TA target site duplication, and no coding capacity. Submariner elements show signs of transposition and are undetectable in the genome of Pandoravirus dulcis, the closest known relative Pandoravirus salinus. We identified a DNA transposon related to Submariner in the genome of Acanthamoeba castellanii, a species thought to host pandoraviruses, which contains remnants of coding sequence for a Tc1/mariner transposase. These observations suggest that the Submariner MITEs of P. salinus belong to the widespread Tc1/mariner superfamily and may have been mobilized by an amoebozoan host. Ten of the 30 MITEs in the P. salinus genome are located within coding regions of predicted genes, while others are close to genes, suggesting that these transposons may have contributed to viral genetic novelty.
Conclusions
Our discovery highlights the remarkable ability of DNA transposons to colonize and shape genomes from all domains of life, as well as giant viruses. Our findings continue to blur the division between viral and cellular genomes, adhering to the emerging view that the content, dynamics, and evolution of the genomes of giant viruses do not substantially differ from those of cellular organisms.
Electronic supplementary material
The online version of this article (doi:10.1186/s12915-015-0145-1) contains supplementary material, which is available to authorized users.
Keywords: Genome evolution, Miniature inverted-repeat transposable element (MITE), Virus
Background
Transposable elements (TEs) are mobile DNA sequences that can insert into new genomic locations, often replicating themselves during the process. Two classes of TEs exist that differ in the molecular mechanism by which they transpose from one genomic location to another – Class I TEs (retrotransposons) transpose via an RNA intermediate, whereas Class II TEs (DNA transposons) transpose via a DNA intermediate [1, 2]. A TE can be autonomous or non-autonomous; transposition enzymes for autonomous TEs are transcribed and translated from the TE’s own sequence, whereas non-autonomous TEs utilize transposition enzymes encoded by other TE loci [1].
Miniature inverted-repeat transposable elements (MITEs) are non-autonomous DNA transposons of relatively short length (100–600 bp) whose transposition requires enzymes encoded by autonomous DNA transposons [3–5]. MITE sequences include terminal inverted repeats (TIRs) and are flanked by short direct repeats (often TA or TAA) called target site duplications (TSDs). MITEs are distinguished from other non-autonomous DNA elements by relatively high copy numbers and length homogeneity [4, 5]. In addition, MITEs in both prokaryotic and eukaryotic genomes are often found close to or within genes, where they may affect gene expression or contribute exonic sequence [4–10].
TEs are widely distributed among both prokaryotic and eukaryotic genomes. TE activity has played a powerful role in the evolution of these groups, providing both the raw material for genetic innovation as well as most of the DNA content in diverse lineages [11, 12]. In contrast, TEs have only rarely been documented in the genomes of viruses. In all previously reported instances, the TEs were restricted to one or two copies per viral genome, and they were interpreted as transient passengers acquired horizontally from their cellular hosts with little to no impact on viral genome evolution [13–23]. Recently, the genomes of several giant DNA viruses within the recently proposed order “Megavirales” [24] have been shown to host other types of mobile genetic elements and repetitive, putatively mobile elements including self-splicing introns, inteins, insertion sequences (ISs), provirophages, and an atypical group of integrative linear plasmids called transpovirons [17, 25–32]. However, to the best of our knowledge, no viral genome has been reported to contain a substantial number of canonical TEs (i.e. Class I or Class II TEs that transpose via typical mechanisms) that proliferated by transposition in the viral genome.
Pandoravirus salinus and Pandoravirus dulcis are related giant viruses that likely infect amoebae of the genus Acanthamoeba [33]. Pandoravirus genomes reach 2.5 Mb, a size exceeding that of some bacterial genomes and comparable to the genomes of some single-celled, parasitic eukaryotes. Pandoravirus genomes are predicted to encode more than 2,500 protein-coding genes, including repetitive open reading frames (ORFs) likely generated by local gene duplications [33]. Protein-coding sequences occupy approximately 80 % of pandoravirus genomes, leaving substantial noncoding intergenic space that could harbor TEs. However, no mobile genetic elements have yet been reported in pandoravirus genomes. Herein, we identify 30 elements in the P. salinus genome with all the hallmarks of a MITE family, providing the first documented case of a virus populated with a canonical TE family that proliferated by transposition within the viral genome. Ten of these 30 MITEs are predicted to contribute coding sequences in the P. salinus genome, while others are in close association with predicted genes, suggesting that TEs were actively involved in shaping the evolution of this viral genome.
Results
Discovery of MITEs in the P. salinus genome
We identified a repeat element in the P. salinus genome with long TIRs, a hallmark of DNA transposons. The element is present in 13 full-length copies (i.e. copies missing fewer than 5 bp of their TIRs), 13 copies >80 % of full-length, and four copies >50 % of full-length in the P. salinus genome. We confirmed that the repeat element has well-defined boundaries by aligning the 13 full-length copies and 60 bp of their flanking sequence (Fig. 1a). Based on an alignment of all 30 copies (Additional file 1: Figure S1), we reconstructed a 243-bp consensus sequence [Repbase ID: Submariner_Ps1] that is nearly palindromic – the TIRs are approximately 100 bp long (Additional file 2: Figure S2). Eleven of the 13 full-length copies are flanked on at least one side by a 5′-TA-3′ dinucleotide (Fig. 1a) likely representing the TSD generated upon integration into the viral genome. Four features of this element suggest that it is a MITE [3, 5]: (1) its small size (243 bp), (2) its TIRs, (3) its 5′-TA-3′ TSDs, and (4) its apparent lack of coding capacity for a transposase. Using the flanking sequences of each MITE copy as BLASTn queries against the P. salinus genome, we identified one paralogous site that lacked the MITE, but contained a copy of the TA dinucleotide at the insertion site (i.e. a paralogous empty site; Fig. 1b, Additional file 3: Figure S3) (sequence divergence between the flanking sequences of the MITE and the paralogous empty site is 11 % over 119 total bp; e-value = 1e-42). These data strongly suggest that the MITE spread within the P. salinus genome via canonical transposition events, producing 5′-TA-3′ TSDs.
Fig. 1.

a ClustalW-generated multiple alignment of the 13 full-length MITEs and their flanking sequence showing partial terminal inverted repeat (TIR) sequences and target site duplication (TSD) sequences. The multiple alignment results indicate well-defined element boundaries because sequence similarity decreases dramatically outside of the MITE. b Pairwise alignment of the flanking sequences of a MITE insertion in P. salinus (bottom sequence) and a paralogous empty site elsewhere in the P. salinus genome (top sequence). TSD sequences (TA) are underlined. The paralogous empty site is evidence of transposition. Numbers of either side of the sequences indicate their coordinates in the P. salinus genome
Proliferation history of the MITEs in the P. salinus genome
Sequence divergences of each MITE copy from the consensus sequence (i.e. the inferred ancestral sequence) range from 8–30 % (Additional file 4: Figure S4). We found no MITE copies within the genome of P. dulcis, the closest known relative of P. salinus; these two viral genomes are, on average, 65–88 % identical in orthologous coding sequence, although many of the non-coding sequences in the P. salinus genome do not have identifiable orthologs in the smaller P. dulcis genome. Together, these data indicate that the MITE may have been active since the divergence of the two pandoraviruses; however, we cannot exclude the possibility that the MITE was active in their common ancestor and subsequently lost from the P. dulcis genome.
Identification of an autonomous DNA transposon related to the P. salinus MITEs in the genome of a potential P. salinus host, the amoeba Acanthamoeba castellanii
MITE transposition requires transposase encoded by autonomous DNA transposons. MITEs and the DNA transposons that mobilize them typically share sequence similarity in their TIRs. We looked for an autonomous DNA transposon that could have facilitated the proliferation of the P. salinus MITEs in both the P. salinus genome as well as the genomes of all species represented in public sequence databases. We found no such DNA transposon in the P. salinus genome using either of two methods — tBLASTn searches against the P. salinus genome using transposase queries representing known DNA transposon superfamilies, or the ‘Anchor’ function of the MITE Analysis Kit, which scans the genome for longer copies with putative coding sequences [34]. However, BLASTn searches using the MITE consensus sequence against the NCBI databases did retrieve one 1604 bp sequence from the genome of A. castellanii with sequence similarity to the MITE TIRs (coordinates AEYA01001964.1: 92260–93863). A. castellanii is a likely host of P. salinus. NCBI-BLAST2 analysis shows that this 1604 bp sequence has 29 bp TIRs, a typical feature of DNA transposons.
To determine whether the 1604 bp sequence encodes proteins associated with transposition, we queried it against the NCBI Conserved Domain Database [35]. We found that it contains a DDE superfamily endonuclease domain (e-value = 5.38e-11), suggesting that the sequence is likely a DNA transposon. We also used this 1604 bp sequence to BLASTx against the proteins encoded by TEs in Repbase to determine whether it shares sequence similarity with any known DNA transposons. We found that it encodes a protein sharing 20–29 % amino acid sequence identity with the putative transposases encoded by four Tc1/mariner DNA transposons described in Acyrthosiphon pisum (pea aphid) and Caenorhabditis briggsae (nematode) — Mariner-2_AP (e-value = 8e-30), Mariner-3_AP (e-value = 5e-29), Mariner-1_AP (e-value = 5e-26), and Mariner44_CB (e-value = 1e-06). These results suggest that the 1604 bp sequence is a DNA transposon of the Tc1/mariner superfamily.
To investigate whether the 1604 bp sequence has been transpositionally active, we looked for paralogous empty sites within the A. castellanii genome. To this end, we used 100 bp of sequence flanking the 1604 bp sequence on either side as queries to BLASTn against the total genomic sequences of A. castellanii. We found one paralogous empty site (Fig. 2a and Additional file 5: Figure S5), confirming transposition activity of the 1604 bp sequence (sequence divergence between the empty site and its paralog containing the 1604 bp sequence is approximately 6 % over 121 bp; e-value = 1e-67). Integration of the new 1604 bp sequence generated a 5′-TA-3′ TSD (Fig. 2a), suggesting that the sequence has a typical TSD of a Tc1/mariner superfamily DNA transposon.
Fig. 2.
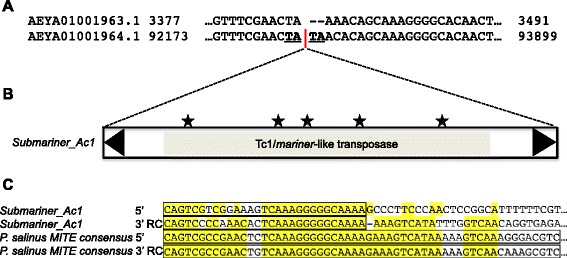
Autonomous DNA transposon in the amoeba Acanthamoeba castellanii that is closely related to the MITEs in P. salinus. a Pairwise alignment of the flanking sequences of the DNA transposon insertion and a paralogous empty site elsewhere in the A. castellanii genome. Red bar indicates the transposon insertion site. Bold and underlined letters (TA) indicate TSD. The paralogous empty site is evidence of transposition. b The structure of the autonomous DNA transposon in A. castellanii. Triangles indicate TIRs. Stars indicate stop codons in the putative transposase sequence. c Alignment of the ends of the consensus sequence of the MITEs in P. salinus and the ends of the autonomous DNA transposon sequence in A. castellanii, referred to as Submariner_Ac1. TIRs for each element are boxed. Columns in the alignment are shaded when nucleotides are conserved in at least three sequences. RC stands for reverse-complement. The sequence similarity between the TIRs of the P. salinus MITE and the A. castellanii DNA transposon Submariner_Ac1 indicates that the P. salinus MITE could have been cross-mobilized in the viral genome by the A. castellanii DNA transposon
Taken together, these data show that the sequence we identified in A. castellanii has all the hallmarks of a Tc1/mariner DNA transposon. We named the sequence Submariner_Ac1. The transposase sequence of Submariner_Ac1 contains many obvious disabling mutations, introducing at least five premature stop codons (Fig. 2b), strongly suggesting that it is no longer active.
Tc1/mariner elements are known to have produced and mobilized MITEs in many species [5, 36–39]. Submariner_Ac1 includes TIRs that share approximately 83 % sequence identity with the TIRs of the P. salinus MITE consensus sequence (Fig. 2c); this level of sequence similarity is typical for autonomous DNA transposons and the MITEs they can mobilize [36, 40, 41]. Based on this sequence similarity, as well as the shared 5′-TA-3′ TSD sequences, we infer that the MITE family identified in P. salinus was likely derived from a Submariner_Ac1-like DNA transposon and subsequently amplified by a Submariner_Ac1-like transposase. Thus, we name the MITE family in P. salinus Submariner_Ps1.
tBLASTx searches using the sequence of Submariner_Ac1 against the A. castellanii genome retrieved one more Submariner-like DNA transposon (coordinates AEYA01001733.1: 913–2735), which we name Submariner_Ac2 (e-value = 1e-125). Like Submariner_Ac1, the transposase sequence of Submariner_Ac2 also contains disabling mutations (two stop codons, one frameshift), suggesting that this element is also inactive. We find no evidence of transposition of Submariner_Ac2 based on searches for paralogous empty sites within the A. castellanii genome. The sequence similarity between the TIRs of Submariner_Ps1 (i.e. the P. salinus MITE family) and Submariner_Ac2 is less than between Submariner_Ps1 and Submariner_Ac1; as described above, the Submariner_Ps1 consensus sequence retrieved only Submariner_Ac1, and not Submariner_Ac2, as a significant BLAST hit. Thus, we inferred that Submariner_Ac2 is less likely to have mobilized the P. salinus MITEs, although we could not completely exclude this possibility.
Submariner sequences in A. castellanii belong to a novel subgroup of Tc1/mariner DNA transposons
DNA transposons are grouped into superfamilies and smaller subclades based, in part, on shared amino acid motifs within the conserved DDE/D catalytic domain of their transposase sequence [42]. The DDE/D motif refers to the acidic amino acid triad that coordinates metal ion binding (most likely Mg2+) during catalysis of typical cut-and-paste transposition [43]. To determine whether Submariner_Ac1 and Submariner_Ac2 are part of any characterized Tc1/mariner subclade, we aligned (1) the putative transposase sequences from Submariner_Ac1 and Submariner_Ac2, (2) the four DNA transposase hits we obtained from Repbase using Submariner_Ac1 as a query (Mariner-1_AP, Mariner-2_AP, Mariner-3_AP, and Mariner44_CB), and (3) two representative transposases from each of five well-established Tc1/mariner subclades (Fot1, Pogo, Tc1, Gizmo, and Mogwai) [44]. To identify other potentially related sequences, we also performed additional BLASTx searches against the NCBI non-redundant protein sequence database using the Submariner_Ac1 sequence as a query; we retained one representative per species from the top 20 hits (five total sequences, four bacterial and one archaeal) to be included in our alignment. Using this alignment, we examined the DDE/D signature in the A. castellanii sequences and found that the residues are located within conserved “DET,” “DNA,” and “PIE” motifs, indicative of Tc1/mariner transposases [36, 44, 45] (Fig. 3a; Additional file 6: Figure S6). The third glutamic acid residue within the conserved “PIE” motif mutated to an N in the apparently inactive Submariner_Ac1 sequence, and the first aspartic acid residue within the conserved “DET” motif mutated to an N in the apparently inactive Submariner_Ac2 sequence (Fig. 3a). In the two Submariner_Ac transposases, the second aspartic acid residue and the glutamic acid were separated by a much longer stretch of amino acids (75 – 86 amino acid positions) than is found in the transposases of well-established Tc1/mariner clades (27 to 31 amino acid positions; Fig. 3a and b) [45]. The Tc1/mariner hits we obtained from Repbase (Mariner-1_AP, Mariner-2_AP, Mariner-3_AP, and Mariner44_CB) also share this long stretch of amino acids between the second aspartic acid and the glutamic acid. Finally, the archaeal and four bacterial sequences we obtained from NCBI also share this long stretch of amino acids. Based on this novel DDE signature, we inferred that the Submariner sequences identified in A. castellanii are part of a novel subgroup of Tc1/mariner transposons with members in all three domains of cellular life. We refer to the subgroup as Submariner.
Fig. 3.
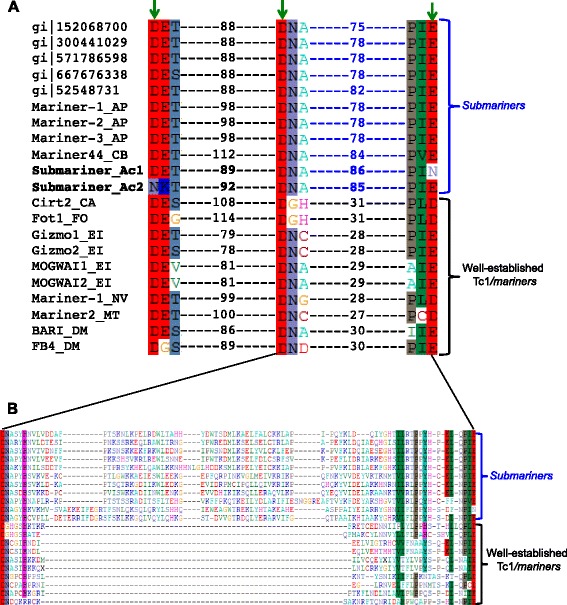
The DDE signature in Submariner transposases. a Sequences are identified by their GenBank accession numbers or Repbase IDs, if applicable. These accession numbers and Repbase IDs correspond to the nucleotide sequences from which the transposase amino acid sequences were deduced. Green arrows indicate the DDE amino acid triad that coordinates metal ion (Mg2+) binding during catalysis of typical cut-and-paste transposition. The DDE residues are shown within their respective conserved motifs (DET, DNA, and PIE). Numbers flanked by dashes indicate the number of amino acid positions that separate the conserved motifs based on a multiple sequence alignment. The 11 Submariner sequences have a much longer stretch of residues between the second D and the E residues than do representatives from the well-established Tc1/mariner clades. gi|152068700, gi|300441029, gi|571786598, and gi|667676338 are bacterial sequences, and gi|52548731 is an archaeal sequence; taxon information is in the text. Mariner-1-3_AP are from Acyrthosiphon pisum (pea aphid) and Mariner44_CB is from Caenorhabditis briggsae (nematode). b Multiple sequence alignment of the amino acids between the second D and the E residues showing the length difference between the Submariners and the other Tc1/mariners. Sequences are in the same order as in (a)
We attempted to corroborate this result using phylogenetic analysis of Tc1/mariner transposase amino acid sequences. However, the divergence between the Submariner sequences and those from the five well-established Tc1/mariner clades, as well as the divergences among the well-established Tc1/mariner clades themselves, were sufficiently great that unambiguous alignment was possible for few amino acid positions (Additional file 6: Figure S6). Consequently, phylogenetic analyses of this alignment resulted in poorly supported trees (not shown).
Submariner_Ps1 proliferated within the P. salinus genome
The paralogous empty site we identified within the P. salinus genome strongly suggests that Submariner_Ps1 transposed within the viral genome (Fig. 1b). We performed additional analyses to exclude the other possibilities, namely that (1) the P. salinus MITEs are artifacts of DNA contamination from the A. castellanii genome (as A. castellanii was used to culture P. salinus), and that (2) the P. salinus MITEs are the result of horizontal transfer into the viral genome multiple times from another organism.
First, to test for contamination, we performed BLASTn searches using every individual copy of Submariner_Ps1 as queries against the A. castellanii genome. Such searches identify only one other DNA element that shares sequence similarity with the Submariner_Ps1 TIRs. This DNA element is a 269-bp-long MITE (coordinates AEYA01002349.1: 6514–6782; e-value = 1e-04). Because there is only one Submariner-like MITE in A. castellanii, and the sequence similarity between this A. castellanii MITE and Submariner_Ps1 is restricted to their TIRs (Additional file 7: Figure S7), we can exclude the possibility that Submariner MITEs in the P. salinus genome are artifacts of DNA contamination from the A. castellanii genome.
Second, to test for multiple horizontal transfer events, we performed BLASTn searches using 100 bp of sequence immediately flanking all Submariner_Ps1 insertions as queries against the A. castellanii, P. dulcis, and P. salinus genomes. Such searches identify three Submariner_Ps1 insertions with flanking sequence on one side that retrieves significant hits from P. dulcis as well as from other locations within the P. salinus genome. Flanking sequence from an additional five Submariner_Ps1 insertions retrieves hits from other locations within the P. salinus genome. No such BLASTn searches retrieve significant hits from A. castellanii (Additional file 8: Table S1).
We also performed BLASTn searches against the NCBI nr database using the sequences of predicted genes present in P. salinus within 2 kb of Submariner_Ps1 insertions. Such searches identify (1) 15 Submariner_Ps1 insertions with an ortholog in P. dulcis on one side of the insertion, (2) two Submariner_Ps1 insertions with orthologs in P. dulcis on both sides of the insertion (coordinates 196891–197139 and 2363527–2363776), and (3) one Submariner_Ps1 insertion with an ortholog in P. dulcis on one side of the insertion and 100 bp flanking sequence that retrieves a significant BLASTn hit from P. dulcis on the other side of the insertion (coordinates 756606–756849; Additional file 8: Table S1). One Submariner_Ps1 insertion with a P. dulcis predicted gene ortholog on one side is flanked on the other side by a predicted gene that retrieves significant hits from both the P. dulcis and A. castellanii genomes. No other such BLAST analyses return significant hits from the A. castellanii genome, although one Submariner_Ps1 insertion is flanked by a predicted gene that retrieves a significant hit from a copepod genome (Additional file 8: Table S1). Taken together, these results show that the majority of sequences flanking the MITE insertions in P. salinus have homologs in P. dulcis and thus can be considered of ancestral viral origin prior to the spread of the MITE in P. salinus. These results are consistent with the idea that Submariner_Ps1 amplified within the viral genome rather than being transferred horizontally into the viral genome multiple times from another organism.
Genomic distribution of Submariner_Ps1 in the P. salinus genome suggests exaptation
The proximity of all Submariner_Ps1 copies to annotated P. salinus ORFs, detailed insertion coordinates, and the ORFs into or near which they insert are summarized in Additional file 9: Table S2. Ten out of the 30 copies of Submariner_Ps1 are part of predicted ORFs, suggesting that these MITEs may have been exonized in the P. salinus genome to form novel proteins (Table 1). In eight cases, the Submariner_Ps1 insertion extends the ORF on either the 5′ (three cases) or 3′ (five cases) end. In the other two cases, the entire predicted ORF is composed of Submariner_Ps1 sequence (Table 1).
Table 1.
MITEs found within annotated genes in the P. salinus genome
| MITE coordinates in P. salinus | Gene associated with the MITE | Gene coordinates in P. salinus | Gene length (bp) | Predicted gene function | Length of overlap (bp) | MITE involved in predicted secondary structure |
|---|---|---|---|---|---|---|
| 148208–148428 | ps_155 | 148230–148322 | 93 | hypothetical protein | All 93 | Yes |
| 196891–197139 | ps_208 | 196674–196934 | 261 | hypothetical protein | 44, C end | Yes |
| 266075–266302 | ps_282 | 266076–266237 | 162 | hypothetical protein | All 162 | Yes |
| 659083–659327 | ps_683 | 658540–659259 | 720 | hypothetical protein | 177, C end | Yes |
| 707659–707892 | ps_736 | 707593–707739 | 147 | hypothetical protein | 80, N end | Yes |
| 1279645–1279868 | ps_1360 | 1276933–1279722 | 2790 | hypothetical protein | 78, N end | Yes |
| 1298951–1299182 | ps_1377 | 1299004–1299717 | 714 | hypothetical protein | 179, N end | Yes |
| 2316942–2317180 | ps_2397 | 2316744–2316953 | 210 | hypothetical protein | 12, C end | No |
| 2363527–2363776 | ps_2438 | 2363321–2363686 | 366 | hypothetical protein | 160, C end | Yes |
| 2373978–2374199 | ps_2448 | 2372753–2373989 | 1237 | 2OG-Fe(II) oxygenase superfamily | 12, C end | No |
Only one of the ten predicted ORFs associated with MITEs has a homolog in any other genome (Table 1). Because of the large evolutionary distance between pandoraviruses and all other known organisms and viruses, genome annotation produced a large number of predicted ORFs with no identifiable homologs in other taxa (i.e. ORFans), consistent with the results from other giant virus genome annotations [46]. In the absence of confirmatory datasets (e.g. transcriptomic or proteomic data), some predicted ORFs are likely to be false positives. To understand if the predicted MITE-associated ORFs in P. salinus encode amino acid sequences that form stable secondary structures, which would suggest that they may be actual protein-coding genes, we used PSIPRED [47]. All ten such translated ORFs are predicted to form some stable secondary structures (e.g. alpha helices and/or beta strands), with the MITE sequences contributing to the secondary structure in eight of the ten cases (Table 1, Additional file 10: Figure S8). In addition, because of the high coding density of the P. salinus genome, all Submariner_Ps1 insertions are necessarily close to predicted ORFs, raising the possibility that they may also contribute to regulatory evolution. Although our results are suggestive, further experimental validation is required to investigate whether any MITE insertions have been exapted as new coding, or otherwise functional, sequence.
Discussion
The discovery of giant viruses forced biologists to radically rethink previously held ideas about the upper limits of viral genome size and complexity [48]. Inspired by the early discovery of Mimivirus [25, 49], targeted searches during the past decade for new, previously undescribed giant viruses have uncovered a spectacular diversity of forms [26, 50], and the mechanisms by which they persist and reproduce within their host cells are the subject of intense research [51, 52]. Because giant viruses are so different from other viruses in genome size, particle size, and enzymatic capacity, their discovery sparked a lively debate about their origins [30, 31, 53–60]. Recent phylogenomic analyses support the independent origins of the three currently known giant virus lineages – pithovirus, the pandoraviruses, and the mimiviruses, all with genomes ≥500 kb – from ancestors within the “Megavirales” with moderately sized genomes, reflecting large-scale accumulation of sequences from multiple donors from all three domains of cellular life [17, 30, 46, 50, 61, 62]. Such genomic expansion was likely facilitated by the evolution of DNA replication machinery capable of replicating larger genomes [63].
Our results are consistent with this view of genomic expansion in giant viruses; the MITE we identify in P. salinus is another example of sequence accumulation underlying genome size increase. However, this particular case of sequence acquisition by a giant virus is notable for several reasons. First, to our knowledge, this is the first example of a predominantly eukaryotic canonical Class II TE (i.e. a Tc1/mariner) colonizing a giant virus, although other TEs (e.g. IS sequences of the bacterial and archaeal IS607 family) have previously been reported in giant virus genomes [17, 30, 31, 46]. Second, we present evidence suggesting that the MITE in P. salinus transposed within the viral genome. In contrast, evidence that other TEs have transposed within viral genomes has been lacking, although previous studies reported this as a possibility [17, 30, 31]. Third, the MITE in P. salinus is present at high copy numbers relative to TEs in other viral genomes and, based on predicted ORFs, some of these copies may have contributed novel protein-coding sequence to the virus.
More generally, comparative genomic analyses across the three domains of cellular life and numerous viral lineages are revealing a complex picture of horizontal transfers among genomes; transfer rates differ among donor/recipient pairs as well as among types of sequences [64–66]. Given such asymmetries, mobile genetic elements that have overcome impediments to colonization across the multiple domains, as well as the viruses, are important models for understanding what limits horizontal transfer across, and outside of, the Tree of Life. IS607 sequences are one such mobile element. These primarily prokaryotic sequences have colonized some eukaryotes as well as giant viruses, although their capacity for transposition outside of prokaryotes remains uncertain [66]. Herein, we demonstrate that Tc1/mariner TEs are another such mobile genetic element. Previously, Tc1/mariner elements and their MITE derivatives had been identified in a wide variety of protozoans [42, 67, 68], plants [69], fungi [37, 70], and metazoans [71, 72], and their related prokaryotic IS sequences had been identified in diverse bacteria and archaea [73, 74]. We report the colonization of a giant virus genome by a MITE derived from an apparently novel Tc1/mariner subgroup with representatives from all three domains of cellular life, expanding the range of this superfamily of TEs even further.
How might the Submariner_Ps1 MITEs have colonized and spread within the P. salinus genome? Based on TIR sequence similarity, as well as the fact that Acanthamoeba is a likely host of P. salinus, it is quite possible that the amoeba-encoded Submariner_Ac1 transposase once mobilized Submariner_Ps1 MITEs in the P. salinus genome. However, the viral MITEs are unlikely to have originated as an internal deletion derivative of Submariner_Ac1 because sequence similarity between the two transposons is largely restricted to their TIRs (Fig. 2c). Thus, Submariner_Ps1 MITEs likely trace their origin to an autonomous transposon related to, but distinct from, Submariner_Ac1. This progenitor element could have occurred in the viral genome or the genome of the viral host (i.e. A. castellanii or another Acanthamoeba). Alternatively, because free-living amoebas ingest a variety of microorganisms through phagocytosis, many of which are resistant to digestion and stably coexist “in sympatry” within the amoeba [75, 76], the progenitor element could have occurred in another amoebal symbiont. Extensive horizontal transfer of sequences among prokaryotic, eukaryotic, and viral microorganisms that stably coexist inside amoebas, as well as the host amoeba itself, has been reported, demonstrating that free-living amoebas serve as “melting pots” for genome evolution [17, 50, 66, 75–77]. Irrespective of the original source of the Submariner_Ps1 MITEs in P. salinus, we show a new combination of ingredients within this “melting pot” — a canonical TE within the genome of a giant virus.
Conclusion
Pandoraviruses were named in reference to the surprises their unusually large genomes likely concealed [26]. Herein, we have shown that the P. salinus genome has been colonized by a MITE derived from the Tc1/mariner superfamily of Class II DNA transposons, and that this MITE was likely mobilized within the viral genome. We have shown that an autonomous Tc1/mariner DNA transposon related to this MITE is present in the genome of a likely pandoravirus host, the amoeba A. castellanii. Our discovery highlights the remarkable ability of DNA transposons to colonize and shape genomes both across, and outside of, the Tree of Life. Our findings continue to blur the division between viral and cellular genomes, adhering to the emerging view that, despite fundamental differences between cellular organisms and viruses (e.g. reproduction by cell division versus virion production) [54], the content, dynamics, and evolution of the genomes of these different biological entities do not substantially differ from one another [78–81].
Materials and methods
Dataset
We downloaded genomic sequences of two pandoraviruses from GenBank [82] (P. salinus and P. dulcis; accession numbers KC977471 and KC977470, respectively). We also downloaded the assembled contigs (assembly version Acas_2.0) for the free-living amoeba A. castellanii (accessions AEYA01000001 to AEYA01002545) from GenBank.
Identification and characterization of repetitive sequences in pandoravirus genomes
We used RepeatScout (version 1.0.5) [83] to identify de novo repeats from the genomic sequences of P. salinus and P. dulcis; the l-mer length was set to 15 and other parameters were set to default values. Only repeats that were >50 bp in length and <50 % low-complexity sequence were included in downstream analysis. We used RepeatMasker (version 3.2.9, [84]) to identify the overall repeat content of each genome based on the corresponding custom repeat library generated with RepeatScout. The search engine for RepeatMasker was Cross_Match [85]. To confirm the boundaries of the repeat element identified in the P. salinus genome, we extracted the sequences of all full-length copies (minus ≤5 bp at each end), along with 60 bp of flanking sequences. We performed multiple sequence alignment of the 13 full-length elements, along with the 60 bp of flanking sequence, using ClustalW implemented in BioEdit (version 7.2.0) [86], and the alignment results were visualized in BioEdit, shading identities and similarities (shade threshold 75 %). We predicted the secondary structure of the repeat element using the mFold web server [87, 88].
Identification of a possible autonomous partner for the MITEs in the P. salinus genome
We looked for an autonomous DNA transposon that could have facilitated the proliferation of P. salinus MITEs in both the P. salinus genome as well as the genomes of all species with representation in public sequence databases. We used two independent methods to search the P. salinus genome. First, we used all the known proteins encoded by DNA transposons as queries to tBLASTn against the DNA sequences of the P. salinus genome, with an e-value cutoff of 1e-5. We excluded helicase, encoded by rolling circle DNA transposons (i.e. Helitrons), because they are not known to generate MITEs. Second, we used the consensus sequence of the P. salinus MITEs as the input for the Anchor function of the MITE Analysis Kit [34, 89] to retrieve longer elements bearing similar terminal sequences and coding sequences whose products share sequence similarity with known proteins encoded by DNA transposons. We checked the output of the MITE Analysis Kit manually to remove false output entries. We obtained the protein sequences encoded by DNA transposons used in these two methods from the TE-encoded protein database, available in the downloaded RepeatMasker package [90].
Next, to search for possible autonomous partners of the P. salinus MITEs in other genomes, we used the consensus sequence of the P. salinus MITEs as the query for homology searches (BLASTn) against the NCBI databases (Nucleotide collection, EST, STS, GSS, WGS, TSA, HTGS, last accessed on 2014 June 1). Finally, to identify other P. salinus MITE-related sequences in the A. castellanii genome, we used every MITE sequence identified in P. salinus as queries for homology searches (BLASTn) against the locally installed most recent assembly of the A. castellanii genome (Acas_2.0), and we manually checked every obtained hit.
Characterization of the possible autonomous partner for the MITEs in the P. salinus genome
We found a 1604 bp sequence representing a possible autonomous partner of the P. salinus MITEs in the A. castellanii genome. To characterize this putative transposon, we (1) used NCBI-BLAST2 to identify its TIR, (2) queried it against the NCBI Conserved Domain Database [35], and (3) queried it against the TE-encoded protein database [90] using BLASTx (e-value ≤1e−5). To identify potential paralogous empty sites, we used 100 bp of its flanking sequences as queries to BLASTn against the genomic sequences of A. castellanii. Based on our results, we named the putative transposon Submariner_Ac1, and we named the related MITE in the P. salinus genome Submariner_Ps1. To look for other related sequences within the A. castellanii genome, we used BLASTn with the Submariner_Ac1 sequence as the query.
To determine whether Submariner_Ac1 and the related Submariner_Ac2 belong to any well-characterized clade of Tc1/mariner DNA transposons, or to a previously uncharacterized clade, we used the complete nucleotide sequence of Submariner_Ac1 for homology searches (BLASTx) against the NCBI non-redundant protein database (nr). We examined the top 20 hits and kept one representative from each species not already represented in our BLASTx results from Repbase; this yielded five total sequences (four bacterial sequences – Beggiatoa sp. PS, gi|152068700; Deltaproteobacterium NaphS2, gi|300441029; Candidatus Magnetoglobus multicellularis str. Araruama, gi|571786598; and Desulfobacula sp. TS, gi|667676338; and one uncultured archaeal sequence, GZfos18F2, gi|52548731). These accession numbers correspond to the nucleotide sequences from which the transposase amino acid sequences were deduced. We aligned these five sequences, Submariner_Ac1 and Submariner_Ac2, the four hits we retrieved from Repbase, and two sequences from each of the five well-characterized clades of Tc1/mariner DNA transposons (Repbase IDs: Cirt2_CA, Fot1_FO, Gizmo1_EI, Gizmo2_EI, MOGWAI1_EI, MOGWAI2_EI, Mariner-1_NV, Mariner2_MT, BARI_DM, and FB4_DM) using PSI-Coffee, an aligner within the T-Coffee multiple alignment package that aligns distantly related protein sequences using homology extension [91, 92]. Based on this alignment, we identified the DDE catalytic amino acid triad, their associated conserved motifs, and their intervening sequences of amino acids.
We then generated a similar alignment, but including a non-Tc1/mariner transposase (Merlin1_CB) as an outgroup. We retained only amino acid positions with alignment scores of “good” (143 amino acid positions) and performed Bayesian phylogenetic analysis using a mixed model of amino acid substitution, implemented in MrBayes 3.2 [93]. We ran the analysis for 10,000,000 generations, sampling every 1000, with three heated chains. Twenty-five percent of the sampled trees were discarded as burn-in and convergence was verified by comparison of the average deviation of split frequencies between two independent runs. The limited phylogenetic signal in this short alignment yielded an unresolved tree. We limited the scope of our analysis to sequences within the Submariner subgroup (Submariner_Ac1, Submariner_Ac2, Mariner-1_AP, Mariner-2_AP, Mariner-3_AP, and Mariner44_CB, and the four bacterial sequences – gi|152068700, gi|300441029, gi|571786598, gi|667676338 – and one archaeal sequence – gi|52548731 – we identified from Genbank) and an outgroup from one of the well-characterized Tc1/mariner clades, performing alignment and phylogenetic analysis as above. In all cases, the distance to the outgroup resulted in low numbers of unambiguously alignable amino acid positions and spurious root placement, demonstrated by the different root attachment points recovered depending on the outgroup sequence used.
Examination of Submariner_Ps1 proliferation dynamics in the P. salinus genome
To characterize the proliferation history of Submariner_Ps1 in the P. salinus genome, we calculated the sequence divergences of Submariner_Ps1 elements from their consensus sequence using RepeatMasker, binned the divergence values, and plotted them as a frequency histogram. To determine whether Submariner_Ps1 proliferation occurred within the P. salinus genome, or whether the multiple Submariner_Ps1 insertions resulted from independent introductions from a different genome (e.g. a viral host), we used 100 bp of immediately flanking sequence from all 30 Submariner_Ps1 insertions to query both amoeba and pandoravirus genomes using BLASTn. We also used the sequences of all of the predicted genes present in P. salinus within 2 kb of each Submariner_Ps1 insertion as queries to BLASTn against the NCBI nr database.
Examination of Submariner_Ps1 exaptation in the P. salinus genome
We summarized the locations of all Submariner_Ps1 copies relative to annotated P. salinus genes to assess whether Submariner_Ps1 insertions were within, or in close proximity to, predicted ORFs. We used PSIPRED [47] to predict the secondary structure of the translated MITE-associated ORFs [94].
Acknowledgements
We thank members of the Mueller lab, D. Sloan, members of the Sloan lab, and anonymous reviewers for helpful discussion of the manuscript. This work was supported by the National Science Foundation (NSF-DEB 1021489 to RLM), the National Institutes of Health (R156M104868 to RLM and R01-GM077582 to CF), and by the Colorado State University Libraries Open Access Research and Scholarship Fund. The funding bodies had no role in the design, collection, analysis, and interpretation of data, manuscript writing, or decision to submit the manuscript for publication.
Abbreviations
- IS
Insertion sequence
- MITE
Miniature inverted-repeat transposable element
- ORF
Open reading frame
- TE
Transposable element
- TIR
Terminal inverted repeat
- TSD
Target site duplication
Additional files
Multiple sequence alignment of all 30 miniature inverted-repeat transposable element (MITE) copies. Sequences are named by their coordinates in the P. salinus genome.
The secondary structure of the P. salinus miniature inverted-repeat transposable element (MITE) consensus sequence [Repbase ID: Submariner_Ps1] predicted by mFold.
Pairwise alignment of a site with a miniature inverted-repeat transposable element (MITE) insertion (bottom sequence) and its paralogous empty site (top sequence) in P. salinus. Target site duplications are underlined in red.
Frequency histogram showing pairwise divergences between individual MITE insertions in P. salinus and the miniature inverted-repeat transposable element (MITE) consensus sequence.
Pairwise alignment of a site with a DNA transposon insertion (bottom sequence) and its paralogous empty site (top sequence) in A. castellanii. Target site duplications are underlined in red.
Multiple sequence alignment of (1) the putative transposase sequences from Submariner_Ac1 and Submariner_Ac2, (2) the four DNA transposase hits we obtained from Repbase using Submariner_Ac1 as a query (Mariner-1_AP, Mariner-2_AP, Mariner-3_AP, and Mariner44_CB), (3) two representative transposases from each of five well-established Tc1/mariner clades (Fot1, Pogo, Tc1, Gizmo, and Mogwai), and (4) the five hits obtained from the NCBI non-redundant protein database (nr) using Submariner_Ac1 as a query (four bacterial sequences — Beggiatoa sp. PS, gi|152068700; Deltaproteobacterium NaphS2, gi|300441029; Candidatus Magnetoglobus multicellularis str. Araruama, gi|571786598; and Desulfobacula sp. TS, gi|667676338, and one uncultured archaeal sequence, GZfos18F2, gi|52548731). Sequences are identified by their GenBank accession numbers or Repbase IDs, if applicable, which correspond to the nucleotide sequences from which the transposase amino acid sequences were deduced. The multiple alignments were generated by PSI-Coffee, an aligner within the T-Coffee multiple alignment package that aligns distantly related protein sequences using homology extension [83, 84]. Red arrows indicate the DDE amino acid triad that coordinates metal ion (Mg2+) binding during catalysis of typical cut-and-paste transposition.
Pairwise alignment of the miniature inverted-repeat transposable element (MITE) in the A. castellanii genome and the MITE in P. salinus (Submariner_Ps1) that has the highest sequence similarity (out of the 30 MITE copies) with the MITE in A. castellanii.
The search for homologs of the P. salinus miniature inverted-repeat transposable element (MITE) flanking sequences in other genomes.
Proximity of P. salinus miniature inverted-repeat transposable elements (MITEs) to annotated P. salinus genes.
Predicted secondary structure of the hypothetical protein encoded by P. salinus gene Ps_1377, one of the predicted P. salinus open reading frames that includes miniature inverted-repeat transposable element (MITE) sequence. In this example, the first 60 amino acids of the predicted protein are MITE-derived and form three stable secondary structures: one β-strand and two α-helices. Secondary structure prediction was done using PSIPRED.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
CS conceived of the study. All authors contributed to study design. CS performed BLAST analyses with input from CF and RLM. RLM and ZW performed the phylogenetic analysis. All authors contributed to manuscript writing and read and approved the final manuscript.
Contributor Information
Cheng Sun, Email: transposable.element@gmail.com.
Cédric Feschotte, Email: cedric@genetics.utah.edu.
Zhiqiang Wu, Email: wu.zhiqiang.1020@gmail.com.
Rachel Lockridge Mueller, Email: rlm@colostate.edu.
References
- 1.Craig NL, Craigie R, Gellert M, Mobile LAM, DNA II. Washington. DC: American Society for Microbiology Press; 2002. [Google Scholar]
- 2.Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, et al. A unified classification system for eukaryotic transposable elements. Nat Rev Gen. 2007;8:973–82. doi: 10.1038/nrg2165. [DOI] [PubMed] [Google Scholar]
- 3.Bureau TE, Wessler SR. Tourist: a large family of small inverted repeat elements frequently associated with maize genes. Plant Cell. 1992;4:1283–94. doi: 10.1105/tpc.4.10.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fattash I, Rooke R, Wong A, Hui C, Luu T, Bhardwaj P, et al. Miniature inverted-repeat transposable elements: discovery, distribution, and activity. Genome. 2013;56:475–86. doi: 10.1139/gen-2012-0174. [DOI] [PubMed] [Google Scholar]
- 5.Feschotte C, Zhang X, Wessler SR. Miniature inverted-repeat transposable elements (MITEs) and their relationship with established DNA transposons. In: Craig N, Craigie R, Gellert M, Lambowitz A, editors. Mobile DNA II. Washington, DC: American Society of Microbiology Press; 2002. pp. 1147–58. [Google Scholar]
- 6.Wessler SR, Bureau TE, White SE. LTR-retrotransposons and MITEs: important players in the evolution of plant genomes. Curr Opin Genet Dev. 1995;5:814–21. doi: 10.1016/0959-437X(95)80016-X. [DOI] [PubMed] [Google Scholar]
- 7.Naito K, Zhang F, Tsukiyama T, Saito H, Hancock CN, Richardson AO, et al. Unexpected consequences of a sudden and massive transposon amplification on rice gene expression. Nature. 2009;461:1130–4. doi: 10.1038/nature08479. [DOI] [PubMed] [Google Scholar]
- 8.Kuang H, Padmanabhan C, Li F, Kamei A, Bhaskar PB, Ouyang S, et al. Identification of miniature inverted-repeat transposable elements (MITEs) and biogenesis of their siRNAs in the Solanaceae: New functional implications for MITEs. Genome Res. 2009;19:42–56. doi: 10.1101/gr.078196.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wei L, Gu L, Song X, Cui X, Lu Z, Zhou M, et al. Dicer-like 3 produces transposable element-associated 24-nt siRNAs that control agricultural traits in rice. Proc Natl Acad Sci. 2014;111:3877–82. doi: 10.1073/pnas.1318131111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Delihas N. Small mobile sequences in bacteria display diverse structure/function motifs. Mol Microbiol. 2008;67:475–81. doi: 10.1111/j.1365-2958.2007.06068.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Levin HL, Moran JV. Dynamic interactions between transposable elements and their hosts. Nat Rev Gen. 2011;12:615–27. doi: 10.1038/nrg3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bennetzen JL, Wang H. The contributions of transposable elements to the structure, function, and evolution of plant genomes. Annu Rev Plant Biol. 2014;65:505–30. doi: 10.1146/annurev-arplant-050213-035811. [DOI] [PubMed] [Google Scholar]
- 13.Miller DW, Miller LK. A virus mutant with an insertion of a copia-like transposable element. Nature. 1982;299:562–4. doi: 10.1038/299562a0. [DOI] [PubMed] [Google Scholar]
- 14.Fraser MJ, Smith GE, Summers MD. Acquisition of host cell DNA sequences by Baculoviruses: relationship between host DNA insertions and FP mutants of Autographa californica and Galleria mellonella nuclear polyhedrosis viruses. J Virol. 1983;47:287–300. doi: 10.1128/jvi.47.2.287-300.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jehle JA, Fritsch E, Nickel A, Huber J, Backhaus H. TC14.7: A novel lepidopteran transposon found in Cydia pomonella granulosis virus. Virology. 1995;207:369–79. doi: 10.1006/viro.1995.1096. [DOI] [PubMed] [Google Scholar]
- 16.Piskurek O, Okada N. Poxviruses as possible vectors for horizontal transfer of retroposons from reptiles to mammals. Proc Natl Acad Sci. 2007;104:12046–51. doi: 10.1073/pnas.0700531104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Filée J, Siguier P, Chandler M. I am what I eat and I eat what I am: acquisition of bacterial genes by giant viruses. Trends Genet. 2007;23:10–5. doi: 10.1016/j.tig.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 18.Marquez CP, Pritham EJ. Phantom, a new subclass of Mutator DNA transposons found in insect viruses and widely distributed in animals. Genetics. 2010;185:1507–17. doi: 10.1534/genetics.110.116673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dupuy C, Periquet G, Serbielle C, Bézier A, Louis F, Drezen JM. Transfer of a chromosomal Maverick to endogenous bracovirus in a parasitoid wasp. Genetica. 2011;139:489–96. doi: 10.1007/s10709-011-9569-x. [DOI] [PubMed] [Google Scholar]
- 20.Piégu B, Guizard S, Spears T, Cruaud C, Couloux A, Bideshi DK, et al. Complete genome sequence of invertebrate iridescent virus 22 isolated from a blackfly larva. J Gen Virol. 2013;94:2112–6. doi: 10.1099/vir.0.054213-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gilbert C, Chateigner A, Ernenwein L, Barbe V, Bézier A, Herniou EA, et al. Population genomics supports baculoviruses as vectors of horizontal transfer of insect transposons. Nat Commun. 2014;5:3348. doi: 10.1038/ncomms4348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thomas J, Schaack S, Pritham EJ. Pervasive horizontal transfer of rolling-circle transposons among animals. Gen Biol Evol. 2010;2:656–64. doi: 10.1093/gbe/evq050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu A-j. Sun X-y, Petherbridge L, Zhao Y-g, Nair V, Cui Z-z. Functional evaluation of the role of reticuloendotheliosis virus long terminal repeat (LTR) integrated into the genome of a field strain of Marek’s disease virus. Virology. 2010;397:270–6. doi: 10.1016/j.virol.2009.11.017. [DOI] [PubMed] [Google Scholar]
- 24.Colson P, De Lamballerie X, Yutin N, Asgari S, Bigot Y, Bideshi DK, et al. “Megavirales”, a proposed new order for eukaryotic nucleocytoplasmic large DNA viruses. Arch Virol. 2013;158:2517–21. doi: 10.1007/s00705-013-1768-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Raoult D, Audic S, Robert C, Abergel C, Renesto P, Ogata H, et al. The 1.2-megabase genome sequence of Mimivirus. Science. 2004;306:1344–50. doi: 10.1126/science.1101485. [DOI] [PubMed] [Google Scholar]
- 26.Legendre M, Bartoli J, Shmakova L, Jeudy S, Labadie K, Adrait A, et al. Thirty-thousand-year-old distant relative of giant icosahedral DNA viruses with a pandoravirus morphology. Proc Natl Acad Sci. 2014;111:4274–9. doi: 10.1073/pnas.1320670111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Desnues C, La Scola B, Yutin N, Fournous G, Robert C, Azza S, et al. Provirophages and transpovirons as the diverse mobilome of giant viruses. Proc Natl Acad Sci. 2012;109:18078–83. doi: 10.1073/pnas.1208835109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Santini S, Jeudy S, Bartoli J, Poirot O, Lescot M, Abergel C, et al. Genome of Phaeocystis globosa virus PgV-16T highlights the common ancestry of the largest known DNA viruses infecting eukaryotes. Proc Natl Acad Sci. 2013;110:10800–5. doi: 10.1073/pnas.1303251110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ogata H, Raoult D, Claverie J-M. A new example of viral intein in Mimivirus. Virol J. 2005;2:8. doi: 10.1186/1743-422X-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Filée J, Chandler M. Gene exchange and the origin of giant viruses. Intervirology. 2010;53:354–61. doi: 10.1159/000312920. [DOI] [PubMed] [Google Scholar]
- 31.Filée J, Chandler M. Convergent mechanisms of genome evolution of large and giant DNA viruses. Res Microbiol. 2008;159:325–31. doi: 10.1016/j.resmic.2008.04.012. [DOI] [PubMed] [Google Scholar]
- 32.Fitzgerald LA, Graves MV, Li X, Feldblyum T, Nierman WC, Van Etten JL. Sequence and annotation of the 369-kb NY-2A and the 345-kb AR158 viruses that infect Chlorella NC64A. Virology. 2007;358:472–84. doi: 10.1016/j.virol.2006.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Philippe N, Legendre M, Doutre G, Couté Y, Poirot O, Lescot M, et al. Pandoraviruses: amoeba viruses with genomes up to 2.5 Mb reaching that of parasitic eukaryotes. Science. 2013;341:281–6. doi: 10.1126/science.1239181. [DOI] [PubMed] [Google Scholar]
- 34.Janicki M, Rooke R, Yang G. Bioinformatics and genomic analysis of transposable elements in eukaryotic genomes. Chrom Res. 2011;19:787–808. doi: 10.1007/s10577-011-9230-7. [DOI] [PubMed] [Google Scholar]
- 35.NCBI’s conserved domain database. http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi.
- 36.Feschotte C, Mouches C. Evidence that a family of miniature inverted-repeat transposable elements (MITEs) from the Arabidopsis thaliana genome has arisen from a pogo-like DNA transposon. Mol Biol Evol. 2000;17:730–7. doi: 10.1093/oxfordjournals.molbev.a026351. [DOI] [PubMed] [Google Scholar]
- 37.Dufresne M, Hua-Van A, El Wahab HA, M’Barek SB, Vasnier C, Teysset L, et al. Transposition of a fungal miniature inverted-repeat transposable element through the action of a Tc1-like transposase. Genetics. 2007;175:441–52. doi: 10.1534/genetics.106.064360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miskey C, Papp B, Mátés L, Sinzelle L, Keller H, Izsvák Z, et al. The ancient mariner sails again: transposition of the human Hsmar1 element by a reconstructed transposase and activities of the SETMAR protein on transposon ends. Mol Cell Biol. 2007;27:4589–600. doi: 10.1128/MCB.02027-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang G, Nagel DH, Feschotte C, Hancock CN, Wessler SR. Tuned for transposition: molecular determinants underlying the hyperactivity of a Stowaway MITE. Science. 2009;325:1391–4. doi: 10.1126/science.1175688. [DOI] [PubMed] [Google Scholar]
- 40.Feschotte C, Swamy L, Wessler SR. Genome-wide analysis of mariner-like transposable elements in rice reveals complex relationships with stowaway miniature inverted repeat transposable elements (MITEs) Genetics. 2003;163:747–58. doi: 10.1093/genetics/163.2.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Feschotte C, Osterlund MT, Peeler R, Wessler SR. DNA-binding specificity of rice mariner-like transposases and interactions with Stowaway MITEs. Nucleic Acids Res. 2005;33:2153–65. doi: 10.1093/nar/gki509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yuan Y-W, Wessler SR. The catalytic domain of all eukaryotic cut-and-paste transposase superfamilies. Proc Natl Acad Sci. 2011;108:7884–9. doi: 10.1073/pnas.1104208108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hickman AB, Chandler M, Dyda F. Integrating prokaryotes and eukaryotes: DNA transposases in light of structure. Crit Rev Biochem Mol Biol. 2010;45:50–69. doi: 10.3109/10409230903505596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Feschotte C. Merlin, a new superfamily of DNA transposons identified in diverse animal genomes and related to bacterial IS1016 sequences. Mol Biol Evol. 2004;21:1769–80. doi: 10.1093/molbev/msh188. [DOI] [PubMed] [Google Scholar]
- 45.Shao H, Tu Z. Expanding the diversity of the IS630-Tc1-mariner superfamily: discovery of a unique DD37E transposon and reclassification of the DD37D and DD39D transposons. Genetics. 2001;159:1103–15. doi: 10.1093/genetics/159.3.1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yutin N, Wolf YI, Koonin EV. Origin of giant viruses from smaller DNA viruses not from a fourth domain of cellular life. Virology. 2014;466:38–52. doi: 10.1016/j.virol.2014.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.The PSIPRED Protein Sequence Analysis Workbench. http://bioinf.cs.ucl.ac.uk/psipred/.
- 48.Claverie J-M, Abergel C. The concept of virus in the post-megavirus era. In: Witzany G, editor. Viruses: Essential Agents of Life. Dordrecht: Springer Netherlands; 2012. pp. 187–202. [Google Scholar]
- 49.La Scola B, Audric S, Robert C, Jungang L, de Lamballerie X, Drancourt M, et al. A giant virus in amoebae. Science. 2003;299:2033. doi: 10.1126/science.1081867. [DOI] [PubMed] [Google Scholar]
- 50.Boyer M, Yutin N, Pagnier I, Barrassi L, Fournous G, Espinosa L, et al. Giant Marseillevirus highlights the role of amoebae as a melting pot in emergence of chimeric microorganisms. Proc Natl Acad Sci. 2009;106:21848–53. doi: 10.1073/pnas.0911354106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fischer MG, Condit RC. Editorial introduction to “Giant Viruses” special issue of Virology. Virology. 2014;466–467:1–2. doi: 10.1016/j.virol.2014.07.047. [DOI] [PubMed] [Google Scholar]
- 52.Chelikani V, Ranjan T, Kondabagil K. Revisiting the genome packaging in viruses with lessons from the “Giants”. Virology. 2014;466:15–26. doi: 10.1016/j.virol.2014.06.022. [DOI] [PubMed] [Google Scholar]
- 53.Legendre M, Arslan D, Abergel C, Claverie J-M. Genomics of Megavirus and the elusive fourth domain of life. Communicative Integr Biol. 2012;5:102–6. doi: 10.4161/cib.18624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Forterre P, Krupovic M, Prangishvili D. Cellular domains and viral lineages. Trends Microbiol. 2014;22:554–8. doi: 10.1016/j.tim.2014.07.004. [DOI] [PubMed] [Google Scholar]
- 55.Boyer M, Madoui M-A, Gimenez G, La Scola B, Raoult D. Phylogenetic and phyletic studies of informational genes in genomes highlight existence of a 4th domain of life including giant viruses. PLoS ONE. 2010;5 doi: 10.1371/journal.pone.0015530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Williams TA, Embley TM, Heinz E. Informational gene phylogenies do not support a fourth domain of life for nucleocytoplasmic large DNA viruses. PLoS ONE. 2011;6 doi: 10.1371/journal.pone.0021080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nasir A, Kim KM, Caetano-Anollés G. Viral evolution: primordial cellular origins and late adaptation to parasitism. Mobile Genetic Elements. 2012;2:247–52. doi: 10.4161/mge.22797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Krupovic M, Koonin EV. Polintons: a hotbed of eukaryotic virus, transposon and plasmid evolution. Nat Rev Microbiol. 2015;13:105–15. doi: 10.1038/nrmicro3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moniruzzaman M, LeCleir GR, Brown CM, Gobler CJ, Bidle KD, Wilson WH, et al. Genome of brown tide virus (AaV), the little giant of the Megaviridae, elucidates NCLDV genome expansion and host–virus coevolution. Virology. 2014;466:60–70. doi: 10.1016/j.virol.2014.06.031. [DOI] [PubMed] [Google Scholar]
- 60.Forterre P. Giant viruses: conflicts in revisiting the virus concept. Intervirology. 2010;53:362–78. doi: 10.1159/000312921. [DOI] [PubMed] [Google Scholar]
- 61.Filee J. Route of NCLDV evolution: the genomic accordion. Curr Opin Virol. 2013;3:595–9. doi: 10.1016/j.coviro.2013.07.003. [DOI] [PubMed] [Google Scholar]
- 62.Yutin N, Koonin EV. Pandoraviruses are highly derived phycodnaviruses. Biol Direct. 2013;8:25. doi: 10.1186/1745-6150-8-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Koonin EV, Krupovic M, Yutin N. Evolution of double‐stranded DNA viruses of eukaryotes: from bacteriophages to transposons to giant viruses. Ann N Y Acad Sci. 2015;1341:10–24. doi: 10.1111/nyas.12728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Keeling PJ, Palmer JD. Horizontal gene transfer in eukaryotic evolution. Nat Rev Gen. 2008;9:605–18. doi: 10.1038/nrg2386. [DOI] [PubMed] [Google Scholar]
- 65.Moran Y, Fredman D, Szczesny P, Grynberg M, Technau U. Recurrent horizontal transfer of bacterial toxin genes to eukaryotes. Mol Biol Evol. 2012;29:2223–30. doi: 10.1093/molbev/mss089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gilbert C, Cordaux R. Horizontal transfer and evolution of prokaryote transposable elements in eukaryotes. Gen Biol Evol. 2013;5:822–32. doi: 10.1093/gbe/evt057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Silva JC, Bastida F, Bidwell SL, Johnson PJ, Carlton JM. A potentially functional mariner transposable element in the protist Trichomonas vaginalis. Mol Biol Evol. 2005;22:126–34. doi: 10.1093/molbev/msh260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pritham EJ, Feschotte C, Wessler SR. Unexpected diversity and differential success of DNA transposons in four species of Entamoeba protozoans. Mol Biol Evol. 2005;22:1751–63. doi: 10.1093/molbev/msi169. [DOI] [PubMed] [Google Scholar]
- 69.Feschotte C, Wessler SR. Mariner-like transposases are widespread and diverse in flowering plants. Proc Natl Acad Sci. 2002;99:280–5. doi: 10.1073/pnas.022626699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Daboussi M-J, Capy P. Transposable elements in filamentous fungi. Annu Rev Microbiol. 2003;57:275–99. doi: 10.1146/annurev.micro.57.030502.091029. [DOI] [PubMed] [Google Scholar]
- 71.Plasterk RH, Izsvák Z, Ivics Z. Resident aliens: the Tc1/mariner superfamily of transposable elements. Trends Genet. 1999;15:326–32. doi: 10.1016/S0168-9525(99)01777-1. [DOI] [PubMed] [Google Scholar]
- 72.Robertson HM. Evolution of DNA transposons in eukaryotes. In: Mobile DNA II, editor. Edited by Craig NL, Craigie R, Gellert M, Lambowitz AM. Washington, DC: ASM Press; 2002. pp. 1093–110. [Google Scholar]
- 73.Filée J, Siguier P, Chandler M. Insertion sequence diversity in Archaea. Microbiol Mol Biol Rev. 2007;71:121–57. doi: 10.1128/MMBR.00031-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Siguier P, Gourbeyre E, Chandler M. Bacterial insertion sequences: their genomic impact and diversity. FEMS Microbiol Rev. 2014;38:865–91. doi: 10.1111/1574-6976.12067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bertelli C, Greub G. Lateral gene exchanges shape the genomes of amoeba-resisting microorganisms. Front Cell Infect Microbiol. 2012;2:110. doi: 10.3389/fcimb.2012.00110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Thomas V, Greub G. Amoeba/amoebal symbiont genetic transfers: lessons from giant virus neighbours. Intervirology. 2010;53:254–67. doi: 10.1159/000312910. [DOI] [PubMed] [Google Scholar]
- 77.Moliner C, Fournier P-E, Raoult D. Genome analysis of microorganisms living in amoebae reveals a melting pot of evolution. FEMS Microbiol Rev. 2010;34:281–94. doi: 10.1111/j.1574-6976.2009.00209.x. [DOI] [PubMed] [Google Scholar]
- 78.Claverie J-M, Ogata H, Audic S, Abergel C, Suhre K, Fournier P-E. Mimivirus and the emerging concept of “giant” virus. Virus Res. 2006;117:133–44. doi: 10.1016/j.virusres.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 79.Colson P, de Lamballerie X, Fournous G, Raoult D. Reclassification of giant viruses composing a fourth domain of life in the new order Megavirales. Intervirology. 2012;55:321–32. doi: 10.1159/000336562. [DOI] [PubMed] [Google Scholar]
- 80.Koonin EV. Virology: Gulliver among the Lilliputians. Curr Biol. 2005;15:R167–9. doi: 10.1016/j.cub.2005.02.042. [DOI] [PubMed] [Google Scholar]
- 81.Claverie J-M, Abergel C. Chapter two – open questions about giant viruses. Adv Virus Res. 2013;85:25–56. doi: 10.1016/B978-0-12-408116-1.00002-1. [DOI] [PubMed] [Google Scholar]
- 82.GenBank database. http://www.ncbi.nlm.nih.gov/genbank/.
- 83.Price AL, Jones NC, Pevzner PA. De novo identification of repeat families in large genomes. Bioinformatics. 2005;21:i351–8. doi: 10.1093/bioinformatics/bti1018. [DOI] [PubMed] [Google Scholar]
- 84.Homepage of the program RepeatMasker. http://www.repeatmasker.org/.
- 85.Source code for the program Cross_Match. http://www.phrap.org.
- 86.Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. In: Nucleic Acids Symposium Series; 1999. [Google Scholar]
- 87.The mfold web server. http://mfold.rna.albany.edu/?q=mfold.
- 88.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–15. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Homepage for MITE Analysis Kit (MAK). http://labs.csb.utoronto.ca/yang/MAK/.
- 90.Transposable element protein database. http://www.repeatmasker.org/RepeatProteinMask.html#database.
- 91.Notredame C, Higgins D, Heringa J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000;302:205–17. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- 92.Kemena C, Notredame C. Upcoming challenges for multiple sequence alignment methods in the high-throughput era. Bioinformatics. 2009;25:2455–65. doi: 10.1093/bioinformatics/btp452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, et al. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61:539–42. doi: 10.1093/sysbio/sys029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.McGuffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server. Bioinformatics. 2000;16:404–5. doi: 10.1093/bioinformatics/16.4.404. [DOI] [PubMed] [Google Scholar]