

Abstract
Background
Restriction site-Associated DNA sequencing (RAD-Seq) is widely applied to generate genome-wide sequence and genetic marker datasets. RAD-Seq has been extensively utilised, both at the population level and across species, for example in the construction of phylogenetic trees. However, the consistency of RAD-Seq data generated in different laboratories, and the potential use of cross-species orthologous RAD loci in the estimation of genetic relationships, have not been widely investigated. This study describes the use of SbfI RAD-Seq data for the estimation of evolutionary relationships amongst ten teleost fish species, using previously established phylogeny as a benchmark.
Results
The number of orthologous SbfI RAD loci identified decreased with increasing evolutionary distance between the species, with several thousand loci conserved across five salmonid species (divergence ~50 MY), and several hundred conserved across the more distantly related teleost species (divergence ~100–360 MY). The majority (>70%) of loci identified between the more distantly related species were genic in origin, suggesting that the bias of SbfI towards genic regions is useful for identifying distant orthologs. Interspecific single nucleotide variants at each orthologous RAD locus were identified. Evolutionary relationships estimated using concatenated sequences of interspecific variants were congruent with previously published phylogenies, even for distantly (divergence up to ~360 MY) related species.
Conclusion
Overall, this study has demonstrated that orthologous SbfI RAD loci can be identified across closely and distantly related species. This has positive implications for the repeatability of SbfI RAD-Seq and its potential to address research questions beyond the scope of the original studies. Furthermore, the concordance in tree topologies and relationships estimated in this study with published teleost phylogenies suggests that similar meta-datasets could be utilised in the prediction of evolutionary relationships across populations and species with readily available RAD-Seq datasets, but for which relationships remain uncharacterised.
Electronic supplementary material
The online version of this article (doi:10.1186/s13104-015-1261-2) contains supplementary material, which is available to authorized users.
Keywords: RAD-sequencing, Teleost phylogeny, Comparative mapping, Orthology
Background
The recent advances in next-generation sequencing (NGS) technologies has meant that genotyping-by-sequencing technologies (such as RAD-Seq) are being utilised in both model and non-model organisms for a variety of applications (e.g. [1–9]). Genome-wide multi-locus data, such as those generated by RAD-Seq, are particularly advantageous for the estimation of evolutionary relationships. This is because unlike estimates obtained by comparing a single orthologous locus across multiple species, methods to address the problem of incomplete lineage sorting using multi-locus datasets are available [4, 10–14].
A particular advantage of RAD-Seq is that the inference of cross-population and cross-species orthologous loci is potentially simplified by the use of the same rare-cutting restriction enzyme (such as SbfI) for the digestion of genomic DNA across all included individuals. Therefore, assuming no polymorphisms in the restriction site, the same genomic regions (i.e. homologous loci) can be sampled and concurrently sequenced across all individuals. The loss or gain of a restriction cut site due to the appearance of new mutations is likely to result in the identification of fewer orthologous RAD loci, particularly between more distantly related species. However, RAD-Seq protocols which involve digestion of genomic DNA using a single infrequent cutter followed by sonication of fragments are likely to be more robust to this issue than other RAD-like protocols (e.g. ddRAD [15]), where repeatable sampling of loci depends on the conservation of two restriction enzyme cut sites a certain distance apart on the genome. Overall, genetic relationships estimated using RAD data have been congruent with those seen in previously published literature (e.g. see Eaton et al. [4], Wang et al. [16]), suggesting that RAD data could prove useful in non-model taxa for which the evolutionary relationships are unknown.
Although RAD-Seq has been successfully applied in several phylogenetic studies (e.g. [4, 5, 16, 17]), these are typically based on sampling, sequencing and analysis by a single laboratory. The reproducibility of RAD loci across studies for the same species, and the ability to identify orthologous RAD loci across closely and distantly related species using cross-laboratory datasets, has not been widely investigated. In silico studies suggest that phylogenetic inference using RAD data may be restricted to relatively closely related species (<100 million years (MY) [18, 19]). Indeed, phylogenetic studies using empirical RAD-Seq datasets are restricted to the estimation of evolutionary relationships between closely related (<100 MY) species (e.g. [5, 20–22]). However, since RAD-Seq datasets from a wide variety of species and studies are now publically available, the utility of RAD-Seq for phylogeny estimation across more distantly related species can now be tested using experimentally-derived datasets. Additionally, while in silico phylogenetic studies have also investigated thresholds for inclusion of RAD loci with missing data (e.g. [23]), these thresholds have not been applied in real cross-laboratory datasets, where ‘missingness’ could arise for both technical as well as biological reasons.
Therefore, the overall aim of this study was to investigate the potential utility of cross-laboratory RAD-Seq data for estimation of phylogenetic relationships across closely and distantly related species, using ten species of teleost fish as an example. The specific aims of the study were to: (1) investigate the reproducibility of RAD data by aligning RAD sequences derived from different laboratories within-species; (2) investigate the performance of cross-laboratory RAD data in the inference of orthologous RAD loci and evolutionary relationships across species; and (3) investigate appropriate thresholds for inclusion of RAD loci for which there is missing data in some species.
Results and discussion
Datasets generated by RAD-Seq using the SbfI restriction enzyme were obtained from previously published studies for ten teleost fish species (five salmonid species and five non-salmonid teleost species). The five salmonid species included were: Atlantic salmon (Salmo salar), rainbow trout (Onchorhynchus mykiss), Chinook salmon (Onchorhynchus tshawytscha), sockeye salmon (Onchorhynchus nerka), and lake whitefish (Coregonus clupeaformis). The five non-salmonid species included were: three-spined stickleback (Gasterosterus aculeatus), Atlantic halibut (Hippoglossus hippoglossus), spotted gar (Lepisosteus oculatus), Baltic sea herring (Clupea harengus) and gudgeon (Gnathopogon sp.) (Table 1). The consensus RAD loci sequences (corresponding to the flanking sequences of the SbfI cleavage sites), which were inferred based on the identification of RAD loci across multiple individuals within the population under investigation, were obtained for each study. Therefore, unlike studies which infer orthologous RAD loci across multiple taxa, insufficient sequencing depth at a given consensus RAD locus within a species is unlikely to be a problem in this study. In the case of Atlantic salmon and rainbow trout, data derived from two and four different studies respectively were utilised (Table 1). Within each dataset, the consensus sequences of the RAD loci were trimmed to 60 base pairs (bp) to be consistent across all studies (see “Methods”).
Table 1.
Descriptions of the RAD sequences and the studies from which they were obtained
| Species | Reference | Consensus sequence availability | Initial number of sequences | Sequence length (bp) | Post-processed number of sequences | Protocol and pipeline details | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| RAD-Seq library preparation protocol | Fragment size selection window (bp) | Sequencing platform | Sequence analysis pipeline | Minimum depth coverage per locus | ||||||
| Chinook salmon (Oncorhynchus tshawytscha) | Brieuc et al. [24]. G3, 4(3) | Online (SE)e | 62,249 | 75 | 62,249 | Baird et al. [25] | 200–500 | Illumina GAII/HiSeq | STACKS | Locus sequenced in 135 (85%) individuals |
| Sockeye salmon (Oncorhynchus nerka) | Everett et al. [26]. BMC Genomics, 13(521) | Provided by authors (SE) | 64,613 | 60 | 64,613 | Baird et al. [25] Etter et al. [27] |
400–800 | Illumina GAII/HiSeq | Custom-written Perl scripts, Bowtie, Novoalign | 10 reads per allele per locus per individual |
| Rainbow trout (Oncorhynchus mykiss) | Hecht et al. [28]. G3, 2(9) | Provided by authors (SE) | 12,073 | 67 | 32,027 | Miller et al. [29] Baird et al. [25] |
200–500 | Illumina GAII/HiSeq 2000 | Perl scripts from Miller et al. (2012), Novoalign | 5 reads per locus per individual |
| Hale et al. [30]. G3, 3(8) | Provided by authors (SE) | 277,469 | 89 | Miller et al. [31] | 300–600 | Illumina HiSeq | Perl scripts from Miller et al. (2012), Novocraft | 5 reads per locus per individual | ||
| Hohenlohe et al. [6]. Molecular Ecology, 22(11) | Online (PE)f | 77,141 | 147–552a | Etter et al. [27] | 330–400 | Illumina HiSeq | STACKS | Locus sequenced in 1/60 (2%) individuals after pooling across individuals | ||
| Miller et al. [31]. Molecular Ecology, 21(2) | Online (SE)g | 40,649 | 68 | Baird et al. [25] Hohenlohe et al. [6] |
200–500 | Illumina HiSeq | Custom-written Perl scripts, Novoalign | Locus sequenced in 3 individuals | ||
| Atlantic salmon (Salmo salar) | Gonen et al. [2]. BMC Genomics, 15(166) | Provided by authors (PE) | 366,219 | 95 | 65,758 | Etter et al. [27] with modifications from Houston et al. [1] | 250–500 | Illumina HiSeq 2000 | RADtools, STACKS | 500 reads per locus across 96 individuals |
| Houston et al. [1]. BMC Genomics, 13(244) | Provided by authors (PE) | 66,073b | 95 | Baird et al. [25] Etter et al. [27] |
250–500 | Illumina GAIIx/HiSeq 2000 | RADtools | 5 reads per allele per locus per individual | ||
| Lake whitefish (Coregonus clupeaformis) | Gagnaire et al. [8]. Evolution, 67(9) | Provided by authors (SE) | 193,258 | 69 | 193,258 | Baird et al. [25] | 200–500 | Illumina HiSeq 2000 | STACKS | Locus is present in at least one mapping parent |
| Three-spined stickleback (Gasterosterus aculeatus) | Roesti et al. [32]. Molecular Ecology, 21(12) | Provided by authors (SE) | 31,118c | 64 or 138d | 31,118 | Baird et al. [25] | 200–500 | Illumina HiSeq 2000 | Novoalign, SAMtools | 12 reads per locus across 284 individuals |
| Atlantic halibut (Hippoglossus hippoglossus) | Palaiokostas et al. [33]. BMC Genomics, 14(566) | Provided by authors (SE) | 83,678 | 96 | 83,678 | Baird et al. [25] Etter et al. [27] with modifications from Houston et al. [1] |
300–550 | Illumina HiSeq 2000 | STACKS | 30 reads per locus per individual |
| Baltic sea herring (Clupea harengus) | Corander et al. [7] Molecular Ecology, 22(11) | Online (SE)h | 63,742 | 95 | 63,742 | Baird et al. [25] Hohenlohe et al. [6] Emerson et al. [34] |
200–500 | Illumina HiSeq 2000 | FLORAGENEX unitag assembler v2.0, FLORAGENEX pipeline | 5 reads per locus per individual |
| Spotted gar (Lepisosteus oculatus) | Amores et al. [35] Genetics, 188(4) | Provided by authors (SE) | 64,483 | 75 | 64,483 | Miller et al. [28] Baird et al. [25] Hohenlohe et al. [6] |
200–500 | Illumina GAIIx | STACKS | Locus sequenced in 85 (90%) individuals |
| Gudgeon (Gnathopogon sp.) | Kakioka et al. [36]. BMC Genomics, 14(32) | Online (SE)i | 44,109 | 70 | 44,109 | Etter et al. [27] | 300–500 | Illumina GAIIx/HiSeq 2000 | STACKS | 3 reads per locus per individual |
SE single-end RAD-Seq, PE paired-end RAD-Seq.
aPaired-end RAD sequencing generated contigs of variable length.
b2 files from two families, sequence counts: 70,207 and 70,739. Subsequently combined into one file with 66,073 common sequences.
c46 files (one per individual). Sequence count range: 25,840 – 42,618. Subsequently combined into one file with 31,118 common sequences.
dTwo separate sequencing studies were implemented, resulting in two different read lengths.
e http://www.g3journal.org/lookup/ suppl/doi:10.1534/g3.113.009316/-/DC1.
f http://datadryad.org/resource/ doi:10.5061/dryad.32b88
g http://onlinelibrary.wiley.com/ doi:10.1111/j.1365-294X.2011.05305.x/
hdoi:10.5061/dryad.jr56h.
Sharing of RAD loci across populations
To investigate RAD data reproducibility across populations (and studies) within species, orthologous RAD loci shared between the two different populations of Atlantic salmon, and between the four different populations of rainbow trout, were identified (Table 1; see Additional file 1 for details). A substantial overlap between RAD loci identified across studies was seen, with 99.5% of Atlantic salmon and 78.8% of rainbow trout sequences being shared across the different studies (percentages are given relative to the study with the fewest number of RAD loci). The higher percentage obtained across the two distinct Atlantic salmon populations may be partly due to the data originating from the same laboratory, and, therefore, more similar library preparation protocols and downstream bioinformatic analyses for data filtering. Overall, the results highlight the ability of RAD-Seq to consistently identify the same RAD loci across studies, despite inevitable technical variation in sample library preparation, sequencing platforms and downstream filtering pipelines. For example, subtle difference in RAD library preparation could affect the reproducibility of loci across studies (see Mastretta-Yanes et al. [37] for a review), including variations in the size selection window used after the sonication step of the protocol. Further, analysis pipelines with relatively strict thresholds for retaining homologous RAD loci across individuals (i.e. the population level consensus sequences utilised in this study), which are required for increased confidence SNP calling and genotyping within a population, could result in a decrease in the number of consensus RAD loci retained per species. This would reduce the number of informative loci available for relationship estimation.
Sharing of RAD loci across species
The correct inference of sequence orthology across species is critical when estimating evolutionary relationships. As such, there is an abundance of literature on best practices for the inference of orthology, typically conditional on the availability of published reference genome sequences (e.g. see [38–40]). In the absence of well-assembled and annotated reference genomes for all included species, sequence similarity is thought to be a reliable way of inferring orthology [18], with higher power to detect orthologous relationships expected with longer sequences. However, the ability to detect orthologous loci based on sequence similarity decreases with increasing evolutionary distance due to the accumulation of mutations. This can be further complicated by major genomic rearrangements, such as the genome duplication that occurred in the Salmonidae [41, 42]. For RAD-Seq specifically, polymorphic variation in the restriction enzyme cut site, variation in methylation status of the locus (if the restriction enzyme is methylation sensitive), or genome rearrangements may further decrease the number of orthologous RAD loci identified [4, 20, 23, 43, 44]. Typical RAD-Seq analysis software (e.g. Stacks [45, 46] and PyRAD [47]) can readily identify homologous RAD loci within populations of individuals, but not necessarily across species when using consensus RAD loci sequences defined at the population level. One way of utilising these software in cross-laboratory and cross-species analyses would be to set the minimum coverage per locus (i.e. stack depth) to one within a given species and then to conduct comparisons across species to identify orthologous loci. In this study, cross-species orthologous loci were identified by pairwise and cross-species BLAST alignments, since BLAST alignment of sequences has been shown to reliably infer orthologous loci across species in the absence of reference genomes as utilised in similar studies (e.g. [26]).
To identify orthologous RAD loci using cross-laboratory datasets, pairwise alignments of consensus RAD sequences across the ten teleost species of varying levels of evolutionary relatedness was conducted. Firstly, pairwise alignments were clustered across salmonid species using strict alignment parameters (95% sequence identity, ≤2 base mismatch, minimum alignment length 50 bp) and, secondly, across all ten teleost species, using more relaxed parameters for alignment (85% sequence identity, ≤10 base mismatch, minimum alignment length 45 bp) (see “Methods” and Additional file 2 for further details).
A large number of orthologous loci were identified between the pairs of salmonid species, ranging from 6,500 to 16,000 (Additional file 3) when using strict alignment parameters. As expected, when alignment parameters were relaxed as described above, the number of putative orthologous RAD loci identified between pairs of salmonid species increased, ranging from 11,000 to 19,500 loci (Additional file 3). This may be due to the increased ability to infer orthology between RAD loci which lie within less conserved regions of the genome of these closely related species (divergence <50 MYA [48]), although a relaxation of alignment parameters is also likely to increase the number of false positive orthologies. Approximately half of the RAD loci were shared between pairs of Oncorhynchus species (rainbow trout, sockeye salmon, Chinook salmon). Sequence clustering based on these pairwise alignments identified a total of 3,050 loci with sequence present in all five salmonid species (‘clusters’) (Table 2). To investigate the effect of including RAD loci that are missing in some species, clusters with at least three sequences from three different salmonid species were identified. A total of 22,710 such RAD loci were identified, of which 78 were removed due to containing sequences which were assigned to multiple clusters (potential paralogous regions), leaving 22,632 clusters for further analysis (Table 2).
Table 2.
Number of RAD locus clusters and interspecific variants identified for each analysis
| Species | Parameters | Analysis pipeline | Minimum taxon coverage | Number of orthologous RAD loci | Number (%) of orthologous RAD loci in genes | Number of variants for relationship estimation | Range of missing interspecific variants in included species | Percentage of missing data in RAxML matrix |
|---|---|---|---|---|---|---|---|---|
| Salmonids | Strict | BLASTN | 5 | 3,050 | 375 (12.3) | 6,959 | NA | 0 |
| Salmonids | Strict | BLASTN | ≥3 | 22,632 | 1,407 (6.2) | 39,890 | 3,135–21,480 | 25.09 |
| All ten species | Relaxed | BLASTN | 10 | 1 | 1 (100.0) | NA | NA | NA |
| All ten species | Relaxed | BLASTN | ≥7 | 137 | 106 (77.4) | 1,440 | 37–745 | 25.50 |
| All ten species | Relaxed | BLASTN | ≥5 | 452 | 321 (71.0) | 4,094 | 371–2,881 | 36.75 |
In contrast, the number of shared RAD loci between pairs of the five distantly related (non-salmonid) species was much lower, with fewer than 500 (<2%) identified in most of the pairwise comparisons (using the ‘relaxed’ alignment parameters described above). For example, the number of orthologous loci in common between lake whitefish and Chinook salmon (~50 MY) was ~16,600, compared to ~300 loci common between Chinook salmon and spotted gar (~360 MY)—an ~55-fold reduction. Of the non-salmonid species pairwise comparisons, stickleback and Atlantic halibut contained the highest number of orthologous RAD loci (~2,700, 9%) as expected due to their closer evolutionary relationship (<100 MY) compared to any other pair of non-salmonid species in the study [42, 49, 50]. This is approximately a six-fold reduction in the number of shared RAD loci compared to lake whitefish and Chinook salmon, where the time since the last most recent common ancestor is almost half that of stickleback and Atlantic halibut.
Only a single RAD locus was identified in all ten species [predicted to occur within the gene coding for Transcription factor 7 (T cell specific, HMG box)]. Therefore, two inclusion thresholds were applied; (1) RAD loci with orthologous sequence in at least seven species (137 clusters); and (2) RAD loci with orthologous sequence in at least five species (4,945 clusters). To prevent bias in the estimation of evolutionary relationships, salmonid species-specific clusters were identified and removed (4,493 clusters), leaving 452 clusters with sequence for a minimum of five species including at least one non-salmonid.
Identification of genic RAD loci
Given the higher degree of conservation of coding (i.e. genic) regions over evolutionary time [51, 52], it is plausible that the majority of orthologous RAD loci in the current study originate from coding regions. Previous studies have suggested that RAD loci obtained from SbfI RAD-Seq analyses may be biased towards gene-rich regions of the genome, in part explained by the GC-rich nature of the SbfI recognition sequence [2, 26, 35, 44, 53]. To test this hypothesis, all RAD loci consensus sequences were repeat-masked and aligned to a custom-made database of known fish gene nucleotide sequences, with significant alignment (E-value <1e−5) being evidence for a genic RAD locus (see “Methods”). In each of the individual salmonid species, approximately 2% of the RAD loci were identified as genic, and ~15% of the cross-species orthologous RAD clusters were predicted to originate from genes (Table 2). For each of the other (non-salmonid) teleost species individually, the percentage of genic RAD loci was higher (ranging from 4 to 50%), and >70% of cross-species orthologous RAD loci were identified as genic (Table 2). Alignment of genic loci across species identified very few (1–3 loci) which contained indels, suggesting high sequence conservation both at the nucleotide and amino acid level across species.
The lower ability to detect genic RAD loci within individual salmonid species (~2%) as compared to the other teleost species (up to 50%) in this study may be explained by the much larger genome sizes of the salmonid species (e.g. Atlantic salmon, ~3 GB; [54]) compared to the generally more compact genomes of the non-teleost species (e.g. stickleback, ~530 MB; [55]). The salmonid genome is known to be highly repetitive, (e.g. large number of transposable elements, repetitive tandem elements, etc.) [41, 42, 56–58]. This could mean that a larger proportion of the genome is non-coding, resulting in the identification of a lower proportion of genic RAD loci over the genome as a whole compared to species with compact, less repetitive genomes. Alternatively, the lower proportion of genic RAD loci predicted within the salmonid species may be attributed to the absence of salmonid gene sequences in the nucleotide database used for alignment, and the closer evolutionary relationship of the other teleost species with those in the database. In the case of stickleback, which has a high-quality, annotated reference genome and was included in the nucleotide database, ~50% of the RAD sequences were identified as genic. Based on the size of the stickleback genome (~530 MB; [55]) and the total length of known stickleback gene sequences (~192 MB; Ensembl 78, [59]), ~36% of the stickleback genome is estimated to be genic.
The large discrepancy in the proportion of cross-species orthologous genic RAD loci between salmonid (~15%) and non-salmonid (>70%) species may be due to the higher genome conservation (both coding and non-coding regions) across the salmonid species, due to their closer evolutionary relatedness. Overall, these results support the hypothesis that SbfI RAD-Seq loci may be biased towards genic regions of the genome [26, 35, 44, 53], and this bias is useful for evolutionary and comparative genomics studies.
Relationship estimation
To our knowledge, the most comprehensive study of teleost phylogeny is that described in Near et al. [49] (232 fish species; nine coding sequences and fossil calibration times). Based on this phylogeny and the salmonid species relationships described in Shedko et al. [48], the expected relationships between the ten teleost species in the current study are given in Figure 1.
Figure 1.
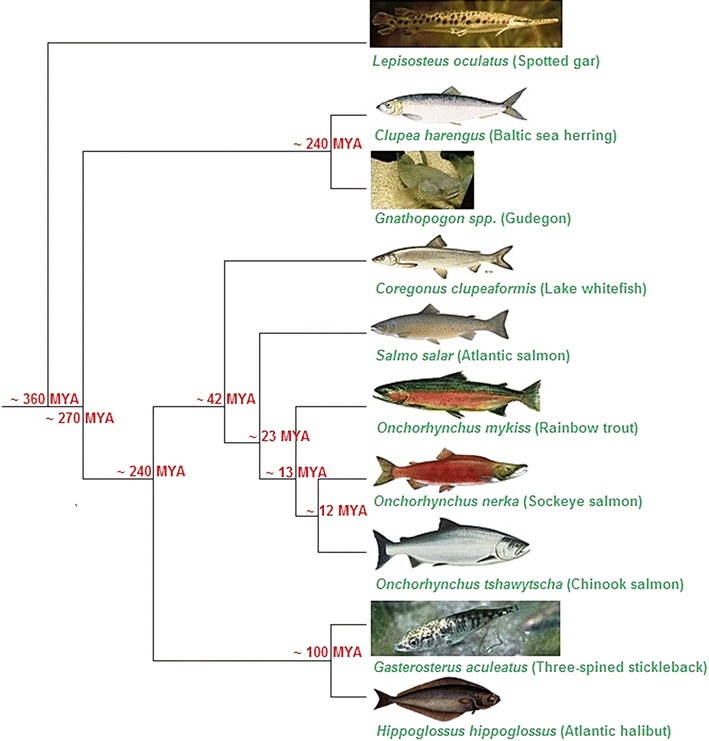
Expected evolutionary relationships as defined by Near et al. [49] and Shedko et al. [48]. Species images were taken from http://en.wikipedia.org/ or are published for open access use. Divergence times and branch lengths not drawn to scale. Divergence estimates for the non-salmonid teleost fish species were obtained from Near et al. [49], and divergence estimates for the salmonid species were obtained from Shedko et al. [48].
To test the utility of the cross-species and cross-laboratory RAD datasets in the construction of phylogenetic trees, multiple alignments of sequences within orthologous RAD clusters was conducted. This allowed the identification of interspecific single nucleotide variants, which were concatenated into a single sequence for each species and used to estimate evolutionary relationships (RAxML software; see Additional file 4 for RAxML parameters). RAxML input files used in all analyses are available at: doi:10.5061/dryad.bg6m0.
Whilst strict filtering thresholds applied in RAD-Seq studies often result in the removal of loci or individuals with excess missing data, recent simulation studies suggest that more relaxed thresholds could be favourable in resolving relationships [4, 23]. In the current study, a comparison was made between phylogenetic tree construction using stringent and more relaxed thresholds for RAD loci missingness across species.
Firstly, for estimating the phylogenetic relationships between the five salmonid species only, dataset 1 included RAD loci present in all five salmonid species (3,050 loci, 6,959 variants; Table 2), whilst dataset 2 included RAD loci present in at least three of the five salmonid species (22,632 loci, 39,890 variants; Table 2). Both datasets were able to recover the expected relationships between the five salmonid species (based on Shedko et al. [48]), with the three Oncorhynchus species forming a monophyletic group relative to Atlantic salmon and lake whitefish (all nodes >96% bootstrap support; Additional file 5, trees 1 and 2).
Likewise, across the ten teleost fish species, evolutionary relationships were estimated using variants derived from RAD loci common to at least seven of the ten species (137 loci, 1,440 variants; Table 2) and compared to the estimates using orthologous RAD clusters common to at least five of the ten species (452 loci, 4,094 variants; Table 2). Overall, tree topologies were consistent with previously published literature (Figures 1, 2; Additional file 5, trees 3 and 4). Monophyly of the Salmonidae and monophyly of the three Onchorhynchus species was predicted with 100% bootstrap support. Across both the salmonid and the teleost datasets, relaxing the threshold for inclusion of RAD loci in the analysis did not change estimated relationships or tree topology. Improvements in node support were also observed, for example, all salmonid species nodes were estimated with 100% support (vs. 98–100%) when the minimum taxon coverage at a RAD locus was reduced from seven to five of the ten species included (e.g. Additional file 5, trees 3 and 4). However, improvements in node support were not seen in all cases, for example, the node placing spotted gar as outgroup was not as strongly supported when the minimum taxon coverage was reduced (48–80%; Additional file 5, trees 3 and 4). Although bootstrap support is generally accepted as a reliable indicator of node accuracy, recent in silico studies suggest that this may not always be the case with RAD-Seq data [18]. Since true node support values obtained using empirical datasets are unknown, the accuracy of the reported bootstrap values cannot be quantified in this study.
Figure 2.
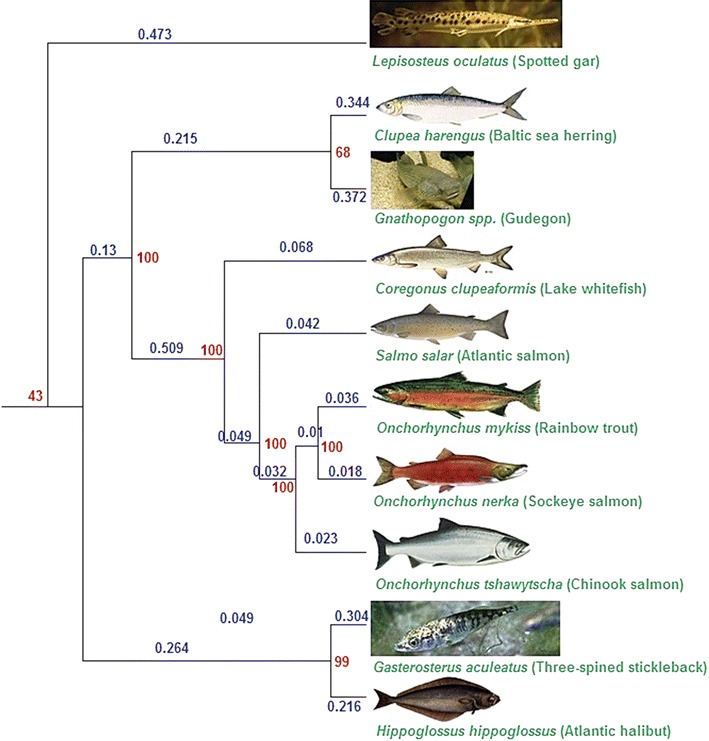
Example tree of all ten fish species obtained in this study using RAxML. Evolutionary relationships obtained using RAD data in this study were congruent with those of Near et al. [49] (teleost species) and Shedko et al. [48] (salmonid species) (Figure 1). Parameters—RAD loci present in at least five of ten species; 452 loci, 4,094 between-species variants. Branch lengths (given as percentages) estimated in RAxML are given along individual branches (in blue), and node bootstrap support values (1,000 bootstrap replicates) are given at individual nodes (in red). Branch lengths are not drawn to scale.
Although tree topologies were generally consistent with published studies, there were some noteworthy differences. For example, in Figure 1 (phylogeny from Near et al. [49] and Shedko et al. [48]), the node connecting stickleback and Atlantic halibut is placed as the sister group to the salmonid species, whereas in Figure 2 (this study, loci with a minimum taxon coverage of 5), the node connecting the Baltic sea herring and gudgeon is placed as sister species to the salmonid lineage, with 100% bootstrap support. However, this was not seen with loci with a minimum taxon coverage of 7 (Additional file 5, tree 3). Recent simulation studies have suggested that the resolution of RAD data is low when estimating relationships between distantly related species (>100 MY) [18–20]. However, although monophyly of the Onchorhynchus species (<13 MY) were predicted with 100% node support, relationships between the species differed depending on the minimum taxon coverage per locus, as well as when using salmonid species specific loci vs loci across all included species (Additional file 5). This is contrary to the expectations of better estimates of relationships between closely related species using RAD datasets as suggested by simulation studies [18–20], and suggests that caution must be applied when interpreting both shallow and deeper evolutionary relationships using this method.
In some cases (for example in the branch separating the salmonid species from the other five teleost species; Additional file 5, trees 3 and 4), branch lengths estimated using loci with a minimum of five species with sequence were approximately double that estimated using loci with a minimum of seven species with sequence. Therefore, while minor variation in the thresholds for inclusion of RAD loci absent in some species is unlikely to affect estimation of evolutionary relationships, it could potentially bias the estimated divergence times between more distantly related species (not estimated in this study). Therefore, the thresholds for inclusion of RAD loci with missing data should be considered and tested before utilising RAD loci for estimating relationships between species.
Conclusion
In this study, RAD-Seq datasets derived from different laboratories were utilised in the estimation of evolutionary relationships between ten teleost fish species. Within species and across populations, a large proportion of shared RAD loci were identified (78–100%), despite variation in laboratory techniques and bioinformatic pipelines. As expected, the number of orthologous RAD loci identified across species decreased as the evolutionary distance increased, ranging from ~3,000 between the most closely related salmonid species to ~450 between distantly related species. Multiple alignments of sequences within orthologous RAD loci allowed the identification of interspecific single nucleotide variants, which were used to estimate evolutionary relationships. These were consistent with previously published phylogenies, even across very distantly related species. Approximately 70% of the orthologous RAD loci used in the analysis of the ten teleost species were predicted to be genic, providing support for previous findings of a the bias of SbfI RAD loci towards genic regions, which is likely to facilitate relationship estimation between distantly related species. Overall, this study has highlighted the potential utility of experimentally-derived cross-laboratory RAD-Seq datasets in the estimation of evolutionary relationships across closely and distantly related species.
Methods
Sequence data
In a typical population genetics RAD-Seq bioinformatic pipeline, sequence reads derived from the flanking regions of the restriction enzyme are collapsed into a single ‘RAD locus’ [25]. For each locus, sequence reads are aligned within and then across individuals, and a single ‘consensus sequence’ is generated. In the case that a particular nucleotide site is polymorphic in a given population, the consensus sequences will show the allele with the highest frequency (>50%). Single-end SbfI RAD consensus sequences (i.e. both monomorphic and polymorphic consensus sequences) were obtained for Atlantic salmon (Salmo salar), rainbow trout (Onchorhynchus mykiss), three-spined stickleback (Gasterosterus aculeatus), gudgeon (Gnathopogon sp.), Chinook salmon (Onchorhynchus tshawytscha), sockeye salmon (Onchorhynchus nerka), spotted gar (Lepisosteus oculatus), lake whitefish (Coregonus clupeaformis), Baltic sea herring (Clupea harengus), and Atlantic halibut (Hippoglossus hippoglossus) (details specific for each study are given in Table 1). RAD-Seq studies using the SbfI restriction enzyme were chosen since this is the most commonly used protocol within aquatic species, and, therefore, had the most publically available data.
For rainbow trout and Atlantic salmon, data from four and two different studies respectively were obtained. For stickleback, consensus RAD sequences were generated within individuals (N = 46) and aligned to the reference genome, and population-level consensus sequences were unavailable (Table 1). For each of these three fish species, a single file of common RAD loci was produced using BLASTN alignments of all sequences (95% identity, ≤2 base mismatch), where common RAD loci were defined if sequence for that locus was observed in more than a certain threshold number of populations/individuals (see Additional file 1).
Data filtering, processing and characterisation
The consensus sequence files from each of the ten species were processed as follows. To avoid bias in alignment parameters due to differences in sequence lengths [60, 61], all sequences were trimmed to 60 bp (the shortest read length amongst the studies). To limit the misleading alignment of sequences to multiple regions due to genomic repetitive elements, low complexity sequences were masked using RepeatMasker [62] (parameters: -s; -lib; -gccalc). To minimise the effect of repeat sequences in potentially duplicated regions of the salmonid species genomes, the Atlantic salmon repetitive element database (http://web.uvic.ca/grasp/salmon_v1.6) was additionally utilised as a library within RepeatMasker.
To investigate the previously reported bias of SbfI RAD-Seq to gene-rich regions of the genome [26, 35, 44, 53], trimmed and repeat-masked sequences for each of the ten species were individually aligned (TBLASTX; BLAST+ version 2.2.25+ ; [63]) to a custom-made database of nucleotide gene sequences. This database comprised gene sequences originating from Atlantic cod (Gadus morhua), puffer fish (Takifugu rubripes), medaka (Oryzias latipes), platyfish (Xiphophorus maculatus), spotted gar (Lepisosteus oculatus), three-spined stickleback (Gasterosterus aculeatus), Tetraodon (Tetraodon nigroviridis), tilapia (Oreochromis niloticus) and zebrafish (Danio rerio) (Ensembl 78 [59]). Alignment significance was taken at E-value <1e−5.
Identification of cross-species orthologous RAD loci
To identify RAD loci conserved across species, pairwise BLASTN analyses of the trimmed and repeat-masked consensus RAD sequences were conducted (‘blastn’ alignment algorithm; BLAST+ version 2.2.25+ ; [63]). The most significant alignment for each sequence (i.e. ‘best hit’) was extracted. Two files of best hits were created: (1) within salmonid species only; and (2) across all ten species (including the salmonid species).
Best hit alignment files were quality-checked and filtered based on the following thresholds: (1) within salmonid species only, using ‘strict’ alignment parameters of ≥95% percentage identity, ≥50 bp alignment length and ≤2 base mismatches; and (2) between all ten species, using more ‘relaxed’ alignment parameters of ≥85% percentage identity, ≥45 bp alignment length and ≤10 base mismatches. The stricter alignment thresholds within salmonids were chosen in an attempt to differentiate between both orthologous and paralogous regions of the salmonid genomes. Alignment parameters remained constant within each analysis (rather than varying parameters according to the evolutionary distance between species) such that: (1) consistency in parameters across all pairwise alignments was maintained, in order to aid comparisons of the number of loci identified between species of differing relatedness; and (2) the identification of misleading alignments (for example between sequences corresponding to conserved regions of the same gene family rather than the same RAD locus) is minimised. To minimise multiple alignments of sequences within salmonid species due to the recent (~90 MYA; [58]) salmonid specific genome duplication [41, 42] or due to uncharacterised repetitive elements across all species, all pairwise alignments were further filtered to retain only unique alignments (i.e. where the subject sequence was the best hit to a single query sequence). Two files of pairwise best hits were created: (1) within salmonids; and (2) across all ten fish.
To identify orthologous RAD loci across groups of species of differing levels of evolutionary relatedness, pairwise alignments were clustered, first within the salmonid species only based on the strict pairwise alignments, and second, across all ten species, based on the relaxed alignment parameters. The clustering pipeline was implemented as follows (also see Additional file 2). Using the two files of filtered pairwise best hits, sequence clusters were inferred if RAD locus sequences across all included species all aligned to each other respectively as the most significant and unique match. To limit the effect of paralogous sequences on inferring clusters across the salmonids and unidentified repetitive elements across all species, clusters containing sequences which were assigned to multiple clusters were removed. Clusters containing more than one RAD locus sequence from a single species were removed.
To analyse the effect of incorporating RAD loci which were ‘absent’ for a given species (i.e. no ortholog identified in the available dataset), clusters were filtered using varying thresholds for sequence absence. Within the salmonid species strict analysis, clusters containing sequences from all five salmonid species and clusters containing sequences from at least three of the five salmonid species were retained. Across all ten species, only a single RAD locus cluster was identified. Therefore, downstream analyses were conducted using clusters with a minimum of seven sequences from at least seven different species or a minimum of five sequences from at least five different species. To prevent bias in the estimation of evolutionary relationships, these clusters were further filtered to remove salmonid species-specific clusters, i.e. clusters that contained sequences originating from salmonid species only. The proportion of clusters within genic regions of the genome was quantified, based on alignment to the custom-made fish nucleotide gene database, as described above.
Reconstructing teleost fish phylogeny using RAD data
To test the utility of cross-laboratory RAD-Seq data to infer teleost species relationships, cross-species orthologous RAD locus clusters described above were used to construct phylogenetic trees. For each identified RAD locus cluster, sequences for each species within the cluster were extracted. If absence of a RAD locus for a given species was permitted (as in salmonid dataset 2 and all fish datasets 1 and 2), species with no sequence for that locus were assigned a string of 60 * ‘N’. Sequences within a cluster were aligned using the MUSCLE software (version 3.8.31 [64]), and the resulting alignments were investigated for the presence of between-species single nucleotide variants. Alleles for each variant for each species across all RAD loci were concatenated into a single sequence. Concatenated variant sequence files were converted into the PHYLIP format [65] for input into the RAxML software (version 8 [66]) (see Additional file 4 for details on RAxML parameters). RAxML employs a maximum likelihood based algorithm for phylogeny inference, and was chosen since it allows for correction of ascertainment bias which may arise when using variants for relationship estimation. RAxML was run using 1,000 bootstraps for all analyses.
Authors’ contributions
SG conceived and designed the study, performed the analysis and wrote the paper. RDH and SCB contributed to the study design and the writing of the paper. All authors read and approved the final manuscript.
Acknowledgements
The author would like to acknowledge the following people for the donation of RAD-Seq data used in this analysis: Dr Pierre-Alexandre Gagnaire (Institut des Sciences de l’Evolution de Montpellier, France), Dr Daniel Berner (Universität Basel, Switzerland), Dr Krista Nichols (Purdue University, US), Dr Matt Hale (Purdue University, US), Dr Ben Hecht (Purdue University, US), Dr Meredith Everett (University of Washington, US), Dr Michaël Bekaert (Institute of Aquaculture, Stirling), Dr Christos Palaiokostas (Institute of Aquaculture, Stirling), Dr Angel Amores (University of Oregon, US), and Dr Julian Catchen (University of Oregon, US). We acknowledge funding from the Biotechnology and Biological Sciences Research Council (BBSRC) (BB/H022007/1) and from the Roslin Institute’s BBSRC Institute Strategic Funding Grant.
Compliance with ethical guidelines
Competing interests The authors declare that they have no competing interests.
Additional files
Inferring consensus RAD sequences within species.
Clustering of pairwise BLASTN alignments into cross-species orthologous RAD loci.
Number and percentage of shared RAD loci identified by pairwise BLASTN alignments.
Parameters for phylogenetic tree construction using RAxML.
Estimated phylogenetic relationships.
Contributor Information
Serap Gonen, Email: Serap.gonen@roslin.ed.ac.uk.
Stephen C Bishop, Email: Stephen.bishop@roslin.ed.ac.uk.
Ross D Houston, Email: Ross.houston@roslin.ed.ac.uk.
References
- 1.Houston RD, Davey JW, Bishop SC, Lowe NR, Mota-Velasco JC, Hamilton A, et al. Characterisation of QTL-linked and genome-wide restriction site-associated DNA (RAD) markers in farmed Atlantic salmon. Bmc Genomics. 2012 doi: 10.1186/1471-2164-13-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gonen S, Lowe NR, Cezard T, Gharbi K, Bishop SC, Houston RD. Linkage maps of the Atlantic salmon (Salmo salar) genome derived from RAD sequencing. BMC Genom. 2014;15(1):166. doi: 10.1186/1471-2164-15-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reitzel AM, Herrera S, Layden MJ, Martindale MQ, Shank TM. Going where traditional markers have not gone before: utility of and promise for RAD sequencing in marine invertebrate phylogeography and population genomics. Mol Ecol. 2013;22(11):2953–2970. doi: 10.1111/mec.12228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eaton DAR, Ree RH. Inferring phylogeny and introgression using RADseq data: an example from flowering plants (Pedicularis: Orobanchaceae) Syst Biol. 2013;62(5):689–706. doi: 10.1093/sysbio/syt032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hipp AL, Eaton DAR, Cavender-Bares J, Fitzek E, Nipper R, Manos PS. A framework phylogeny of the American Oak clade based on sequenced RAD data. PLoS One. 2014;9(4):e93975. doi: 10.1371/journal.pone.0093975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hohenlohe PA, Bassham S, Etter PD, Stiffler N, Johnson EA, Cresko WA. Population genomics of parallel adaptation in threespine stickleback using sequenced RAD tags. PLoS Genet. 2010 doi: 10.1371/journal.pgen.1000862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Corander J, Majander KK, Cheng L, Merila J. High degree of cryptic population differentiation in the Baltic Sea herring Clupea harengus. Mol Ecol. 2013;22(11):2931–2940. doi: 10.1111/mec.12174. [DOI] [PubMed] [Google Scholar]
- 8.Gagnaire PA, Normandeau E, Pavey SA, Bernatchez L. Mapping phenotypic, expression and transmission ratio distortion QTL using RAD markers in the Lake Whitefish (Coregonus clupeaformis) Mol Ecol. 2013;22(11):3036–3048. doi: 10.1111/mec.12127. [DOI] [PubMed] [Google Scholar]
- 9.Chen AL, Liu CY, Chen CH, Wang JF, Liao YC, Chang CH, et al. Reassessment of QTLs for late blight resistance in the tomato accession L3708 using a restriction site associated DNA (RAD) linkage map and highly aggressive isolates of Phytophthora infestans. PLoS One. 2014 doi: 10.1371/journal.pone.0096417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lynch M. The age and relationships of the major animal phyla. Evolution. 1999;53(2):319–325. doi: 10.2307/2640769. [DOI] [PubMed] [Google Scholar]
- 11.Leache AD, Chavez AS, Jones LN, Grummer JA, Gottscho AD, Linkem CW. Phylogenomics of Phrynosomatid lizards: conflicting signals from sequence capture versus restriction site associated DNA sequencing. Genome Biol Evol. 2015;7(3):706–719. doi: 10.1093/gbe/evv026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maddison WP, Knowles LL. Inferring phylogeny despite incomplete lineage sorting. Syst Biol. 2006;55(1):21–30. doi: 10.1080/10635150500354928. [DOI] [PubMed] [Google Scholar]
- 13.Gontcharov AA, Marin B, Melkonian M. Are combined analyses better than single gene phylogenies? A case study using SSU rDNA and rbcL sequence comparisons in the Zygnematophyceae (Streptophyta) Mol Biol Evol. 2004;21(3):612–624. doi: 10.1093/molbev/msh052. [DOI] [PubMed] [Google Scholar]
- 14.Castresana J. Topological variation in single-gene phylogenetic trees. Genome Biol. 2007;8(6):216. doi: 10.1186/gb-2007-8-6-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE. Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. Plos One. 2012 doi: 10.1371/journal.pone.0037135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang XQ, Zhao L, Eaton DAR, Li DZ, Guo ZH. Identification of SNP markers for inferring phylogeny in temperate bamboos (Poaceae: Bambusoideae) using RAD sequencing. Mol Ecol Resour. 2013;13(5):938–945. doi: 10.1111/1755-0998.12136. [DOI] [PubMed] [Google Scholar]
- 17.Jones JC, Fan SH, Franchini P, Schartl M, Meyer A. The evolutionary history of Xiphophorus fish and their sexually selected sword: a genome-wide approach using restriction site-associated DNA sequencing. Mol Ecol. 2013;22(11):2986–3001. doi: 10.1111/mec.12269. [DOI] [PubMed] [Google Scholar]
- 18.Rubin BER, Ree RH, Moreau CS. Inferring phylogenies from RAD sequence data. PLoS One. 2012;7(4):e33394. doi: 10.1371/journal.pone.0033394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cariou M, Duret L, Charlat S. Is RAD-seq suitable for phylogenetic inference? An in silico assessment and optimization. Ecol Evol. 2013;3(4):846–852. doi: 10.1002/ece3.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cruaud A, Gautier M, Galan M, Foucaud J, Sauné L, Genson G, et al. Empirical assessment of RAD sequencing for interspecific phylogeny. Mol Biol Evol. 2014 doi: 10.1093/molbev/msu063. [DOI] [PubMed] [Google Scholar]
- 21.Viricel A, Pante E, Dabin W, Simon-Bouhet B. Applicability of RAD-tag genotyping for interfamilial comparisons: empirical data from two cetaceans. Mol Ecol Resour. 2014;14(3):597–605. doi: 10.1111/1755-0998.12206. [DOI] [PubMed] [Google Scholar]
- 22.Pante E, Abdelkrim J, Viricel A, Gey D, France SC, Boisselier MC, et al. Use of RAD sequencing for delimiting species. Heredity. 2015;114(5):450–459. doi: 10.1038/hdy.2014.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang H, Knowles LL. Unforeseen consequences of excluding missing data from next-generation sequences: simulation study of RAD sequences. Syst Biol. 2014 doi: 10.1093/sysbio/syu046. [DOI] [PubMed] [Google Scholar]
- 24.Brieuc MSO, Waters CD, Seeb JE, Naish KA. A dense linkage map for Chinook salmon (Oncorhynchus tshawytscha) reveals variable chromosomal divergence after an ancestral whole genome duplication event. G3 (Bethesda) 2014;4:447–460. doi: 10.1534/g3.113.009316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA, et al. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One. 2008 doi: 10.1371/journal.pone.0003376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Everett MV, Miller MR, Seeb JE. Meiotic maps of sockeye salmon derived from massively parallel DNA sequencing. BMC Genomics. 2012;13:521. doi: 10.1186/1471-2164-13-521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Etter PD, Preston JL, Bassham S, Cresko WA, Johnson EA. Local de novo assembly of RAD paired-end contigs using short sequencing reads. Plos One. 2011;6:e18561. doi: 10.1371/journal.pone.0018561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hecht BC, Thrower FP, Hale MC, Miller MR, Nichols KM. Genetic architecture of migration-related traits in rainbow and steelhead trout, Oncorhynchus mykiss. G3 (Bethesda) 2012;2:1113–1127. doi: 10.1534/g3.112.003137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miller MR, Dunham JP, Amores A, Cresko WA, Johnson EA. Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers. Genome Res. 2007;17:240–248. doi: 10.1101/gr.5681207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hale MC, Thrower FP, Berntson EA, Miller MR, Nichols KM. Evaluating adaptive divergence between migratory and nonmigratory ecotypes of a Salmonid fish, Oncorhynchus mykiss. G3 (Bethesda) 2013;3:1273–1285. doi: 10.1534/g3.113.006817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miller MR, Brunelli JP, Wheeler PA, Liu SX, Rexroad CE, Palti Y, et al. A conserved haplotype controls parallel adaptation in geographically distant salmonid populations. Mol Ecol. 2012;21:237–249. doi: 10.1111/j.1365-294X.2011.05305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roesti M, Moser D, Berner D. Recombination in the threespine stickleback genome patterns and consequences. Mol Ecol. 2013;22:3014–3027. doi: 10.1111/mec.12322. [DOI] [PubMed] [Google Scholar]
- 33.Palaiokostas C, Bekaert M, Khan MGQ, Taggart JB, Gharbi K, McAndrew BJ, et al. Mapping and validation of the major sex-determining region in Nile Tilapia (Oreochromis niloticus L.) using RAD sequencing. Plos One. 2013;8:e68389. doi: 10.1371/journal.pone.0068389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Emerson KJ, Merz CR, Catchen JM, Hohenlohe PA, Cresko WA, Bradshaw WA, Holzapfel CM (2010) Resolving postglacial phylogeography using high-throughput sequencing. PNAS 107(37). doi:10.1073/pnas.1006538107 [DOI] [PMC free article] [PubMed]
- 35.Amores A, Catchen J, Ferrara A, Fontenot Q, Postlethwait JH. Genome evolution and meiotic maps by massively parallel DNA sequencing: spotted gar, an outgroup for the teleost genome duplication. Genetics. 2011;188(4):799–808. doi: 10.1534/genetics.111.127324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kakioka R, Kokita T, Kumada H, Watanabe K, Okuda N. A RAD-based linkage map and comparative genomics in the gudgeons (genus Gnathopogon, Cyprinidae) BMC Genomics. 2013;14:32. doi: 10.1186/1471-2164-14-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mastretta-Yanes A, Arrigo N, Alvarez N, Jorgensen TH, Piñero D, Emerson BC. Restriction site-associated DNA sequencing, genotyping error estimation and de novo assembly optimization for population genetic inference. Mol Ecol Resour. 2015;15(1):28–41. doi: 10.1111/1755-0998.12291. [DOI] [PubMed] [Google Scholar]
- 38.Dewey CN. Positional orthology: putting genomic evolutionary relationships into context. Brief Bioinform. 2011;12(5):401–412. doi: 10.1093/bib/bbr040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kristensen DM, Wolf YI, Mushegian AR, Koonin EV. Computational methods for gene orthology inference. Brief Bioinform. 2011;12(5):379–391. doi: 10.1093/bib/bbr030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schmitt T, Messina DN, Schreiber F, Sonnhammer ELL. Letter to the Editor: SeqXML and OrthoXML: standards for sequence and orthology information. Brief Bioinform. 2011;12(5):485–488. doi: 10.1093/bib/bbr025. [DOI] [PubMed] [Google Scholar]
- 41.Volff JN. Genome evolution and biodiversity in teleost fish. Heredity. 2005;94(3):280–294. doi: 10.1038/sj.hdy.6800635. [DOI] [PubMed] [Google Scholar]
- 42.Berthelot C, Brunet F, Chalopin D, Juanchich A, Bernard M, Noël B, et al. The rainbow trout genome provides novel insights into evolution after whole-genome duplication in vertebrates. Nat Commun. 2014 doi: 10.1038/ncomms4657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Poland JA, Rife TW. Genotyping-by-sequencing for plant breeding and genetics. Plant Genome. 2012;5(3):92–102. doi: 10.3835/plantgenome2012.05.0005. [DOI] [Google Scholar]
- 44.Arnold B, Corbett-Detig RB, Hartl D, Bomblies K. RADseq underestimates diversity and introduces genealogical biases due to nonrandom haplotype sampling. Mol Ecol. 2013;22(11):3179–3190. doi: 10.1111/mec.12276. [DOI] [PubMed] [Google Scholar]
- 45.Catchen JM, Amores A, Hohenlohe P, Cresko W, Postlethwait JH. Stacks: building and genotyping loci de novo from short-read sequences. G3: Genes, Genomes. Genetics. 2011;1(3):171–182. doi: 10.1534/g3.111.000240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA. Stacks: an analysis tool set for population genomics. Mol Ecol. 2013;22(11):3124–3140. doi: 10.1111/mec.12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eaton DAR. PyRAD: assembly of de novo RADseq loci for phylogenetic analyses. Bioinformatics. 2014;30(13):1844–1849. doi: 10.1093/bioinformatics/btu121. [DOI] [PubMed] [Google Scholar]
- 48.Shedko SV, Miroshnichenko IL, Nemkova GA. Phylogeny of salmonids (Salmoniformes: Salmonidae) and its molecular dating: analysis of mtDNA data. Russ J Genet. 2013;49(6):623–637. doi: 10.1134/S1022795413060112. [DOI] [PubMed] [Google Scholar]
- 49.Near TJ, Eytan RI, Dornburg A, Kuhn KL, Moore JA, Davis MP, et al. Resolution of ray-finned fish phylogeny and timing of diversification. Proc Natl Acad Sci. 2012;109(34):13698–13703. doi: 10.1073/pnas.1206625109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Broughton RE, Betancur RR, Li C, Arratia G, Ortí G. Multi-locus phylogenetic analysis reveals the pattern and tempo of bony fish evolution. PLoS Curr. 2013 doi: 10.1371/currents.tol.2ca8041495ffafd0c92756e75247483e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cooper GM, Brown CD. Qualifying the relationship between sequence conservation and molecular function. Genome Res. 2008;18(2):201–205. doi: 10.1101/gr.7205808. [DOI] [PubMed] [Google Scholar]
- 52.Bergmiller T, Ackermann M, Silander OK. Patterns of evolutionary conservation of essential genes correlate with their compensability. PLoS Genet. 2012 doi: 10.1371/journal.pgen.1002803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bruneaux M, Johnston SE, Herczeg G, Merila J, Primmer CR, Vasemagi A. Molecular evolutionary and population genomic analysis of the nine-spined stickleback using a modified restriction-site-associated DNA tag approach. Mol Ecol. 2013;22(3):565–582. doi: 10.1111/j.1365-294X.2012.05749.x. [DOI] [PubMed] [Google Scholar]
- 54.Davidson WS, Koop BF, Jones SJM, Iturra P, Vidal R, Maass A, et al. Sequencing the genome of the Atlantic salmon (Salmo salar) Genome Biol. 2010;11(9):403. doi: 10.1186/gb-2010-11-9-403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jones FC, Grabherr MG, Chan YF, Russell P, Mauceli E, Johnson J, et al. The genomic basis of adaptive evolution in threespine sticklebacks. Nature. 2012;484(7392):55–61. doi: 10.1038/nature10944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Koop BF, Davidson WS (2008) Genomics and the genome duplication in salmonids. Fisheries for Global Welfare and Environment, 5th World Fisheries Congress 2008
- 57.Guyomard R, Boussaha M, Krieg F, Hervet C, Quillet E. A synthetic rainbow trout linkage map provides new insights into the salmonid whole genome duplication and the conservation of synteny among teleosts. BMC Genet. 2012 doi: 10.1186/1471-2156-13-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Macqueen DJ, Johnston IA. A well-constrained estimate for the timing of the salmonid whole genome duplication reveals major decoupling from species diversification. Proc Biol Sci. 2014;281(1778):20132881. doi: 10.1098/rspb.2013.2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Flicek P, Amode MR, Barrell D, Beal K, Billis K, Brent S, et al. Ensembl 2014. Nucleic Acids Res. 2014;42(D1):D749–D755. doi: 10.1093/nar/gkt1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rognes T. ParAlign: a parallel sequence alignment algorithm for rapid and sensitive database searches. Nucleic Acids Res. 2001;29(7):1647–1652. doi: 10.1093/nar/29.7.1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Agostino M (2012) Introduction to the BLAST Suite and BLASTN reference. In: Practical Bioinformatics. Garland Science, New York, pp 47–71
- 62.Smit A, Hubley R, Green P (1996–2010) RepeatMasker Open-3.0
- 63.Zhang Z, Schwartz S, Wagner L, Miller W. A greedy algorithm for aligning DNA sequences. J Comput Biol. 2000;7(1–2):203–214. doi: 10.1089/10665270050081478. [DOI] [PubMed] [Google Scholar]
- 64.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Felsenstein J (2005) PHYLIP (Phylogeny Inference Package) version 3.6. Distributed by the author. Department of Genome Sciences, University of Washington, Seattle
- 66.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]