

Abstract
Coordinated post-translational modifications (PTMs) of nucleosomal histones emerge as a key mechanism of gene regulation by defining chromatin configuration. Patterns of histone modifications vary in different cells and constitute core elements of cell-specific epigenomes. Recently, in addition to canonical histone proteins produced during the S phase of cell cycle, several non-canonical histone variants have been identified and shown to express in a DNA replication-independent manner. These histone variants generate diversity in nucleosomal structures and add further complexity to mechanisms of epigenetic regulation. Cell-specific functions of histone variants remain to be determined. Several recent studies reported an association between some point mutations in the non-canonical histone H3.3 and particular types of brain and bone tumors. This suggests a possibility of differential physiological effects of histone variants in different cells and tissues, including bone. In this review, we outline the roles of histone variants and their PTMs in the epigenetic regulation of chromatin structure and discuss possible mechanisms of biological effects of the non-canonical histone mutations found in bone tumors on tumorigenesis in differentiating bone stem cells.
Introduction
Bone growth and development are highly regulated processes involving various types of bone cells with diverse functions.1,2 In addition to factors controlling bone cell proliferation and differentiation to support bone remodeling in adults, growth-stage-specific factors are critical for adequate bone growth and regeneration. Action of these factors involves modification of chromatin organization and modulation of transcriptional activity of bone cell genes. In fact, dysfunction of some DNA-binding transcriptional regulators has been clinically and experimentally proven to lead to serious defects in bone development, growth and remodeling.3,4,5
Apparently, as a general principle of cell differentiation, the bone lineage cell fate decision depends on the function of a number of DNA-binding transcriptional regulators in a spatio-temporal manner. Although genetic and biochemical characterization of such transcriptional factors has been a cutting edge aspect in research on gene regulation in bone as well as other types of cells over the last few decades, recent progress by means of the genome-wide association study (GWAS) opened new frontiers in analysis of regulatory mechanisms controlling gene activity, particularly epigenetic events.
Although new biologically significant types of histone modifications will likely emerge, currently known histone modifications and their combinations are sufficient to provide a mechanistic insight how chromatin configuration is regulated in living cells. As the chromatin configuration depends on epigenomic regulation, histone decorations represent major parts of epigenetic machinery. Considering dynamic genomic processes that support skeletal cell proliferation and differentiation, investigation of factors and mechanisms regulating histone modifications in bone and muscle cells became one of the promising research subjects for skeletal biology.
Transcriptional events are closely associated with chromatin configuration
Finely tuned expression of particular sets of genes is a fundamental mechanism in regulation of cell proliferation and differentiation. In eukaryotic cells, key mechanisms controlling gene expression operate at the level of transcription. Multiple transcriptional factors and their complexes have been isolated and characterized, and experiments on assembly of purified transcriptional factors in vitro were successful to recapitulate formation of transcriptional complexes in vivo. These studies had shown that three classes of transcriptional factors are indispensable for transcriptional events in vitro, and this further was confirmed at whole-genome levels in living cells and tissues by recent GWAS approach.6,7
First, RNA polymerase-associated general transcriptional factors constitute basal transcriptional complexes that exhibit low DNA affinity with limited, if any, promoter specificity and are essential for transcription of all genes in the cell. The second class includes DNA-binding transcriptional factors that recognize specific promoter DNA elements and may recruit RNA polymerase complexes to particular genomic loci, thereby regulating transcription in a promoter context-dependent manner. Although majority of the general or basal transcriptional factors are expressed in all cells as so-called housekeeping genes, the expression of DNA-binding transcriptional regulatory factors is tightly controlled in spatio-temporal and cell type-specific manners.3,4,5 The complexity of combined functions and specificity of expression of DNA-binding transcriptional regulatory factors attribute to a high precision of mechanisms governing gene expression.
The third class of transcriptional factors was initially dubbed as transcriptional co-regulators as these factors were shown to form regulatory complexes with already known transcriptional factors, as well as with yet uncharacterized protein subunits.5,8 Later, it had been found that this class of factors was more diverse, in comparison with proteins of the other two classes, in mechanisms of their involvement in transcriptional regulation. It includes factors directly involved in transcriptional events, such as the mediator complex subunits that were shown to physically bridge between general transcriptional factors of the basal RNA polymerase II complexes and promoter-specific DNA-binding regulatory proteins.8 Another group of regulatory factors were found in nuclear complexes that affect transcription indirectly by changing chromatin conformation. Molecular characterization and functional dissection of subunits have revealed that such complexes define state of chromatin in genomic loci through chromatin remodeling and histone modifications.9,10,11
A compact state of chromatin with higher density of nucleosomal arrays is inhibitory for transcriptional events, whereas relaxed chromatin conformation with sparser nucleosomal arrays favors transcription (Figure 1).
Figure 1.

Chromatin conformation underlies gene regulation. During the cytodifferentiation of skeletal cells, genes governing cell fates are activated or inactivated in a bone cell type-specific manner. Chromatin remodelers and histone chaperones control accessibility of genomic regulatory elements for DNA-binding transcriptional factors through rearrangement of nucleosomal arrays and histone mobilization. Specific combination of post-translational histone modifications directs activation or repression of chromatin.
Chromatin is remodeled by rearrangement of nucleosomal array through mobilization and sliding of nucleosomes along the DNA together with deposition and replacement of nucleosomal core histone proteins. The chromatin remodelers act using energy of ATP provided by activity of ATPase subunits that define molecular identity of a remodeler complex.12,13 Currently, four types of such complexes have been identified and named after their corresponding ATPase subunits as SWI/SNF-, ISWI-, Mi-2- and INO80-remodeling complexes.12,13
Although their overt actions in transcriptional regulation have not been proven by the standard in vitro assays, histone chaperones are thought to regulate chromatin remodeling and histone protein mobilization processes.14,15,16 This assumption has been well supported by genome-wide co-localization of chromatin remodelers and histone chaperones in intact animals.17,18 These findings suggest that histone chaperones function as indirect transcriptional co-regulators.16,19
Covalent modifications of histone proteins may affect chromatin conformation and, therefore, its transcriptional activity. The number of known histone modifiers and histone modifications is still growing. Currently, at least 13 types of covalent modifications have been identified at around 60 different amino acid residues of histone proteins.20,21 Mounting evidence from diverse experimental settings indicates that specific combination patterns of histone modifications closely correlate with the state of chromatin, suggesting that histone modifications control functional activity of genomic loci. Accordingly, histone modifiers can be viewed as global genomic factors that control chromatin environment and indirectly regulate downstream transcriptional events.10,11
Histone modifications are major epigenetic elements defining chromatin configuration
Nucleosomal histone proteins are substrate for various reversible post-translational modifications (PTMs) at specific amino acid residues, mostly situated in their N-terminal tails protruding from the octamer core (Figure 2). Upstream PTMs are shown to influence or even direct subsequent histone modifications. Concurrent and sequential histone PTMs are orchestrated and tightly controlled by various regulatory factors. PTMs in histone proteins alter physicochemical properties of the nucleosomes and their interaction with the DNA and therefore may significantly affect the state of chromatin and its transcriptional activity. Furthermore, specific sets of chromatin modifications have been recognized as the second genetic code named as the ‘histone code' to govern gene expressions through defining chromatin configuration.9,10,11,22,23,24 With over 100 of different PTMs found in non-histone proteins, the currently known 13 different types of histone PTMs20,21 might represent just the most common and frequent ones, and new types of histone modifications are expected to be identified in foreseeable future.
Figure 2.
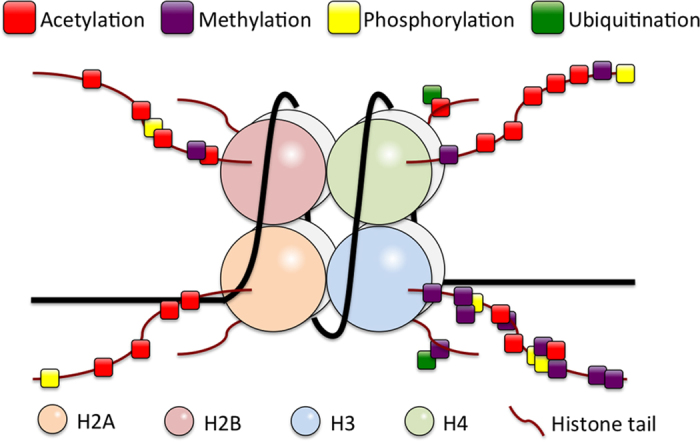
Histone modifications. Post-translational modifications (PTMs) occur in the N- or C- terminal tails of histones protruded from the octamer core wrapped by DNA. Histone PTMs are reversible through action of corresponding modifying and de-modifying enzymes. Shown are four best-characterized histone PTMs controlling functional states of chromatin.
Acetylation is the first identified and best-characterized histone modification to be functionally linked to transcriptional activation. Negatively charged acetyl groups reduce overall positive charge of nucleosomes, loosen their interaction with the DNA and induce electric repulsion between acetylated histones.22,23,24 Resulting relaxation of nucleosomal arrays renders genomic DNA more accessible for DNA-binding transcriptional factors and stabilizes their interaction with target DNA sequences of genomic regulatory elements. Consistently, histone deacetylation reverses these changes in the state of chromatin and leads to reduction in its transcriptional activity. A large body of independent observations suggests that acetylation/deacetylation is a downstream consequence of other types of preceding histone modifications, whereas histone methylation is considered to be the most upstream PTM of nucleosomal histones.9,10,11,22,23,24
Unlike the case with histone acetylation, biological effect of histone methylation on chromatin reorganization depends on positions of methylated lysine residues in histone proteins. Although besides lysine about a dozen of arginine residues can also be potentially methylated, four specific lysine residues in the N-terminus of histone H3 appear to be the most frequent and functionally consequential epigenetic marks in the genome.9,10,11,22,23,24
Mono-, di- and, particularly, tri-methylation at the lysine 4 and 36 in histone H3 proteins (H3K4me1-3 and H3K36me1-3) are believed to activate the chromatin through induction of other histone modifications, including histone acetylation, although methylated H3K36 is associated with chromatin inactivation under some chromatin environment.25 This has been supported by consistent co-localization of methylated H3K4 and H3K36 with euchromatin, particular at transcriptionally active gene regions, and tri-methylated H3K4 (H3K4me3) is shown to be a characteristic feature of fully activated state of chromatin.9,10,11,22,23,24 GWAS analysis of chromatin modification landscapes has shown that H3K4me3 is also enriched in genomic regions exhibiting ‘poised' transcription state.26 In the sharp contrast, methylation at H3K9 and H3K27 triggers chromatin inactivation or heterochromatinization, and tri-methylated H3K9me3 and H3K27me3 are considered to be epigenetic hallmarks of silenced genomic loci.9,10,11,22,23,24
Ser/Tyr phosphorylation is a classical PTM also found at several sites in histone proteins. Phosphorylation of H2A histone variant H2A.X at Ser139 residue is crucial in early response to double-strand DNA breaks and initiation of downstream DNA repair reactions. Ser139-phosphoryl H2A.X, known as γH2A.X, also appears to be involved in the sex chromosome inactivation.27 Recently, methylation of the glutamine residue at position 104 in the human histone H2A (H2AQ104me) has been found to facilitate RNA polymerase I—mediated transcription of rRNA through recruitment of FACT, a histone chaperone complex.28 This suggests an existence of unidentified sites and novel amino acid residues in histones as potential substrate for methylation.
Considerably, bulkier modifications such as glycosylation (O-GlcNAcylation), mono- and poly-ubiquitinylation, sumoylation or ADP-ribosylation of histones have been also found to modulate transcription though induction of chromatin rearrangement.20,21,29,30 However, many of the functions and detailed mechanisms of molecular effects of these and some other recently identified histone PTMs remain to be determined.
Histone variants as functional elements of epigenetic machinery
Highly conserved from yeast to human, histones H2A, H2B, H3 and H4 are recognized as canonical histone proteins, and two copies of each are assembled into a canonical octamer complex. A histone octamer constitutes a protein core that is wrapped around in 1.7 turns by ∼147 bp of DNA to form a nucleosome, the basic repeating structural unite of eukaryotic chromatin.
The genes for canonical histones (H2A.1, H2A.2, H2B, H3.1, H3.2 and H4) are present in multiple copies organized in gene clusters, transcription of which is highly coordinated with DNA replication. In the S phase, nascent histone octamers stabilize daughter DNA into nucleosomal arrays for genomic DNA packing.31,32,33
In addition to the canonical histone proteins, several non-canonical histone protein variants have been identified (Figure 3). Significantly, genes encoding the non-canonical histones are not located in the canonical histone gene clusters, and expression of the non-canonical histone variants is not coupled with DNA synthesis but resembles the pattern of housekeeping gene expression.31,32
Figure 3.
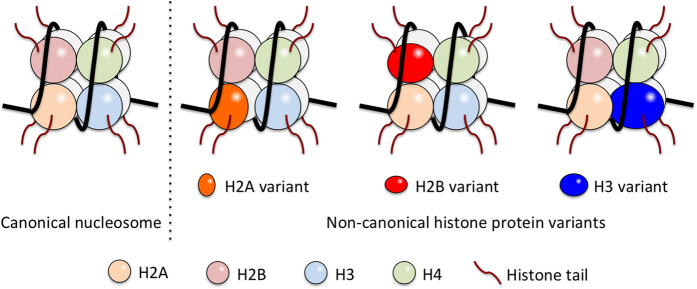
Nucleosomes are composed of canonical and non-canonical histones. Canonical histones (H2A, H2B, H3 and H4) are produced only during the S-phase of the cell cycle to organize replicated DNA into the chromatin structure. Non-canonical histone variants express throughout cell life span and replace related canonical histones to form non-canonical nucleosomes. Known non-canonical variants for H2A are H2A.B, H2A.X, H2A.Z and macroH2A; for H2B is TSH2B; for H3 are H3.3, H3.4, H3.5, CENP-A/CenH3, H3.X and H3.Y.
Initially, once formed core nucleosomal octamers were viewed as comparatively stable protein complexes. Further studies revealed that individual canonical histone proteins could be removed from core nucleosomal octamers and replaced with their non-canonical variants, causing transition of chromatin state. GWAS data show that non-canonical histones are not ubiquitous throughout the chromatin but concentrated at specific genomic loci in response to chromatin activation or silencing.31,32 These suggest that the non-canonical histones are mainly used for replacement of canonical histones pre-integrated into nucleosomal particles to trigger rearrangement of chromatin configuration, whereas the canonical histones are used for organization of fundamental chromatin structures during DNA replication.
Thus, in addition to various histone PTMs, non-canonical histone variants contribute to the dynamics and complexity of epigenetic mechanisms in regulation of genomic functions.
Histone variants for H2A, H2B and H3
Several non-canonical protein variants and their corresponding genes have been identified for canonical nucleosomal histones H2A, H2B and H3, whereas existence of non-canonical variants for histone H4 remains unclear.
Some histone variants appear to be ubiquitously expressed, whereas others may exhibit high cell specificity of expression. Histone TSH2B has been found only in spermatocytes and is classified as a testis-specific variant of histone H2B. Incorporation of TSH2B is believed to destabilize nucleosomes and promote histone octamer replacement with protamine in transition from histone-based chromatin into nucleoprotamine complex during mammalian sperm maturation.34,35 In contrast, majority of non-canonical variants of histones H2A and H3 have been found in fractions of nucleosomes from all examined cell types.31,32,36
Reflecting diverse roles of non-canonical histones in modulation of chromatin conformation, four and six non-canonical variants have been identified for human H2A and H3 histones, respectively.31,32,36 The H2A histone variants include H2A.B (H2A.Bbd), H2A.X, H2A.Z (H2A.Z.1/H2AZ.2) and macroH2A (mH2A.1/mH2A.2). Among the human non-canonical H3 histone variants, H3.3 is widely expressed and enriched in nucleosomes of active genes; CENP-A (CenH3) is a centromere-specific variant; H3.4 (H3T) and H3.5 are testis-specific variants; H3.X and H3.Y are low-level expressed variants that together with the H3.5 were found only in primates.31,32,36
The canonical variants of histone H2A (H2A.1 and H2A.2) and histone H3 (H3.1 and H3.2) differ only in a single amino acid residue. However, neither of these variants confers any apparent functional or structural diversity to nucleosomes, and no evidence for significance of the single amino acid difference between these respective variants has been yet obtained.
The sequence of non-canonical histone H3.3 differs in four and five amino acid residues with the canonical H3.2 and H3.1 proteins, respectively, whereas the same H3.3 protein is produced by the two independent genes, H3F3A and H3F3B, in humans and mice. Although some H3.3 amounts can be found in the pericentromeric and telomeric regions, high accumulation of H3.3 is observed at transcriptionally activated and poised genomic loci, suggesting that H3.3 is a key non-canonical histone variant regulating chromatin activation.14,26,36,37 The protein sequences of non-canonical H3.4, H3.4, H3.X and H3.Y variants are also very close to that of canonical H3.1/H3.1 histones, but their specific roles in modulation of chromatin configuration remain to be determined.
A specific marker and essential component of centromeric chromatin, CENP-A, is the most divergent non-canonical H3 variant exhibiting only about 50% homology with other H3 histone variants. Consistently, the structure of CENP-A-containing nucleosomes is quite distinct from that of other types of nucleosomes. Atomic force microscopy and small angle X-ray analysis data indicate that CENP-A-containing nucleosomes induce a right-handed wrap of DNA instead of the left-handed wrap of canonical nucleosomes and are half the volume (‘hemisomes') of canonical nucleosomes, presumably consisting of histone tetramers. The unique characteristics of CENP-A-containing chromatin suggest that CENP-A histones are pivotal for formation, maintenance and functions of centromeres.36,38
H3.3 is epigenetic regulator
Experimental inactivation of any of the two mouse H3.3 genes, H3f3a or H3f3b, resulted in developmental defects in early stages of the embryogenesis.39,40 Severe abnormalities were also observed in Drosophila, Xenopus and zebrafish when expression levels of their H3.3 genes were genetically manipulated.18,41 Thus, appropriate deposition of H3.3 histones in chromatin appears to be physiologically crucial already at early stages of development, particularly in higher organisms.18,39,40,41
Apparently, H3.3 replaces canonical H3.1/2 during chromatin rearrangement outside of the S phase of cell cycle.14,39,42,43 This is believed to impact chromatin configuration through induction of specific PTMs of other histones at the sites with abundant H3.3 proteins. In Drosophila, a constitutive H3K4 methylation-mimicking mutation in the canonical H3.1 gene ameliorated impaired reproduction in fly mutants with deficiency in H3.3 expression.44,45 A substitution of Lys27 with Arg in the mouse H3.3 (K3.3K27R mutation) perturbed the embryogenesis and caused a marked decrease in the levels of total H3K27me3, a hallmark for transcriptionally repressed state.46,47 These findings suggest that H3.3 protein per se is dispensable for proper expression of genes controlling germ cell development, but H3.3 presence is required for induction and coordination of certain PTMs, including methylation at H3K4 and H3K27.9,10,11,22,23,24
Consistent with detection of H3.3 in pericentromeric and telomeric regions, H3.3 depletion was shown to result in karyotypic abnormalities apparently caused by mislocalization of CENP-A and aberrant centromeres.48,49,50 These suggest that H3.3 is required for proper chromosome maintenance and segregation.
As methylation is the most upstream epigenetic PTM, and coordinated interplay between methylation at H3K4/H3K36 and H3K9/H3K27 is fundamental in the mechanisms of transcriptional activation and silencing,9,10,11,22,23,24 nucleosomal deposition of H3.3 appears to have a critical role in orchestration of consequent epigenetic modifications in the adjacent chromatin, and H3.3 functions as epigenetic regulator of transition between alternative chromatin conformations. Although, in contrast to canonical H3.1 and H3.2, the expression of non-canonical H3.3 is not limited to particular cell cycle stages,31,32 it is evident that H3.3 protein mobilization to chromatin is highly regulated in the spatio-temporal manner. However, mechanisms of targeting of H3.3 deposition at specific genomic loci remain largely unclear.
Two histone chaperon complexes mediate H3.3 nucleosomal integration
Reflecting high spatio-temporal specificity of chromatin integration and accumulation of histone variants, it can be postulated that multiple regulators assist in their deposition and eviction. Special auxiliary proteins, histone chaperones, have been shown to have a key role in dynamics of histone variant exchange. Many histone chaperones act as parts of multi-subunit nuclear protein complexes.51 In genome-wide ChIP analysis, several of such complexes have been shown to specifically co-localize with particular histone variants. The chromatin-assembly factor 1 (CAF-1) and anti-silencing function 1are involved in the S phase-dependent incorporation of canonical H3.14 The Holiday junction recognition protein, HJURP, has been identified as a highly specific chaperone for the H3 histone variant CENP-A.36,52
Two distinct protein complexes, HIRA (histone regulator A)-and DAXX (death-associated protein 6)-ATRX (alpha thalassemia/mental retardation syndrome X-linked protein)-containing complexes, have been shown to function as H3.3-specific chaperones, in which HIRA and DAXX protein subunits directly interact with H3.3 molecules.14,35,53,54 Although both complexes are able to directly interact with H3.3-containing nucleosomes, each complex is likely to exert specific effects on chromatin configuration, as their locations are not overlapping in living cells. Although HIRA has been found associated with H3.3-containing nucleosomes in transcriptionally active genomic loci, DAXX was co-localized with H3.3 in transcriptionally inactive regions and pericentromeric heterochromatin. Functional distinction and specificity of HIRA and DAXX–ATRX has been further delineated by disruption of their genes in cultured cells and genetic model animals. HIRA depletion in mice and Xenopus resulted in embryonic lethality, confirming impact of H3.3-dependent pathways on global gene activation.55 ATRX gene disruption in mice led to aneuploidy and centromere instability in the mutant oocytes and muscle cells.17,56
Thus, the facts that H3.3 is a common target for functionally different histone chaperone complexes and that H3.3 involves in organization of functionally opposite chromatin states reflect profound biological significance of H3.3 histone variant in regulation of various genomic functions.
High frequency H3.3 point mutations in pediatric glioma
Considering fundamental roles of epigenetic factors in modulation of chromatin state and functions, it is not surprising that aberrant expression or malfunction of epigenetic regulators may induce and facilitate tumor development. Aberrant PTM patterns of histones have been reported in many types of cancers.57,58,59 Supporting these observations, several compounds known to modulate activity of epigenetic factors have been indeed successful to attenuate development of certain types of cancers.58,59,60
In 2012, it has been reported that over a third of investigated pediatric glioblastoma patients had point mutations in one of the H3.3 gene, H3F3A but not in the H3F3B.61,62 In tumor cells of these patients, lysine 27 was replaced by methionine (K27M mutation), and in some of the patients glycine 34 was replaced by arginine or valine (G34R/V).61,62 Both mutated residues are located in the N-tail of the H3.3 protein protruding out of the nucleosomal octamer. Significantly, K27 in the H3.3 molecule represents a functionally critical target for repressive methylation and can also be acetylated.40,63,64 Although the H3.3 G34 is not a substrate for any known PTM, its mutations may affect epigenetic modifications at the adjacent H3.3 K36, an important target for activating methylation.37,40,64 These point mutations were found only in one of the H3F3A alleles in the glioma cell samples with the other allele remained intact (wild type). The occurrence of the two mutations appeared to be mutually exclusive, and topology of tumors seemed to associate with the position of the mutated residue. Gliomas with H3.3 K27M were predominantly found in midline brain locations, whereas H3.3 G34R/V tumors were mainly located in cerebral hemisphere areas. The H3.3 K27M pediatric glioblastoma was diagnosed at earlier ages and had shorter overall survival rates in comparison with glioblastomas bearing mutations at the H3.3 G34.61,62 These mutations were further co-related with other mutations previously found in pediatric glioma.40,65
About a third of the K27M tumors contained mutations in the ATRX/DAXX, H3.3-specific chaperone complex proteins involved in chromatin inactivation at telomeric and pericentromeric regions, and, about two-thirds bore mutations in the p53, the best-known tumor-suppressor protein (TP53). Co-occurrence of mutations in ATRX/DAXX and p53 was even higher in the G34R/V glioma cells.61,62 Furthermore, pediatric glioblastomas with wild-type H3.3 bear mutations in the IDH1/2 resulting in an overproduction of 2-hydoxyglutarate, a metabolite inhibiting H3K27 and K36 demethylases.66 These observations have established that aberrations in H3.3-mediated pathways may trigger tumorigenesis in the neuronal stem cells and that proper regulation of H3.3 methylation at K27 and K36 is pivotal for normal proliferation/differentiation of neural cells.
Two different H3.3 point mutations lead to distinct yypes of bone tumors
The discovery of high frequency H3.3 protein mutations in pediatric glioblastomas persuaded search for H3.3 mutations in various types of cancer cells besides glioma, and several independent research group consortia had been organized to coordinate their studies. In late 2013, a European team reported that H3.3 mutations are prevalent in two types of skeletal tumors, chondroblastoma and giant cell tumor of bone.67
Several striking differences immerged between character of H3.3 mutations in the brain and bone tumors. Although only one (H3F3A) of the two H3.3 genes was found mutated in the samples from pediatric glioma patients,61,62 both H3.3, H3F3A and H3F3B genes bore mutations in the bone tumor cells.67 Overall, 73 out of the 77 tested samples (95%) of chondroblastoma carried substitution of lysine 36 with methionine (K36M) predominantly in the H3F3B gene (68 cases versus 5 cases of the mutation in the H3F3A). Overall, 92% (49/53) of the tested giant cell tumors were found to carry a point mutation in the glycine 34 codon: G34W in 48 and G34L in 1 out of 49 tested tumor samples. Interestingly, all of the G34W/L mutations were located in in the H3F3A gene. Thus, these two H3.3 mutations exhibit remarkable tumor type specificity: G34/W/L for chondroblastoma and K36M for giant cell tumor of bone.
Other types of skeletal tumors exhibited considerably lower frequencies of H3.3 mutations. Only 2 out of 102 osteosarcoma samples had G34R mutation in either of the H3.3 genes; 1 out of 75 analyzed conventional chondrosarcomas bore K36M mutation in the H3F3A; and 1 out of 15 clear-cell chondrosarcomas had K36M mutation encoded in the H3F3B. No H3.3 mutation was detected in the assessed samples of chondromyxoid fibroma (0/43), chordoma (0/25) and soft tissue/synovial chondroma (0/7).
Unlike in pediatric brain tumors, no H3.3 K27M mutation was detected in samples of all types of bone tumors. Furthermore, no genetic association between H3.3 and p53 tumor-suppressor gene mutations has been observed in tested bone tumor cells.67 These strongly suggest that molecular basis of the onset and development of bone tumors triggered by H3.3 mutations is independent from that of brain tumors.
Tumorigenesis through epigenetic alterations
Methylation of H3.3 K27 is catalyzed by the enhancer of zest homolog 2 (EZH2), a SET domain-containing lysine-N-methyltransferase that functions as a catalytic subunit of the Polycomb repressive complex group 2 (PRC2). PRC2 is a multiunit nuclear protein complex shown to be essential for suppression of oncogenic genes.63,64,68,69 Direct interaction of EZH2 with mono- or di-methylated K27 (K27me1/me2) is required for further di- and tri-methylation, respectively.61,62 Brain tumors carrying H3.3 K27M mutation display reduced levels of global H3K27me2 and H3K27me3 in all endogenous wild-type H3 protein variants, suggesting that K27M is inhibitory for EZH2 enzymatic activity.40,63,64 As PRC2 complex malfunction or loss of the EZH2 enzymatic activity has been shown to associate with development of various tumors,68,69 K27M mutation is likely to facilitate tumorigenesis through dysregulation in EZH2-mediated regulatory processes.
In comparison with the H3.3 K27M mutation, molecular basis for tumorigenesis associated with the H3.3 G34W/L mutation in skeletal cells or H3.3 G34R/V mutation in brain cells is less clear. Nevertheless, it has been reported that both H3.3 G34W and G34L mutations interfere with the action of the SETD2 methyltransferase, the only known enzyme responsible for H3K36 tri-methylation (Figure 4), and result in global reduction in K36me3 in all endogenous histone H3 variants.70,71 Similarly, H3.3 G34R/V mutations found in pediatric glioblastoma are associated with a decrease in global H3K36me3 levels.63,64 Therefore, it is conceivable that H3.3 G34 substitution with a bulky, hydrophobic or charged amino acid residue would perturb nucleosomal environment and/or H3.3 tail conformation in a way that disrupts accessibility of the neighboring K36 to SETD2 and inhibits H3.3 K36 tri-methylation, thereby causing reduction in global H3K36m3 levels (Figure 5). Supporting this idea, an overexpression of H3.3 K36M mutant resulted in decreased global levels of tri-methylation of the endogenous wild-type H3K36.71 Hypo-tri-methylation of H3.3K36me2 residues might affect their normal interactions with downstream epigenetic factors such as histone demethylases10,11,22,23,24,72 (Figure 5). Furthermore, functional interaction between SETD2-mediated H3K36 tri-methylation and PRC2-mediated H3K27 tri-methylation could be postulated, as H3K36 and H3K27 tri-methylations appear to be mutually exclusive, and the balance between these two residues methylation is maintained under the normal condition.69 Although no direct experimental evidence is so far available to assess the impact of G34W/L mutation on the global H3K36me3 levels seen in the bone tumors, the effects of H3.3 K36M mutation observed in the chondrosarcoma67 suggest that dysregulation of tri-methylation at the H3K36 is a key epigenetic event triggering skeletal tumorigenesis, conceivably through aberrant tri-methyltion-associated events at H3K36 and H3K27.
Figure 4.

Tri-methylation of H3K36 by SETD2. The lysine 36 residue (K36) in the N-tails of H3 and H3.3 can be mono-, di- and tri-methylated by histone methyltransferase (HMT) enzymes. The SETD2 is the only known HMT responsible for tri-methylation of di-methylated K36 in both H3 and H3.3. H3K36me3 may provide a docking site for a downstream epigenetic effector (labeled as Y).
Figure 5.

H3.3 G34 mutations disrupt methylation and downstream functions of H3.K36. Point mutations at G34 appear to prevent tri-methylation of di-methylated K36 by SETD2 (a), thereby preventing downstream epigenetic events mediated by other factors (b).
Role of tri-methylated H3K36 in skeletal cell development and its clinical implication
A large body of evidence obtained in various experimental settings has long established that H3K36 methylation associates with gene activation.9,10,11,22,23,24 However, recent findings illuminated H3K36 involvement in other various genetic processes, including transcriptional suppression, alternative splicing, DNA repair and recombination.48,49,73,74 Therefore, molecular events underlying skeletal tumorigenesis may be more complex than just aberrant expression of oncogenes and/or tumor suppressors.
The HIRA- and DAXX/ATRA-containing chaperone complexes are known to assist post-replicative chromatin mobilization of H3.3,14,35,53,54 but the scope of their functions in skeletal cells is still unknown. A causal link between these histone chaperone activities and bone tumor development presents an intriguing possibility.
The fact that H3.3 G34W/L and K36M mutations were not found in the osteoclast lineage cells and were restricted to the stromal cells67 reveals physiological impact of SETD2 and H3K36me3 on differentiation of mesenchymal stem cells that give rise to multiple types of skeletal cells. In this regard, tri-methylation of H3K36 appears to be a critical event in the chondro-osteoblastic cell lineage decision (Figure 6). Therefore, understanding of molecular pathways and mediators defining timing and extend of this PTM may provide a novel insight into skeletal cell biology and lead to identification of therapeutic targets for prevention and treatment of skeletal tumors.
Figure 6.

Pivotal roles of tri-methylated H3.3 K36 in cell lineage fate and proliferation of mesenchymal stem cells in bone. Tri-methylation at H3.3 K36 appears critic for cell fate decision and/or cytodifferentiation of mesenchymal stem cells into osteoblasts and chondrocytes. Aberrant methylation at H3.3 K36 is assumed to trigger tumorigenesis during cell lineage determination processes through disruption of spatio-temporal expression of lineage-specific genes.
Footnotes
The authors declare no conflict of interest.
References
- Zaidi M. Skeletal remodeling in health and disease. Nat Med 2007; 13: 791–801. [DOI] [PubMed] [Google Scholar]
- Harada S, Rodan GA. Control of osteoblast function and regulation of bone mass. Nature 2003; 423: 349–355. [DOI] [PubMed] [Google Scholar]
- Teitelbaum SL, Ross FP. Genetic regulation of osteoclast development and function. Nat Rev Genet 2003; 4: 638–649. [DOI] [PubMed] [Google Scholar]
- Karsenty G, Kronenberg HM, Settembre C. Genetic control of bone formation. Annu Rev Cell Dev Biol 2009; 25: 629–648. [DOI] [PubMed] [Google Scholar]
- Karsenty G. Transcriptional control of skeletogenesis. Annu Rev Genomics Hum Genet 2008; 9: 183–196. [DOI] [PubMed] [Google Scholar]
- Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature 2012; 489: 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulon A, Chow CC, Singer RH, Larson DR. Eukaryotic transcriptional dynamics: from single molecules to cell populations. Nat Rev Genet 2013; 14: 572–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conaway RC, Conaway JW. The Mediator complex and transcription elongation. Biochim Biophys Acta 2013; 1829: 69–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Dent SY. Chromatin modifiers and remodellers: regulators of cellular differentiation. Nat Rev Genet 2014; 15: 93–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld MG, Lunyak VV, Glass CK. Sensors and signals: a coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes Dev 2006; 20: 1405–1428. [DOI] [PubMed] [Google Scholar]
- Kato S, Yokoyama A, Fujiki R. Nuclear receptor coregulators merge transcriptional coregulation with epigenetic regulation. Trends Biochem Sci 2011; 36: 272–281. [DOI] [PubMed] [Google Scholar]
- Morrison AJ, Shen X. Chromatin remodelling beyond transcription: the INO80 and SWR1 complexes. Nat Rev Mol Cell Biol 2009; 10: 373–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luger K, Dechassa ML, Tremethick DJ. New insights into nucleosome and chromatin structure: an ordered state or a disordered affair? Nat Rev Mol Cell Biol 2012; 13: 436–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagami H, Ray-Gallet D, Almouzni G, Nakatani Y. Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell 2004; 116: 51–61. [DOI] [PubMed] [Google Scholar]
- Burgess RJ, Zhang Z. Histone chaperones in nucleosome assembly and human disease. Nat Struct Mol Biol 2013; 20: 14–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avvakumov N, Nourani A, Côté J. Histone chaperones: modulators of chromatin marks. Mol Cell 2011; 41: 502–514. [DOI] [PubMed] [Google Scholar]
- Baumann C, Viveiros MM, De La Fuente R. Loss of maternal ATRX results in centromere instability and aneuploidy in the mammalian oocyte and pre-implantation embryo. PLoS Genet 2010; 6: e1001137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szenker E, Lacoste N, Almouzni G. A developmental requirement for HIRA-dependent H3.3 deposition revealed at gastrulation in Xenopus. Cell Rep 2012; 1: 730–740. [DOI] [PubMed] [Google Scholar]
- Sawatsubashi S, Murata T, Lim J, Fujiki R, Ito S, Suzuki E et al. A histone chaperone, DEK, transcriptionally coactivates a nuclear receptor. Genes Dev 2010; 24: 159–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan M, Luo H, Lee S, Jin F, Yang JS, Montellier E et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 2011; 146: 1016–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suganuma T, Workman JL. Signals and combinatorial functions of histone modifications. Annu Rev Biochem 2011; 80: 473–499. [DOI] [PubMed] [Google Scholar]
- Strahl BD, Allis CD. The language of covalent histone modifications. Nature 2000; 403: 41–45. [DOI] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD. Translating the histone code. Science 2001; 293: 1074–1080. [DOI] [PubMed] [Google Scholar]
- Bannister AJ, Schneider R, Kouzarides T. Histone methylation: dynamic or static? Cell 2002; 109: 801–806. [DOI] [PubMed] [Google Scholar]
- Lee CH, Wu J, Li B. Chromatin remodelers fine-tune H3K36me-directed deacetylation of neighbor nucleosomes by Rpd3S. Mol Cell 2013; 52: 255–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delbarre E, Jacobsen BM, Reiner AH, Sørensen AL, Küntziger T, Collas P. Chromatin environment of histone variant H3.3 revealed by quantitative imaging and genome-scale chromatin and DNA immunoprecipitation. Mol Biol Cell 2010; 21: 1872–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Attikum H, Gasser SM. Crosstalk between histone modifications during the DNA damage response. Trends Cell Biol 2009; 19: 207–217. [DOI] [PubMed] [Google Scholar]
- Tessarz P, Santos-Rosa H, Robson SC, Sylvestersen KB, Nelson CJ, Nielsen ML et al. Glutamine methylation in histone H2A is an RNA-polymerase-I-dedicated modification. Nature 2014; 505: 564–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakabe K, Wang Z, Hart GW. Beta-N-acetylglucosamine (O-GlcNAc) is part of the histone code. Proc Natl Acad Sci USA 2010; 107: 19915–19920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiki R, Hashiba W, Sekine H, Yokoyama A, Chikanishi T, Ito S et al. GlcNAcylation of histone H2B facilitates its monoubiquitination. Nature 2011; 480: 557–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber CM, Henikoff S. Histone variants: dynamic punctuation in transcription. Genes Dev 2014; 28: 672–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banaszynski LA, Allis CD, Lewis PW. Histone variants in metazoan development. Dev Cell 2010; 19: 662–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, Fujiyama-Nakamura S, Kimura S, Lim J, Kamoshida Y, Shiozaki-Sato Y et al. Epigenetic silencing of core histone genes by HERS in Drosophila. Mol Cell 2012; 45: 494–504. [DOI] [PubMed] [Google Scholar]
- Montellier E, Boussouar F, Rousseaux S, Zhang K, Buchou T, Fenaille F et al. Chromatin-to-nucleoprotamine transition is controlled by the histone H2B variant TH2B. Genes Dev 2013; 27: 1680–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govin J, Dorsey J, Gaucher J, Rousseaux S, Khochbin S, Berger SL. Systematic screen reveals new functional dynamics of histones H3 and H4 during gametogenesis. Genes Dev 2010; 24: 1772–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipescu D, Szenker E, Almouzni G. Developmental roles of histone H3 variants and their chaperones. Trends Genet 2013; 29: 630–640. [DOI] [PubMed] [Google Scholar]
- Wen H, Li Y, Xi Y, Jiang S, Stratton S, Peng D et al. ZMYND11 links histone H3.3K36me3 to transcription elongation and tumour suppression. Nature 2014; 508: 263–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbert PB, Henikoff S. Histone variants--ancient wrap artists of the epigenome. Nat Rev Mol Cell Biol 2010; 11: 264–275. [DOI] [PubMed] [Google Scholar]
- Bush KM, Yuen BT, Barrilleaux BL, Riggs JW, O'Geen H, Cotterman RF et al. Endogenous mammalian histone H3.3 exhibits chromatin-related functions during development. Epigenetics Chromatin 2013; 6: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen BT, Knoepfler PS. Histone H3.3 mutations: a variant path to cancer. Cancer Cell 2013; 24: 567–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox SG, Kim H, Garnett AT, Medeiros DM, An W, Crump JG. An essential role of variant histone H3.3 for ectomesenchyme potential of the cranial neural crest. PLoS Genet 2012; 8: e1002938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad K, Henikoff S. The histone variant H3.3 marks active chromatin by replication-independent nucleosome assembly. Mol Cell 2002; 9: 1191–1200. [DOI] [PubMed] [Google Scholar]
- Ray-Gallet D, Woolfe A, Vassias I, Pellentz C, Lacoste N, Puri A et al. Dynamics of histone H3 deposition in vivo reveal a nucleosome gap-filling mechanism for H3.3 to maintain chromatin integrity. Mol Cell 2011; 44: 928–941. [DOI] [PubMed] [Google Scholar]
- Sakai A, Schwartz BE, Goldstein S, Ahmad K. Transcriptional and developmental functions of the H3.3 histone variant in Drosophila. Curr Biol 2009; 19: 1816–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hödl M, Basler K. Transcription in the absence of histone H3.3. Curr Biol 2009; 19: 1221–1226. [DOI] [PubMed] [Google Scholar]
- Voigt P, Tee WW, Reinberg D. A double take on bivalent promoters. Genes Dev 2013; 27: 1318–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santenard A, Ziegler-Birling C, Koch M, Tora L, Bannister AJ, Torres-Padilla ME. Heterochromatin formation in the mouse embryo requires critical residues of the histone variant H3.3. Nat Cell Biol 2010; 12: 853–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CJ, Conti M, Ramalho-Santos M. Histone variant H3.3 maintains a decondensed chromatin state essential for mouse preimplantation development. Development 2013; 140: 3624–3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunleavy EM, Almouzni G, Karpen GH. H3.3 is deposited at centromeres in S phase as a placeholder for newly assembled CENP-A in G□ phase. Nucleus 2011; 2: 146–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacoste N, Woolfe A, Tachiwana H, Garea AV, Barth T, Cantaloube S et al. Mislocalization of the centromeric histone variant CenH3/CENP-A in human cells depends on the chaperone DAXX. Mol Cell 2014; 53: 631–644. [DOI] [PubMed] [Google Scholar]
- Filipescu D, Müller S, Almouzni G. Histone H3 variants and their chaperones during development and disease: contributing to epigenetic control. Annu Rev Cell Dev Biol 2014; 30: 615–646. [DOI] [PubMed] [Google Scholar]
- Foltz DR, Jansen LE, Bailey AO, Yates JR, Bassett EA, Wood S et al. Centromere-specific assembly of CENP-a nucleosomes is mediated by HJURP. Cell 2009; 137: 472–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drané P, Ouararhni K, Depaux A, Shuaib M, Hamiche A. The death-associated protein DAXX is a novel histone chaperone involved in the replication-independent deposition of H3.3. Genes Dev 2010; 24: 1253–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg AD, Banaszynski LA, Noh KM, Lewis PW, Elsaesser SJ, Stadler S et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell 2010; 140: 678–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts C, Sutherland HF, Farmer H, Kimber W, Halford S, Carey A et al. Targeted mutagenesis of the Hira gene results in gastrulation defects and patterning abnormalities of mesoendodermal derivatives prior to early embryonic lethality. Mol Cell Biol 2002; 22: 2318–2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh MS, Price O'Dea T, Ouazia D, McKay BC, Parise G, Parks RJ et al. Compromised genomic integrity impedes muscle growth after Atrx inactivation. J Clin Invest 2012; 122: 4412–4423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet 2012; 13: 343–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookes E, Shi Y. Diverse epigenetic mechanisms of human disease. Annu Rev Genet 2014; 48: 237–268. [DOI] [PubMed] [Google Scholar]
- Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M. Epigenetic protein families: a new frontier for drug discovery. Nat Rev Drug Discov 2012; 11: 384–400. [DOI] [PubMed] [Google Scholar]
- Helin K, Dhanak D. Chromatin proteins and modifications as drug targets. Nature 2013; 502: 480–488. [DOI] [PubMed] [Google Scholar]
- Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DT, Konermann C et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 2012; 22: 425–437. [DOI] [PubMed] [Google Scholar]
- Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet 2012; 44: 251–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis PW, Müller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA et al. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 2013; 340: 857–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan KM, Han J, Fang D, Gan H, Zhang Z. A lesson learned from the H3.3K27M mutation found in pediatric glioma: a new approach to the study of the function of histone modifications in vivo? Cell Cycle 2013; 12: 2546–2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Network CGAR. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008; 455: 1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 2011; 19: 17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behjati S, Tarpey PS, Presneau N, Scheipl S, Pillay N, Van Loo P et al. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat Genet 2013; 45: 1479–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracken AP, Helin K. Polycomb group proteins: navigators of lineage pathways led astray in cancer. Nat Rev Cancer 2009; 9: 773–784. [DOI] [PubMed] [Google Scholar]
- Hock H. A complex Polycomb issue: the two faces of EZH2 in cancer. Genes Dev 2012; 26: 751–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmunds JW, Mahadevan LC, Clayton AL. Dynamic histone H3 methylation during gene induction: HYPB/Setd2 mediates all H3K36 trimethylation. EMBO J 2008; 27: 406–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfister SX, Ahrabi S, Zalmas LP, Sarkar S, Aymard F, Bachrati CZ et al. SETD2-dependent histone H3K36 trimethylation is required for homologous recombination repair and genome stability. Cell Rep 2014; 7: 2006–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba A, Ohtake F, Okuno Y, Yokota K, Okada M, Imai Y et al. PKA-dependent regulation of the histone lysine demethylase complex PHF2-ARID5B. Nat Cell Biol 2011; 13: 668–675. [DOI] [PubMed] [Google Scholar]
- Guo R, Zheng L, Park JW, Lv R, Chen H, Jiao F et al. BS69/ZMYND11 reads and connects histone H3.3 lysine 36 trimethylation-decorated chromatin to regulated pre-mRNA processing. Mol Cell 2014; 56: 298–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey A, Listovsky T, Guilbaud G, Sarkies P, Sale JE. Histone H3.3 is required to maintain replication fork progression after UV damage. Curr Biol 2014; 24: 2195–2201. [DOI] [PMC free article] [PubMed] [Google Scholar]