

Abstract
Mechanistic understanding of the preferential homing of circulating tumor cells to bone and their perturbation on bone metabolism within the tumor–bone microenvironment remains poorly understood. Alteration in both transforming growth factor β (TGFβ) signaling and sphingolipid metabolism results in the promotion of tumor growth and metastasis. Previous studies using MDA-MB-231 human breast cancer-derived cell lines of variable metastatic potential were queried for changes in sphingolipid metabolism genes to explore correlations between TGFβ dependence and bone metastatic behavior. Of these genes, only sphingosine kinase-1 (SPHK1) was identified to be significantly increased following TGFβ treatment. Induction of SPHK1 expression correlated to the degree of metastatic capacity in these MDA-MB-231-derived cell lines. We demonstrate that TGFβ mediates the regulation of SPHK1 gene expression, protein kinase activity and is critical to MDA-MB-231 cell viability. Furthermore, a bioinformatic analysis of human breast cancer gene expression supports SPHK1 as a hallmark TGFβ target gene that also bears the genetic fingerprint of the basal-like/triple-negative breast cancer molecular subtype. These data suggest a potential new signaling axis between TGFβ/SphK1 that may have a role in the development, prognosis or the clinical phenotype associated with tumor-bone metastasis.
Introduction
Bone metastases are common in patients with advanced solid tumors such as breast, lung and prostate cancers as well as melanoma often causing debilitating bone pain, hypercalcemia and nerve compression syndromes.1,2 In addition, once primary tumors have spread to bone, they are considered incurable. Transforming growth factor β (TGFβ) has a central role in bone metastasis, but also in the regulation of normal bone homeostasis.2 Bone is the most abundant source of TGFβ in the body and it drives a vicious feed-forward cycle of skeletal metastases that has provided the rationale to test pharmacological inhibitors of TGFβ or the Smad signaling pathway in early-phase clinical trials.2,3
Sphingolipids comprise a family of membrane lipids important for the regulation of membrane fluidity and lipid sub-domain structure of lipid bi-layers.4 Regulators of sphingosine metabolism are capable of producing and liberating a number of bioactive sphingolipid species or metabolites including complex ceramides, sphingosine 1-phosphate (S1P) and glucosylceramides. Many of these bioactive signaling molecules have roles in the pathogenesis of cancer and its therapy.5 Regulatory functions of these sphingolipid-derived signaling molecules include alteration of cellular proliferation, survival, migration, chemotaxis, senescence, inflammation and angiogenesis.5,6,7,8 Recent studies suggest that the relative balance of sphingosine metabolism away from pro-apoptotic ceramide generation, but toward production of S1P by sphingosine kinase (SphK) is a potentially important survival and metastatic rheostat in many cancer cell types.8 To this end, cancer therapeutics targeting various aspects of these pathways as a means of increasing intracellular ceramide generation, or blockade of S1P production or signaling are under active development.9,10 Pre-clinical efficacy of sphingosine kinase-1 (SphK1) inhibitors has been demonstrated by several groups both in the setting of human breast cancer xenograft or syngeneic mouse tumor metastasis models.9,10,11,12 Furthermore, treatment with FTY720 (fingolimod), a potent functional antagonist of S1P signaling recently Food and Drug Administration approved for relapsing multiple sclerosis, displays significant prevention of tumor growth and metastasis at non-bone sites in numerous pre-clinical models including the Balb/C mouse flank-inoculated JygMC(A) cell breast cancer metastasis model.13 Intriguingly, the importance of S1P production and signaling to normal bone homeostasis has also been recently described. Specifically, S1P serves as an osteoclast–osteoblast coupling factor,14 as well as a central promoter of chemotaxis and motility of osteoclast precursors to and from the bone surface in vivo.15,16 In these studies, it was also found that antagonism of S1P signaling by FTY720 results in the blockade of ovariectomy-induced bone turnover and rescue of bone mineral density in these mice. Therefore, these data provide a compelling role for regulation of sphingolipid/S1P metabolism in not only tumor metastasis but also in physiological bone homeostasis, where pharmacologic intervention of these pathways appears tractable.
Recent evidence reveals direct regulatory activities between TGFβ/Smad signaling and alterations in sphingosine metabolism. Early in vitro studies in fibroblasts demonstrated that TGFβ stimulated both SPHK1 mRNA and SphK1 kinase activity.17 Subsequently, numerous observations have further demonstrated the convergence and, in many cases, the interdependence of bioactive sphingolipids and TGFβ signaling pathways for chemotaxis, connective tissue growth factor (CTGF) production, extracellular matrix (ECM)/collagen production, and cell survival.18,19,20,21,22,23 Interestingly, no disclosure of cross-talk between these two pathways has been alluded to in either physiological bone homeostasis or cellular behavior within the tumor–bone microenvironment.
Given the critical known importance of both TGFβ and SphK/S1P signaling in both cancer and bone biology, we sought to query regulatory connections between these pathways using previously described microarray studies of MDA-MB-231 human breast cancer sublines of varying metastatic capacity and aggressiveness.24 To this end, we identified the TGFβ/SphK1 signaling axis as a marker with tumor-bone metastatic potential.
Results
Identification of TGFβ-induced SPHK1 mRNA as a determinant in MDA-MB-231 metastatic behavior
We performed a systematic analysis of previously disclosed microarray data generated by Kang et al.24 using human MDA-MB-231-derived breast cancer cell lines with variable metastatic potential to identify any patterns related to sphingolipid/S1P metabolism or S1PR gene expression profiles. A total of 12 genes considered important to regulation of sphingosine/S1P metabolism and signaling were chosen as part of this analysis and are highlighted in italics (Figure 1a). Among the 12 genes, only 10 mRNAs were identified as detectible within the Affymetrix microarray data and thus used in our analysis. SP1R3 and SGPP2 were not detectible, presumably due to low or no expression of these genes in these cell lines. All correlative patterns were explored in an unbiased manner between low, median and high bone-metastatic MDA-MB-231 sublines, including both in vivo-selected sublines and in vitro-selected single-cell progeny (SCP)-derived expression data.24 TGFβ1-induced SPHK1 mRNA expression was shown to be significantly higher in SCPs with high bone-metastatic capacity when compared with low-metastatic capacity SCPs (Figure 1b). This association indicates that the capacity for TGFβ1-induced SPHK1 mRNA induction may present an inherent component and/or a biomarker of TGFβ-mediated osteolytic bone metastasis. Interestingly, the in vivo-selected sublines all showed high induction of SPHK1 by TGFβ1 regardless of metastatic capability. No significant relationship in baseline gene expression characteristics versus observed in vivo metastatic behavior of the sublines analyzed was evident for any of the 10 genes.
Figure 1.
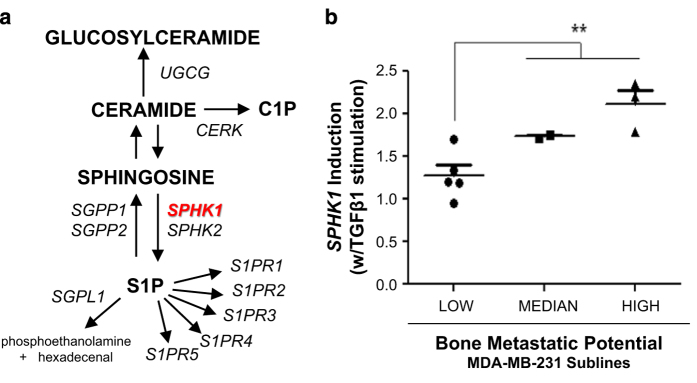
Query of sphingolipid metabolism and S1P signaling receptor genes from low, median and highly metastatic MDA-MB-231 sublines. Previously described microarray data obtained from various MDA-MB-231 sublines of varying metastatic capacity were queried for predictive value to the expression of genes involved in sphingolipid/S1P metabolism and signaling. Technical details of these studies have been described previously.24 (a) Displays a simplified schematic of sphingolipid/S1P metabolism and S1PR receptor genes (italicized) chosen for this retrospective analysis. TGFβ1-induced SPHK1 expression was identified and provided significant predictive value between low, median and high metastatic behavior of the various MDA-MB-231 (SCP-derived) sublines in mouse metastasis models (b). **P<0.01 (Student's t-test, low vs median/high).
Human cancer cell line screening and characterization of TGFβ1-induced SPHK1 expression
To further characterize the TGFβ1-mediated regulation of sphingolipid/S1P metabolism genes, several human cancer cell lines with differential but known in vivo bone-metastatic behavior were screened via quantitative PCR (qPCR)±TGFβ1 stimulation for various periods of time. We observed that only the osteolytic cell lines, MDA-MB-231 (breast) and 1205Lu (melanoma) increased SPHK1 and S1P lyase 1 (SGPL1) expression as a function of time in response to TGFβ1 (Figure 2). Robust upregulation of SGPL1 was noted in PC-3 prostate cancer cells and to a lesser extent in MDA-MB-231 and 1205Lu cell lines indicating that multiple pathways for control of S1P levels may lie downstream of TGFβ. No regulation of any S1P metabolizing genes by TGFβ1 was observed in either ‘osteoblastic' or ‘mixed-phenotype' cancer cell lines tested (Figure 2). Furthermore, we show that all three human TGFβ isoforms (TGFβ1, TGFβ2 and TGFβ3) and Activin A significantly increase the expression of SPHK1 in MDA-MB-231 cells, but not BMP2 or the hypoxia mimetics L-MIM and DMOG (Figure 3a). Additional experiments revealed that TGFβ1 and Activin A-induced SPHK1 expression in MDA-MB-231 cells is both time and dose dependent and displays similar induction kinetics to the highly induced and well-characterized TGFβ/Smad target gene, PMEPA125 (Figure 3b, Supplementary Figure S1). We demonstrate that SPHK1 upregulation is both RNA Polymerase II and type I TGFβ receptor (TβR1/Alk5) dependent as pre-treatment with Polymerase II inhibitor (actinomycin D) or TβR1/Alk5 inhibitor (SD-208) completely abolishes SPHK1 mRNA stimulation by TGFβ1, while pre-treatment with protein synthesis inhibitor cycloheximide had little effect on fold SPHK1 induction from baseline (Figure 4).
Figure 2.

qPCR screen for TGFβ1-mediated regulation of sphingosine/S1P metabolizing enzymes in human cancer cell lines. Human cancer cell lines grown in monolayer cultures were treated with 5 ng ml−1 hTGFβ1 for 2, 8 or 24 h followed by RNA isolation and Taqman-based qRT-PCR of SPHK1, sphingosine kinase-1; SPHK2, sphingosine kinase-2; SGPP1, sphingosine 1-phosphate phosphatase -1; SGPP2, sphingosine 1-phosphate phosphatase -2; and SGPL1, sphingosine 1-phosphate lyase. All mRNA values were normalized to GAPDH mRNA and plotted as fold induction versus the untreated control group (value=1). Only detectable/measurable mRNAs for each gene are displayed on their respective graphs.
Figure 3.

Effects of various TGFβ isoforms/family members and kinetics of SPHK1 expression in MDA-MB-231 cells. MDA-MB-231 cells were treated with recombinant hTGFβ1 (5 ng ml−1), hTGFβ2 (5 ng ml−1), hTGFβ3 (5 ng ml−1), hActivin-A (50 ng ml−1) or hBMP-2 (50 ng ml−1) for 8 h followed by qRT-PCR analysis (a). Cells treated with hypoxia mimetics L-mimosine (L-MIM) and dimethyloxalyl glycine (DMOG) were harvested after 24 h (a). Both time and dose response kinetic experiments for SPHK1 and PMEPA1 were treated for approximately 8 h followed by qRT-PCR analysis (b). All data were normalized to GAPDH mRNA, analyzed using the Δ/Δ CT method and plotted as fold induction versus the untreated control group (value=1x). All data are plotted as mean fold-induction±s.e.m., n=3 per treatment group. *P<0.05 and **P<0.01 versus control group (one-way ANOVA, Bonferroni-corrected).
Figure 4.

TβR1/Alk5 and RNA Polymerase II dependency for TGFβ1-mediated induction of SPHK1 expression. MDA-MB-231 cells were pre-treated for 30 min with either 10 μM SD-208 (TβR1/Alk5 inhibitor), 5 μg ml−1 actinomycin D (RNA Pol-II inhibitor) or 1 μM cycloheximide (protein synthesis inhibitor) followed by continued treatment with each agent in combination with 5 ng ml−1 TGFβ1 for 8 h. Cells were harvested, RNA was isolated and qRT-PCR analysis of SPHK1 and PMEPA1 expression was performed. All data were normalized to GAPDH mRNA, analyzed using the Δ/Δ CT method and plotted as fold induction versus their respective control groups (value=1x). All data are plotted as mean fold induction±s.e.m., n=3 per treatment group. *P<0.05 and **P<0.01 versus control group (one-way ANOVA, Bonferroni-corrected).
Regulation of TGFβ1-induced SphK1 protein, immunoprecipitation kinase activity and sphingosine/S1P content in MDA-MB-231 cells
In order to determine whether the observed increased expression of SPHK1 mRNA results in significant changes to SphK1 protein, kinase activity and/or sphingosine/S1P changes in parental MDA-MB-231 cells, several experiments were performed. Numerous commercial anti-SphK1 and anti-phospho SphK1 antibodies were screened to identify antibodies for detection and immunoprecipitation (IP) of SphK1 protein from lysates obtained of TGFβ1-treated monolayer cell cultures. Several groups have described multiple SphK1 protein isoforms via western blotting.26 We report similar results in our studies where 2–3 predominant isoforms (between 41 and 53 kDa) can be detected by western blotting in a time-dependent manner upon treatment with either TGFβ1 or Activin A with peak protein accumulation occurring at 24 h (Figure 5, Supplementary Figure S1).
Figure 5.

TGFβ1-mediated increase in SphK1 protein and kinase activity in MDA-MB-231 cells. MDA-MB-231 cells were treated for 2, 8 or 24 h with hTGFβ1 (5 ng ml−1). After TGFβ1 treatment, cells were lysed and protein separated via SDS-PAGE and western blotted for SphK1 and α-tubulin. For kinase assays, cell lysate was prepared and used for immunoprecipitation of SphK1 using a fixed combination of specific SphK1 (Abgent/ECM) antibodies. SphK1 kinase activity was assessed by the Sphingosine Kinase Activity Assay Kit (Echelon) in the absence or presence of the substrate sphingosine (600 μM). Sphk1 kinase activity was plotted as a bar graph function of ATP depletion using values interpolated from the ATP standard curve. All data are plotted as percent activity±s.e.m., n=3 per treatment group. *P<0.05 and **P<0.01 versus comparator group (one-way ANOVA, Tukey's HSD). Cellular sphingosine and S1P content were quantified via LC-MS/MS.
We measured sphingosine-dependent kinase activity via SphK1 IP and the SphK1 Kinase Activity Assay Kit (Echelon Inc.). Using this IP kinase assay, kinase activity from cell lysates was significantly increased upon TGFβ1 treatment at 2 and 8 h and returned to baseline by 24 h (Figure 5). Although we cannot rule out the potential cross-contamination of SphK2 activity in these IP kinase experiments, no SPHK2 mRNA increases were observed upon TGFβ1 treatment. Interestingly, the kinetics of mRNA and SphK1 protein increases do not mirror the IP kinase profile, suggesting that TGFβ1 may independently control SphK1 kinase activity via other post-transcriptional mechanisms not queried by our current studies. In corroboration with our findings, several other groups have documented the ability of TGFβ to acutely increase both SphK1 activity and S1P production within hours of stimulation in a MAPK-dependent manner.27,28 However, we do not observe significant ERK activation at these time points upon TGFβ1-stimulation of MDA-MB-231 cells (data not shown). To determine whether TGFβ has the potential to alter S1P levels, we measured sphingosine and S1P content in MDA-MB-231 cells±TGFβ1 at 8 h post stimulation. Upon TGFβ1 treatment, no significant change in cellular S1P or sphingosine content was detectable versus unstimulated cells (Figure 5). Further experiments with additional time points and/or measurement of the extracellular sphingolipid content may be required to fully address this question. Upregulation of SGPL1 is also observed acutely upon TGFβ1 stimulation (Figure 2). This upregulation may have significant effects on measurable S1P content as S1P lyase activity utilizes S1P as substrate to generate phosphoethanolamine and fatty aldehyde. To our knowledge, this is the first description that TGFβ regulates the SGPL1 gene or that SPHK1 and SGPL1 may be co-regulated gene products (Figure 2).
TGFβ-stimulated MDA-MB-231 conditioned medium enhances RAW264.7 monocyte motility and is sensitive to SphK inhibition
To assess a functional consequence of TGFβ stimulation of SphK1 expression/activity, we tested whether increased SphK1 activity would result in enhanced RAW264.7 mouse monocyte motility in transwell migration assays. Other groups have previously shown the ability of S1P/S1PR agonism to promote the motility/migration of RAW264.7 cells, therefore, we set out to validate these findings using S1P (S1PR agonist), SEW2871 (S1PR1 agonist) and known monocyte chemotactic factor SDF-1α prior to performing our own studies.29 Indeed, we also demonstrate robust dose-dependent enhancement of RAW264.7 cell transwell migration with all three agents and thereby validated published data (Figure 6). Subsequently, we observed that MDA-MB-231 conditioned medium (231-CM; complete medium that has been incubated for 8 h with confluent monolayer culture of MDA-MB-231 cells) significantly enhances RAW264.7 cell transwell migration. Combined treatment of TGFβ1 with MDA-MB-231 cells (231-CM+TGFβ1 Tx) during the media conditioning period further significantly increases RAW264.7 migratory capacity beyond 231-CM only (Figure 6). Although Activin A treatment (231-CM+ActA Tx) also increased migration, it did not achieve statistical significance (Figure 6). To control for the direct migratory effects of TGFβ1 being present in the conditioned media, we found that adding TGFβ1 protein directly to 231-CM (231-CM+TGFβ1) did not enhance RAW264.7 cell migration versus 231-CM alone (Supplementary Figure S2A).
Figure 6.

RAW264.7 monocyte cell transmigration enhancement by S1PR agonism and TGFβ1-treated MDA-MB-231 conditioned media. RAW264.7 cells were seeded into upper Transwell chambers where differing concentrations of S1P, SEW2871 or SDF-1α resided in lower chambers. Similarly, RAW264.7 cells were seeded into upper chambers where differing MDA-MB-231 conditioned media(s) from untreated or compound/TGF pre-treated studies resided in lower chambers. Numbers of migrating cells through the Transwell membranes were fixed, stained and counted for each independent chamber. Data are plotted as mean±s.e.m. for total number of cell migrated per membrane field, n=3 membranes for each treatment. *P<0.05 and **P<0.01 versus comparator group (one-way ANOVA, Tukey's HSD).
To assess whether SphK inhibition alters the observed TGFβ1-mediated RAW264.7 migratory enhancement, SphK inhibitors DMS and 2-(p-hydroxyanilino)-4-(p-chlorophenyl) thiazole (SKI II), were added just prior to TGFβ1 treatment of MDA-MB-231 cells. In doing so, we observed a dose-dependent and significant blunting of RAW264.7 cell transwell migration but not complete abolition of the migratory enhancement (Figure 6). To control for any deleterious direct effects of these SphK inhibitors on RAW264.7 cell migration alone, control experiments were conducted. They revealed no significant effects of these compounds on baseline RAW264.7 cell migration or 231-CM mediated migratory activity (Supplementary Figure S2B) suggesting that these effects are a TGFβ1-dependent process. These data suggest that SphK activity may have a significant but only a partial role in these phenomena. From a mechanistic standpoint, these studies were not designed to confirm S1P dependence for these observations, but do provide substrate for further inquiry into this question. Several possibilities beyond S1P could be hypothesized including changes in other TGFβ-mediated secreted products, other SphK1 lipid substrates or alterations in the emerging area of extracellular vesicle/exosome sloughing.30,31
Effects of SPHK1 genetic manipulation or SphK1 inhibition on MDA-MB-231 cell viability
To determine whether alteration of SphK1 status, expression or activity is an important determinant in a mouse model of osteolytic bone metastasis, several strategies were employed to generate both mutant SPHK1 transgenic and knockout MDA-MB-231 sublines for in vivo metastases evaluation. Although SphK1 small interfering RNA- and short hairpin RNA-expressing plasmids mediated significant silencing, these effects were short lived and no stably expressing viable MDA-MB-231 cell clones could be generated for long-term evaluation. Alternatively, we generated, selected and cloned overexpressing sublines of MDA-MB-231 harboring either wild-type SphK1 or mutant SphK1; constitutively active (1–363 aa) isoform or a dominant-negative/catalytically inactive (G82D) isoform. Although several dozen clonal sublines were obtained upon chronic exposure to the selection agent, no expression or presence of the transgene could be confirmed and validated in these clones. Finally, we employed zinc-finger nuclease (ZNFn) technology (Sigma-Aldrich) custom designed to ‘knock-out' the SPHK1 gene from parental MDA-MB-231 cells. Therefore, we co-transfected the ZNFn targeting plasmid cassette along with an enhanced green fluorescent protein (eGFP) expression plasmid, followed by FACS-based sorting of eGFP-positive cells. Two sorted ‘pools' of MDA-MB-231 cells were obtained (P2-high eGFP, P3-moderate eGFP) and genomic PCR profiling revealed appropriate ZNFn-mediated genomic PCR products (∼0.150–0.160 kb) at the SPHK1 genomic locus versus a vendor-provided positive control PCR template confirming that this strategy was successful (Figure 7a). Cell culture-based expansion of P2 and P3 pools, namely P2.e and P3.e, or single cell cloning by limiting dilution did not allow us to identify or isolate any MDA-MB-231 clonal sublines with the appropriate ‘KO' genetic modification, despite successful genomic PCR evidence to the contrary upon initial cell sorting (Figure 7a).
Figure 7.

ZNF nuclease-mediated targeting of SPHK1 in MDA-MB-231 and cytotoxic sensitivity to SphK inhibitors. MDA-MB-231 cells were transiently-transfected with a custom SphK1 ZNF nuclease genomic targeting cassette along with CMV–eGFP expressing plasmid followed by FACS-mediated cell sorting of cell pools of varying fluorescence intensity (P1, P2 and P3) and subsequent culture-expanded pools (P2.e and P3.e). Genomic PCR was performed to assess genomic modification of the SPHK1 locus in various transfected and expanded MDA-MB-231 cell pools. Positive genomic alteration is noted by a black arrow corresponding to a PCR products band at ∼150–160 nt by gel electrophoresis (a). To assess MDA-MB-231 cell viability, cells were seeded and treated with varying concentrations of DMS, SKI II, FTY720 and SEW2781 for 7 days followed by cell viability determination by Cell Titer Glo analysis. Table IC50 calculations were based on the concentration response values analyzed by four-parameter non-linear regression curve fitting using GraphPad Prism (ver6.0) from two independent studies (b).
Because of the lack of cell viability following genetic manipulation of SphK1 activity in MDA-MB-231, we assessed the sensitivity of these cells to small molecule SphK1/S1PR modulators in the presence or absence of TGFβ1. We hypothesized that the presence of TGFβ1-mediated increase in SPHK1 expression may drive enhanced resistance to SphK1/S1PR modulator treatment. To test this hypothesis, we treated cells±TGFβ1 for 7 days in two independent studies with varying concentrations of FTY720 (S1PR functional antagonist/SphK inhibitor), SKI II (SphK inhibitor), DMS (SphK inhibitor) or SEW2871 (S1PR1 agonist) and measured cell viability as a function of cellular ATP content using a CellTiter-Glo assay. Cell viability curves were plotted, fitted and calculated IC50 concentration values determined for each compound tested (Figure 7b). The IC50 values generated support the hypothesis that MDA-MB-231 cells require SphK activity to maintain cellular viability with a relative rank order potency—DMS>FTY720>SKI II for each of the SphK inhibitors tested. SEW2871 displayed little to no activity in these studies consistent with its distinct mechanism of action as an S1PR functional agonist versus the comparator compounds being SphK inhibitors. However, no significant differences were observed for IC50 values when TGFβ1 is present or absent in the treatment media (Figure 7b).
SPHK1 expression shares a co-expression pattern with TGFβ target genes and the basal-like/TNBC molecular subtype
To explore the potential clinical relevance of SPHK1 expression and its connection to TGFβ signaling and/or human breast cancer, we utilized co-expression data derived as a part of The Cancer Genome Atlas (TCGA) project, which has comprehensively analyzed primary breast cancers by genomic DNA copy number, mRNA arrays, microRNA expression sequencing and reverse-phase protein arrays.32 For our analysis, we exclusively focused on the query of gene co-expression profiles specifically from data generated from 526 patient tumor samples with mRNA data generated using the Agilent mRNA microarray platform. Co-expression profiles and correlations from TCGA project data were obtained using cBioPortal for Cancer Genomics (http://cbioportal.org), which provides a Web resource for analysis and visualization of multi-dimensional cancer genomics data.33,34 We prospectively identified three broad lists of genes to query against this breast cancer genomics data set: (1) TGFβ/Activin signaling complex genes, (2) well-characterized TGFβ pathway target genes and the (3) PAM50 breast cancer subtype gene profiling panel.35 As a control gene, we included SPHK2 in this analysis as its regulation was not shown to be TGFβ dependent in our studies, and its purported function often opposes SPHK1 in several contexts.36
The results of our co-expression analysis provide a compelling argument that the SPHK1 gene signature shares an expression profile with two classes of genes in the TGCA human breast cancer samples: (1) SPHK1 expression positively correlates with numerous well-characterized TGFβ target genes (CTGF, LOX, MMPs and PMEPA1), and (2) the PAM50 gene panel suggests that SPHK1 expression is positively correlated with triple-negative/basal-like subtype genes (KRT17 and FOXC1), whereas negatively correlated with the estrogen receptor gene program (ESR1 and FOXA1). To further explore the subtype selectivity, we added a fourth gene set that was recently described as a list of highly correlated up and downregulated genes identified from a large meta-analysis of seven independent clinical studies of triple-negative breast cancer (TNBC).37 This list compiled from the meta-analysis included 20 overexpressed and 20 under-expressed genes in TNBC. Indeed, SPHK1 expression positively correlates with a majority of overexpressed genes (MTHFD1L and FOXC1), while having a negative correlation for a majority of the under-expressed genes (GATA3 and SLC39A6). By comparison, SPHK2 displays no positive co-expression to the TGFβ signaling complex or its known target genes. Interestingly, SPHK2 does show some positive and negative co-expression to several PAM50 and TBNC genes, but is the exact inverse of the SPHK1 co-expression profile. This analysis demonstrates that SPHK1 expression shares the hallmark signature of a TGFβ target gene and is correlative with the basal-like/TNBC molecular subtype expression profile.
Discussion
Bone metastasis is the ultimate step of malignancy for many solid tumor types resulting in severe symptoms that markedly reduce quality of life and ultimately results in death of the patient. Bone metastases are classified as either being ‘osteolytic', ‘osteoblastic' or of mixed phenotype due to either excessive bone destruction (osteolytic) or bone formation (osteoblastic) as determined by radiographic analysis. Mechanistic understanding of these cellular dynamics within the tumor–bone compartment remains poorly understood. It is believed that the enzymes, receptors and/or chemical mediators involved in these processes may serve as important next-generation drug targets for future cancer therapy in this space.
The aim of our work was to identify functional correlates between expression of sphingolipid metabolism/signaling genes and TGFβ stimulation and/or metastatic capacity. We retrospectively queried microarray data sets from previously described MDA-MB-231 sublines of low, median or high metastatic nature.24 Only a single significant correlation was detected by this analysis and revealed that significant enhancement of TGFβ1-mediated induction of SPHK1 mRNA occurs in median/highly metastatic MDA-MB-231 (SCP derived) sublines versus comparator sublines with low metastatic capacity (Figure 1). This finding was compelling as SPHK1 is a well-documented classic oncogene, prognostic indicator in several human cancer types and is one of the only two SPHK genes in the human genome encoding the proteins, SphK1 and SphK2, which are capable of producing the important signaling molecule S1P.9,38 Therefore, we sought to further characterize the nature of TGFβ-mediated regulation of SPHK1 in parental MDA-MB-231 cells. Although several groups have documented this signaling axis in other cellular settings including fibroblasts and mesangioblasts, no demonstration of this has been made in cancer or tumor cell lines to our knowledge.17,39 Screening of a small panel of human cancer cell lines of differing solid tumor origin and bone metastatic behavior reveals that this regulation occurs only in osteolytic MDA-MB-231 breast cancer and 1205Lu melanoma cells (Figure 2). We found that all TGFβ isoforms (TGFβ1, TGFβ2 and TGFβ3) as well as Activin A are capable of driving significant induction of SPHK1 expression (Figure 3). This upregulation is both time and dose dependent, and closely mimics the kinetics of a prototypical TGFβ/Smad target gene, PMEPA1 (Figure 3). We demonstrated that SPHK1 induction was dependent on the TβR1/Alk5 activity and requires de novo transcription through RNA Pol-II, but not de novo protein synthesis as cycloheximide treatment has little effect on this induction (Figure 4). Moreover, we were able to detect significantly increased SphK1 protein and activity in TGFβ-treated cells (Figure 5). The temporal increase in SphK1 protein versus measureable SphK activity did not correlate suggesting additional post-translational mechanisms by which TGFβ may independently regulate SphK1 activity as has been shown in other cellular contexts.27,28 Despite the observed increase in SphK activity, we were not able to detect the concomitant production of S1P by LC-MS/MS at the peak kinase activity time point of 8 h with multiple experiments (Figure 5). A more rigorous sphingolipodomic approach may be needed to appropriately address the kinetics of TGFβ-mediated alterations in phosphorylated sphingolipid metabolites and/or S1P content upon stimulation.
To determine whether SPHK1 upregulation by TGFβ in parental MDA-MB-231 cells results in a functional consequence to cell growth or metastatic aggressiveness, we initiated a series of attempts to alter SPHK1 status through in vitro-based ZNFn-mediated knockout or mutant transgene-mediated manipulation of the SphK1 signaling pathway (Figure 7). We believe we successfully ‘knocked out' the SPHK1 gene but were unable to expand these cells (Figure 7). Furthermore, stable overexpression and clonal selection of cells harboring either constitutively active or dominant-negative SphK1 isoforms were also not successful. Our conclusion is that manipulation of SPHK1 genetic status or activity is not compatible with MDA-MB-231 cell viability. Unfortunately, these ‘mutant' or ‘KO' SphK1 MDA-MB-231 clonal cell lines would have been critical to validate our hypothesis that SphK1 expression/activity alters in vivo metastatic potential and/or MDA-MB-231 osteolytic behavior in mouse intracardiac-injection models of bone metastasis.40,41 In support of our genetic findings, we also showed that treatment with small molecule SphK1 inhibitors significantly impairs cell viability (Figure 7). Overall, these data suggest that SphK1 is critical for MDA-MB-231 cell health and that ‘balanced' SphK1 activity is required for sustainable growth in culture.
Finally, we sought to profile SPHK1 expression using available Agilent microarray data derived from invasive human breast cancer/tumor biopsies obtained as a part of TCGA Project.32 The past decade of research on SphK1 has revealed its unique tumorigenic and prognostic role in human breast cancer, as well as other cancers.42,43,44,45,46,47,48 SphK1 has been shown to promote the growth and responsiveness in estrogen receptor (ER)-positive breast cancer cells, but it may also directly regulate estrogen receptor signaling to mediate endocrine or tamoxifen resistance.49,50,51,52 Moreover, a microarray analysis of 43 sphingolipid metabolism genes from over ∼960 breast cancer/tumor samples identified SPHK1 gene expression to be of high prognostic significance in breast cancer patients.42 Specifically, survival analysis from this study revealed overall worse clinical outcomes and diminished time-to-metastasis (5-year period) for patients with higher expression of SPHK1. To avoid confounding effects due to ER status in these findings, the authors restricted the analysis to the roughly ∼750 ER+ patient tumor samples and still found that high SPHK1 expression in these tumors predicts a negative outcome.42 Additional gene expression analysis from a small ∼112 BrCa patient cohort found increased SPHK1 expression in ER-negative versus ER-positive breast cancer.53 Furthermore, the prognostic significance of perturbed sphingolipid metabolism in ER-negative BrCa was also identified through a pooled analysis of ER-negative tumors, which identified the glyco- and sphingo-lipid pathway as dysregulated and served as a prognostic signature of this BrCa subtype.54 Our own gene co-expression analyses from the TCGA data support many of these previously published data (Figure 8). Using SPHK2 as a ‘negative control' gene, we found that SPHK1 shares the hallmark signature of a TGFβ/Activin target gene. This was exemplified by positive correlative expression with such genes as CTGF, NOX4, LOX, PMEPA1 and TGFBI. Query of the PAM50 gene panel, often used for molecular subtyping of BrCa, revealed a gene signature with a bias toward the basal-like/TNBC molecular subtype (e.g., FOXC1, KRT5 and KRT17). To explore the connection to the TNBC phenotype further, we used a cassette of 40 genes recently identified as either overexpressed (e.g., FOXC1 and FAM171A1) or under-expressed (e.g., GATA3 and FOXA1) from a large meta-analysis of seven independent ER-negative BrCa patient tumor profiling studies.37 Indeed, we found very strong correlation in both classes of TNBC genes, where SPHK1 expression positively correlates with TNBC overexpressed genes, but negatively correlates with TNBC under-expressed genes. Interestingly, SPHK2 expression does not share this signature, but alternatively displays the inverse relationship suggesting potential merit to a purported yin–yang hypothesis between SphK1 and SphK2.36 Overall, we believe that this analysis provides an additional piece to the growing body of evidence for the prognostic value of SPHK1 expression in breast cancer progression, subtype profiling or patient/treatment tailoring approaches.
Figure 8.

Evaluation of the SPHK1 gene expression signature in human breast cancer. Agilent microarray data compiled as part of The Cancer Genome Atlas (TCGA) Project were queried for correlative gene co-expression profiles to previously identified TGFβ/Activin signaling, PAM50 and triple-negative breast cancer (TBNC) gene panels and molecular signatures. Analysis was performed using the cBioPortal (http://cbioportal.org) gateway and repository for cancer genomics data—specifically utilizing the TCGA (Nature) invasive breast cancer patient data.32,33,34,35,37
An emerging pharmacopeia is developing from both industry and academic groups with the therapeutic goal of manipulating the activity of TGFβ and/or SphK-S1P signaling for several disease indications including cancer, fibrosis and autoimmune disease.2,6,9,55 Here, we provide evidence that SphK1 regulation may, in part, lie downstream of perturbed TGFβ signaling in the context of tumor–bone metastasis. These findings are intriguing as they may provide evidence for a new TGFβ/SphK1 axis in breast cancer. Certainly, additional research will be needed to validate the functional importance of this potential connection, but may constitute an opportunity for novel drug discovery, cancer patient tailoring or therapeutic strategy.
Materials and methods
Cell culture and reagents
The human cancer cell lines MDA-MB-231 (breast), MCF7 (breast), PC-3 (prostate), ZR-75-1 (breast), C4-2B (prostate) and mouse monocyte RAW264.7 cells were purchased and cultured accordingly from the American Type Culture Collection (Manassas, VA, USA). 1205Lu melanoma cells were a kind gift from Dr M Herlyn of the Wistar Institute (Philadelphia, PA, USA). All cell lines were grown at 37 °C with 5% CO2 in a humidified tissue culture chamber. MCF7, RAW264.7 and MDA-MB-231 cells were cultured in Dulbecco's Modified Eagle Medium (DMEM, high glucose) containing 10% fetal bovine serum (FBS). PC-3 cells were cultured in RPMI supplemented with 10% FBS. C4-2B cells were cultured in T-Medium containing 10% FBS. 1205Lu melanoma cells were cultured in W489 medium comprised of three parts MDCB153 (Sigma-Aldrich, St Louis, MO, USA) and one part Leibovitz15 supplemented with 4% FBS. No antibiotics were present in the cell media(s) that were used for the experiments. Recombinant hTGFβ1, hTGFβ2, hTGFβ3, hActivin-A and hBMP-2 were all purchased from R&D Systems, Inc. (Minneapolis, MN, USA). Human SDF-1α protein was acquired from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). SD-208 was obtained from Scios, Inc. (San Francisco, CA, USA). N,N-dimethylshingosine (DMS) was obtained from BioMol (Plymouth Meeting, PA, USA). S1P and SKI II were obtained from Echelon Biosciences Inc. (Salt Lake City, UT, USA). L-mimosine, dimethyloxaloglycine, actinomycin D and cycloheximide were obtained from Sigma-Aldrich. FTY720 and SEW2781 were obtained from Cayman Chemical Co. (Ann Arbor, MI, USA). All small organic compounds were re-suspended in dimethylsulfoxie (DMSO) or methanol at 10 mM stock concentrations and stored at −80 °C until use. All recombinant proteins were reconstituted in phosphate-buffered saline (PBS), 0.1% bovine serum albumin and stored at −80 °C until use.
Cell treatment, RNA isolation and quantitative RT-PCR
All cell-based experiments evaluating gene expression were performed by seeding cells at ∼80% confluence in six-well tissue culture plates or 100 mm tissue culture dishes and incubated overnight (∼12 h) in 1% FBS-containing media(s) prior to stimulation. All cell stimulation or treatment experiments were performed at full confluence in 1% FBS-containing media(s) in the presence/absence of a 30 min pre-treatment with chemical compounds (e.g., cycloheximide, SD-208, actinomycin D) prior to addition of the stimulus (e.g., TGFβ1, Activin A). Cell harvest and RNA isolation was performed using the GenElute Total RNA Miniprep Kit (Sigma-Aldrich). Complementary DNA (cDNA) synthesis was performed using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA). qPCR of all samples was performed in triplicate and analyzed using Taqman Gene Expression Assays with Universal PCR Master Mix (Applied Biosystems) by the Δ/Δ-CT method. Taqman primer-pair/probes used for each gene are listed in Supplementary Table S1.
Western blotting and SphK1 IP kinase assays
Cells were seeded at ∼80% confluence in 100 mm tissue culture dishes and incubated overnight (∼12 h) in 1% FBS-containing media(s) prior to stimulation. Cells were stimulated for various times with TGFβ1 (5 ng ml−1) or Activin A (50 ng ml−1) in 1% FBS serum. To harvest, cells were washed once with cold PBS, drained and lysed in ∼500 μl of RIPA cell lysis buffer plus COMPLETE protease inhibitors and PhosSTOP phosphatase inhibitors (Roche Diagnostics, Nutley, NJ, USA). Protein quantification was performed with the Bradford Assay (Bio-Rad, Hercules, CA, USA). Protein samples were separated by 10% SDS-PAGE and transferred onto Hybond-P membrane (GE Healthcare Life Sciences, Waukesham, WI, USA). Membranes were blocked in tris-buffered saline-T-milk (5%) for 1 h., incubated overnight with 1° antibody, followed by incubation for 1 h. with 2° horseradish peroxidase (HRP)-conjugated antibody. Protein detection was performed by ECL using Immobilon Western Chemiluminescent HRP substrate (Millipore, Billerica, MA, USA). Primary antibodies used for blotting were anti-SphK1 (Cat# AP7237c; Abgent Inc., San Diego, CA, USA) and anti-α-tubulin (Cat# T5168; Sigma-Aldrich). Anti-mouse IgG and anti-rabbit IgG secondary HRP-conjugated antibodies were acquired from Sigma-Aldrich.
To perform IP kinase assays, cells were stimulated with TGFβ1 as described previously, whereas cell lysis for IP was performed specifically using M-PER Mammalian Protein Extract Reagent (Pierce Biotechnology, Rockford, IL, USA). Approximately 150 μg of cell extract (∼1 ml) was used per IP using a 1:1 combination (5 μg total) of two SphK1 antibodies: anti-SphK1 (Cat# AP7237c; Abgent Inc.) and anti-SphK1 (Cat# SP1621; ECM Biosciences, Versailles, KY, USA). Primary antibody capture for each IP was performed using Protein-A Sepharose4B beads (Invitrogen, Camarillo, CA, USA). Measurement of SphK1 activity on bead-captured SphK1 protein was performed in the absence or presence of 600 μM sphingosine using the Sphingosine Kinase Activity Assay kit acquired from Echelon Inc. (Salt Lake City, UT. USA). Recombinant full-length ‘Active' SphK1 (Cat# E-K068) was obtained from Echelon Inc. and used as a positive control. Kinase activity values were determined based on ATP depletion and interpolated from an ATP standard curve. Recombinant ‘active' SphK1 protein was purchased and used as a positive control for kinase activity in this assay (Echelon Inc.).
Lipid extraction and sample preparation for LC-MS/MS analysis
Liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis of sphingosine and S1P was performed for N=6 per treatment group using a TSQ Quantum Ultra-triple quadrupole mass spectrometer (Thermo Fisher, San Jose, CA, USA) equipped with an electrospray ionization (ESI) probe and interfaced with an Agilent 1100 HPLC (Agilent Technologies, Wilmington, DE, USA). Lipid extracts were separated with an Xbridge C8 (Waters, Milford, MA, USA). Mobile phase A was: MeOH/H2O/formic acid (55:45:0.4% by v/v), and mobile phase B was: MeOH/acetonitrile/formic acid (50:50:0.4% by v/v). Mass spectrometric analyses were performed online using ESI tandem mass spectrometry in the positive multiple reaction monitoring mode. Samples were extracted using the one-phase extraction method (methanol-dichloromethane) with internal standards (IS) and 0.1% diethylamine. Pure synthetic standards of sphingosine (C17:0 and C18:0) and S1Ps (C17:0 and C18:0) were obtained from Avanti Polar Lipids (Alabaster, AL, USA). Sphingosine and S1P were quantified by the ratio of analyte to IS and calibration curve obtained by serial dilution of sphingosine and S1P.
RAW264.7 mouse monocyte migration assays
All transwell experiments were performed using 24 mm Transwell with 8.0 μM pore polycarbonate membrane inserts (Cat# 3428; Corning, Tewksbury, MA, USA). Briefly, 1.5 × 105 RAW264.7 cells were seeded into upper Transwell chambers, whereas differing concentrations of S1P, SEW2871, SDF-1α or various types of MDA-MB-231-treated and/or conditioned media resided in lower chambers. MDA-MB-231 conditioned media experiments used media removed from the cell monolayer±TGFβ1 (5 ng ml−1) or Activin A (50 ng ml−1) for ∼8 h. Pre-treatment of MDA-MB-231 with SphK inhibitors, DMS and SKI II, began 30 min prior to the addition of TGFβ1. RAW264.7 cells seeded into upper Transwell chambers were allowed to migrate for ∼3 h before removal of media and membrane inserts. Membranes were swabbed, formalin-fixed, placed in hematoxylin stain and washed prior to counting by light microscopy. Numbers of migrating RAW264.7 cells passing through insert membranes were counted for each independent chamber. Data were plotted as the mean±s.e.m. of the total number of cells per membrane field. N=3 membranes were used for each treatment and control groups. One-way analysis of variance, Tukey's HSD (*P<0.05, **P<0.01) versus comparator group was used to evaluate statistical significance.
SPHK1 ZNFn genomic modification and cell viability assays
Genomic modification of the human SPHK1 in MDA-MB-231 cells was performed using the CompoZr Knockout Zinc-Finger Nucleases Kit (Cat# CKOZFN1568-1kt; Sigma-Aldrich). This kit contains all necessary pZNF plasmids, primers and ZNF Control DNA for analysis of mutation detection (via CEL-I assay). The manufacturer's protocols were followed as described, but included addition of pGreen Lantern plasmid (Life Technologies, Grand Island, NY, USA) upon transient transfection of MDA-MB-231 cells. CEL-I survey or mutation detection assay products were two PCR products (0.156 bp and 0.164 bp) and were revealed by agarose gel electrophoresis. Cell sorting and isolation by eGFP fluorescence was performed on a BD FACSAria (BD BioSciences, San Jose, CA, USA) by technical staff at the Flow Cytometry Facility at the IU Simon Cancer Center.
To evaluate MDA-MB-231 cell viability in the presence of SphK1 inhibitors FTY720, SKI II, DMS and the S1PR1 agonist SEW2781, two independent cell viability experiments were performed. Briefly, MDA-MB-231 cells were seeded at ∼600 cells per well in a 96-well plate containing 1% FBS DMEM media, ±5 ng ml−1 TGFβ1 and variable SphK1 inhibitor concentrations (1% DMSO final concentration) and incubated together with cells for 7 days. At day 7, MDA-MB-231 cell viability was assessed using a Cell Titer Glo Luminescent Viability Assay from Promega (Madison, WI, USA). Regression curve fitting and IC50 determinations were performed using GraphPad Prism Software (La Jolla, CA, USA).
TCGA data mining and BrCa tumor gene co-expression analysis
Comprehensive detail concerning data generated by the TCGA Project specifically related to breast cancer patient samples used in our analysis can be found.32 For our analysis, we exclusively focused on bioinformatic query of patient tumor-gene co-expression profiles specifically from 526 patient tumor samples with mRNA data generated using the Agilent mRNA microarray platform. Co-expression data and profiles were obtained using cBioPortal for Cancer Genomics (http://cbioportal.org), which provides a web resource for analysis and visualization of multi-dimensional cancer genomics data.33,34 Pearson's correlations were computed first. For genes with a Pearson's correlation greater than 0.3 or less than −0.3, the Spearman's correlation was then also computed. Only gene pairs having both correlation values greater than 0.3 or less than −0.3 were considered meaningful (designated by either green or red colors in the custom heat map). Green color designations were given to co-expression patterns that were positively correlated with SPHK1 or SPHK2. Red color designations were given to co-expression patterns that are negatively correlated with SPHK1 or SPHK2. All guidelines for the use of TCGA data and visualization data tools provided by cBioPortal were followed accordingly and can be found at the following link (http://cancergenome.nih.gov/publications/publicationguidelines).
Supplementary Material
Acknowledgments
We would like to acknowledge Yong Wei, Princeton University, NJ, USA for providing microarray data and guidance from previously disclosed research.24 This work was supported by grants to TAG (U01CA143057 from the NCI Tumor Microenvironment Network; R01CA69158), the Susan G Komen Foundation, the Indiana Economic Development Grant, the Jerry and Peggy Throgmartin Endowment of the IU Simon Cancer Center, IU Simon Cancer Center Breast Cancer Program and a generous donation from the Withycombe family. KRS would also like to acknowledge the support by the Devault Foundation/IU Simon Cancer Center in providing a fellowship grant to support this work.
Footnotes
TAG received commercial research grants from AstraZeneca and Exelexis and is a consultant/advisory board member of Novartis. KRS, JKM, DFE and HHB are employees of Eli Lilly and Company. The remaining authors declare no conflict of interest.
References
- Sethi N, Kang Y. Unravelling the complexity of metastasis - molecular understanding and targeted therapies. Nat Rev Cancer 2011; 11: 735–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buijs JT, Stayrook KR, Guise TA. The role of TGFβ in bone metastasis: novel therapeutic perspectives. BoneKey Rep 2012; 1: 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buijs JT, Stayrook KR, Guise TA. TGFβ in the bone microenvironment: role in breast cancer metastasis. Cancer Microenviron 2011; 4: 261–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futerman AH, Hannun YA. The complex life of simple sphingolipids. EMBO Rep 2004; 5: 777–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kok JW, Sietsma H. Sphingolipid metabolism enzymes as targets for anticancer therapy. Curr Drug Targets 2004; 5: 375–382. [DOI] [PubMed] [Google Scholar]
- Takabe K1, Paugh SW, Milstien S, Spiegel S. "Inside-out" signaling of sphingosine-1-phosphate: therapeutic targets. Pharmacol Rev 2008; 60: 181–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox TE, Finnegan CM, Blumenthal R, Kester M. The clinical potential of sphingolipid-based therapeutics. Cell Mol Life Sci 2006; 63: 1017–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modrak DE, Gold DV, Goldenburg DM. Sphingolipid targets in cancer therapy. Mol Cancer Ther 2006; 5: 200–208. [DOI] [PubMed] [Google Scholar]
- Pyne S, Bittman R, Pyne NJ. Sphingosine kinase inhibitors and cancer: seeking the golden sword of Hercules. Caner Res 2011; 71: 6576–6582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truman JP, Garcia-Barros A, Obeid LM, Hannun YA. Evolving concepts in cancer therapy through targeting sphingolipid metabolism. Biochim Biophys Acta 2014; 1841: 1174–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoon JW, White MD, Driver JL, Burow ME, Beckman BS. Sphingosine kinase isoforms as a therapeutic target in endocrine therapy resistant luminal and basal-A breast cancer. Exp Biol Med 2012; 237: 832–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagahashi M, Ramachandran S, Kim EY, Allegood JC, Rashid OM, Yamada A et al. Sphingosine-1-phosphate produced by sphingosine kinase 1 promotes breast cancer progression by stimulating angiogenesis and lymphangiogenesis. Cancer Res 2012; 72: 726–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azuma H, Takahara S, Ichimaru N, Wang JD, Itoh Y, Otsuki Y et al. Marked prevention of tumor growth and metastasis by a novel immunosuppressive agent, FTY720, in mouse breast cancer models. Cancer Res 2002; 62: 1410–1419. [PubMed] [Google Scholar]
- Pederson L1, Ruan M, Westendorf JJ, Khosla S, Oursler MJ. Regulation of bone formation by osteoclasts involves Wnt/BMP signaling and the chemokine sphingosine-1-phosphate. Proc Natl Acad Sci USA 2008; 105: 20764–20769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii M, Egen JG, Klauschen F, Meier-Schellersheim M, Saeki Y, Vacher J et al. Sphingosine-1-phosphate mobilizes osteoclast precursors and regulates bone homeostasis. Nature 2009; 458: 524–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii T, Shimazu Y, Nishiyama I, Kikuta J, Ishii M. The role of sphingosine 1-phosphate in migration of osteoclast precursors; an application of intravital two-photon microscopy. Mol Cells 2011; 31: 399–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka M, Shegogue D, Pei H, Bu S, Bielawska A, Bielawski J et al. Sphingosine kinase 1 (SPHK1) is induced by transforming growth factor-beta and mediates TIMP-1 up-regulation. J Biol Chem 2004; 279: 53994–54001. [DOI] [PubMed] [Google Scholar]
- Xin C, Ren S, Kleuser B, Shabahang S, Eberhardt W, Radeke H et al. Sphingosine-1-phosphate cross-activates the Smad signaling cascade and mimics transforming growth factor-beta-induced cell responses. J Biol Chem 2004; 279: 35255–35262. [DOI] [PubMed] [Google Scholar]
- Gomez-Brouchet A, Pchejetski D, Brizuela L, Garcia V, Altié MF, Maddelein ML et al. Critical role for sphingosine kinase-1 in regulating survival of neuroblastoma cells exposed to amyloid-beta peptide. Mol Pharmacol 2007; 72: 341–349. [DOI] [PubMed] [Google Scholar]
- Xin C, Ren S, Eberhardt W, Pfeilschifter J, Huwiler A. The immunomodulator FTY720 and its phosphorylated derivative activate the Smad signalling cascade and upregulate connective tissue growth factor and collagen type IV expression in renal mesangial cells. Br J Pharmacol 2006; 147: 164–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igarashi J, Shoji K, Hashimoto T, Moriue T, Yoneda K, Takamura T et al. Transforming growth factor-beta1 downregulates caveolin-1 expression and enhances sphingosine 1-phosphate signaling in cultured vascular endothelial cells. Am J Physiol Cell Physiol 2009; 297: C1263–C1274. [DOI] [PubMed] [Google Scholar]
- Kono Y, Nishiuma T, Nishimura Y, Kotani Y, Okada T, Nakamura S et al. Sphingosine kinase 1 regulates differentiation of human and mouse lung fibroblasts mediated by TGF-beta1. Am J Respir Cell Mol Biol 2007; 37: 395–404. [DOI] [PubMed] [Google Scholar]
- Radeke HH, von Wenckstern H, Stoidtner K, Sauer B, Hammer S, Kleuser B. Overlapping signaling pathways of sphingosine 1-phosphate and TGF-beta in the murine Langerhans cell line XS52. J Immunol 2005; 174: 2778–2786. [DOI] [PubMed] [Google Scholar]
- Kang Y, Siegal PM, Shu W, Drobnjak M, Kakonen SM, Cordon-Cardo C et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003; 3: 537–549. [DOI] [PubMed] [Google Scholar]
- Itoh S, Thorikay M, Kowanetz M, Moustakas A, Itoh F, Heldin CH et al. Elucidation of Smad requirement in transforming growth factor-beta type I receptor-induced responses. J Biol Chem 2003; 278: 3751–3761. [DOI] [PubMed] [Google Scholar]
- Yagoub D, Wilkins MR, Lay A, Kaczorowski DG, Hatoum D, Bajan S et al. Sphingosine kinase 1 isoform-specific interactions in breast cancer. Mol Endocrinol 2014; 28: 1899–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitson SM, Moretti PAB, Zebol JR, Lynn HE, Xia P, Vadas MA et al. Activation of sphingosine kinase 1 by ERK1/2-mediated phosphorylation. EMBO J 2003; 22: 5491–5500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song JH, Kim M, Park SW, Chen SW, Pitson SM, Lee HT. Isoflurane via TGF-beta1 release increases caveolae formation and organizes sphingosine kinase signaling in renal proximal tubules. Am J Physiol Renal Physiol 2010; 298: 1041–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu SL, Pang H, Xu JZ, Wu XH. C/EBPβ mediates osteoclast recruitment by regulating endothelial progenitor cell expression of SDF-1α. PLoS ONE 2014; 9: e91217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendler F, Bota-Rabassedas N, Franch-Marro X. Cancer becomes wasteful: emerging roles of exosomes(†) in cell-fate determination. J Extracell Vesicles 2013; 2: 22390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta S, Warshall C, Bandyopadhyay C, Dutta D, Chandran B. Interactions between exosomes from breast cancer cells and primary mammary epithelial cells leads to generation of reactive oxygen species which induce DNA damage response, stabilization of p53 and autophagy in epithelial cells. PLoS ONE 2014; 9: e97580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012; 490: 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Bulent AA, Dogrusoz U, Dresdner G, Gross B, Sumer SO et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013; 6: pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross B, Sumer SO, Bulent AA et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012; 2: 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol 2009; 27: 1160–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maceyka M, Sankala H, Hait NC, Le Stunff H, Liu H, Toman R et al. SphK1 and SphK2, sphingosine kinase isoenzymes with opposing functions in sphingolipid metabolism. J Biol Chem 2005; 280: 37118–37129. [DOI] [PubMed] [Google Scholar]
- Al-Ejeh F, Simpson PT, Sanus JM, Klein K, Kalimutho M, Shi W et al. Meta-analysis of the global gene expression profile of triple-negative breast cancer identifies genes for the prognostication and treatment of aggressive breast cancer. Oncogenesis 2014; 21: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hait HC, Oskeritziana CA, Paugh SW, Milstien S, Spiegel S. Sphingosine kinases, sphingosine 1-phosphate, apoptosis and diseases. Biochim Biophys Acta 2006; 1758: 2016–2026. [DOI] [PubMed] [Google Scholar]
- Donati C, Cencetti F, De Palma C, Rapizzi E, Brunelli S, Cossu G et al. TGFbeta protects mesoangioblasts from apoptosis via sphingosine kinase-1 regulation. Cell Signal 2009; 21: 228–236. [DOI] [PubMed] [Google Scholar]
- Mohammad KS, Chen CG, Balooch G, Stebbins E, McKenna CR, Davis H et al. Pharmacologic inhibition of the TGF-beta type I receptor kinase has anabolic and anti-catabolic effects on bone. PLoS ONE 2009; 4: e5275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammad KS, Javelaud D, Fournier PG, Niewolna M, McKenna CR, Peng XH et al. TGF-beta-RI kinase inhibitor SD-208 reduces the development and progression of melanoma bone metastases. Cancer Res 2011; 71: 175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruckhäberle E, Rody A, Engels K, Gaetje R, von Minckwitz G, Schiffmann S et al. Microarray analysis of altered sphingolipid metabolism reveals prognostic significance of sphingosine kinase 1 in breast cancer. Breast Cancer Res Treat 2008; 112: 41–52. [DOI] [PubMed] [Google Scholar]
- Shida D, Takabe K, Kapitonov D, Milstien S, Spiegel S. Targeting SphK1 as a new strategy against cancer. Curr Drug Targets 2008; 9: 662–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KR, Johnson KY, Crellin HG, Ogretmen B, Boylan AM, Harley RA et al. Immunohistochemical distribution of sphingosine kinase 1 in normal and tumor lung tissue. J Histochem Cytochem 2005; 53: 1159–1166. [DOI] [PubMed] [Google Scholar]
- Kawamori T, Osta W, Johnson KR, Pettus BJ, Bielawski J, Tanaka T et al. Sphingosine kinase 1 is up-regulated in colon carcinogenesis. FASEB J 2006; 20: 386–388. [DOI] [PubMed] [Google Scholar]
- Van Brocklyn JR, Jackson CA, Pearl DK, Kotur MS, Snyder PJ, Prior TW. Sphingosine kinase-1 expression correlates with poor survival of patients with glioblastoma multiforme: roles of sphingosine kinase isoforms in growth of glioblastoma cell lines. J Neuropathol Exp Neurol 2005; 64: 695–705. [DOI] [PubMed] [Google Scholar]
- Malavaud B, Pchejetski D, Mazerolles C, de Paiva GR, Calvet C, Doumerc N et al. Sphingosine kinase-1 activity and expression in human prostate cancer resection specimens. Eur J Cancer 2010; 46: 3417–3424. [DOI] [PubMed] [Google Scholar]
- Song LB, Li W, Yu CP, Xia JT, Zhang L, Weng GX et al. Sphingosine kinase 1 Is associated with gastric cancer progression and poor survival of patients. Clin Cancer Res 2009; 15: 1393–1399. [DOI] [PubMed] [Google Scholar]
- Antoon JW, Meacham WD, Bratton MR, Slaughter EM, Rhodes LV, Ashe HB et al. Pharmacological inhibition of sphingosine kinase isoforms alters estrogen receptor signaling in human breast cancer. J Mol Endocrinol 2011; 46: 205–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia P, Sukocheva O, Wang LJ, Verrier E, Vadas MA. Restoring endocrine response in breast cancer cells by inhibition of the sphingosine kinase-1 signaling pathway. Endocrinology 2009; 150: 4484–4492. [DOI] [PubMed] [Google Scholar]
- Watson C, Long JS, Orange C, Tannahill CL, Mallon E, McGlynn LM et al. High expression of sphingosine 1-phosphate receptors, S1p1 and S1p3, sphingosine kinase 1 and Erk-1/2 is associated with development of tamoxifen resistance in ER-positive breast cancer patients. Am J Pathol 2010; 177: 2205–2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukocheva O, Wang L, Verrier E, Vadas MA, Xia P. Restoring endocrine response in breast cancer cells by inhibition of the sphingosine kinase-1 signaling pathway. Endocrinology 2009; 150: 4484–4492. [DOI] [PubMed] [Google Scholar]
- Ruckhaberle E, Karn T, Denkert C, Loibl S, Ataseven B, Reimer T et al. Predictive value of sphingosine kinase 1 expression in neoadjuvant treatment of breast cancer. J Cancer Res Clin Oncol 2013; 139: 1681–1689. [DOI] [PubMed] [Google Scholar]
- Iwamoto T, Bianchini G, Booser D, Qi Y, Coutant C, Shiang CY et al. Gene pathways associated with prognosis and chemotherapy sensitivity in molecular subtypes of breast cancer. J Natl Cancer Inst 2011; 103: 264–272. [DOI] [PubMed] [Google Scholar]
- Pyne NJ, Pyne S. Sphingosine 1-phosphate and cancer. Nat Rev Cancer 2010; 10: 489–503. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.