

Abstract
The final cytokinesis event involves severing of the connecting intercellular bridge (ICB) between daughter cells. FIP3-positive recycling endosomes (FIP3 endosomes) and ESCRT complexes have been implicated in mediating the final stages of cytokinesis. Here we analyse the spatiotemporal dynamics of the actin cytoskeleton, FIP3-endosome fusion and ESCRT-III localization during cytokinesis to show that the ICB narrows by a FIP3-endosome-mediated secondary ingression, whereas the ESCRT-III complex is needed only for the last scission step of cytokinesis. We characterize the role of FIP3 endosomes during cytokinesis to demonstrate that FIP3 endosomes deliver SCAMP2/3 and p50RhoGAP to the ICB during late telophase, proteins required for the formation of the secondary ingression. We also show that the FIP3-endosome-induced secondary ingression is required for the recruitment of the ESCRT-III complex to the abscission site. Finally, we characterize a FIP3-endosome-dependent regulation of the ICB cortical actin network through the delivery of p50RhoGAP. These results provide a framework for the coordinated efforts of actin, FIP3 endosomes and the ESCRTs to regulate cytokinesis and abscission.
Cytokinesis begins with a primary ingression mediated by a contraction of an actomyosin ring1, leading to the formation of the ICB, which is further resolved by thinning of the ICB from ~2μm to ~100 nm (secondary ingression)2, followed by the ICB plasma membrane fusion (abscission)2–4. Recent studies have shown that secondary ingression and abscission involve depolymerization of ICB actin and microtubules2–4, fusion of FIP3 endosomes2,5–7 and recruitment of the ESCRT complexes8–10. However, although it was shown that midbody accumulation of ALIX and TSG101 (ESCRT-I) recruits CHMP4B (ESCRT-III) during late cytokinesis3,8,10, the roles of FIP3 endosomes and ESCRT complexes during the formation of the secondary ingression and abscission remain to be fully understood.
Here, we demonstrate that secondary ingression is mediated by FIP3-endosome fusion and occurs before ESCRT-III recruitment to the abscission site, and that ESCRT-III is required only for stabilization of the formed secondary ingression and final membrane scission. Furthermore, we use time-lapse microscopy and organelle proteomics to identify secretory carrier membrane protein 2/3 (SCAMP2/3) and p50RhoGAP as membrane-traffic- and actin-regulating proteins required for abscission, which are delivered to the ICB by FIP3 endosomes. We support these findings by demonstrating that FIP3-endosome delivery of SCAMP2/3 or p50RhoGAP regulates actin depolymerization within the ICB and recruitment of ESCRT-III to the abscission site.
RESULTS
ESCRT-III recruitment to the midbody coincides with the accumulation of FIP3 endosomes
Many reports have shown that FIP3- and Rab11-positive endosomes accumulate at the ICB and are required for abscission2,6,7,11,12, and that the movement of FIP3 from centrosomes to the ICB marks the progression of cells from early to late telophase (Supplementary Fig. S1A). A recent study suggested that endosomes are not required for abscission, on the basis of an observation that RAB8 endosomes are not present at the ICB during late cytokinesis4. As it is unclear whether RAB8 actually marks FIP3 endosomes, we compared RAB8 and FIP3 during cytokinesis and show that there is little co-localization between RAB8 and FIP3 during early telophase, as RAB8 endosomes are delivered to the ICB before FIP3-endosome accumulation (Fig. 1a). Subsequently, during late telophase, FIP3 endosomes occupy the ICB, whereas RAB8 endosomes are no longer present within the ICB (Fig. 1b). Interestingly, RAB8 and FIP3 co-localize on a small population of endosomes outside the ICB (Fig. 1b). This demonstrates that during cytokinesis RAB8 and FIP3 mark different endocytic populations within the ICB (Fig. 1a,b).
Figure 1.
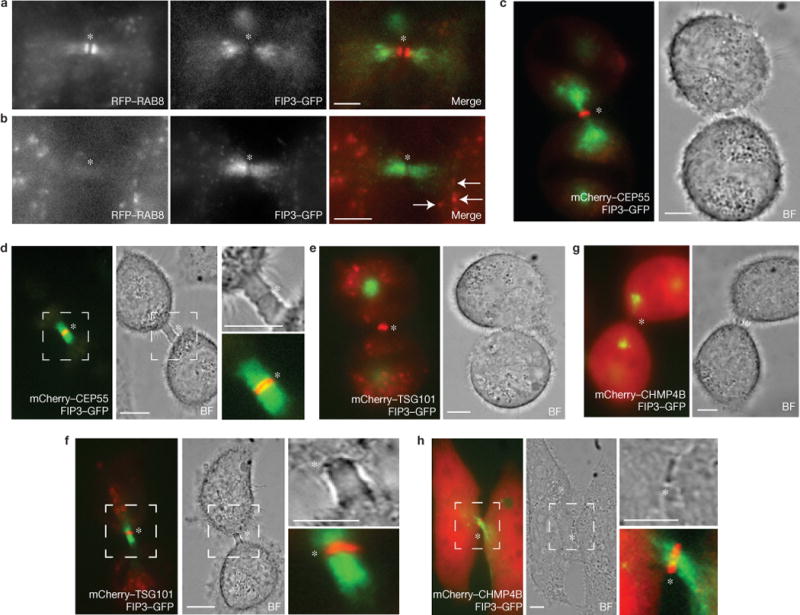
Spatiotemporal distribution of FIP3 endosomes, RAB8 endosomes, CEP55 and ESCRT complexes during cytokinesis. (a–h) Fixed-cell imaging of HeLa FIP3–GFP cells co-expressing RFP–RAB8 (a,b) and live-cell imaging of mCherry–CEP55 (c,d), mCherry–TSG101 (e,f) and mCherry–CHMP4B (g,h). HeLa FIP3–GFP cells were imaged in early cytokinesis (a,c,e,g) or late cytokinesis (b,d,f,h). The arrows in b correspond to FIP3 and RAB8 co-localization outside the intercellular bridge. The outlined regions (d,f,h) mark enlarged areas. The asterisks mark the midbodies. BF, bright-field micrograph. Scale bars, 5μm.
Various ESCRT complex components have been documented to accumulate at the midbody, culminating in CHMP4B (ESCRT-III) recruitment during late telophase3,4,8,10. To establish the timing of FIP3-endosome and ESCRT transport to the ICB, we co-imaged FIP3 and ESCRT complex components CEP55, TSG101 and CHMP4B during cytokinesis. As previously reported3,8,13, CEP55 and TSG101 arrive at the midbody during early telophase, with FIP3 endosomes still residing outside the ICB (Fig. 1c–f). In contrast, CHMP4B arrives at the midbody during late telophase (Fig. 1g–h), demonstrating that ESCRT-III and FIP3 endosomes are both recruited to the ICB during the final stages of cytokinesis, before the formation of the secondary ingression.
CHMP4B is recruited to the abscission site after formation of the secondary ingression
Previously it was suggested that FIP3 endosomes mediate the generation of the secondary ingression within the ICB before abscission2,12. Interestingly, two recent studies proposed that ESCRT-III oligomers also play a role in the formation of the secondary ingression3,4. To further define the involvement of FIP3 endosomes and ESCRT-III during abscission, we used time-lapse microscopy to image the formation of the secondary ingression in HeLa cells co-expressing FIP3–GFP and mCherry–CHMP4B. We observed that the secondary ingression is able to form without CHMP4B moving to the site of ICB narrowing (Figs 2a–e and 3a,b). Furthermore, FIP3–GFP fluorescence disappears during the formation of the secondary ingression (42 cells imaged; Fig. 2b,g). The disappearance of FIP3–GFP might be caused by the fusion of FIP3–GFP endosomes, or due to endosomes exiting the ICB. To investigate this, we imaged FIP3 endosomes during mid-telophase (Supplementary Movie S1) or late telophase (Supplementary Movie S2). Consistent with previous reports12,14, during mid-telophase FIP3 endosomes are actively delivered to and from the ICB. In contrast, during late telophase, FIP3 endosomes are largely stationary and remain within the ICB. Together with previous reports2, this suggests that FIP3 endosomes fuse with the ICB plasma membrane during the formation of the secondary ingression.
Figure 2.
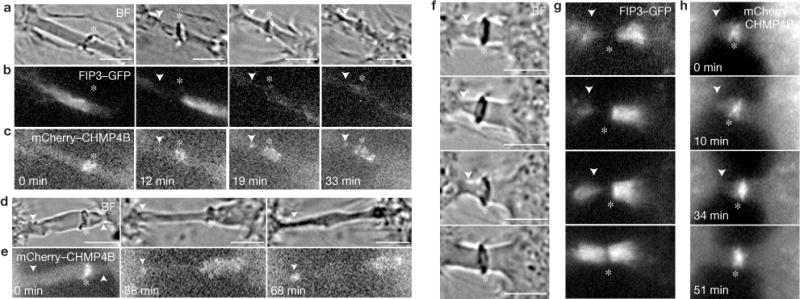
The secondary ingression forms before the recruitment of CHMP4B. Mitotic HeLa FIP3–GFP cells expressing mCherry–CHMP4B were imaged using time-lapse microscopy. (a–e) Sequential time-lapse images show abscission occurring either close (<2μm; a–c) or far away (>6μm) from the midbody (d–e). (f–h) Sequential time-lapse images show failed secondary ingression. In all images, the arrowheads mark secondary ingression and abscission sites. The asterisks mark the midbodies. Scale bars, 5 μm.
Figure 3.
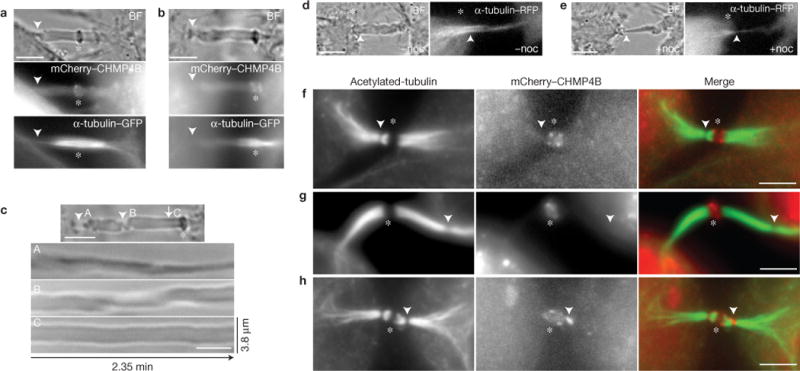
Clearance of microtubules from the ICB is required for secondary ingression. (a–c) Mitotic HeLa cells expressing α-tubulin–GFP and mCherry–CHMP4B were analysed by time-lapse microscopy. The arrowheads mark the secondary ingression sites. The kymographs in c (from the cell shown in b) depict the ICB dynamics at the regions with (C) or without (A and B) detectable microtubules. (d,e) Mitotic HeLa cells expressing α-tubulin–GFP and mCherry–CHMP4B were imaged before or after 10min incubation with nocodazole (noc; 7.5 μg ml−1). The arrowheads mark the point where the secondary ingression site will be induced. (f–h) HeLa cells expressing mCherry–CHMP4B were fixed and stained with anti-acetylated-tubulin antibodies. The arrowheads mark the regions of microtubule severing/depletion. In all images (a–h), the asterisks mark the midbodies. Scale bars, 5 μm.
This demonstrates that the formation of the secondary ingression is not dependent on CHMP4B and probably is mediated by another mechanism, possibly fusion of FIP3 endosomes. After secondary ingression, CHMP4B can be observed accumulating at the site of ICB thinning (Fig. 2c,e), marking the location where abscission occurs (5 cells imaged). Note, we never observed abscission without the delivery of CHMP4B to the secondary ingression site. Furthermore, we often see cases where secondary ingression was initiated, but was not followed by the recruitment of CHMP4B (Fig. 2f–h; >30 cells imaged). In these cases, the ICB regressed to its original width and did not undergo abscission (Fig. 2f–h), suggesting that whereas ESCRT-III is not required for the initial formation of secondary ingression, it is required for the scission of the ICB.
The factors that initiate ESCRT-III recruitment to the abscission site remain elusive, but it was suggested that CHMP4B oligomers extend from the midbody to the abscission site3,4. In 27% of cells (6 of 22 cells imaged) with CHMP4B at the abscission site, we observe possible continuous CHMP4B spirals connecting the midbody to the abscission site (Supplementary Fig. S1G). However, in 73% of cells (16 of 22 cells imaged), CHMP4B forms distinct puncta at the abscission site (Fig. 2c,e and Supplementary Figure 1H).
Microtubule depolymerization within the ICB is required for abscission1,3,4,14, and may determine the site of the secondary ingression2. To confirm that depolymerization of microtubules is sufficient to cause secondary ingression, we imaged telophase cells before and after the addition of nocodazole (7.5 μg ml−1). Consistent with the requirement of microtubule depolymerization for the formation of secondary ingression, the addition of nocodazole induced rapid thinning of the ICB (Fig. 3d,e).
As the secondary ingression forms before CHMP4B recruitment to the abscission site (Fig. 2), we compared microtubule depolymerization with FIP3 and CHMP4B recruitment to the abscission site. Consistent with previous work3,4, localized depolymerization of microtubules (microtubule gaps) is observed in ~40% of dividing HeLa cells (Fig. 3f–h and Supplementary Fig. S1B), with few of these sites containing CHMP4B (Fig. 3h and Supplementary Fig. S1C). In most cells, localized microtubule gaps did not contain any detectable CHMP4B (Fig. 3f,g and Supplementary Fig. S1C), suggesting that ESCRT-III may not be absolutely required for localized microtubule depolymerization. To investigate this possibility, we depleted CHMP4B (Supplementary Fig. S1F) and analysed the presence of microtubule gaps within the central spindle during telophase. As reported previously15,16, CHMP4B depletion increased the number of multinucleated cells (Supplementary Fig. S1D). Interestingly, CHMP4B knockdown had no effect on the formation of microtubule gaps (Supplementary Fig. S1E). Taken together, these results suggest that ESCRT-III is required only for the final abscission step, and not microtubule depolymerization or secondary ingression formation.
In addition to cells with microtubule gaps, some cells exhibited depolymerization occurring at the minus ends of the microtubules (retracting microtubules) within the ICB (Fig. 3a,b and Supplementary Fig. S1B). In these cases, the formation of the secondary ingression always occurred behind the minus ends of the microtubules (Fig. 3a,b). Furthermore, kymographs (Fig. 3c) show increased ICB plasma membrane dynamics behind the minus ends of the microtubules (Fig. 3c(A,B)), whereas the plasma membrane is stable where ICB microtubules are present (Fig. 3c(C)). Thus, microtubule clearing during abscission may result from either of two processes—localized severing3,4 or minus-end retraction (Fig. 3a–c).
SCAMP2 and SCAMP3 are required for cytokinesis
As ESCRT-III and FIP3 endosomes accumulate at the secondary ingression site2, it raises a possibility that FIP3 endosomes deliver proteins that mediate the recruitment of ESCRT complexes. Recent work determined that SCAMP3 interacts with TSG101 (refs 17–19). As SCAMPs reside in endosomes20, they potentially could regulate ESCRT complex recruitment to the abscission site. Thus, we examined the localization of different SCAMPs and have shown that SCAMP1–3 localize to FIP3 endosomes during cytokinesis (Fig. 4a–g and Supplementary Fig. S2). Moreover, SCAMP1–3 localization at the ICB is blocked after FIP3 knockdown (Fig. 4h–k and Supplementary Fig. S2). Knockdown of SCAMP2 or SCAMP3 caused an increase in the number of multinuclear cells, whereas SCAMP1 depletion showed little effect (Fig. 5a,b). As SCAMP isoforms can have complementary functions21–23, we also evaluated combinatorial knockdowns of SCAMPs. Co-depletion of SCAMP2 and SCAMP3 resulted in an increase in division failures (Fig. 5b), and in the time required for cells to complete cytokinesis (Fig. 5c–e).
Figure 4.
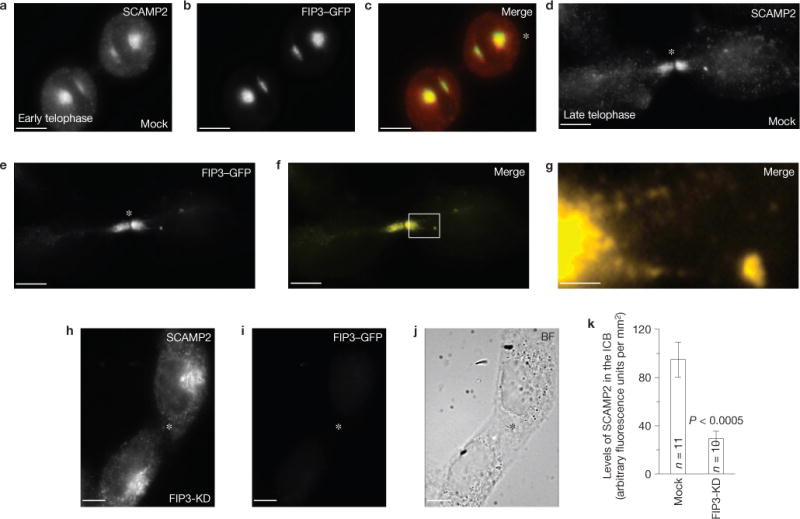
SCAMP1, 2 and 3 localize to the ICB during cytokinesis and their localization is dependent on FIP3 endosomes. (a–i) Mock-(a–g) or FIP3-siRNA-treated (h,i; FIP3-KD) HeLa FIP3–GFP cells were fixed and stained with anti-SCAMP2 antibodies. The asterisks mark the midbodies. The image in g shows a magnification of the area outlined in f. (k) Quantification of SCAMP2 localization within the ICB in mock-transfected or FIP3-siRNA-treated cells. The data shown are the means and s.d. of the anti-SCAMP2 fluorescence intensity from 21 randomly chosen cells in telophase. Scale bars, 5 μm (a–f,h–i) and 1μm (g).
Figure 5.
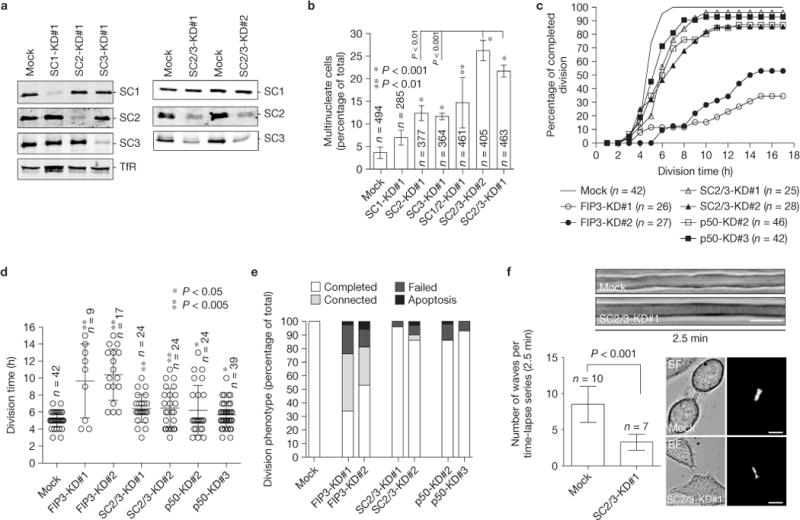
SCAMP2 and SCAMP3 are required for successful completion of cytokinesis. (a) Western blot analysis probed for the indicated proteins in single and double SCAMP knockdowns (KD). (b) Quantification of multinucleate HeLa cells in single or double SCAMP knockdowns. The data shown are means and s.d. from three independent experiments. n indicates the number of cells counted. The asterisks indicate a statistically significant difference from the mock-transfected cells. (c–e) Mock, FIP3, SCAMP2/3 or p50RhoGAP siRNA-treated (KD) HeLa cells were imaged by time-lapse microscopy for 24 h (time-lapse 1h). The data shown are the percentage of cells that completed division (c), the average time required for cells to divide (d) and the division phenotype (e). n indicates the number of cells counted. The error bars represent s.d. of the mean. The asterisks indicate a statistically significant difference from the mock-transfected cells. (f) Mock-or SCAMP2/3-siRNA-treated cells were imaged by time-lapse microscopy to analyse the dynamics of the ICB during late telophase, as determined by FIP3–GFP localization within the ICB (images on right). Shown are representative kymographs used to quantify the dynamics of the ICB. The histogram depicts means and s.d. of the data. n is the number of randomly chosen cells in late cytokinesis. Scale bars, 5μm. Uncropped images of blots are shown in Supplementary Fig. S7.
Originally SCAMPs were described as proteins that regulate exocytotic membrane fusion21–23. To determine whether SCAMP2/3 may regulate FIP3-endosomal fusion, we analysed the effect of SCAMP2/3 knockdown on wave generation of the ICB plasma membrane. It was previously shown that during late cytokinesis, the ICB plasma membrane becomes very dynamic and that generation of the short-lived (few seconds) ICB plasma membrane protrusions, or waves24, occurs in a FIP3-endosome-dependent manner before the formation of the secondary ingression2. Consistently, SCAMP2/3 knockdown resulted in a significant reduction in the number of plasma membrane waves (Fig. 5f). Together, these results suggest that SCAMP2/3 are required for abscission, possibly by regulating FIP3-endosome fusion and/or mediating ESCRT-III assembly by recruiting TSG101 to the abscission site.
FIP3 endosomes deliver p50RhoGAP to the ICB during late telophase
Interestingly, the effect of SCAMP2/3 knockdown was not as pronounced as depletion of FIP3 (Fig. 5c,f), indicating that FIP3 endosomes may also deliver other proteins that regulate cytokinesis. To identify these cargo proteins, we used proteomics to analyse immuno-isolated FIP3 endosomes (Supplementary Fig. S3; see Methods). This led to the identification of 441 proteins that are present only in immuno-isolated FIP3 endosomes (Supplementary Table S1). Proteins known to bind ribosomes, mitochondria and DNA/RNA were eliminated as contaminants, leaving the remaining (103) proteins as candidates (Supplementary Fig. S3 and Table S1). Many of the candidate proteins have known roles in cytokinesis, but were not previously associated with FIP3 endosomes. We also identified several proteins that were reported to reside in endosomes, but were never implicated in regulating cell division. One such protein was p50RhoGAP (also known as ARHGAP1), which has been shown to reside in endosomes during interphase25,26. To confirm that p50RhoGAP is present on FIP3 endosomes, we generated anti-p50RhoGAP antibody (Supplementary Fig. S4A–D) and have shown that endogenous p50RhoGAP and FIP3 co-localize within the ICB during late cytokinesis (Fig. 6a–f). FIP3 knockdown inhibited targeting of p50RhoGAP to the ICB, while having no effect on p50RhoGAP total cellular levels (Fig. 6g–j). Similarly, RFP-tagged p50RhoGAP co-localized with FIP3–GFP-containing organelles in live telophase cells (Supplementary Fig. S4E–H). Interestingly, we could also observe a small pool of p50RhoGAP that is recruited to the midbody during early telophase and does not co-localize with FIP3 endosomes (Fig. 6b,c), suggesting that a small portion of p50RhoGAP can also be recruited to the ICB in a FIP3-endosome-independent manner.
Figure 6.
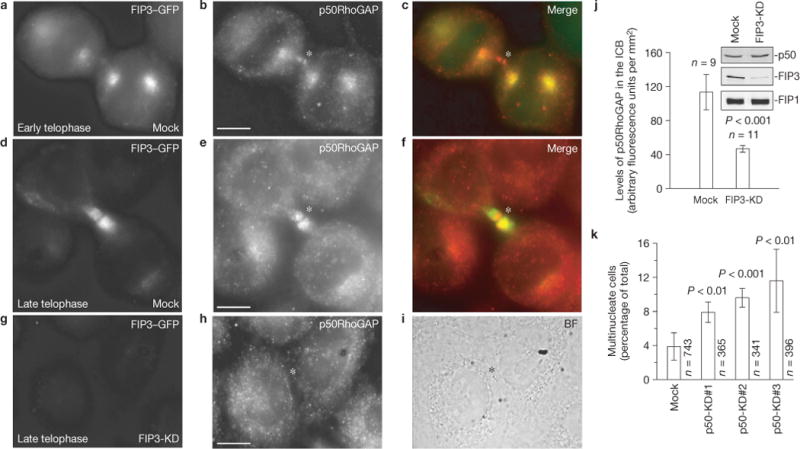
Localization of p50RhoGAP during cytokinesis. (a–f) Dividing HeLa cells expressing FIP3–GFP were stained with anti-p50RhoGAP antibody. (g–i) FIP3 was depleted by RNA-mediated interference (KD) and cells were stained with anti-p50RhoGAP antibodies. The asterisks mark the midbodies. Scale bars, 5μm. (j) Quantification of p50RhoGAP levels at the ICB in mock- or FIP3-siRNA-treated cells. The data shown are the means and s.d. of 20 randomly picked cells in late telophase. (k) Cells transduced with three different p50RhoGAP siRNAs were analysed for defects in cytokinesis by counting the number of multinucleate cells. The data shown are the means and s.d. of three independent experiments. n is the total number of cells counted. Uncropped images of blots are shown in Supplementary Fig. S7.
To determine whether p50RhoGAP is required for cytokinesis, we transfected cells with three different p50RhoGAP short interfering RNAs (siRNAs) to deplete endogenous p50RhoGAP (Supplementary Fig. S4A) and have shown that depletion of p50RhoGAP increased the number of multinucleate cells (Fig. 6k). To further confirm that p50RhoGAP is required for cytokinesis, we analysed the division of p50RhoGAP-depleted cells by time-lapse microscopy. Although p50RhoGAP knockdown did not have any effect on the formation and initial ingression of the cleavage furrow, depletion of p50RhoGAP increased the time required for cells to complete cytokinesis (Fig. 5c–e), and also increased the number of cells that failed abscission (Fig. 5e). FIP3 knockdown again had a much more pronounced effect on cytokinesis than p50RhoGAP knockdown (Fig. 5c–e and Supplementary Fig. S4A), suggesting that FIP3 endosomes function by delivering multiple cytokinesis regulators, which include, but are probably not limited to, p50RhoGAP and SCAMP2/3.
p50RhoGAP-dependent depolymerization of actin within the ICB is required for abscission
Although actin cytoskeleton plays a key role in the progression of the cleavage furrow27 and maintenance of cell shape during cytokinesis28, depolymerization of the ICB cortical actin during late telophase is required for successful completion of cytokinesis. This raises a possibility that FIP3 endosomes may deliver p50RhoGAP to the ICB to inactivate Rac/Rho GTPases and induce cortical actin disassembly. To investigate this, we imaged filamentous actin (F-actin) in dividing cells using a previously characterized F-actin-binding protein utrophin–GFP (ref. 29; Supplementary Fig. S5). In early anaphase to early telophase, F-actin is enriched at the cleavage furrow (Supplementary Fig. S5A–C). Similarly, GFP-tagged myosin IIA is also enriched at the furrow during its formation and initial primary ingression (Supplementary Fig. S5D). However, on completion of primary ingression, myosin-IIA–GFP is no longer detectable within the ICB (Supplementary Fig. S5D). During late telophase, F-actin is also gradually removed from the ICB (Supplementary Fig. S5A,C) and is no longer detectable within the forming secondary ingression (Supplementary Fig. S5B). To determine whether FIP3 endosomes are required for the ICB actin depolymerization, we imaged F-actin within the ICB using utrophin–RFP after knockdown of either SCAMP2/3 or p50RhoGAP. Mock-transfected cells in late cytokinesis exhibit little cortical F-actin within the ICB, as compared with the cell body (Supplementary Fig. S6A,D). In contrast, SCAMP2/3 or p50RhoGAP knockdown resulted in significant increases in the total cortical F-actin levels within the ICB (Supplementary Fig. S6B–D).
To further confirm that FIP3, SCAMP2/3 and p50RhoGAP are required for actin disassembly at the ICB, we also visualized F-actin in fixed HeLa cells with rhodamine–phalloidin. We analysed the levels of F-actin during anaphase and telophase, and consistent with the utrophin–RFP data, early telophase F-actin was enriched at the ICB (Fig. 7a–f). In contrast, during late telophase (as determined by the presence of FIP3 endosomes within the ICB), the levels of F-actin were significantly decreased (Fig. 7g–j). The depletion of SCAMP2/3, p50RhoGAP or FIP3 using siRNAs led to statistically significant increases in the F-actin levels at the ICB during late telophase (Fig. 7k–s). These data imply that FIP3-endosome-dependent delivery of SCAMP2/3 and p50RhoGAP to the ICB is required for the depolymerization of the cortical F-actin network before abscission.
Figure 7.
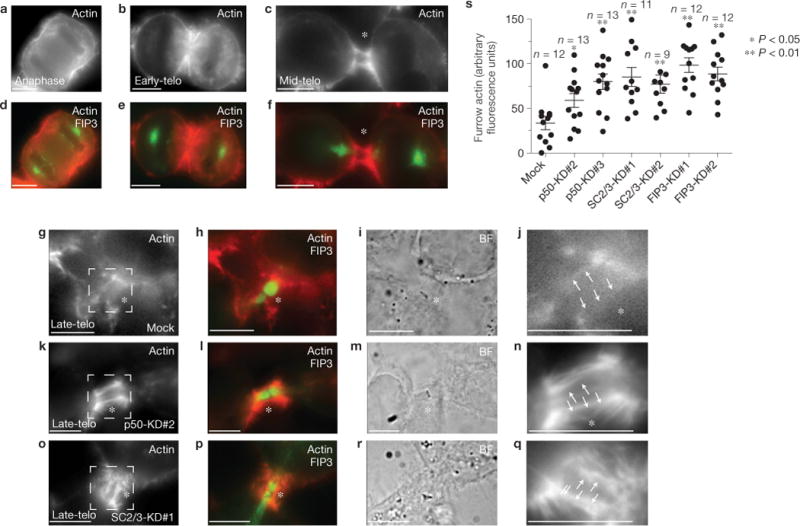
FIP3-mediated p50RhoGAP and SCAMP2/3 delivery is required for actin depolymerization during late telophase. (a–s) Mock- (a–j,s), p50RhoGAP- (k–n,s), SCAMP2/3- (o–q,s) or FIP3- (s) siRNA-treated HeLa cells were fixed and stained with rhodamine–phalloidin. a,d show a cell in anaphase. b,e show a cell in early telophase. c,f show a cell in mid-telophase. g–q show cells in late telophase, as determined by the presence of FIP3–GFP in the ICB. j,n and q are high-magnification images of the areas outlined in g,k and o. (s) The quantification of rhodamine–phalloidin fluorescence intensity within the ICB during late telophase. The data shown are the means and s.d. n indicates the number of randomly chosen cells. The arrows in j,n and q point to the region within the ICB that was used for quantification of the rhodamine–phalloidin fluorescence intensity. Scale bars, 5μm
SCAMP2/3 and p50RhoGAP are required for the recruitment of CHMP4B to the abscission site
Our data demonstrate that FIP3, SCAMP2/3 and p50RhoGAP are required for actin depolymerization within the ICB, and as disassembly of the cortical acto-myosin network is probably required for the thinning of the ICB, we investigated whether SCAMP2/3 and p50RhoGAP are also required for the formation of the secondary ingression. We used bright-field time-lapse microscopy to image secondary ingressions in late telophase cells (n = 91), and over 45% had a clearly identifiable stable secondary ingression (Fig. 8a,c and Supplementary Movie S3). Depletion of p50RhoGAP or SCAMP2/3 resulted in a decrease in the frequency of the secondary ingression (Fig. 8c). In contrast, CHMP4B knockdown had no effect on the ability of cells to form the secondary ingression (Fig. 8b,c and Supplementary Movies S4 and S5). Thus, consistent with our previous observations, p50RhoGAP and SCAMP2/3 are required for the initial formation of the secondary ingression, whereas CHMP4B is probably required only for final scission of the ICB.
Figure 8.
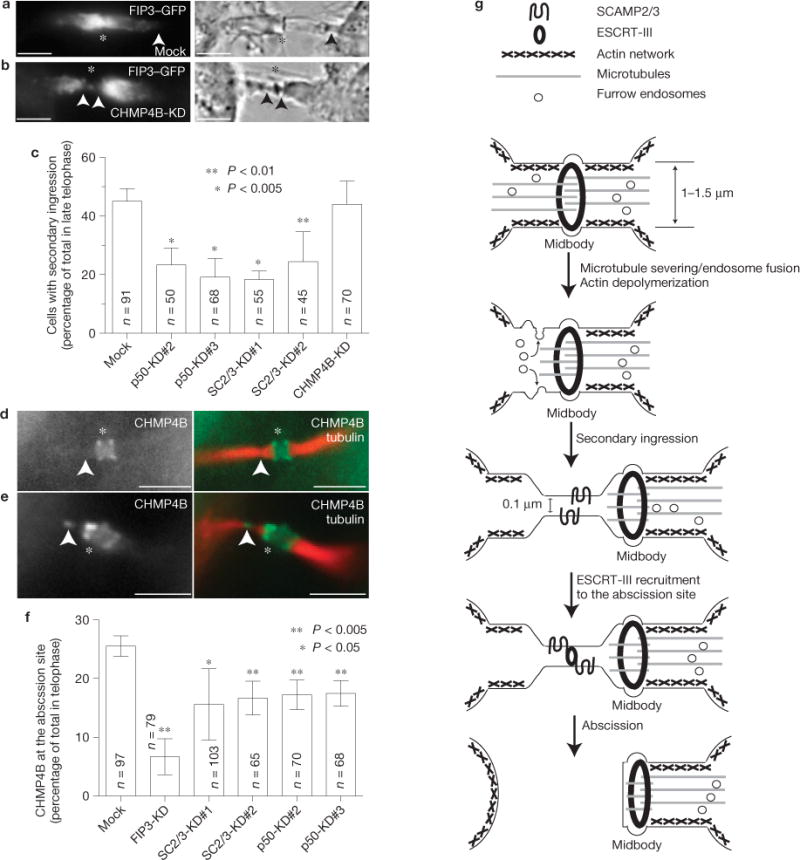
FIP3-mediated SCAMP2/3 and p50RhoGAP delivery recruits CHMP4B to the secondary ingression and abscission site. (a–c) HeLa cells expressing FIP3–GFP were treated with p50RhoGAP, SCAMP2/3 or CHMP4B siRNAs (KD). The presence or absence of the stable secondary ingression was then determined by time-lapse imaging of cells in telophase. The asterisks mark the midbody; the arrowheads mark secondary ingression. (c) Quantification of the number of telophase cells with secondary ingression. The data shown are means and s.d. derived from three separate experiments. n indicates the number of telophase cells counted. The asterisks indicate a statistically significant difference from the mock. (d–f) HeLa cells expressing YFP–CHMP4B were treated with p50RhoGAP, SCAMP2/3 or FIP3 siRNAs. Cell were then fixed and stained for acetylated tubulin. The asterisks mark the midbody; the arrowheads mark the microtubule gap/abscission site. (f) Quantification of the number of telophase cells with CHMP4B at the abscission site. The data shown are means and s.d. derived from three separate experiments. n indicates the number of telophase cells counted. The asterisks indicate a statistically significant difference from the mock. (g) Proposed abscission model (see text for details). Scale bars, 5μm.
Although CHMP4B is recruited to the abscission site during the final stages of cytokinesis, it remains unclear whether the formation of this secondary ingression induces ESCRT-III recruitment. It has been shown that the ESCRT-III complex preferentially associates with >100nm diameter high-curvature membranes, a diameter similar to that of the secondary ingression30. Furthermore, it was previously shown that the SCAMP3 associates with TSG101 (ref. 17), a protein known to recruit the ESCRT-III complex. To determine whether FIP3 endosomes are required for ESCRT-III recruitment, we analysed the localization of CHMP4B–YFP during late telophase after FIP3, SCAMP2/3 or p50RhoGAP knockdown. In most cells, CHMP4B formed a double ring flanking the midbody (Fig. 8d), which was previously described3,4. Only in ~25% of cells, were we able to detect CHMP4B present at the secondary ingression sites, suggesting that these cells are at the final stage of abscission (Fig. 8e,f). In FIP3-, SCAMP2/3- or p50RhoGAP-knockdown cells we could observe a significant decrease in the number of cells with CHMP4B located at the abscission site (Fig. 8f). As a whole, the data indicate that FIP3-endosome-dependent delivery of SCAMP2/3, p50RhoGAP and probably other regulatory proteins is required for the ICB actin depolymerization, the formation of the secondary ingression and the subsequent translocation of CHMP4B from the midbody to the abscission site within the ICB.
DISCUSSION
Recent studies have identified FIP3 endosomes, actin, microtubules and ESCRT complexes as regulators of the abscission. However, the exact mechanisms and linkages between these players remain elusive. Here, we show that FIP3 endosomes have an essential role in the formation of the secondary ingression, placement of the ESCRT-III machinery and regulating actin cytoskeleton disassembly within the ICB.
It has been established that depolymerization of microtubules within the ICB is a key step in allowing the final separation of daughter cells, and that the microtubule-severing enzyme spastin plays an important role in localized clearing of microtubules at the future abscission site4,31. This study also identifies a previously uncharacterized alternative mechanism of microtubule depolymerization during cytokinesis by way of minus-end microtubule depolymerization, which occurs away from midbody-associated ESCRT-III. This mechanism seems to be more common in cells forming long ICBs and may explain why this mechanism was not observed in MDCK cells, where ICBs are less extended3. It is likely that the mechanism of microtubule clearance is cell type dependent. For example, epithelial cells may have a higher dependence on spastin-mediated severing3, and highly motile cells, such as fibroblasts, could rely on minus-end depolymerization. This could also explain why spastin knockdown only delays, but does not block, abscission2,31. Thus, the combination of several microtubule depolymerization mechanisms probably contributes to clearing the ICB of microtubules during abscission.
The sequential building of the ESCRT machinery in the midbody has been well documented3,8,10. Although the involvement of ESCRT complexes in cytokinesis is clear, how this fits with the role of FIP3 endosomes remains undetermined. Here, we compare spatiotemporal dynamics of secondary ingression formation, ESCRT recruitment and FIP3-endosome delivery. We show that FIP3 endosomes are present throughout the entire ICB, including the site of secondary ingression. Although time-lapse analysis confirmed that CHMP4B is required for ICB membrane scission, the data also show that CHMP4B is recruited to the abscission site after the formation of the secondary ingression. Furthermore, CHMP4B depletion does not have any effect on microtubule gap formation or the initiation of the secondary ingression, suggesting that the ESCRT complexes do not mediate the formation of the secondary ingression. A question arises of what regulates the movement of CHMP4B from the midbody to the site of the secondary ingression. Recent data using yeast ESCRT complexes found that ESCRT-II and CHMP6 act together to direct ESCRT-III polymerization to regions of highly curved membranes in vitro, with a preference towards >100 nm liposomes30. Consistent with that, ESCRT-dependent intraluminal multivesicular body generation and viral budding32–37 generate vesicles ranging in size from 50 to 75 nm, well below the 2μm diameter of the ICB. On the basis of our findings, we propose that the FIP3-endosome-induced secondary ingression is required to establish a region of highly curved membrane, thus enabling the recruitment of ESCRT-III. The formation of the secondary ingression before the arrival of CHMP4B also alleviates potential size restrictions placed on the configuration of ESCRT machinery during scission.
Another barrier for ESCRT-mediated abscission is the presence of a plasma-membrane-associated cortical actin network. This cortical F-actin network serves to structurally form, maintain and hold the shape of the ICB during early telophase. Recently, the importance of the actin–myosin cortical network in stabilizing and positioning the cleavage furrow during cytokinesis has been demonstrated38. This actin cortical network needs to be disassembled for abscission to take place. Consistently, a recent study shows that the inhibition of ICB actin disassembly during late cytokinesis results in abscission defects39. Here we also observe that the formation of the secondary ingression is preceded by the depolymerization of the cortical actin within the ICB. The identification of several Rac/Rho and actin regulators within immuno-isolated FIP3 endosomes in our proteomic studies raised an interesting possibility that FIP3 endosomes may regulate the depolymerization of the cortical actin network. Consistent with that, inhibition of FIP3-endosome fusion by SCAMP2/3 knockdowns also blocked actin disassembly within the ICB and inhibited cytokinesis. In addition, knockdown of the FIP3-endosome cargo protein, p50RhoGAP, also blocks actin disassembly within the ICB, inhibits CHMP4B recruitment to the secondary ingression sites and leads to late cytokinesis defects. Similarly, recent work39 demonstrated that Rab35 endosomes also regulate actin disassembly within the ICB by regulating levels of phosphatidylinositol-4,5-bisphosphate (PtdIns(4, 5)P2). As Rab35 seems to mark endosomes that are distinct from FIP3 endosomes, there may be at least two independent endocytic pathways of actin regulation, Rab11 or Rab35 dependent40.
Here we show that SCAMP2/3 may be functioning to regulate FIP3-endosome fusion, as signified by the inhibitory effects of knockdown on membrane dynamics. However, it seems that the SCAMP roles also manifest post-fusion, assisting in organizing CHMP4B to the abscission site. Although the exact mechanisms of SCAMP2/3 recruitment of CHMP4B are unclear, SCAMP3 might help organize ESCRT assembly at the abscission site by way of binding to TSG101 through its PSAP motif17. The interaction of SCAMPs with phosphoinositides41 may control PtdIns(4, 5)P2 distribution in a manner that promotes the ESCRT-III recruitment and actin depolymerization. Indeed, SCAMP2/3 knockdown inhibits the recruitment of CHMP4B to the microtubule gaps, the putative future abscission sites.
In summary we propose that FIP3 endosomes regulate abscission by delivering actin-regulating and ESCRT-III recruitment proteins to the ICB. The fusion of these endosomes with the plasma membrane results in localized depolymerization of the cortical actin cytoskeleton, leading to the formation of the secondary ingression (Fig. 8c). The thinning of the ICB to less than 100 nm, as well as delivery of the TSG101-binding protein SCAMP3, then allows for the recruitment of the ESCRT machinery, and abscission of daughter cells (Fig. 8c). Proteomic identification of other putative FIP3-endosome proteins can now serve as a starting point to unravel FIP3-endosome control of the complex ICB remodelling events that must occur to allow for abscission.
METHODS
Materials, antibodies and expression constructs
Reagents were obtained from Invitrogen, Sigma-Aldrich, Clontech or Fisher, unless otherwise specified. Mouse monoclonal anti-HA (catalogue number sc-7392) antibody, used as per the manufacture’s recommendation, was purchased from Santa Cruz Biotechnology and rabbit anti-CHMP4B antibody (catalogue number ab105767), used as per the manufacture’s recommendation, was obtained from Abcam. Rabbit anti-GFP, SCAMP1 (western blotting: 0.1 μgml−1; immunofluorescence analysis: 1 μgml−1), SCAMP2 (serum used at 1:1,000 for western blotting, 1:500 for immunofluorescence analysis), SCAMP3 (western blotting: 0.1 μgml−1; immunofluorescence analysis: 1 μgml−1) andSCAMP4 (western blotting: 1 μgml−1; immunofluorescence analysis: 5 μgml−1) antibodies were previously described1,12,21,22. Rabbit polyclonal anti-p50RhoGAP antibodies were generated using recombinant purified GST–p50RhoGAP as previously described12 and used at 0.5 μgml−1. Secondary antibodies conjugated with Alexa488 or Texas red were purchased from Jackson ImmunoResearch.
The following DNA constructs were previously described: FIP3–GFP (refs 6, 7), mCherry–CEP55 (ref. 8), mCherry-TSG101 (ref. 8), YFP–CHMP4B (ref. 8) and p50RhoGAP–GFP (ref. 25). HA–TPD52 (ref. 42), pmEGFP–α-tubulin-C1 (Addgene Plasmid 21039; ref. 43) and pmRFP–α-tubulin-C1 (Addgene Plasmid 21040; ref. 43) were obtained from the Addgene plasmid repository. RFP–RAB8 was a gift from C. Westlake. p50RhoGAP–GFP and p50RhoGAP-RFP were gifts from M. Geiszt. mCherry–CHMP4B was constructed by generating a PCR fragment of CHMP4B from CHMP4B–YFP using the forward primer 5′-AAACTCGAGTCGGTGTTCGGGAAGCTGTTC-3′ and the reverse primer 5′-TTGAATTCTTACATGGATCCAGCCCAGTT-3′. The CHMP4B PCR fragment and Clontech pmCherry-C1 plasmid were digested with Xho1 and EcoRI restriction enzymes (New England Biolabs) and subsequently re-ligated.
Nocodazole drug treatment
All live HeLa cells were staged and imaged before drug treatment. Nocodazole, (Sigma-Aldrich) at a 7.5 μgml−1 final concentration, was added to live HeLa cells for 10 min before obtaining images.
Light microscopy
For immunofluorescence microscopy, cells were fixed with 4% paraformaldehyde, and then permeabilized in phosphate-buffered saline (PBS) containing 0.4% saponin, 0.2% BSA and 1% fetal bovine serum. Cells were stained by standard immunofluorescence procedures and imaged with an inverted Zeiss Axiovert 200M deconvolution microscope. Images were acquired and processed using Intelligent Imaging Innovations three-dimensional rendering and exploration software.
For time-lapse microscopy, cells were placed on collagen-coated 35 mm glass bottom dishes (MatTek) for 24 h. Cells were then mounted on a culture dish heater (DH-35; Warner Instruments) fitted with a TC-344B dual automatic temperature controller (Warner Instruments), and imaged at 37 °C using a × 63 oil-immersion lens.
Fluorescent intensity data were determined either by a line scan through the fluorescent region or the total fluorescence within an outlined area by using the Intelligent Imaging Innovations three-dimensional rendering and exploration software.
To determine the ratio of utrophin fluorescent intensity, aline was drawn through the entire ICB, or a 5 μm line was drawn away from the cell body/ICB border along the plasma membrane. The data were then expressed as arbitrary fluorescence units per square micrometre and the ratio between the ICB and cell body fluorescence intensity was calculated. Eight to twelve randomly chosen cells were analysed for every condition.
Transfection and RNA interference
To knockdown FIP3, p50RhoGAP, SCAMP1, SCAMP2, SCAMP3 and VAMP8, HeLa cells were transfected with 2 nM of specific siRNA using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. Transfected cells were incubated for 48 h, trypsinized and plated on collagen-coated 35-mm glass-bottom dishes for 24 h, and then processed for imaging or western blotting. siRNAs targeting SCAMPs (refs 17,21) (SCAMP1 siRNA1: 5′-UCAUCUCACUAGUUAAUGTT-3′; SCAMP2 siRNA1: 5′-CACUGUAGCCAACUUGCAUTT-3′, siRNA2: 5′-CGGACCCAGUGGAUGUAATT-3′; SCAMP3 siRNA1: 5′-CAGCUACUCGACAG-AACAATT-3′, siRNA2: 5′-AACGGAUCCACUCCUUAUATT-3′), FIP3 (siRNA1: 5′-GGCGUGUGCUGGAGCUGGA-3′, siRNA2: 5′-GGCAGUGAGGCGGAGC-UGU-3′; ref. 7), p50RhoGAP (siRNA1: 5′-ACUUACAGGCCCACGCUCUTT-3′; ref. 25), and CHMP4B {Carlton, 2012 no. 203} (siRNA1: 5′-AUCGAUAAAGUUG-AUGAGUUATT-3′) were previously described. siRNAs targeting p50RhoGAP (siRNA2 and siRNA3) were designed by Thermo Scientific using the human p50RhoGAP sequence (siRNA2: 5′-GGCGGAAGAUCAUUGUGUU-3′, and siRNA3: 5′- UAACCUGGCUGUUGUUUUC-3′).
Immuno-isolation of FIP3–GFP endosomes from telophase cells
Magnetic Dyna-beads (Invitrogen) conjugated to protein A were crosslinked to either anti-rabbit IgG or anti-GFP, according to the manufacturer’s protocol. HeLa cells stably expressing FIP3–GFP were grown on 300 cm2 tissue culture flasks (Light Labs) and synchronized using a thymidine/nocodazole double block, and collected by mitotic shake off after a 90 min incubation at 37 °C (Supplementary Fig. S3A). It was previously shown that under these conditions, most of these cells are in late telophase12 (Supplementary Fig. S3B). Cells were pelleted and resuspended in 10 mM HEPES at pH7.4. After a 10 min incubation, the cells were mechanically homogenized using a glass–glass homogenizer. The resulting lysate was centrifuged at 5,000g for 5 min to remove cell debris. Salts were added to a final concentration of 150 mM NaCl and 1 mM MgCl2 to the remaining microsomal preparation. To pre-clear the microsomal preparation, Dyna-beads conjugated to IgG were added for 1 h while rotating at room temperature. Pre-cleared microsomal preparations were split into two different groups. For the control group, Dyna-beads conjugated to rabbit IgG were added. For the sample group, Dyna-beads conjugated to rabbit polyclonal anti-GFP antibody were added and incubated for 1 h while rotating at room temperature. Beads from each group were washed five times with 5 ml of buffer (PBS with 10 mM HEPES at pH 7.4, 150 mM NaCl and 1 mM MgCl2), and bound organelles were eluted by 0.2 M glycine at pH 2.5. After elution, the pH of the beads was neutralized with 1 M Tris at pH 11.
FIP3–GFP endosome proteomics
Control (IgG) and sample (FIP3–GFP) immuno-isolates were analysed in three independent proteomic studies (details provided below). For each sample, the proteins were precipitated using methanol–chloroform44, solubilized using RapiGest (Waters, Corp.) and digested with trypsin (Promega).
In study 1, two aliquots of each IgG and FIP3–GFP digests were subjected to micro liquid chromatography–tandem mass spectrometry45. Specifically, peptides were separated with a microcapillary column (100 μm) using C18 reverse-phase resin (5 μ, 125 Å, Aqua, Phenomenex) with a 2 h gradient (5%–65%). Tandem mass spectra obtained by data-dependent acquisition using a Thermo Electron LTQ mass spectrometer were analysed using normalized Sequest46 and an IPI human protein database (v3.45,71983 sequences, released 10 June 2008) that had been concatenated with a randomized database. The resulting peptide identifications were assembled into a protein list using DTASelect47 with requirements for at least two peptides per protein, peptides that were fully digested by trypsin and at least 5 amino acids long.
In study 2, three aliquots of IgG and FIP3–GFP digest were subjected to micro liquid chromatography–tandem mass spectrometry as in the first study. A newer version of Sequest (v.2.7) and separate human protein and randomized databases were used to analyse the tandem mass spectra. Triplicate (IgG or FIP3–GFP) spectral matches were evaluated using Percolator48. Unique peptide matches of at least 6 amino acids with false discovery rate (q) values ≤0.01 were assembled into protein lists using DTASelect with requirements for a minimum of two fully tryptic peptides.
In study 3, two immuno-isolates (IgG and FIP3–GFP telophase) were treated and digested as above. Each sample was subjected to MudPIT analysis as previously described49. Specifically, peptides were separated with a microcapillary column (100 μm) using C18 reverse-phase resin (5 μ, 125Å, Aqua, Phenomenex), and strong cation exchange resin (Agilent Technologies) using eight increasing salt (ammonium acetate) pulses, each followed by a 2 h gradient (5%–65%). Tandem mass spectra obtained by data-dependent acquisition using a Thermo Electron LTQ mass spectrometer were analysed as in the second study.
Only proteins identified in the anti-GFP antibody sample, but not the IgG control, were considered for further analysis (Supplementary Table S1). Proteins identified in both, IgG control and FIP3-endosome samples (separated by study), are listed in Supplementary Tables S2–S4. Multiple previously identified proteins that have been shown to associate with FIP3 endosomes and function during cytokinesis, namely centralspindlin50, dynein51, kinesin-1 (ref. 51), and VAMP8 (ref. 52), were also identified in our studies, supporting the validity of our proteomic analysis.
Quantification
Multinucleation was counted by staining cells with Hoerscht and counting the number of cells with an aberrant number of nuclei. At least 100 randomly chosen cells were counted for every condition. The data shown are the means and s.d. of at least three independent experiments.
To establish division times in cells treated with FIP3, p50RhoGAP or SCAMP2/3 siRNAs, HeLa cells were plated on 35-mm glass-bottom dishes and 10 μm mini-stacks were acquired every hour for 24 h on an inverted Zeiss Axiovert 200M microscope equipped with an incubator with a heating stage environmental chamber (Pecon). The resulting images were analysed using ImageJ software (NIH) and the division time was calculated on the basis of cell morphology. At least 25 randomly chosen cells were used for analysis.
To establish the frequency of CHMP4B–YFP localization to the abscission site during late telophase, the cells were treated with FIP3, p50RhoGAP or SCAMP2/3 siRNAs. Cells were then fixed and stained with anti-acetylated-tubulin antibodies. Cells with CHMP4B–YFP present at the abscission site outside the midbody were then counted. Only cells that were connected by an ICB and that had CHMP4B–YFP at the midbody were analysed. Finally, only cells expressing low levels of CHMP4B–YFP were used for analysis. The experiment was repeated three times and the data shown are the means and s.d.
To determine the role of CHMP4B, SCAMP2/3 or p50RhoGAP on secondary furrow ingression, FIP3–GFP-expressing HeLa cells were treated with various siRNAs and plated on collaged-coated coverslips. At 24h later, randomly chosen cells in late telophase (as determined by the presence of FIP3–GFP within the ICB) were analysed by bright-field time-lapse microscopy. To ensure that we are not analysing short-lived plasma membrane waves (as described in ref. 24), only cells that contained a stationary ICB thinning that was maintained during the entire time-lapse series (50 time points, total series time 5 min) were scored as cells containing the secondary ingression. The experiment was repeated three times and the data shown are the means and s.d.
To analyse p50RhoGAP or SCAMP2/3 levels within the ICB, cells were stained with anti-p50RhoGAP or anti-SCAMP antibodies. At least 20 (for each analysis) randomly picked cells in late telophase were imaged using the same exposure times (1.5s for p50RhoGAP, 1 s for FIP3, 1.2s for SCAMPs). The fluorescence intensity within the ICB was then analysed using Intelligent Imaging Innovations software. All data were expressed as arbitrary fluorescence units per square micrometre.
To analyse F-actin within the ICB, cells were either stained with rhodamine–phalloidin or transiently transduced with utrophin–RFP. At least ten (for each analysis) randomly picked cells in late telophase were imaged using the same exposure times. Only cells with a clearly defined ICB were used for this analysis. The fluorescence intensity within the ICB was then analysed using Intelligent Imaging Innovations software. All data were expressed as arbitrary fluorescence units per square micrometre.
Statistical analysis was performed using GraphPad Prism using Student’s t-test with a two-tailed, 95% confidence interval to generate P values.
Supplementary Material
Acknowledgments
We thank C. Pearson and C. Willenborg (University of Colorado Denver, USA) for critically reading the manuscript. We also thank J. Martin-Serrano (King’s College London, UK) for mCherry–CEP55, mCherry–TSG101 and YFP-CHMP4B; M. Geiszt (Semmelweis University, Hungary) for GFP-p50RhoGAP; C. Westlake (NCI-Frederick, USA) for RAB8 complementary DNA; A. Bresnick (Albert Einstein College of Medicine, USA) for myosin-IIA-GFP; and W. Bement (University of Wisconsin Madison, USA) for utrophin-GFP and utrophin-RFP. This work was supported in part by a grant from NIH-NIDDK (DK064380 to R.P.).
Footnotes
Note: Supplementary Information is available in the online version of the paper
AUTHOR CONTRIBUTIONS
JA.S. and R.P. conceived the project and wrote the manuscript with the help of D.C. and C.C.W. Proteomic round one was performed and analysed by G.C.S. and J.W. Proteomic round two was performed and analysed by JA.S. and J.W. Proteomic round three was performed and analysed by J.A.S. and C.Z. All remaining experiments and data analysis were performed by J.A.S. and R.P.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Eggert US, Mitchison TJ, Field CM. Animal cytokinesis: from parts list to mechanisms. Annu Rev Biochem. 2006;75:543–566. doi: 10.1146/annurev.biochem.74.082803.133425. [DOI] [PubMed] [Google Scholar]
- 2.Schiel JA, et al. Endocytic membrane fusion and buckling-induced microtubule severing mediate cell abscission. J Cell Sci. 2011;124:1411–1424. doi: 10.1242/jcs.081448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Elia N, Sougrat R, Spurlin TA, Hurley JH, Lippincott-Schwartz J. Dynamics of endosomal sorting complex required for transport (ESCRT) machinery during cytokinesis and its role in abscission. Proc Natl Acad Sci USA. 2011;108:4846–4851. doi: 10.1073/pnas.1102714108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guizetti J, et al. Cortical constriction during abscission involves helices of ESCRT-III-dependent filaments. Science. 2011;331:1616–1620. doi: 10.1126/science.1201847. [DOI] [PubMed] [Google Scholar]
- 5.Baluska F, Menzel D, Barlow PW. Cytokinesis in plant and animal cells: endosomes ‘shut the door’. Dev Biol. 2006;294:1–10. doi: 10.1016/j.ydbio.2006.02.047. [DOI] [PubMed] [Google Scholar]
- 6.Fielding AB, et al. Rab11-FIP3 and FIP4 interact with Arf6 and the exocyst to control membrane traffic in cytokinesis. EMBO J. 2005;24:3389–3399. doi: 10.1038/sj.emboj.7600803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wilson GM, et al. The FIP3-Rab11 protein complex regulates recycling endosome targeting to the cleavage furrow during late cytokinesis. Mol Biol Cell. 2005;16:849–860. doi: 10.1091/mbc.E04-10-0927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carlton JG, Agromayor M, Martin-Serrano J. Differential requirements for Alix and ESCRT-III in cytokinesis and HIV-1 release. Proc Natl Acad Sci USA. 2008;105:10541–10546. doi: 10.1073/pnas.0802008105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lindas AC, Karlsson EA, Lindgren MT, Ettema TJ, Bernander R. A unique cell division machinery in the Archaea. Proc Natl Acad Sci USA. 2008;105:18942–18946. doi: 10.1073/pnas.0809467105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morita E, et al. Human ESCRT and ALIX proteins interact with proteins of the midbody and function in cytokinesis. EMBO J. 2007;26:4215–4227. doi: 10.1038/sj.emboj.7601850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Horgan CP, Walsh M, Zurawski TH, McCaffrey MW. Rab11-FIP3 localises to a Rab11-positive pericentrosomal compartment during interphase and to the cleavage furrow during cytokinesis. Biochem Biophys Res Commun. 2004;319:83–94. doi: 10.1016/j.bbrc.2004.04.157. [DOI] [PubMed] [Google Scholar]
- 12.Simon GC, et al. Sequential Cyk-4 binding to ECT2 and FIP3 regulates cleavage furrow ingression and abscission during cytokinesis. EMBO J. 2008;27:1791–1803. doi: 10.1038/emboj.2008.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gromley A, et al. Centriolin anchoring of exocyst and SNARE complexes at the midbody is required for secretory-vesicle-mediated abscission. Cell. 2005;123:75–87. doi: 10.1016/j.cell.2005.07.027. [DOI] [PubMed] [Google Scholar]
- 14.Montagnac G, et al. ARF6 Interacts with JIP4 to control a motor switch mechanism regulating endosome traffic in cytokinesis. Curr Biol. 2009;19:184–195. doi: 10.1016/j.cub.2008.12.043. [DOI] [PubMed] [Google Scholar]
- 15.Carlton JG, Caballe A, Agromayor M, Kloc M, Martin-Serrano J. ESCRT-III governs the Aurora B-mediated abscission checkpoint through CHMP4C. Science. 2012;336:220–225. doi: 10.1126/science.1217180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morita E, et al. Human ESCRT-III and VPS4 proteins are required for centrosome and spindle maintenance. Proc Natl Acad Sci USA. 2010;107:12889–12894. doi: 10.1073/pnas.1005938107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aoh QL, Castle AM, Hubbard CH, Katsumata O, Castle JD. SCAMP3 negatively regulates epidermal growth factor receptor degradation and promotes receptor recycling. Mol Biol Cell. 2009;20:1816–1832. doi: 10.1091/mbc.E08-09-0894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Falguieres T, Castle D, Gruenberg J. Regulation of the MVB pathway by SCAMP3. Traffic. 2012;13:131–142. doi: 10.1111/j.1600-0854.2011.01291.x. [DOI] [PubMed] [Google Scholar]
- 19.Mota LJ, Ramsden AE, Liu M, Castle JD, Holden DW. SCAMP3 is a component of the Salmonella-induced tubular network and reveals an interaction between bacterial effectors and post-Golgi trafficking. Cell Microbiol. 2009;11:1236–1253. doi: 10.1111/j.1462-5822.2009.01329.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Castle A, Castle D. Ubiquitously expressed secretory carrier membrane proteins (SCAMPs) 1–4 mark different pathways and exhibit limited constitutive trafficking to and from the cell surface. J Cell Sci. 2005;118:3769–3780. doi: 10.1242/jcs.02503. [DOI] [PubMed] [Google Scholar]
- 21.Liao H, et al. Nonredundant function of secretory carrier membrane protein isoforms in dense core vesicle exocytosis. Am J Physiol Cell Physiol. 2008;294:C797–C809. doi: 10.1152/ajpcell.00493.2007. [DOI] [PubMed] [Google Scholar]
- 22.Liu L, et al. SCAMP2 interacts with Arf6 and phospholipase D1 and links their function to exocytotic fusion pore formation in PC12 cells. Mol Biol Cell. 2005;16:4463–4472. doi: 10.1091/mbc.E05-03-0231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang J, Castle D. Regulation of fusion pore closure and compound exocytosis in neuroendocrine PC12 cells by SCAMP1. Traffic. 2011;12:600–614. doi: 10.1111/j.1600-0854.2011.01170.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Byers B, Abramson DH. Cytokinesis in HeLa: post-telophase delay and microtubule-associated motility. Protoplasma. 1968;66:413–435. doi: 10.1007/BF01255868. [DOI] [PubMed] [Google Scholar]
- 25.Sirokmany G, et al. Sec14 homology domain targets p50RhoGAP to endosomes and provides a link between Rab and Rho GTPases. J Biol Chem. 2006;281:6096–6105. doi: 10.1074/jbc.M510619200. [DOI] [PubMed] [Google Scholar]
- 26.Zhou YT, Chew LL, Lin SC, Low BC. The BNIP-2 and Cdc42GAP homology (BCH) domain of p50RhoGAP/Cdc42GAP sequesters RhoA from inactivation by the adjacent GTPase-activating protein domain. Mol Biol Cell. 2010;21:3232–3246. doi: 10.1091/mbc.E09-05-0408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pollard TD. Mechanics of cytokinesis in eukaryotes. Curr Opin Cell Biol. 2010;22:50–56. doi: 10.1016/j.ceb.2009.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Robinson DN, Cooley L. Stable intercellular bridges in development: the cytoskeleton lining the tunnel. Trends Cell Biol. 1996;6:474–479. doi: 10.1016/0962-8924(96)84945-2. [DOI] [PubMed] [Google Scholar]
- 29.Burkel BM, von Dassow G, Bement WM. Versatile fluorescent probes for actin filaments based on the actin-binding domain of utrophin. Cell Motil Cytoskeleton. 2007;64:822–832. doi: 10.1002/cm.20226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fyfe I, Schuh AL, Edwardson JM, Audhya A. Association of the endosomal sorting complex ESCRT-II with the Vps20 subunit of ESCRT-III generates a curvature-sensitive complex capable of nucleating ESCRT-III filaments. J Biol Chem. 2011;286:34262–34270. doi: 10.1074/jbc.M111.266411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Connell JW, Lindon C, Luzio JP, Reid E. Spastin couples microtubule severing to membrane traffic in completion of cytokinesis and secretion. Traffic. 2009;10:42–56. doi: 10.1111/j.1600-0854.2008.00847.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Demirov DG, Ono A, Orenstein JM, Freed EO. Overexpression of the N-terminal domain of TSG101 inhibits HIV-1 budding by blocking late domain function. Proc Natl Acad Sci USA. 2002;99:955–960. doi: 10.1073/pnas.032511899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Garrus JE, et al. TSG101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell. 2001;107:55–65. doi: 10.1016/s0092-8674(01)00506-2. [DOI] [PubMed] [Google Scholar]
- 34.Martin-Serrano J, Yarovoy A, Perez-Caballero D, Bieniasz PD. Divergent retroviral late-budding domains recruit vacuolar protein sorting factors by using alternative adaptor proteins. Proc Natl Acad Sci USA. 2003;100:12414–12419. doi: 10.1073/pnas.2133846100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martin-Serrano J, Zang T, Bieniasz PD. Role of ESCRT-I in retroviral budding. J Virol. 2003;77:4794–4804. doi: 10.1128/JVI.77.8.4794-4804.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nickerson DP, West M, Henry R, Odorizzi G. Regulators of Vps4 ATPase activity at endosomes differentially influence the size and rate of formation of intralumenal vesicles. Mol Biol Cell. 2010;21:1023–1032. doi: 10.1091/mbc.E09-09-0776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Von Schwedler UK, et al. The protein network of HIV budding. Cell. 2003;114:701–713. doi: 10.1016/s0092-8674(03)00714-1. [DOI] [PubMed] [Google Scholar]
- 38.Sedzinski J, et al. Polar actomyosin contractility destabilizes the position of the cytokinetic furrow. Nature. 2011;476:462–466. doi: 10.1038/nature10286. [DOI] [PubMed] [Google Scholar]
- 39.Dambournet D, et al. Rab35 GTPase and OCRL phosphatase remodel lipids and F-actin for successful cytokinesis. Nat Cell Biol. 2011;13:981–988. doi: 10.1038/ncb2279. [DOI] [PubMed] [Google Scholar]
- 40.Kouranti I, Sachse M, Arouche N, Goud B, Echard A. Rab35 regulates an endocytic recycling pathway essential for the terminal steps of cytokinesis. Curr Biol. 2006;16:1719–1725. doi: 10.1016/j.cub.2006.07.020. [DOI] [PubMed] [Google Scholar]
- 41.Liao H, et al. Secretory carrier membrane protein SCAMP2 and phosphatidylinositol 4, 5-bisphosphate interactions in the regulation of dense core vesicle exocytosis. Biochemistry. 2007;46:10909–10920. doi: 10.1021/bi701121j. [DOI] [PubMed] [Google Scholar]
- 42.Thomas DD, Frey CL, Messenger SW, August BK, Groblewski GE. A role for tumor protein TPD52 phosphorylation in endo-membrane trafficking during cytokinesis. Biochem Biophys Res Commun. 2010;402:583–587. doi: 10.1016/j.bbrc.2010.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Steigemann P, et al. Aurora B-mediated abscission checkpoint protects against tetraploidization. Cell. 2009;136:473–484. doi: 10.1016/j.cell.2008.12.020. [DOI] [PubMed] [Google Scholar]
- 44.Wessel D, Flugge UI. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal Biochem. 1984;138:141–143. doi: 10.1016/0003-2697(84)90782-6. [DOI] [PubMed] [Google Scholar]
- 45.McCormack AL, et al. Direct analysis and identification of proteins in mixtures by LC/MS/MS and database searching at the low-femtomole level. Anal Chem. 1997;69:767–776. doi: 10.1021/ac960799q. [DOI] [PubMed] [Google Scholar]
- 46.MacCoss MJ, Wu CC, Yates JR., 3rd Probability-based validation of protein identifications using a modified SEQUEST algorithm. Anal Chem. 2002;74:5593–5599. doi: 10.1021/ac025826t. [DOI] [PubMed] [Google Scholar]
- 47.Cociorva D, Tabb DL, Yates JR. Validation of tandem mass spectrometry database search results using DTASelect. Curr Protoc Bioinformatics/editorial board. 2007 doi: 10.1002/0471250953.bi1304s16. Unit 13.4 http://dx.doi.org/10.1002/0471250953.bi1304s16. [DOI] [PubMed]
- 48.Brosch M, Yu L, Hubbard T, Choudhary J. Accurate and sensitive peptide identification with Mascot Percolator. J Proteome Res. 2009;8:3176–3181. doi: 10.1021/pr800982s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lowenthal MS, Howell KE, Wu CC. Proteomic analysis of the stacked Golgi complex. Methods Mol Biol. 2008;432:37–49. doi: 10.1007/978-1-59745-028-7_3. [DOI] [PubMed] [Google Scholar]
- 50.Glotzer M. The molecular requirements for cytokinesis. Science. 2005;307:1735–1739. doi: 10.1126/science.1096896. [DOI] [PubMed] [Google Scholar]
- 51.Glotzer M. The 3Ms of central spindle assembly: microtubules, motors and MAPs. Nat Rev Mol Cell Biol. 2009;10:9–20. doi: 10.1038/nrm2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Low SH, et al. Syntaxin 2 and endobrevin are required for the terminal step of cytokinesis in mammalian cells. Dev Cell. 2003;4:753–759. doi: 10.1016/s1534-5807(03)00122-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.