

Abstract
The present study describes the development of a good manufacturing practice (GMP)-grade liposomal nanotherapy containing prednisolone phosphate for the treatment of inflammatory diseases. After formulation design, GMP production was commenced which yielded consistent, stable liposomes sized 100 nm ± 10 nm, with a prednisolone phosphate (PLP) incorporation efficiency of 3-5%. Pharmacokinetics and toxicokinetics of GMP-grade liposomal nanoparticles were evaluated in healthy rats, which was compared to daily and weekly administration of free prednisolone phosphate, revealing a long circulatory half-life with minimal side effects. Subsequently, non-invasive multimodal clinical after liposomal nanotherapy’s intravenous administration revealed anti-inflammatory effects on the vessel wall of atherosclerotic rabbits. The present program led to institutional review board approval for two clinical trials with patients with atherosclerosis.
Keywords: Nanomedicine, GMP-grade, formulation design, prednisolone phosphate, atherosclerosis
Graphical abstract
Pharmaceutical development of a GMP-grade liposomal nanotherapy for the treatment of inflammatory diseases
Introduction
Anti-inflammatory therapies offer a range of potential benefits in a variety of conditions, including rheumatoid arthritis, inflammatory bowel disease and cardiovascular diseases [1,2]. The disadvantage of such anti-inflammatory treatments is the chance of systemic side effects such as immunosuppression. A viable strategy to improve the effect of anti-inflammatory compounds at the target site and reduce systemic effects is targeted delivery to diseased sites [3]. In oncological and inflammatory diseases nanoparticle therapeutics have shown to facilitate selective delivery of drugs through the enhanced permeability and retention (EPR) effect, referring to the non-specific extravasation of nanoparticles at sites with a highly permeable microvasculature [4]. This can result in enhanced local drug accumulation at the pathological site and improved efficacy combined with reduced systemic side effects [5]. Atherosclerosis is a chronic inflammatory disease that builds up in medium and large-sized arteries over time [6]. The most deleterious consequences of atherosclerosis, myocardial infarction and stroke, have profound morbidity and mortality [7]. New developments focus on modulating the inflammatory component of atherosclerosis [8]. Similar to nanoparticle EPR targeting of tumors, the angiogenically activated vasa-vasorum is rich of leaky neovessels, which facilitates nanoparticle accumulation in atherosclerotic plaques [9]. We recently demonstrated that a polyethylene glycol (PEG)-modified liposomal nanoparticle, containing a magnetic resonance imaging (MRI) contrast agent and the glucocorticoid prednisolone phosphate, effectively targeted atherosclerotic plaques in a rabbit model of atherosclerosis [10]. A single intravenous administration resulted in a strong vessel wall inflammation reduction in atherosclerotic rabbits as measured with both 18F-fluorodeoxyglucose positron emission tomography/computed tomography (18F-FDG PET/CT) and dynamic contrast enhanced (DCE)-MRI. The goal of the current study was to develop and consequently produce this anti-inflammatory nanoparticle therapy according to GMP, to establish its safety profile in rats and evaluate efficacy in a rabbit model of atherosclerosis by clinical multimodal imaging, thereby paving the way for clinical trials.
Methods
Liposomal nanoparticle preparation for biodistribution studies in rats
Liposomes were prepared by hydrating a lipid film created by rotary evaporation with phosphate buffered saline (PBS) at an initial total lipid concentration of 20 μmol/ml. [3H]-cholesteryl oleylether was added as a non-degradable liposome lipid phase marker. The liposomes were sized by multiple extrusion using a medium pressure extruder equipped with two stacked polycarbonate membrane filters, one with a pore size of 200 nm on top of one with 100 nm pores. Components that were not incorporated in liposomes were removed by size exclusion gel chromatography. Radioactivity of the liposomal dispersions was assayed in a liquid scintillation cocktail and counted in a Philips PW 4700 liquid scintillation counter (Philips, Best, The Netherlands). Lipid content of the liposomal dispersion was determined by assessing the radioactivity of the liposomes before and after preparation. The mean particle size of the liposomes was determined by dynamic light scattering and ranged between 140 and 160 nm with a polydispersity index of below 0.15.
Manufacturing of GMP-grade LN-PLP
The liposomal nanoparticle prednisolone phosphate (LN-PLP) formulation was composed of dipalmitoyl phosphatidyl choline (DPPC), cholesterol, and PEG2000 distearoyl phosphatidylethanolamine (PEG-DSPE) in a 62%, 33%, and 5% molar ratio. Starting at a concentration of 100 mg/mL, water-soluble PLP was chosen as the active ingredient, to be encapsulated in the aqueous interior of the liposome in a starting concentration of 100 mg/mL. The manufacturing method involved the following steps: PLP was dissolved in Water for Injections while the lipids were dissolved in absolute ethanol at 65 °C. The alcoholic lipid solution was injected in the aqueous PLP solution and mixed under heating to 65 °C forming a multilamellar vesicle dispersion. This dispersion was downsized to the desired particle size of approximately 100 nm in diameter by repeated homogenization cycles using an Avestin C55 high-shear homogenizer (Avestin, Mannheim, Germany). Unencapsulated PLP was removed by ultrafiltration using membranes with a molecular weight cut off of 30 kD and replaced with clean dispersion buffer. Finally, the final dispersion liposomal prednisolone phosphate (LN-PLP) was sterile filtered, collected in vials and stored between 2 and 8 °C. ML-PLP’s size, polydispersity, encapsulated drug content, free drug content, lipid content, impurities and degradation products, residual ethanol, pH, osmolarity, sterility, and endotoxin content were analyzed. Shelf life stability studies indicated that LN-PLP remained stable on storage for at least 2 years if kept between 2 and 8 °C.
Biodistribution studies in rats
Male Wistar rats (n=60 total) with an approximate body weight of 200 g were used (outbred, SPF-quality). Single-dose i.v. injections of liposomal preparations containing 5 μmol total lipid and approximately 50 kBq of radio-activity, were administered via the tail vein in the different groups (n = 4 per group for different PEG length, PEG density, membrane phospholipids and prednisolone derivates). Blood samples of 100 μl were collected at 5 minutes and 1, 4, 8, 24 and 48 hours post-injection. Radioactivity in blood samples was determined by adding tissue solubilizer and hydrogen peroxide. After overnight incubation the samples were assayed in a scintillation cocktail and counted for radioactivity with a Philips PW 4700 liquid scintillation counter. To obtain the percentage of injected dose values in the circulation, the radioactivity of the blood samples was multiplied with a factor 10 times the total mass of blood in grams (calculated as 7% of the total body weight) and divided by the injected dose.
Toxicokinetic study in Sprague-Dawley rats with LN-PLP
The pharmacokinetic and toxicokinetic program was outsourced to LAB-research (Montreal, Canada), and conducted to determine the toxicity and toxicokinetics of LN-PLP and to compare it to free PLP, following i.v. administration to Sprague-Dawley rats (n = 120) for 28 days. The LN-PLP, free PLP and saline were administered to groups as shown in supporting information table 1. Clinical signs, body weights, food consumption, ophthalmoscopy and clinical pathology were evaluated in the animals. Blood was collected from animals on 2 occasions at 2 time points (control group), at 6 time points (free PLP groups) and at 7 time points (LN-PLP groups) relative to the first and last doses (Day 28 for Groups 2 and 3 and Day 22 for Groups 4-6). Plasma concentrations of PLP and its active metabolite PL were measured using a validated bioanalytical rat assay [11] after the first (Day 1) and last (Day 22/28) i.v. administration of LN-PLP or free PLP. After the final blood collection, all animals were euthanized and subjected to a gross pathology examination, with tissue collection for histopathological examination. All rat experiments were carried out humanely and followed protocols approved by the LAB-research Animal Welfare Body.
Pharmacokinetics and efficacy in rabbits
Rabbit model and study design
A total of 32 male New Zealand white rabbits (Charles River Laboratories, Wilmington, Massachusetts, USA) were included. Twelve healthy rabbits were used for evaluation of pharmacokinetics. Blood was withdrawn through a catheter placed in the central ear artery by taking samples before and after injection of the different therapeutic compounds at different time points. Serum samples were frozen at -80°C and shipped to the Analytical Biochemical Laboratory in Assen, The Netherlands for analysis.
The remaining twenty rabbits were used for a therapeutic efficacy study after inducing atherosclerosis by a well-established method [12]. In short, 3-4 month old rabbits (2.5 – 3.0 kg) were fed a 0.3% cholesterol-enriched diet (Research Diet Inc., New Brunswick, New Jersey, USA). After two weeks on diet, rabbits were subjected to a femoral angioplasty, during which a 4-F Fogarty embolectomy catheter (Edwards Lifesciences, California, USA) was introduced up to the thoracic aorta and subsequently inflated and pulled back to the iliac bifurcation. This process was repeated 3 times to denude the aorta of endothelial cells and speed the process of creating atherosclerotic lesions. After 4 weeks aortic balloon injury was repeated from the contralateral extremity and rabbits were subsequently switched to a lower cholesterol diet of 0.15% 2 weeks after the last procedure, for a total of 24 weeks on diet. Both imaging and surgical procedures were performed under anaesthesia by an intramuscular injection of ketamine (20 mg/kg, Vedco, Missouri) and Xylazine (7 mg/kg; Vedco, Missouri, USA).
All animals underwent baseline imaging with anatomical MRI, DCE-MRI and 18F-FDG-PET/CT, and were subsequently injected with LN-PLP at 1 mg/kg or 10 mg/kg PLP or saline. Scanning with the same imaging modalities was repeated after 2 and 7 days. Animals were sacrificed one day after the final scan to provide time for 18F decay. All rabbit experiments were carried out humanely and were approved by the Icahn School of Medicine at Mount Sinai Institute Animal Care and Use Committee.
Image acquisition
Anatomical and DCE-MRI
Rabbits were imaged on a 3.0 T whole body MRI system (Philips Achieva, Best, The Netherlands), using an 8-channel knee coil (Philips Achieva, Best, The Netherlands). T1- and T2-weighted images were acquired from the left renal artery to the iliac bifurcation. DCE-MRI was performed using a multi-slice T1-weighted black blood turbo spin echo sequence as described previously [12].
FDG-PET/CT Imaging
Rabbits fasted 4 hours prior to 37 – 74 MBq (1-2 mCi) /kg of 18F-FDG injection through the marginal ear vein. After allowing the radiotracer to circulate for 3 hours to eliminate blood-pool activity, imaging was performed on a combined PET/CT scanner (Discovery LS, GE Healthcare, Milwaukee, Wisconsin) according to methods described previously [12]. A single-bed position in 3D mode during 10 minutes was used to scan from the celiac artery to the iliac bifurcation (~15.5 cm). Using a Fourier iterative algorithm, image reconstruction resulted in a slice thickness of 4.25 mm.
Image analysis
Vessel wall area (VWA) was quantitated by tracing vessel wall contours manually on 5 contiguous T2-weighted MRI slices using VesselMass (Leiden, The Netherlands). An average of 5 slices per rabbit was used for data analysis. DCE-MRI data was analysed using non model based approaches [12]. The area under the curve (AUC) was calculated after tracing contours manually. All DCE-MRI analysis was performed using a custom-built software program in MATLAB (Math Works Inc, Natick, Massachussetts, USA). 18F-FDG PET/CT data analysis was performed with OsiriX (Pixmeo, Geneva, Switzerland) by drawing contiguous regions of interest (ROI) from the left renal artery to the iliac bifurcation on axial slices. The ROIs obtained information on standard uptake value (SUV)max and SUVmean. Consequently the average of the SUVmax and SUVmean ROIs per rabbit were used for data analysis.
Statistical analysis
Changes from baseline to end of the therapeutic efficacy study were assessed using a paired Student t test. Statistical analyses were performed using Prism version 5.0 (GraphPad software, La Jolla California). Data are reported as mean ± SD. P values < 0.05 were considered statistically significant.
Results
Liposomal nanoparticle prednisolone phosphate formulation design
The anti-inflammatory nanotherapy developed for this program is based on platform technology we previously thoroughly evaluated in preclinical studies [10,13,14]. We set out a developmental route for the production of a GMP-grade liposomal formulation of prednisolone (Figure 1A). Prerequisites included the stable inclusion of a high payload of a water-soluble prednisolone derivative, a reproducible size with a narrow size distribution and an excellent inter-batch reproducibility. Before large-scale production of this formulation at GMP-grade was initiated, the pharmacokinetics of differently formulated radiolabeled liposomal nanoparticles were determined after a single i.v. injection in male Wistar rats. PEG-coating consisting of PEG with a molecular mass of 2000 at a molar content of 7.5% relative to the phospholipid molar content had the optimal pharmacokinetic profile (Figure 1B,C). Subsequently, the drug inclusion stability was evaluated as a function of lipid composition as well as the choice for a specific prednisolone derivative (Figure 1D,E). A liposomal nanoparticle (LN) composed of DPPC: cholesterol: PEG-DSPE at a molar ratio 1.85: 1.0: 0.15 and containing prednisolone phosphate in the aqueous interior was found to be optimal and chosen for subsequent studies.
Fig. 1.
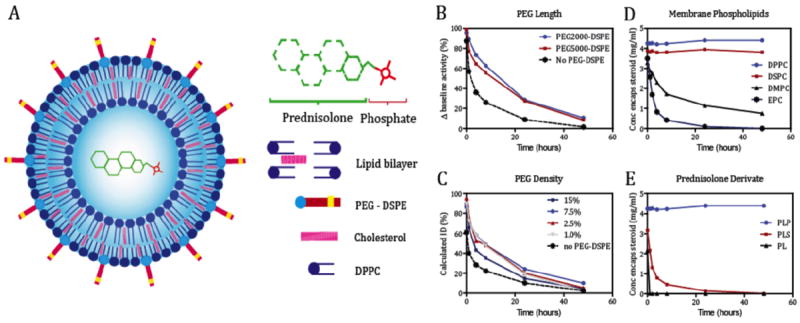
A manufacturing method for this product that would eventually allow for the scale up and production of GMP-grade clinical trial material was developed. We selected a high-throughput method involving injection of an organic lipid solution into the aqueous dispersion medium containing prednisolone phosphate, after which high-shear homogenization was performed to reduce size and size distribution of the LN. Unencapsulated prednisolone phosphate was removed by ultrafiltration and washing with dispersion buffer. The process was concluded by sterile filtration and filling. The production of GMP-grade lipsomes resulted in formulations with a size ranging from 91 to 110 nm (mean size: 100 nm ± 10 nm), with a PLP incorporation efficiency of 3-5%. The zeta potential was consistently – 5 mV. The final PLP content varied between 1.5 and 2.5 mg/mL. Free drug content was always below 5% of the total PLP content. The total lipid content varied between 42 mg/mL and 57 mg/mL. Neither in vitro (buffer, 37 °C) nor in vivo (in blood circulation) release of encapsulated drug from the liposomes was observed.
Pharmacokinetics and toxicokinetics of LN-PLP in rats and rabbits
After establishing the composition of the liposomal nanoparticles we performed an extensive pharmacokinetic and toxicokinetic multiple-dose study in healthy Sprague-Dawley rats (n = 120) comparing weekly administration of 0.5, 2 and 8 mg/kg GMP-grade LN-PLP with daily administration of 15 and 60 mg/kg free PLP. AUCs of active PL in the weekly 0.5 and 2 mg/kg LN-PLP dosing groups were comparable to active PL in the daily 15 and 60 mg/kg free PLP dosing groups respectively. Clearance of PLP encapsulated in LN was dramatically reduced as compared with free PLP (clearance of LN-PLP and free PLP were 0.56 and 7053 mL hour-1 kg-1 respectively, p < 0.001). The plasma concentrations of PLP (encapsulated in LN-PLP) showed a gradual decrease over the course of days whereas very short half-lives in the free PLP groups were observed (t½ for LN-PLP 39.8 hours compared to 0.1 hours for free PLP, p < 0.001). No evidence of systemic accumulation of PLP was observed following repeated dosing of LN-PLP (Cmax dose from 30 after 1 dose, to 35 μg/mL/mg/kg after multiple doses, p = 0.2966). A summary of pharmacokinetic parameters is provided in Table 1. We observed no changes in body weight or blood chemistry indicative of toxic effects of LN-PLP. Post-mortem organ size and weight assessment showed no effects of treatment allocation. Full overview of rat toxicokinetics is provided in Table 2.
Table 1.
Pharmacokinetic summary in multiple dose study in Sprague Dawley rats
| Part A: PK of PLP after administration in its liposomal encapsulated or free form in rats | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| PLP | Weekly LN-PLP (in mg/kg) | Daily PLP (in mg/kg) | |||||||||
| 0.5 | 2 | 8 | 15 | 60 | |||||||
| M | F | M | F | M | F | M | F | M | F | ||
| Day 1 | Clearance (mL hour-1 kg-1) | 0.70 | 1.09 | 0.59 | 0.64 | 0.49 | 0.62 | 8543 | 3968 | 7053 | nc |
| T½ (hours) | 34.7 | 22.9 | 39.1 | 33.8 | 45.7 | 33.9 | 0.08 | 0.42 | 0.11 | nc | |
| Cmax (μg/mL) | 14.2 | 13.8 | 60.4 | 63.6 | 248 | 262 | 6.48 | 5.91 | 31.8 | 38.9 | |
| Cmax/dose (μg/mL)/(mg/kg) | 28.4 | 27.5 | 30.2 | 31.8 | 31.0 | 32.7 | 0.43 | 0.39 | 0.53 | 0.65 | |
|
| |||||||||||
| Day 22/28 | Clearance (mL hour-1 kg-1) | 0.59 | 0.96 | 0.50 | 0.51 | 0.30 | 0.48 | 6885 | 4104 | 6125 | NR |
| T½ (hours) | 0.25 | 0.25 | 0.25 | 0.25 | 0.25 | 0.25 | 0.08 | 0.08 | 0.08 | 0.08 | |
| Cmax (μg/mL) | 17.3 | 14.5 | 72.5 | 76.3 | 300 | 288 | 7.94 | 5.92 | 37.2 | 40.1 | |
| Cmax/dose (μg/mL)/(mg/kg) | 34.6 | 29.1 | 36.2 | 38.1 | 37.5 | 36.0 | 0.53 | 0.40 | 0.62 | 0.67 | |
|
| |||||||||||
| Part B: Pharmacokinetics of the active metabolite PL after administration of PLP in liposomal encapsulated or its free form in rats | |||||||||||
| PL | Weekly LN-PLP (in mg/kg) | Daily PLP (in mg/kg) | |||||||||
| 0.5 | 2 | 8 | 15 | 60 | |||||||
| M | F | M | F | M | F | M | F | M | F | ||
|
| |||||||||||
| Day 1 | T½ (hours) | nc* | 0.40 | 0.37 | 0.38 | 0.37 | |||||
| Cmax (μg/mL) | 0.34 | 0.17 | 1.18 | 0.67 | 6.27 | 3.66 | 7.99 | 10.2 | 37.9 | 45.4 | |
| Cmax/dose (μg/mL)/(mg/kg) | 0.70 | 0.34 | 0.59 | 0.34 | 0.78 | 0.46 | 0.53 | 0.68 | 0.63 | 0.76 | |
|
| |||||||||||
| Day 22/28 | T½ (hours) | nc* | 0.35 | 0.43 | 0.37 | 0.39 | |||||
| Cmax (μg/mL) | 0.23 | 0.08 | 1.17 | 0.50 | 6.87 | 3.59 | 9.80 | 10.0 | 40.9 | 41.3 | |
| Cmax/dose (μg/mL)/(mg/kg) | 0.47 | 0.17 | 0.58 | 0.25 | 0.86 | 0.45 | 0.65 | 0.67 | 0.68 | 0.69 | |
Cmax, maximal concentration; F, female rats; LN, liposomal nanoparticle; M, male rats; nc, not calculated; PL, prednisolone (active metabolite); PLP, prednisolone phosphate (prodrug); T½; terminal half-live.
not calculated because plasma concentrations result of indirect release.
Table 2.
Post-mortem evaluation of organs and tissues in Sprague Dawley rats
| Anatomical location Abnormality | Saline | LN-PLP (in mg/kg) | Free PLP (in mg/kg) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0.5 | 2 | 8 | 15 | 60 | |||||||||
| M | F | M | F | M | F | M | F | M | F | M | F | ||
| Adrenal Cortex | Minimal atrophy | - | - | - | - | 1 | - | 4 | 1 | - | - | 1 | - |
| Bone marrow | Fatty change | 5 | 7 | 8 | 8 | 8 | 10 | 10 | 10 | 9 | 9 | 9 | 9 |
| Heart | Minimal myocardial degeneration | - | - | - | - | - | - | - | - | - | - | 2 | - |
| Injection site | Hemorrhage and mild inflammation 3 | 2 | - | - | - | - | - | 2 | 2 | - | 6 | 3 | |
| Kidney | Minimal infarct | - | - | - | - | - | 1 | - | - | - | 1 | ||
| Minimal nephropathy | - | - | - | - | - | - | - | 4 | - | 1 | - | 1 | |
| Liver | Hepatocellar vacuolation | - | - | - | 1 | - | 3 | 4 | 2 | 4 | 7 | 5 | 9 |
| Mild infarct or necrosis | - | - | 1 | - | - | - | - | - | 2 | - | 1 | - | |
| Lungs | Minimal hemorrhage | 4 | 2 | - | - | - | - | 1 | - | - | - | 5 | 2 |
| Mild inflammation | 1 | 3 | - | - | - | - | - | - | - | - | 1 | - | |
| Mesenteric | Minimal lymphoid depletion | - | - | - | - | - | - | 8 | 6 | - | - | 3 | 7 |
| Spleen | Minimal lymphoid depletion | - | - | - | - | - | - | 10 | 5 | - | - | 1 | 2 |
| Thymus | Mild lymphoid depletion | - | - | - | - | - | - | 9 | 9 | - | 2 | 9 | 10 |
| Mild hemorrhage | - | - | 2 | - | 1 | 1 | - | - | - | - | - | - | |
| Thyroid | Mild hyperplasia | 1 | 4 | - | - | - | - | 3 | 2 | - | - | 2 | 1 |
| Brown fat | Macrovacuolar change | - | - | 5 | - | 8 | - | 9 | 6 | 9 | 4 | 9 | 8 |
| Prostate | Minimal atrophy | - | na | - | na | - | na | - | na | - | na | 2 | na |
| Seminal vesicle | Minimal atrophy | - | na | - | na | - | na | - | na | - | na | 2 | na |
| Uterus | Minimal dilatation | na | 4 | na | - | na | - | na | 1 | na | - | na | - |
F, female rats; LN, liposomal nanoparticle; M, male rats; na, not assessed (due to gender); PL, prednisolone (active metabolite); PLP, prednisolone phosphate (prodrug); T½; terminal half-live.
Pharmacokinetics of LN-PLP was also assessed in 12 healthy male New Zealand white rabbits, in which we compared a single injection of LN-PLP at a dose of 1 or 10 mg/kg with a single injection of free PLP at a dose of 1 mg/kg or 10 mg/kg. Pharmacokinetics in these animals showed similar pharmacokinetics of LN-PLP and free PLP as compared to the rats, with an increased circulation half-life and AUC (t½ 39.9 hours for LN-PLP versus 0.06 hours for free PLP, p < 0.001 and AUC0-t 19246 3.65 μg * h/mL for LN-PLP versus 3.65 μg * h/mL for free PLP, p < 0.001). The rabbit pharmacokinetic parameters are provided in Table 3.
Table 3.
Pharmacokinetics in rabbits
| PK of PLP after administration in its liposomal encapsulated or free form in rabbits | ||||
|---|---|---|---|---|
| PLP | Single dose LN-PLP (mg/kg) | Single dose free PLP (mg/kg) | ||
| 1 | 10 | 1 | 10 | |
| Clearance (mL hour-1 kg-1) | 1.35 | 0.53 | * | 5494 |
| Terminal half –life (T½) (hours) | 19.4 | 39.9 | * | 0.06 |
| Cmax (μg/mL) | 29.9 | 342.0 | 1.69 | 20.3 |
|
| ||||
| PK of the active metabolite PL after administration of PLP in liposomal encapsulated or its free form in rabbits | ||||
| PL | Single dose LN-PLP (mg/kg) | Single dose free PLP (mg/kg) | ||
| 1 | 10 | 1 | 10 | |
|
| ||||
| Clearance (mL hour-1 kg-1) | nc* | nc* | 3831 | 2808 |
| Terminal half –life (T½) (hours) | nc* | nc* | 0.29 | 0.25 |
| Cmax (μg/mL) | 1.84 | 12.1 | 0.54 | 9.56 |
Half-life and clearance rate for 1mg/kg free PLP could not be determined due to undetectable PLP plasma levels after the first time point (t=0.08 hours).
Cmax, maximal concentration; LN, liposomal nanoparticle; PL, prednisolone (active metabolite); PLP, prednisolone phosphate (prodrug); T½; terminal half-live.
nc, not calculated because plasma concentrations result of indirect release;
Therapeutic efficacy in an atherosclerotic rabbit model
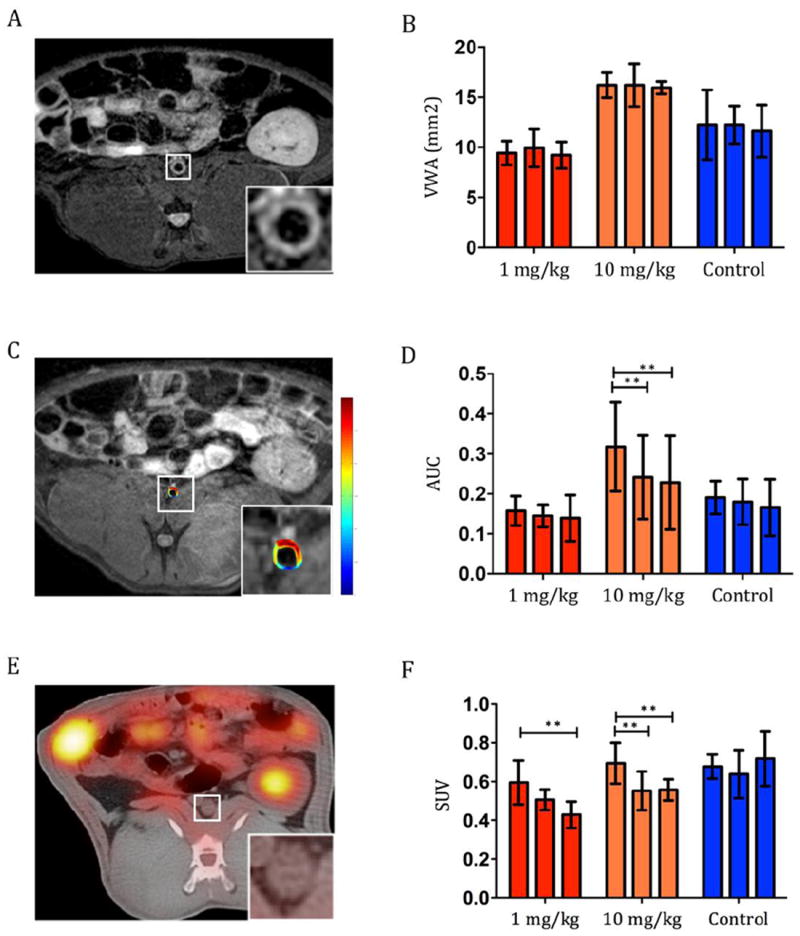
To evaluate effects of GMP-grade LN-PLP on experimental atherosclerosis we performed a therapeutic efficacy study in 20 male New Zealand white rabbits. Rabbits were fed a high-cholesterol diet and underwent balloon angioplasties of the abdominal aorta to induce atherosclerotic lesions [12]. After development of atherosclerotic lesions, 6 months after initiating the high-cholesterol diet, all rabbits underwent conventional T1- and T2-weighted MRI and DCE-MRI to obtain baseline values of plaque anatomy, VWA, and measures of plaque permeability, respectively, as the DCE-MRI value AUC has been extensively validated as a tool to estimate plaque microvascular density [15,16]. In addition, 18F-FDG PET/CT was performed to assess metabolic activity within the vessel wall of rabbits [17,18]. Rabbits were divided into 3 groups, receiving a single dose of 1 mg/kg LN-PLP (n = 4), 10 mg/kg LN-PLP (n = 8) or saline (n = 8) i.v., respectively. After 2 and 7 days, no changes in VWA were detected (Figure 2A,B). The AUC significantly declined 2 and 7 days after treatment in the 10 mg/kg LN-PLP group (p = 0.008 and p = 0.017 respectively), whereas no difference was observed in the 1 mg/kg LN-PLP and the saline group (Figure 2C,D). 18F-FDG PET/CT measurements showed significant decreases in both the SUVmax at 10 mg/kg at 2 and 7 days and after 7 days in the 1 mg/kg group (Figure 2E,F) (p < 0.05 for both), indicating decreased metabolic activity within atherosclerotic plaques after treatment with LN-PLP.
Fig. 2.
Discussion
We here present the development of an anti-inflammatory nanotherapy consisting of a liposomal nanoparticle containing prednisolone phosphate into a GMP-grade product ready for clinical investigations. After optimizing LN-PLP’s pharmaceutical design, we executed a preclinical evaluation program that included, pharmacokinetic and toxicologic evaluation in rats as well as therapeutic efficacy in a rabbit model of atherosclerosis. The aim of the preclinical program was to establish the safety and efficacy profile that, if positive, would warrant LN-PLP’s clinical evaluation in patients with advanced atherosclerosis. To facilitate the latter, we used clinical scanners and FDA-approved protocols to determine the efficacy of LN-PLP in atherosclerotic plaques of rabbits in our preclinical studies.
Our study was motivated by previous work in which we observed a strong anti-inflammatory effect on atherosclerotic aortic rabbit plaques after a single intravenous administration of a liposomal nanoparticle formulation [10]. This formulation contained both prednisolone phosphate in the aqueous interior of the liposomes and a high phospholipid bilayer payload of gadolinium (Gd), a paramagnetic contrast agent. The latter was included to enable studying nanoparticle plaque targeting by MRI.
For clinical translation the inclusion of Gd may not be feasible, as there is an association with long circulating Gd with nephrogenic systemic fibrosis [19]. We therefore omitted Gd in the current GMP-grade LN-PLP formulation. Encouragingly, we observed the previously reported anti-inflammatory effect in the vessel wall in the current study with GMP-produced LN-PLP, as is demonstrated with our FDG-PET/CT and DCE-MRI results. At the same time only minimal and mild adverse effects of LN-PLP were seen, which are fully attributable to low levels of systemic exposure to PLP.
The dose levels of LN-PLP selected for the toxicology program in rats were selected from an earlier pharmokinetic study that was performed to evaluate a feasible dosing scheme that lead to similar exposure levels (areas under the plasma concentration-time curves) of LN-PLP to free PLP. The dose levels of LN-PLP selected for the efficacy studies in the rabbit atherosclerosis model were based on our previous experience with LN-PLP in a preclinical arthritis model[20].
Based on our observations, we received IRB-approval for the testing of LN-PLP in human subjects with atherosclerosis in two different trials. The aim of the first trial, called DELIVER (NCT01647685), was to study nanoparticle accumulation in human plaque tissue that was collected from patients who were scheduled for endarterectomy and were pretreated with LN-PLP. The second trial, SILENCE (NCT01601106), was a small phase 2a efficacy trial, for which recently developed imaging protocols served as the primary endpoints. These imaging biomarkers were identical to the ones used to measure efficacy on the atherosclerotic rabbits in the current study. The results of the aforementioned trials are presented in a separate publication in this issue.
In conclusion, we have presented a liposomal formulation of prednisolone phosphate with a good safety profile that exerts a potent anti-inflammatory effect on the vessel wall of atherosclerotic rabbits. The nanotherapy is produced according to GMP standards and can be readily used in human subjects. In addition to its indication for atherosclerosis we also foresee LN-PLP’s use for other inflammatory conditions such as rheumatoid arthritis.
Supplementary Material
Acknowledgments
Author disclosures and funding:
This work was supported by a European Framework Program 7 grant (ESS, GS: FP7-Health 309820: Nano-Athero); the Dutch network for Nanotechnology NanoNext NL, in the subprogram “Drug Delivery”; the National Heart, Lung, and Blood Institute, National Institutes of Health, as a Program of Excellence in Nanotechnology (PEN) Award, Contract #HHSN268201000045C (Z.A.F.), NIH grants R01 HL118440 (W.J.M.M.), R01 HL125703 (W.J.M.M.), R01 EB009638 (Z.A.F.), NWO Vidi 91713324 (W.J.M.M.). Additionally, this work was supported by a grant from Enceladus Pharmaceuticals (Amsterdam, The Netherlands). M.E.L was partially supported by the International Atherosclerosis Society (USA) and by the Foundation “De Drie Lichten” (The Netherlands).
Footnotes
All other authors declare that they have no conflict of interest and no relationships with industry relevant to this study.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tabas I, Glass CK. Anti-inflammatory therapy in chronic disease: challenges and opportunities. Science. 2013;339(6116):166–72. doi: 10.1126/science.1230720. [Internet]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23307734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105(9):1135–1143. doi: 10.1161/hc0902.104353. [Internet]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11877368. [DOI] [PubMed] [Google Scholar]
- 3.Petros RA, DeSimone JM. Strategies in the design of nanoparticles for therapeutic applications. Nat Rev Drug Discov. 2010;9(8):615–627. doi: 10.1038/nrd2591. [Internet]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20616808. [DOI] [PubMed] [Google Scholar]
- 4.Farokhzad OC, Langer R. Impact of nanotechnology on drug delivery. ACS Nano. 2009;3(1):16–20. doi: 10.1021/nn900002m. [Internet]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19206243. [DOI] [PubMed] [Google Scholar]
- 5.Wagner V, Dullaart A, Bock A-K, Zweck A. The emerging nanomedicine landscape. Nat Biotechnol. 2006;24(10):1211–7. doi: 10.1038/nbt1006-1211. [Internet]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17033654. [DOI] [PubMed] [Google Scholar]
- 6.Falk E. Pathogenesis of atherosclerosis. J Am Coll Cardiol. 2006;47(8 Suppl):C7–12. doi: 10.1016/j.jacc.2005.09.068. [Internet]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16631513. [DOI] [PubMed] [Google Scholar]
- 7.Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473(7347):317–325. doi: 10.1038/nature10146. [Internet]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21593864. [DOI] [PubMed] [Google Scholar]
- 8.Mulder WJM, Jaffer FA, Fayad ZA, Nahrendorf M. Imaging and Nanomedicine in Inflammatory Atherosclerosis. Sci Transl Med. 2014;6(239):239sr1. doi: 10.1126/scitranslmed.3005101. [Internet]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24898749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lobatto ME, Fuster V, Fayad ZA, Mulder WJM. Perspectives and opportunities for nanomedicine in the management of atherosclerosis. Nat Rev Drug Discov. 2011;10(11):835–852. doi: 10.1038/nrd3578. [Internet]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22015921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lobatto ME, Fayad ZA, Silvera S, Vucic E, Calcagno C, Mani V, et al. Multimodal clinical imaging to longitudinally assess a nanomedical anti-inflammatory treatment in experimental atherosclerosis. Mol Pharm. 2010;7(6):2020–9. doi: 10.1021/mp100309y. [Internet]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21028895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Derendorf H, Rohdewald P, Hochhaus G, Möllmann H. HPLC determination of glucocorticoid alcohols, their phosphates and hydrocortisone in aqueous solutions and biological fluids. J Pharm Biomed Anal. 1986;4(2):197–206. doi: 10.1016/0731-7085(86)80042-5. [Internet]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16867616. [DOI] [PubMed] [Google Scholar]
- 12.Lobatto ME, Calcagno C, Metselaar JM, Storm G, Stroes ES, Fayad ZA, et al. Imaging the efficacy of anti-inflammatory liposomes in a rabbit model of atherosclerosis by non-invasive imaging. Methods Enzymol. 2012;508:211–228. doi: 10.1016/B978-0-12-391860-4.00011-2. [Internet]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22449928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Banciu M, Schiffelers RM, Metselaar JM, Storm G. Utility of targeted glucocorticoids in cancer therapy. J Liposome Res. 2008;18(1):47–57. doi: 10.1080/08982100801893978. [Internet]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18348071. [DOI] [PubMed] [Google Scholar]
- 14.Van den Hoven JM, Van Tomme SR, Metselaar JM, Nuijen B, Beijnen JH, Storm G. Liposomal drug formulations in the treatment of rheumatoid arthritis. Mol Pharm. 2011;8(4):1002–15. doi: 10.1021/mp2000742. [Internet]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21634436. [DOI] [PubMed] [Google Scholar]
- 15.Kerwin WS, Oikawa M, Yuan C, Jarvik GP, Hatsukami TS. MR imaging of adventitial vasa vasorum in carotid atherosclerosis. Magn Reson Med. 2008;59(3):507–14. doi: 10.1002/mrm.21532. [Internet]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18306402. [DOI] [PubMed] [Google Scholar]
- 16.Calcagno C, Mani V, Ramachandran S, Fayad ZA. Dynamic contrast enhanced (DCE) magnetic resonance imaging (MRI) of atherosclerotic plaque angiogenesis. Angiogenesis. 2010;13(2):87–99. doi: 10.1007/s10456-010-9172-2. [Internet]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20526859. [DOI] [PubMed] [Google Scholar]
- 17.Tawakol A, Migrino RQ, Bashian GG, Bedri S, Vermylen D, Cury RC, et al. In vivo 18F-fluorodeoxyglucose positron emission tomography imaging provides a noninvasive measure of carotid plaque inflammation in patients. J Am Coll Cardiol. 2006;48(9):1818–24. doi: 10.1016/j.jacc.2006.05.076. [Internet]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17084256. [DOI] [PubMed] [Google Scholar]
- 18.Rudd JHF, Myers KS, Bansilal S, Machac J, Pinto CA, Tong C, et al. Atherosclerosis inflammation imaging with 18F-FDG PET: carotid, iliac, and femoral uptake reproducibility, quantification methods, and recommendations. J Nucl Med. 2008;49(6):871–8. doi: 10.2967/jnumed.107.050294. [Internet]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18483100. [DOI] [PubMed] [Google Scholar]
- 19.Chang Y, Lee GH, Kim T-J, Chae K-S. Toxicity of magnetic resonance imaging agents: small molecule and nanoparticle. Curr Top Med Chem. 2013;13(4):434–45. doi: 10.2174/1568026611313040004. [Internet]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23432006. [DOI] [PubMed] [Google Scholar]
- 20.Metselaar JM, Wauben MHM, Wagenaar-Hilbers JPA, Boerman OC, Storm G. Complete remission of experimental arthritis by joint targeting of glucocorticoids with long-circulating liposomes. Arthritis Rheum. 2003;48(7):2059–66. doi: 10.1002/art.11140. [Internet]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12847701. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.