

SUMMRAY
Telomerase is required for long-term telomere maintenance and protection. Using single budding yeast mother cell analyses we found that, even Early after Telomerase Inactivation (ETI), yeast mother cells show transient DNA Damage Response (DDR) episodes, stochastically altered cell cycle dynamics, and accelerated mother cell aging. The acceleration of ETI mother cell aging was not explained by increased reactive oxygen species (ROS), Sir protein perturbation, or deprotected telomeres. ETI occurred well before the population senescence and caused late after telomerase inactivation (LTI). ETI phenotypes were morphologically distinct from LTI senescence, genetically uncoupled from telomere length, and were rescued by elevating dNTP pools. Our combined genetic and single-cell analyses show that well before critical telomere shortening, telomerase is continuously required to respond to transient DNA replication stress in mother cells, and that a lack of telomerase accelerates otherwise normal aging.
INTRODUCTION
Telomeres, protective DNA-protein complexes at the ends of eukaryotic chromosomes, buffer against the loss of sequence during DNA replication and distinguish normal chromosome ends from potentially dangerous double-strand breaks. Telomeres are composed of sequence-specific DNA binding proteins bound to highly repetitive DNA sequences and are increasingly recognized as genomic regions prone to replication stress (Miller et al., 2006; Sfeir et al., 2009; Drosopoulos et al., 2012). Without the telomeric DNA-elongating enzyme telomerase, progressive telomere shortening eventually causes the collapse of the protective DNA-protein complex (deprotection), but this occurs only after many cell divisions, Late after Telomerase Inactivation (LTI). In LTI cells, telomere deprotection shares many properties with classic DNA damage (Nautiyal et al., 2002; Fagagna et al., 2003) and induces a DNA Damage Response (DDR) and a permanent G2/M cell cycle arrest (senescence).
Previously, responses to telomerase deletion have generally been reported only after a significant delay (in S. cerevisiae, after ~50–80 divisions). Thus, it was thought that cells sense altered telomere properties that signal senescence only when telomeres become critically short and deprotected. Hence, responses and phenotypes of cells Early after Telomerase Inactivation (ETI) have not been extensively investigated. However, it was previously shown that, in ETI cells, very short telomeres appear at low frequencies that fuse to an induced double-strand break (DSB) (~10−4 to 10−3). These rare fusions became molecularly detectable when telomerase was inactivated by either deletion of the telomerase RNA template TLC1 (tlc1Δ) or by replacing the reverse transcriptase subunit, EST2, with the mutant est2-D530A, which assembles a telomerase ribonucleoprotein enzyme complex lacking telomeric DNA polymerization activity (Chan and Blackburn, 2003; Lingner et al., 1997). These fusogenic telomeres arose in ETI cells well before any signs of bulk population senescence and even if the telomeres had been pre-lengthened. Therefore, even the short-term absence of telomerase activity causes cells to experience a low but detectable genomic instability.
In a process distinct from the permanent bulk population cell-cycle arrest resulting from critically short telomeres in senescent LTI cells, an individual wild type (WT) yeast mother cell will cease divisions after it has produced approximately 25 daughter cells. As of yet, there has been very little evidence suggesting interaction between the pathways that regulate these two kinds of aging, hereafter referred to as “LTI senescence” and “mother cell aging/lifespan”, respectively. Despite the identification of multiple genes that regulate mother cells lifespans (Bishop and Guarente, 2007; Johnson et al., 1999; Kaeberlein, 2010), the mechanisms causing mother cell aging of even WT yeast remain poorly understood.
Here we report experiments employing single cell methodologies supporting a model in which budding yeast mother cells lacking telomerase activity are less able to resolve replication stress inherent to telomeres. These cells show induction of a signaling pathway indicative of transient DNA replication stress, altered cell cycle dynamics even in young mother cells, and accelerated aging (reduced lifespan), independently of telomere length. Our results demonstrate that this occurs well before the onset of LTI senescence and that the accelerated aging of ETI mother cells resembles the normal mother cell aging process.
RESULTS
Mother Cells Lacking Active Telomerase Show Increased Heterogeneity of Cell Cycle Durations and Reduced Lifespans
We analyzed the properties of individual haploid ETI mother cells, well before any signs of cellular LTI senescence, freshly isolated from sporulation of heterozygous telomerase-competent diploids. Following genotyping, cells were taken from logarithmically growing cell cultures (~ 25–30 generations after telomerase loss), in which the overwhelming majority of cells were robustly-growing newborn or very young mother cells. These cells were placed in a microfluidic device, and the budding cycles and lifespans of individual mother cells were continuously monitored for two days by repeated microscopic imaging (Xie et al., 2012; Zhang et al., 2012).
First, even the youngest ETI mother cells (tlc1Δ or est2-D530A) immediately showed higher frequencies of stochastically longer, and more heterogeneous cell cycle durations than WT (Figure 1A, 1B, 1C; note especially between times 0 to 5 hours as marked on Y axes). As the durations of the last two budding cycles were highly heterogeneous in both WT and ETI mothers, they were discarded from all cell cycle duration analyses discussed here. This cell cycle heterogeneity was consistent with observations of bulk ETI population budding kinetics, as manifested by cells lingering in the large-budded state (G2/M), enriched for cells with short spindles and unsegregated chromosomes (Figure S1A, S1B, S1C). Secondly, we analyzed the mother cell aging of individual ETI cells and found that the lack of telomerase activity reduced ETI mother cell lifespan. Mean budding lifespan for tlc1Δ was 12.6 (7 replicates), and 7.6 generations for est2-D530A (3 replicates), compared to 22.1 for WT mother cells (Figure 1A, 1B, 1C, 1E). Furthermore, the catalytically-inactive telomerase est2-D530A point mutant showed even longer cell cycle durations than tlc1Δ ETI mother cells, and the lifespan reduction was even more severe. Hence, lack of telomerase enzymatic activity, rather than the absence of an assembled telomerase ribonucleoprotein complex, causes increased cell cycle heterogeneity and faster mother cell aging.
Figure 1. ETI Mother Cells Show a Non-progressive Cell Cycle Length Phenotype and Reduced Lifespan that is Rescued by SML1 Deletion.

Mother cell budding profiles for (A) WT, (B) tlc1Δ, (C) est2-D530A, (D) tlc1Δsml1Δ, showing cell cycle durations and heterogeneity (see exponential color scale, cell cycles with durations 1.4 hours or less were colored in purple). The x-axis displays individual mother cells shown as vertical bars with budding events indicated as horizontal white divisions. Mean lifespan for each genotype is presented in the upper left corner of the plot. (E) ETI mother cells showed reduced replicative lifespans compared to WT cells (number of cells (n): tlc1Δ, 354; est2-D530A, 117; WT, 234. p-value for difference between tlc1Δ and WT < 1e-37). (F) Deletion of Sml1 restores lifespan of ETI mother cells to WT levels (n: tlc1Δsml1Δ, 77; sml1Δ, 39). (G) The heterogeneity of cell cycle lengths in ETI cells did not progressively worsen relative to WT as mother cells aged. Fold increase in cell cycle variability from 1st and 2nd to 3rd and 4th last cell cycles compared for each genotype (shown below each set). The variance of 1st and 2nd cell cycles of tlc1Δ and est2530A is significantly greater than that of WT (F-test p-value < 1e-16 and 1e-13 respectively).
Heterogeneous Cell Cycles do not Progressively Worsen With Shortening Telomeres
If the extended, heterogeneous cell cycle lengths of ETI mother cells were due solely to telomere shortening, we would have expected the phenotype to worsen progressively with each successive cell division. However, this was not the case. First, as individual mother cells progressed from being very young to old, ETI mother cells did not show any significant progressive increase in mean duration or heterogeneity of mother cell cycle lengths relative to WT (Figure 1G). Second, during the individual ETI mother cell lineages, a young mother cell whose initial cell cycle was long had no greater probability of having subsequent longer cell cycles or a shorter lifespan than one with an initial short cell cycle (Figure S2), supporting a stochastic and episodic, rather than progressive, nature of the occurrence of longer cell cycles. These highly stochastic episodes of cell cycle heterogeneity and lack of any progressive worsening of this phenotype as ETI mother cells aged, are not the predicted result of progressive telomere shortening.
SML1 Deletion Rescues Mother Cell Lifespan of ETI Cells Independently of Telomere Length
Because we observed an extended G2/M phase in bulk population analyses (Figure S1), which is often the result of DDR activation, we determined if mutations affecting the DDR affected the above ETI phenotypes. Responses to various forms of DNA damage, including that sensed at critically short telomeres in LTI senescence, involve a cascade of phosphorylation events, with early upstream steps occurring at the source of DNA damage through PIKK family member kinases Mec1 (ATR) and/or Tel1 (ATM). Strains lacking only Mec1 are inviable, but this mec1Δ lethality can be rescued by deletion of SML1 (Zhao et al., 1998). Sml1 inhibits ribonucleotide reductase (RNR), which catalyzes the rate limiting step in dNTP production (Reichard, 1988). Deletion of Sml1 increases RNR activity and elevates dNTP pools, obviating the need for certain DDR components under healthy growing conditions, and can be protective against some forms of DNA damage (Andreson et al., 2010; Jossen and Bermejo, 2013). Strikingly, deletion of SML1 in ETI tlc1Δ strains efficiently rescued the ETI-induced heterogeneity of budding cycle durations (Figure 1D) as well as the shortening of mother cell lifespan (Figure 1F). However, SML1 deletion alone produced no change in the rates of bulk telomere shortening in ETI cells, nor in the subsequent onset of LTI senescence (Figure 2, S3). We also confirmed that the deletion of SML1 alone caused no significant effect on mother cell lifespans and telomere length compared to WT (Figure 1F, S4B). Hence, the dramatic rescue of ETI cell cycle heterogeneity and accelerated mother cell aging by SML1 deletion cannot be explained by increased telomere length or by slower rates of telomere shortening.
Figure 2. SML1 Deletion Rescues Mother Cell Lifespan of ETI Cells Independently of Telomere Length.
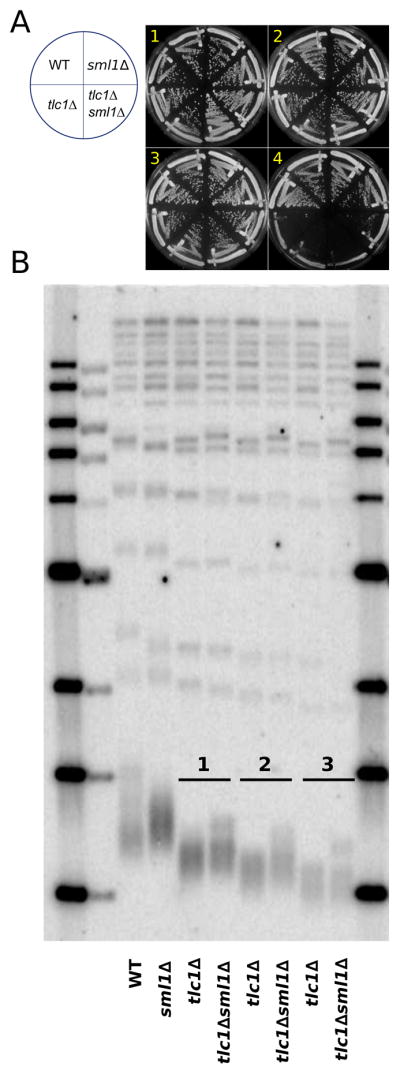
(A) SML1 deletion had no significant effect on the rate of bulk population senescence in ETI cells passaged on solid media to induce LTI-senescence. (B) Southern blot analysis of telomeric DNA restriction fragment lengths of cells taken from serial streaks shown in (A), using TG(1-3) repeat telomeric probe. ETI (tlc1Δ) and ETI sml1Δ(tlc1Δsml1Δ) displayed similar rates of telomere shortening and the lower end of the telomere length distributions were similar.
ETI Mother Cells Age with Terminal Cellular and Mitochondrial Morphologies Distinct from LTI Senescence but Similar to those of Normal Mother Cell Aging
We tested further whether budding cessation due to mother cell aging in ETI or WT cells was distinguishable from the G2/M arrest caused by LTI senescence by examining cell and mitochondrial morphology at the end of the lifespans (terminal morphology). Typical WT mother cell aging produces terminal cells that are mostly small-budded with minimal or no mitochondrial fluorescence signal from a mitochondrially localized GFP (mtGFP) (Figure 3Ai) and a smaller population of elongated cells with brighter mitochondrial fluorescence (Figure 3Aii). In contrast, cells terminally arrested due to LTI senescence accumulate with a swollen, large-budded (“dumbbell”) morphology and with mitochondrial fluorescence that gradually forms very bright dots (Figure 3Aiii) (Nautiyal et al., 2002). We created and analyzed two populations of tlc1Δ sml1Δ cells. The first population was isolated as soon as possible after genotyping (ETI) and was enriched for mother cells that would reach their aging limit prior to LTI senescence. The second population was passaged for approximately 10 additional generations prior to microfluidics analysis, thus enriching for cells that would undergo LTI senescence (critically short telomeres) before the mother cells reached their aging limit (Figure 3B). Terminally aged ETI tlc1Δ and ETI tlc1Δ sml1Δ mother cells accumulated mostly in two dominant terminal morphologies, which resembled the two dominant WT terminal morphology phenotypes (Xie et al., 2012), and only very rarely in the dumbbell morphology (Figure 3A, 3C). In contrast, in terminal LTI tlc1Δ sml1Δ mother cells, terminal dumbbell morphologies became the major type observed, indicating that a large proportion of the population had entered LTI senescence (Figure 3Aiii, 3C). These results support terminal cellular and mitochondrial morphology as an accurate distinction between LTI senescence and normal mother cell aging, and provide further evidence that ETI mother cells cease divisions as a result of mother cell aging rather than LTI senescence.
Figure 3. ETI Mother Cells Age with Terminal Cellular and Mitochondrial Morphologies Distinct from LTI Senescence but Similar to those of Normal Mother Cell Aging.

(A) Three possible terminal death morphologies were observed in WT, ETI, and/or LTI (see text for definition) mother cells: small budded (type i), elongated (type ii), and a G2/M large-budded (“dumbbell” shape) (type iii). Mitochondrial volume was measured using mitochondrially localized GFP (mtGFP). (B) ETI and LTI populations of tlc1Δsml1Δ cells were prepared to distinguish cell death from normal mother cell aging and LTI senescence. tlc1Δsml1Δ LTI strains senesced and showed reduced lifespan as expected. (C) ETI cells terminally arrest in a manner similar to WT mother cells and distinct from LTI senescence. Most of the cells in ETI tlc1Δ and ETI tlc1Δ sml1Δ show type i or type ii death morphologies (>95%), similar to terminal WT mother cells. In LTI cells, a major fraction (~70%) displayed type iii morphology (p-value < 1e-4 compared to ETI tlc1Δ sml1Δ by Fisher’s exact test) indicative of senescence induced by critically short telomeres.
Mutation of Specific DDR Components Exacerbates ETI Cell Cycle and Lifespan Phenotypes
We investigated other proteins previously implicated in yeast telomere maintenance and in the DDR for effects on mother cell aging. Maintenance of yeast telomeres at normal length requires DDR kinases Mec1 and Tel1 (Sabourin and Zakian, 2008; Takata et al., 2004) and the replication stress-specific DDR adaptor protein Mrc1 (Grandin et al., 2005). First, we found that cell cycle heterogeneity and mother cell lifespan were similar in WT, tel1Δ, and mec1Δsml1Δ strains (Figure 1A, 4A, 4C, S4C, S5A). Because tel1Δ in haploid cells reduces telomerase action on telomeres, telomeres decline to a short length that is then stably maintained (Greenwell et al., 1995; Lustig and Petes, 1986). The tel1Δ cells used here were isolated immediately after sporulation of heterozygous parent diploids and analyzed when telomeres were still shortening from near-WT lengths. Therefore, having telomeres that are shortening but eventually stably maintained is not alone sufficient to alter cell cycle duration and lifespan.
Figure 4. TEL1 Deletion Exacerbates ETI Cell Cycle and Lifespan Phenotypes but not Senescence or Telomere Shortening Rates.

Mother cell budding profiles for tel1Δ (A) and tlc1Δ tel1Δ (B). (C) tel1Δ (n=38) strain lifespan does not differ from WT. (D) tlc1Δ tel1Δ (n=83) mutation worsens the lifespan reduction caused by ETI mutations in mother cells (p-value < 1e-4, compared with tlc1Δ alone). (E) ETI and ETI tel1Δ mutants displayed similar rates of senescence when passaged on solid media. (F) Southern blot analysis of telomeric DNA restriction fragment lengths of cells taken from plates after serial streaks shown in (E).
Next, we examined how mutations of Mec1 and Tel1 affect the ETI phenotypes. Because sml1Δ, as shown above, efficiently rescues the accelerated aging of ETI mother cells, it is difficult to determine if Mec1 has a role in this process, due to the necessity of deleting SML1 for viability in mec1Δ strains. However, ETI tlc1Δ tel1Δ double mutant mother cells had even greater cell cycle heterogeneity and shorter budding lifespan (mean 9.8 generations, 2 replicates) than control ETI tlc1Δ single mutant mother cells (Figure 1B, 4B, 4D). As shown previously, freshly isolated ETI haploid cells that are also mutated for Tel1 or Mec1 (tlc1Δmec1Δsml1Δ or tlc1Δtel1Δ) have a rate of initial telomere-shortening and progression to LTI population senescence similar to tlc1Δ single mutants (Chan and Blackburn, 2003) (Figure 4E, 4F). Hence, the exacerbation of the ETI cell cycle heterogeneity and lifespan reduction phenotypes caused by lack of Tel1 is not explained by faster telomere shortening or accelerated population senescence.
Because sml1Δ rescues the cell cycle and lifespan phenotypes of tlc1Δ mother cells, and is known to facilitate DNA replication by increasing nucleotide levels (Chabes et al., 2003), we suspected that ETI cells may be more vulnerable to telomeric DNA replication stress. Therefore, we mutated the DDR adaptor protein Mrc1, which is required specifically for the DNA replication stress checkpoint (Alcasabas et al., 2001; Osborn and Elledge, 2003) and has a minor role in telomere length maintenance (Tsolou and Lydall, 2007). Mutation of 17 potential PIKK family kinase consensus phosphorylation sites on Mrc1 (mrc1AQ) allows full cell viability but disables the DNA replication stress response (Osborn and Elledge, 2003). Despite the lack of any mother cell lifespan or cell cycle effect of mrc1AQ alone (Figure 5A, 5C), tlc1Δmrc1AQ double mutant ETI mother cells showed even greater cell cycle length heterogeneity than the tlc1Δ single mutant ETI cells (Figure 5B). Consistent results were also seen in the G2/M durations in bulk populations (Figure S1D) and mean lifespan was markedly reduced to 8.8 generations (2 replicates), compared with 12.6 generations for the control tlc1 Δ ETI strains (Figure 1B, 5D). These effects were not explainable by reduced telomere length or accelerated senescence, as the mrc1AQ mutant allele produced stable telomeres only slightly shorter than WT and had no effect on the kinetics of telomere shortening or bulk population senescence (Figure 5E, 5F). We also tested the epistasis relationship of tel1Δ and mrc1AQ in the ETI context. ETI triple mutant tlc1Δ tel1Δ mrc1AQ cells showed the same lifespan shortening as the double ETI mutants (Figure S5B). We conclude that Tel1 and Mrc1 checkpoint functions act in the same pathway and that lack of either one acts synthetically with the ETI mother cell phenotypes.
Figure 5. MRC1 Mutation Exacerbates ETI Cell Cycle and Lifespan Phenotypes but not Senescence or Telomere Shortening Rates.

Mother cell budding profiles for mrc1AQ(A) and tlc1Δ mrc1AQ (B). (C) mrc1AQ (n=40) strain lifespan does not differ from WT. (D) tlc1Δmrc1AQ (n=90) mutation worsens the lifespan reduction caused by ETI mutations in mother cells (p-value < 2e-7, compared with tlc1Δ). (E) ETI and ETI mrc1AQ mutants displayed similar rates of senescence when passaged on solid media. (F) Southern blot analysis of telomeric DNA restriction fragment lengths of cells taken from plates after serial streaks shown in (E).
In the DDR cascade, downstream of Tel1 or Mec1, the DDR adaptor protein Rad9 can act semi-redundantly with the adaptor protein Mrc1. Mrc1 is specifically involved in the replication stress response while Rad9 is mostly important for responding to DNA breaks and other DNA damage. In contrast to tlc1 Δ mrc1AQ ETI cells, tlc1 Δ rad9 Δ ETI mother cell cycle durations and lifespans were not significantly different from tlc1Δ ETI cells, consistent with bulk population analyses (Figure S1, and data not shown). The ETI tlc1Δ rad9Δ mother cells had a mean lifespan of 16.5 generations (2 replicates), while the control tlc1Δ strain had a mean lifespan of 13.7 generations (Figure S5C). Thus, rad9Δ did not significantly affect the accelerated aging phenotypes of ETI mother cells. These results confirmed the specificity of the Mrc1 checkpoint function in the ETI mother cell phenotypes and indicate the involvement of a DNA replication stress response, rather than a response to other forms of DNA damage, which requires Rad9. In summary, disrupting the DDR via tel1Δ or mrc1AQ mutations, but not by mec1Δ sml1Δ or rad9Δ, strongly exacerbated the cell cycle abnormalities and acceleration of mother cell aging in ETI cells, independently of telomere length and without accelerating LTI senescence.
ETI Mother Cell Phenotypes are Not Caused by Deprotected Telomeres
Previous results showed that short, fusogenic telomeres occur spontaneously at very low frequencies in ETI cells (Chan and Blackburn, 2003). These fusogenic telomeres derive from rare individual deprotected telomeres and can be detected by PCR assays upon their fusion to an induced DNA double-stranded break. We tested whether the amount of such fusogenic telomeres correlated with the severity of our ETI mother cell phenotypes using the same system (Chan and Blackburn, 2003) for semi-quantitative PCR analyses. In agreement with the published work, we found that single mutant ETI (tlc1Δ) and tel1Δ strains each showed detectable but low amounts of fusions resulting from a deprotected telomere fusing to an induced DSB and that tlc1Δ tel1Δ strains showed a synergistic increase (Figure 6A, 6B). However, in tlc1Δmrc1AQ ETI cells (Figure 6A), the mrc1AQ mutation produced no further significant increase over a tlc1Δ single mutant. Furthermore, sml1Δ did not reduce (and possibly increased) the number of fusogenic telomeres detected (Figure 6A, 6B). This complete non-concordance in these various ETI genotypes with the phenotypes we have observed here in ETI mother cells argues strongly against deprotected telomeres as a cause for the exacerbated cell cycle heterogeneity and accelerated mother cell aging.
Figure 6. Genotype Dependence of Telomere Fusions and Transient DNA Damage Response Episodes in Mother Cells.

(A) Semi-quantitative PCR of DNA species resulting from the fusion of a deprotected telomere with an induced double-strand break in genetic backgrounds containing ETI and mrc1AQ mutant combinations. (B) Same as in (A), but with genetic backgrounds containing ETI and tel1Δ mutations. (C) Two representative profiles of RNR3-GFP peaks occurring in still-dividing individual mother cells. Cell divisions (green diamonds) and RNR3-GFP reporter levels (blue circles) were plotted throughout an individual mother cell’s lifespan. Spline fitting is shown as red lines. (D) Frequencies of RNR3-GFP induction peaks in still-dividing cells such as those shown in (C). p-values < 0.01 (*) and p-values < 0.001 (**) by Fisher’s exact test are indicated. (E) Two representative mother cell profiles shown, as in (C), with cells displaying terminal RNR3-GFP induction peaks. (F) Frequencies of RNR3-GFP induction peaks in terminal mother cells, such as those shown in (E).
Further evidence that ETI phenotypes are not caused by deprotected telomeres, which induce a robust DDR (Nautiyal et al., 2002; Fagagna et al., 2003), came from comparing the genetic dependencies of ETI cell phenotypes versus DNA damage sensitivity. As previously reported, mec1Δ sml1Δ and rad9Δ mutations made cells highly sensitive to treatment with various classic DNA damaging agents (HU, UV, phleomycin or MMS) (Figure S4D). This is in dramatic contrast to the experiments described above, in which mec1Δ sml1Δ and rad9Δ did not exacerbate the ETI phenotypes. Hence, the genotype dependencies of ETI mother cell phenotypes are quite distinct from the dependencies of responses to classic DNA damaging agents.
Altered Recombination Levels are Not Responsible for ETI Mother Cell Phenotypes
Recombination is another process that has been implicated in maintaining yeast telomeres and occurs when telomeres lose protection, such as in LTI cells (McEachern and Blackburn, 1996; Basenko et al., 2011). Following the onset of LTI senescence, Rad52-dependent recombination at telomeres allows a small fraction (~10−4) of senescing LTI yeast cells to survive and continue dividing (Lundblad and Blackburn, 1993). Also, DNA replication stress can be relieved by mechanisms involving recombination. We therefore asked if recombination plays any role in the ETI accelerated mother cell cycle kinetics and aging response. Deletion of RAD52 alone causes no changes in telomere length maintenance and telomeres in tlc1Δrad52Δ strains shorten no faster than with tlc1Δ alone (Lundblad and Blackburn, 1993). However, rad52Δ alone caused increased mother cell cycle duration heterogeneity (data not shown) and an acceleration of mother cell budding aging (Park et al., 1999). Notably, these rad52Δ phenotypes were not substantially rescued by SML1 deletion (mean lifespan, rad52Δ: 9.4, n=130 versus rad52Δsml1Δ: 13.2, n=70) (Figure S6A and data not shown). Furthermore, the mean lifespan of ETI tlc1Δrad52Δsml1Δ mother cells was even lower than rad52Δsml1Δ: 8.2 versus 13.2 (Figure S6A). Hence, lack of Rad52 function appears to act additively to the effect of TLC1 deletion. This epistasis relationship indicates that absence of telomerase activity and of Rad52 each causes acceleration of mother cell aging, but by two distinct mechanisms.
ETI Phenotypes are Not Caused By Relocalization of Sir Proteins
Another pathway previously implicated in yeast mother cell aging involves changes in Sir protein concentration and localization. For example, Sir2 overexpression has been shown to increase mother cell lifespan (Kaeberlein et al., 1999). However, several lines of evidence argue that Sir2 sequestration in ETI cells does not explain their accelerated aging. First, all our ETI strains mated normally, implying that the mating type loci were still silenced and arguing against a large relocalization of Sir proteins. Second, localized puncta of Sir3-GFP, indicative of telomere-bound Sir complex proteins (Martin et al., 1999) were not significantly different between ETI and WT mother cells (Figure S6B). Third, although a single induced unrepairable DNA break has been reported to cause Rad9-dependent delocalization of Sir2 from telomeres (Martin et al., 1999; Mills et al., 1999), as described above, rad9Δ neither exacerbated nor significantly rescued the accelerated aging in ETI mother cells. Together, these findings indicate that altered sequestration of the Sir complex is not the mechanism causing the accelerated aging of ETI cells.
Lifespan Reduction of ETI Mother Cells is Not Caused by Increased Reactive Oxygen Species (ROS)
We re-examined the previously described transcriptional profile datasets (Nautiyal et al., 2002; Table S2, passage 1) of ETI tlc1Δ cells. In an unbiased approach, we compared the large available number of yeast gene expression profiles, measured under different environmental conditions and genetic backgrounds (Edgar et al., 2002), to that of ETI cells (Tables S3, S4, S5). The top hit (Pearson correlation = 0.495, p-value < 1e-254) was treatment with diamide, a thiol-oxidizing agent that causes oxidative stress. Because intracellular ROS have long been theorized to play a role in aging and ROS levels can be elevated as a result of DNA damage (Rowe et al., 2008; Salmon et al., 2004), we tested if oxidative stress caused the ETI mother cell phenotypes.
We assessed oxidative stress in ETI cells by quantifying ROS levels in our strains. If the accelerated aging of ETI cells is caused by higher intracellular ROS levels, it would be predicted that ETI tlc1Δ mother cells would have higher ROS than WT and that ETI tlc1Δ sml1Δ cells would have lower ROS levels than ETI single tlc1Δ mutants. However, ETI cells did not have significantly higher levels of ROS than WT (Figure S6C). Furthermore, sml1Δ and tlc1Δ sml1Δ strains showed even higher levels of ROS, the opposite effect from that predicted if ETI causes faster aging of cells via higher intracellular ROS level. We also tested the effects of anti-oxidants by treatment with N-Acetyl-L-Cysteine (NAC). However, NAC equally and only modestly lengthened mother cell lifespans of both ETI and WT mother cells (Figure S6D). Hence, we conclude that even though the transcriptional profile changes in ETI cells include features of an oxidative stress response, increased ROS levels and oxidative stress are not a primary cause of accelerated mother cell aging elicited by ETI.
ETI Cells Show Transient RNR3 Upregulation during Mother Cell Divisions
Given the connections between DNA damage and cell cycle regulation, we turned to a more detailed analysis to establish if DDR signaling was induced in ETI mother cells. Notably, phosphorylation of DDR components such as Rad53 and Mrc1 is only detectable in LTI cells and not in ETI cells (Grandin et al., 2005) (Figure S4A). Therefore, we employed a more sensitive single cell monitoring method to detect evidence for DDR signaling. We examined DDR activation during mother cell aging using a GFP-tagged allele of RNR3, a gene that is strongly induced as a downstream component of the DDR. We monitored GFP intensity during mother cell lifespan assays and quantified RNR3-GFP peaks (at least 1.3 fold above background) using strains containing RNR3-GFP and all relevant combinations of tlc1Δ, sml1Δ, mrc1AQ, and tel1Δ mutations. Peaks were classified as occurring before the last two cell divisions or during/after the last two divisions, referred to here as “still-dividing” and “terminal” peaks respectively (Figure 6C, 6E). Terminal peaks were scored as peaks per mother cell, and still-dividing peaks were scored as peaks per cell cycle as mother cells underwent a different average number of cycles depending on genotype (Figure 6D, 6F). In WT mother cells (2 replicates), a transient RNR3-GFP peak appeared at a low frequency during the cell cycles of still-dividing mother cells (0.0062, 95% confidence limits 0.0031 – 0.0111; n=11 events in 1,766 cell cycles) and terminal peaks occurred in 25.0% of mother cells (24/96 cells, 95% confidence limits 0.1736 – 0.3456). In contrast, in tlc1Δ ETI mother cell lineages (3 replicates), RNR3-GFP peaks occurred at significantly greater frequency in still-dividing mother cells (0.0177, 95% confidence limits 0.0123 – 0.0252; n=30 events out of 1,696 cell cycles, p-value < 0.0025 compared with WT), and in 27.7% of terminal mother cells (44/159 cells, 95% confidence limits 0.213 – 0.351). Hence, in still-dividing mother cells, ETI elicits an increased number of transient episodes of DDR signaling.
Notably, relative to tlc1Δ single mutants, RNR3-GFP peaks in tlc1Δ sml1Δ ETI mother cells (3 replicates) were significantly diminished in frequency in the still-dividing mother cells (0.0104, 95% confidence limits 0.0071 – 0.0152; n=27 events in 2,587 cell cycles, p-value < 0.05 compared with tlc1Δ), and terminal peaks occurred in only 13.1% of mother cells (22/168 cells, 95% confidence limits 0.087 – 0.191, p-value < 0.01 compared with tlc1Δ). This result is explainable as sml1Δ raises nucleotide pools, making replication fork stalling less likely to occur (Andreson et al., 2010; Jossen and Bermejo, 2013), hence reducing the possibility of eliciting a DNA replication stress response.
ETI tlc1Δ tel1Δ mother cells (2 replicates) showed fewer RNR3 peaks than WT (or tlc1Δ ETI) in still-dividing cells (0.006, 95% confidence limits 0.0034 – 0.0124; n=10 events in 1,505 cell cycles, p-value < 0.006 compared with tlc1Δ) and peaks occurred in 12.0% of terminal mother cells (15/125 cells, 95% confidence limits 0.0730 – 0.1897, p-value < 0.009 compared with tlc1Δ). This finding indicates that abrogating Tel1 greatly exacerbated the ETI mother cell aging phenotypes (Figure 4B, 4D), while reducing RNR3 induction events. We propose that the optimal response to the replication stress in tlc1Δ ETI cells requires Tel1 checkpoint function to activate DDR signaling, monitored here as downstream RNR3 induction.
Interestingly, the tlc1Δmrc1AQ ETI mother cells showed significantly more RNR3 peaks than WT cells in both still-dividing (0.0193, 95% confidence limits 0.0127 – 0.0289; n=23 events in 1,192 cell cycles) and terminal mothers (32.4 %, 35/108 cells, 95% confidence limits 0.243 – 0.417), and also trends to more RNR3 peaks than tlc1Δ (3 replicates). Because the mrc1AQ mutation exacerbates the ETI cell cycle duration and lifespan reduction phenotypes, this finding indicates that telomeric replication stress requires Mrc1 checkpoint function in order to elicit an appropriate response in the absence of telomerase, but not for induction of the RNR3 reporter.
As the Rad9 adaptor protein is semi-redundant with Mrc1 in the DDR cascade, we investigated whether Rad9 is required for induction of RNR3 in the absence of Mrc1 checkpoint function. However, when combined with the tlc1Δ mutation, the mrc1AQ rad9Δ double mutation induced rapid lethality of the bulk ETI cell population, precluding mother cell analyses. Remarkably, this loss of viability in tlc1Δmrc1AQrad9Δ cells was also completely rescued by sml1Δ (data not shown). We conclude that if ETI mother cells lack either Te11 or Mrc1 checkpoint function, the response to, or repair of, telomeric DNA replication stress-induced damage is compromised.
The Degree of Heterogeneity of Cell Cycle Durations and Mitochondrial Changes in Young ETI Mother Cells each Quantitatively Predict Lifespan
Strikingly, for each mother cell genotype described above, the frequency and degree of lengthened cell cycles in young mother cells predicted the degree of reduction in mean lifespan (Figure S7A, S7B). Furthermore, for both WT and ETI mother cells, the extent of the mitochondrial fluorescence quantified at a given early time point in the budding lineage (4 hours) predicted the lifespan of that particular mother cell (Figure S7C, S7D). The finding that these relationships held across multiple genotypes suggests that responses that occur in even the youngest mother cells are caused by the same problem that eventually regulates the lifespan of the cell.
DISCUSSION
Here we have shown that lack of active telomerase affects yeast mother cells much earlier than expected, well before any effect on cells that can be attributed to critical telomere shortness. Notably, early telomerase inactivation in yeast mother cells caused increased heterogeneity of the cell cycle and accelerated aging. These phenotypes were rescued by increasing nucleotide pool levels and were sensitive to inactivation of specific DDR components. By several criteria, the ETI mother cell aging phenotype is consistent with an acceleration of normal mother cell aging processes and not senescence caused by loss of telomere protective function. These criteria included terminal cell and mitochondrial morphologies characteristic of aging WT mother cells and distinct from those in senescent cells.
Previously, it was thought that telomeres had to become critically short in order to elicit a cellular DDR. In contrast, our results suggest that independent of critical telomere shortness, ETI cells initiate signaling that accelerates an otherwise normal mother cell aging pathway. In the ETI setting, across multiple genotypes, the premature onset of mother cell aging is anticipated by the frequency and severity of stochastically slower cell cycling events occurring in even young mother cells. In addition, we have shown that it is a lack of telomerase activity, rather than the lack of an assembled telomerase complex, that is the proximate cause of the response. Therefore, the action of telomerase on telomeres appears to be the most likely molecular property whose alteration causes these effects in ETI cells.
Our combined findings provide evidence for the model shown in Figure 7. In this model, during mother cell divisions in ETI cells, lack of telomerase activity may eliminate a potential bypass mechanism for replication stress in the telomere. This causes transient episodes of a much milder DDR than the robust and sustained DDR elicited by critically short telomeres (Nautiyal et al., 2002; Fagagna et al., 2003). We propose that deletion of Sml1, via its known phenotype of increasing dNTP levels, alleviates this replication stress at an upstream level, preventing DNA damage signaling. This explains why Sml1 deletion suppressed both the transient DDR signaling and the accelerated aging in ETI mother cells. We propose that such replication stress arises often in telomeres and is sensed by Tel1, which is required to cause the observed transient rises in RNR3 levels in ETI cells. This response promotes either repair or tolerance of the replication stress that allows ETI cells to bypass it and enter the next cell cycle. In ETI cells lacking the checkpoint functions of Tel1 or Mrc1 (tlc1Δtel1Δ or tlc1Δ mrc1AQ), cells cannot activate the appropriate response to this replication stress, thus exacerbating the ETI phenotypes. Without Mrc1 checkpoint function, compensation by the semi-redundant adaptor Rad9 occurs to some degree, but the telomeric replication stress is not fully resolved and further damage can ensue. However, in the absence of telomerase, even a fully functional DDR is not sufficient to fully alleviate telomeric replication stress and prevent accelerated mother cell aging.
Figure 7. Proposed Model.

Top left panel: Proposed signaling interactions that regulate aging in response to telomeric DNA stress. Deleterious effects shown in red. Top right panel: In ETI cells lacking Sml1 (tlc1Δsml1Δ), telomerase cannot alleviate replication stress. However, due to elevated dNTP levels, replication stress is prevented and aging is not accelerated. Eliminated or reduced signaling is shown in grey. Bottom left panel: In ETI cells lacking Tel1 (tlc1Δtel1Δ), as well as lacking telomerase rescue, the DDR response is unavailable to alleviate replication stress, as indicated by the elimination of RNR3 signaling, and ETI-accelerated aging is exacerbated. Bottom right panel: in ETI cells lacking Mrc1 function (tlc1Δmrc1AQ), telomerase rescue is unavailable and the DDR response to telomeric replication stress is partially hindered. Rad9 is able to partially compensate and induce RNR3 induction, but other downstream DDR targets cannot be induced, thus exacerbating the ETI-accelerated aging.
We determined that neither ROS, recombination, deprotected (fusogenic) telomeres, redistribution of SIR protein complexes, nor a DDR similar to that in response to classic DNA damaging agents, can account for the accelerated mother cell aging of ETI cells. Deletion of SML1 rescued the cell cycle, lifespan, and DDR (RNR3) induction phenotypes in dividing ETI mother cells and is known to suppress replication fork stalling (Andreson et al., 2010; Jossen and Bermejo, 2013). The majority of mutant phenotypes known to be suppressed by sml1Δ are related to DNA replication, including replication fork progression. This suggests that suppression by sml1Δ is very specific, and occurs through elevated nucleotide pools, via the release of inhibition of the RNR complex. Furthermore, the transcriptional profile of ETI tlc1Δ cells indicates that they upregulate RNR2, 3, and 4 gene expression (Nautiyal et al., 2002) (Table S2). Taken together with previous findings, our results suggest that the higher nucleotide pools in sml1Δ cells prevent telomeric replication stress from occurring, thus suppressing the ETI phenotype by preventing any need for DDR activation or telomerase intervention.
Telomerase is predicted to be recruited to backtracked replication forks resulting from stalling, which has been proposed to occur at measurable frequencies in telomeric DNA (Miller et al., 2006; Drosopoulos et al., 2012). Such backtracked forks will expose single-stranded leading strand TG(1-3) repeat sequence DNA, which is the substrate for telomerase elongation. Also, telomerase could aid in the repair of a broken telomeric fork generated when a stalled replisome collapses (Chang et al., 2009). The resulting shortened telomere is a known preferred telomerase substrate (Miller et al., 2006). Other known interactions between DNA polymerase and telomerase actively engaged on telomeres may normally be required to optimize fork movement or fork restarting in telomeres. For example, the telomere-binding Cdc13-Stn1-Ten1 complex interacts via its Cdc13 subunit with a subunit of the telomerase complex (Est1) and also interacts (via Stn1) with a subunit of DNA polymerase alpha (Grossi et al., 2004).
The causal mechanism underlying mother cell aging remains unknown even for WT yeast despite extensive identification of genetic and environmental modifiers of this process. Our findings indicate that telomerase functionality is required throughout the divisions of yeast mother cells in a more continuous mode than previously thought. The findings reported here indicate that telomerase activity is required to alleviate normal telomeric replication stress and allow mother cell aging to occur with wild type kinetics. In addition, mutations known to inhibit telomerase activity or telomere maintenance have been implicated in the premature onset of diseases of aging and reduced lifespan in humans and mice (Codd et al., 2013, Armanios and Blackburn, 2012) and replication stress has been shown to induce aging in mouse cells (Flach et al., 2014). Therefore, this early requirement for active telomerase in preventing premature mother cell aging in yeast suggests a new possibility: that loss of telomerase activity may have telomere length independent consequences that accelerate aging and cause aging related diseases in other eukaryotes.
Experimental Procedures
Yeast Strain Construction
All strains used in this study are listed in Table S1. Plasmid and oligo sequences are available upon request. Complete disruption of ORFs was carried out using PCR-mediated gene disruption (Rose, M. D. et al., 1990). mrc1AQ mutant strains were made either via a loop-in, loop-out of a plasmid containing the mutant, followed by PCR verification, or via plasmid transformation of the mutant into an mrc1Δ strain.
Growth of Mutants for Monitoring Early Loss of Telomerase
ETI cells were produced by two methods: sporulation of diploid heterozygote strains (tlc1Δ/TLC1, est2-D530A/EST2), or by loss of a covering plasmid in a haploid telomerase-deficient background strain struck on solid media. Colonies underwent two days of growth at 30 C, were genotyped, and grown overnight (5–10 generations) in YPD prior to experimentation.
Microfluidics Technique Analyses of Mother Cells
Mother cells were monitored for two days by repeated microscopic imaging as described (Xie et al., 2012; Zhang et al., 2012). Microposts contained within the microfluidic device were used to clamp mother cells in place while daughter cells were washed away by hydrodynamically-controlled flow of the surrounding liquid medium. Cell cycle durations analyzed here excluded the first cycle observed and the terminal two divisions.
Southern Blotting Analysis of Telomere Length
Genomic DNA was prepared from cells from serial streaks on solid media after the indicated number of passages. Genomic DNA was then digested with XhoI and run on 0.8% agarose gels. DNA was transferred from the gels to Hybond N+ membranes and probed with γ32P end-labeled WT telomeric repeat oligonucleotide (TGTGGTGTGTGGGTGTGGTGT) as described previously (Rose, M. D. et al., 1990; Sambrook J and Russel D. W., 2001) and visualized using a phosphoimager.
Mitochondrial and SIR3 Quantification
Cells containing mito-tagged (mt)GFP were placed on a microfluidics chip as described above. Images were taken every 2 hours, and mtGFP intensity was measured relative to WT to determine volume of mitochondria. For Sir3-GFP foci, eleven images were taken for the Z stack, projected to a single image using the maximum value of the column, and the fluorescence intensity of the foci was measured using customized software Cellseg 5.4.
Statistical Analysis
Lifespans were compared using the Wilcoxon Rank-Sum test. Significance for the variation of cell cycle length was determined by F-tests. We used Fisher’s exact test to determine the significance of frequency of RNR3 peaks in different genotypes.
Quantification of RNR3 Peaks
Fluorescence was measured every 30 min during lifespan tracking. Frequency of RNR3 peaks (at least 1.3 fold over background) for “still-dividing” mother cells was calculated as the number of peaks, divided by all cell cycles occurring within that genotype and, for “terminal” mother cells as the percentage of mother cell lifespans that contained a peak during or after the last two divisions.
See Extended Experimental Procedures for supplemental experiments: bulk population budding and chromosome segregation kinetics, NAC treatment, and ROS straining.
Supplementary Material
Acknowledgments
This work was supported by NIH grant GM26259 to EHB and NIH grants (GM070808 & AG043080) and a Packard Fellowship in Science and Engineering to HL, and by the NIH Center for Systems and Synthetic Biology (P50 GM081879). Z.X. thanks the Postdoctoral Fellowship from PKU-THU Center for Life Science and Special Financial Grant from the China Postdoctoral Science Foundation.
Footnotes
Author Contributions
Z.X. performed the microfluidic lifespan assays, cell counting and scoring, image segmentation and fluorescence quantification, K.A.J. performed strain constructions, sample genotyping, bulk population studies, telomere length (southern blot) experiments, and PCR-based telomere deprotection assays, D.L.S. performed strain constructions, cell cycle and budding analyses, and DNA-damage assays. E.H.B., Z.X. and H. L. analyzed the statistics, Y.Z. performed the Rad52 mutant lifespan assays, Z.L. synthesized the mtGFP plasmids, J.Z. & H.L. performed the bioinformatics analyses, R.T. aided in strain preparation, E.H.B., H.L., K.A.J. & Z.X. conceived the plans, interpreted and analyzed the data, and wrote the paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alcasabas AA, Osborn AJ, Bachant J, Hu F, Werler PJH, Bousset K, Furuya K, Diffley JFX, Carr AM, Elledge SJ. Mrc1 transduces signals of DNA replication stress to activate Rad53. Nat Cell Biol. 2001;3:958–965. doi: 10.1038/ncb1101-958. [DOI] [PubMed] [Google Scholar]
- Andreson BL, Gupta A, Georgieva BP, Rothstein R. The ribonucleotide reductase inhibitor, Sml1, is sequentially phosphorylated, ubiquitylated and degraded in response to DNA damage. Nucleic Acids Res. 2010;38:6490–6501. doi: 10.1093/nar/gkq552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armanios M, Blackburn EH. The telomere syndromes. Nat Rev Genet. 2012;13:693–704. doi: 10.1038/nrg3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basenko E, Topcu Z, McEachern MJ. Recombination Can either Help Maintain Very Short Telomeres or Generate Longer Telomeres in Yeast Cells with Weak Telomerase Activity. Eukaryot Cell. 2011;10:1131–1142. doi: 10.1128/EC.05079-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop NA, Guarente L. Genetic links between diet and lifespan: shared mechanisms from yeast to humans. Nat Rev Genet. 2007;8:835–844. doi: 10.1038/nrg2188. [DOI] [PubMed] [Google Scholar]
- Chabes A, Georgieva B, Domkin V, Zhao X, Rothstein R, Thelander L. Survival of DNA damage in yeast directly depends on increased dNTP levels allowed by relaxed feedback inhibition of ribonucleotide reductase. Cell. 2003;112:391–401. doi: 10.1016/s0092-8674(03)00075-8. [DOI] [PubMed] [Google Scholar]
- Chan SW, Blackburn EH. Telomerase and ATM/Tel1p Protect Telomeres from Nonhomologous End Joining. Mol Cell. 2003;11:1379–1387. doi: 10.1016/s1097-2765(03)00174-6. [DOI] [PubMed] [Google Scholar]
- Chang M, Luke B, Kraft C, Li Z, Peter M, Lingner J, Rothstein R. Telomerase is essential to alleviate pif1-induced replication stress at telomeres. Genetics. 2009;183(3):779–91. doi: 10.1534/genetics.109.107631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codd V, Nelson CP, Albrecht E, Mangino M, Deelen J, Buxton JL, Hottenga JJ, Fischer K, Esko T, Surakka I, et al. Identification of seven loci affecting mean telomere length and their association with disease. Nat Genet. 2013;45:422–427. doi: 10.1038/ng.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drosopoulos WC, Kosiyatrakul ST, Yan Z, Calderano SG, Schildkraut CL. Human telomeres replicate using chromosome-specific, rather than universal, replication programs. J Cell Biol. 2012;197:253–266. doi: 10.1083/jcb.201112083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- d’Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, von Zglinicki T, Saretzki G, Carter NP, Jackson SP. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426:194–198. doi: 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]
- Flach J, Bakker ST, Mohrin M, Conroy PC, Pietras EM, Reynaud D, Alvarez S, Diolaiti ME, Ugarte F, Forsberg EC, Le Beau MM, Stohr BA, Méndez J, Morrison CG, Passegué E. Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature. 2014;512(7513):198–202. doi: 10.1038/nature13619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandin N, Bailly A, Charbonneau M. Activation of Mrc1, a mediator of the replication checkpoint, by telomere erosion. Biol Cell. 2005;97:799–814. doi: 10.1042/BC20040526. [DOI] [PubMed] [Google Scholar]
- Greenwell PW, Kronmal SL, Porter SE, Gassenhuber J, Obermaier B, Petes TD. TEL1, a gene involved in controlling telomere length in S. cerevisiae, is homologous to the human ataxia telangiectasia gene. Cell. 1995;82:823–829. doi: 10.1016/0092-8674(95)90479-4. [DOI] [PubMed] [Google Scholar]
- Grossi S, Puglisi A, Dmitriev PV, Lopes M, Shore D. Pol12, the B subunit of DNA polymerase α, functions in both telomere capping and length regulation. Genes Dev. 2004;18:992–1006. doi: 10.1101/gad.300004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson FB, Sinclair DA, Guarente L. Molecular Biology of Aging. Cell. 1999;96:291–302. doi: 10.1016/s0092-8674(00)80567-x. [DOI] [PubMed] [Google Scholar]
- Jossen R, Bermejo R. The DNA damage checkpoint response to replication stress: A Game of Forks. Front Genet. 2013;4:26. doi: 10.3389/fgene.2013.00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeberlein M. Lessons on longevity from budding yeast. Nature. 2010;464:513–519. doi: 10.1038/nature08981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999;13:2570–2580. doi: 10.1101/gad.13.19.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingner J, Hughes TR, Shevchenko A, Mann M, Lundblad V, Cech TR. Reverse Transcriptase Motifs in the Catalytic Subunit of Telomerase. Science. 1997;276:561–567. doi: 10.1126/science.276.5312.561. [DOI] [PubMed] [Google Scholar]
- Lundblad V, Blackburn EH. An alternative pathway for yeast telomere maintenance rescues est1-senescence. Cell. 1993;73:347–360. doi: 10.1016/0092-8674(93)90234-h. [DOI] [PubMed] [Google Scholar]
- Lustig AJ, Petes TD. Identification of yeast mutants with altered telomere structure. Proc Natl Acad Sci U S A. 1986;83:1398–1402. doi: 10.1073/pnas.83.5.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SG, Laroche T, Suka N, Grunstein M, Gasser SM. Relocalization of telomeric Ku and SIR proteins in response to DNA strand breaks in yeast. Cell. 1999;97:621–633. doi: 10.1016/s0092-8674(00)80773-4. [DOI] [PubMed] [Google Scholar]
- McEachern MJ, Blackburn EH. Cap-prevented recombination between terminal telomeric repeat arrays (telomere CPR) maintains telomeres in Kluyveromyces lactis lacking telomerase. Genes Dev. 1996;10:1822–1834. doi: 10.1101/gad.10.14.1822. [DOI] [PubMed] [Google Scholar]
- Miller KM, Rog O, Cooper JP. Semi-conservative DNA replication through telomeres requires Taz1. Nature. 2006;440:824–828. doi: 10.1038/nature04638. [DOI] [PubMed] [Google Scholar]
- Mills KD, Sinclair DA, Guarente L. MEC1-Dependent Redistribution of the Sir3 Silencing Protein from Telomeres to DNA Double-Strand Breaks. Cell. 1999;97:609–620. doi: 10.1016/s0092-8674(00)80772-2. [DOI] [PubMed] [Google Scholar]
- Nautiyal S, DeRisi JL, Blackburn EH. The genome-wide expression response to telomerase deletion in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 2002;99:9316–9321. doi: 10.1073/pnas.142162499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborn AJ, Elledge SJ. Mrc1 is a replication fork component whose phosphorylation in response to DNA replication stress activates Rad53. Genes Dev. 2003;17:1755–1767. doi: 10.1101/gad.1098303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park PU, Defossez PA, Guarente L. Effects of Mutations in DNA Repair Genes on Formation of Ribosomal DNA Circles and Life Span inSaccharomyces cerevisiae. Mol Cell Biol. 1999;19:3848–3856. doi: 10.1128/mcb.19.5.3848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichard P. Interactions Between Deoxyribonucleotide and DNA Synthesis. Annu Rev Biochem. 1988;57:349–374. doi: 10.1146/annurev.bi.57.070188.002025. [DOI] [PubMed] [Google Scholar]
- Rowe LA, Degtyareva N, Doetsch PW. DNA damage-induced reactive oxygen species (ROS) stress response in Saccharomyces cerevisiae. Free Radic Biol Med. 2008;45:1167–1177. doi: 10.1016/j.freeradbiomed.2008.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabourin M, Zakian VA. ATM-like kinases and regulation of telomerase: lessons from yeast and mammals. Trends Cell Biol. 2008;18:337–346. doi: 10.1016/j.tcb.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon TB, Evert BA, Song B, Doetsch PW. Biological consequences of oxidative stress-induced DNA damage in Saccharomyces cerevisiae. Nucleic Acids Res. 2004;32:3712–3723. doi: 10.1093/nar/gkh696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sfeir A, Kosiyatrakul ST, Hockemeyer D, MacRae SL, Karlseder J, Schildkraut CL, de Lange T. Mammalian Telomeres Resemble Fragile Sites and Require TRF1 for Efficient Replication. Cell. 2009;138:90–103. doi: 10.1016/j.cell.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata H, Kanoh Y, Gunge N, Shirahige K, Matsuura A. Reciprocal Association of the Budding Yeast ATM-Related Proteins Tel1 and Mec1 with Telomeres In Vivo. Mol Cell. 2004;14:515–522. doi: 10.1016/s1097-2765(04)00262-x. [DOI] [PubMed] [Google Scholar]
- Tsolou A, Lydall D. Mrc1 protects uncapped budding yeast telomeres from exonuclease EXO1. DNA Repair. 2007;6:1607–1617. doi: 10.1016/j.dnarep.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Zhang Y, Zou K, Brandman O, Luo C, Ouyang Q, Li H. Molecular phenotyping of aging in single yeast cells using a novel microfluidic device. Aging Cell. 2012 doi: 10.1111/j.1474-9726.2012.00821.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Luo C, Zou K, Xie Z, Brandman O, Ouyang Q, Li H. Single Cell Analysis of Yeast Replicative Aging Using a New Generation of Microfluidic Device. PLoS ONE. 2012;7:e48275. doi: 10.1371/journal.pone.0048275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Muller EG, Rothstein R. A suppressor of two essential checkpoint genes identifies a novel protein that negatively affects dNTP pools. Mol Cell. 1998;2:329–340. doi: 10.1016/s1097-2765(00)80277-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.