

Abstract
The nuclear lamina is a proteinaceous structure located underneath the inner nuclear membrane (INM), where it associates with the peripheral chromatin. It contains lamins and lamin-associated proteins, including many integral proteins of the INM, chromatin modifying proteins, transcriptional repressors and structural proteins. A fraction of lamins is also present in the nucleoplasm, where it forms stable complexes and is associated with specific nucleoplasmic proteins. The lamins and their associated proteins are required for most nuclear activities, mitosis and for linking the nucleoplasm to all major cytoskeletal networks in the cytoplasm. Mutations in nuclear lamins and their associated proteins cause about 20 different diseases that are collectively called laminopathies’. This review concentrates mainly on lamins, their structure and their roles in DNA replication, chromatin organization, adult stem cell differentiation, aging, tumorogenesis and the lamin mutations leading to laminopathic diseases.
Keywords: nuclear lamina, nuclear envelope, chromatin, transcription, filament assembly, stem cells, cancer
-
The lamin molecule
Domain organization of lamins
Lamins are divided to type A and type B
Post-translational processing of lamin molecules
Lamin molecules in evolution
-
The supramolecular assembly of lamins
From lamin monomer to lamin dimer
From dimers to filaments
The roles of the different domains in the assembly of lamins
Laminopathic mutations affect lamin filament assembly
Lamin assembly in vivo
-
Lamin-binding proteins
Lamins, chromatin and epigenesis
Lamin binding to DNA
Lamin binding to chromatin
Lamins affect chromatin organization and epigenesis
Lamins are involved in many nuclear functions
Lamins determine the shape and stiffness of the nucleus
Lamins and DNA replication
Lamins in transcription and splicing
-
Lamins and aging
Lamins and laminopathies
Mutations in lamins and their associated proteins causing ‘laminopathies’
Animal models for laminopathies
Molecular models for laminopathies
-
Lamins and stem cells
The Notch pathway
The Wnt/β-catenin pathway
Other pathways
-
Lamins and cancer
Lamins as biomarkers for cancer
Lamins and cancer regulating pathways
Lamins and cancer related aneuploidy
Lamin and viruses
The lamin molecule
Domain organization of lamins
Lamins are type V intermediate filaments (IFs) and like all members of the IF family of proteins, they have the tripartite domain organization of a central α helical rod domain, flanked by a short head and a longer tail domains (Fig. 1A). The rod domain is made of four coiled-coil segments: 1A, 1B, 2A and 2B, each of which is made of heptad repeats. The coiled-coil segments are linked by three linkers termed L1, L12 and L2, which are conserved in length and sequence among lamins [1, 2]. The end segments of the central rod domain (∼30 amino acids on both sides) are similar among lamins, as well as in all IF proteins. When mutated or deleted, they affect the assembly of dimers into higher-order structures [1]. Coil 1B in all lamins is similar to invertebrate cytoplasmic IFs and is 42 amino acids (6 heptads) longer than vertebrate cytoplasmic IFs [2]. The lamin tail domain contains a ∼120-residue immunoglobulin (Ig) fold [3]. Between the rod domain and the Ig fold there is an unfolded region containing a nuclear localization signal (NLS). The carboxy tail domain of all B-type lamins and lamin A (but not lamin C) proteins contains a CAAX motif, which undergoes post-translational modifications (see below, Fig. 2).
Figure 1.
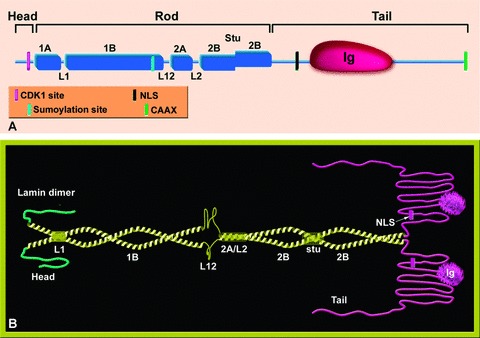
Schematic representation of the structure of lamin proteins. (A) The lamin monomer. The lamin is divided into three domains, head, rod and a globular tail. The rod domain is composed of four coiled-coil regions (1A, 1B, 2A, 2B) that are connected through three short linkers (L1, L12, L2). Marked on the scheme are the Ig globular domain in the tail and the stutter (a discontinuity of the heptad repeat) in coil 2B. Also shown by colour code are the positions of the CDK-1 recognition site (absent in Ce-lamin), the sumoylation site in human lamin A, the nuclear localization signal (NLS) and the CAAX motif. (B) A model of lamin dimers. A pair of parallel coiled-coil rods forms the lamin dimer (yellow). The non-α-helical head and tail domains are coloured green and pink, respectively. The different sub-domains are indicated. In coil 2B the stutter leads to a local unwinding.
Figure 2.
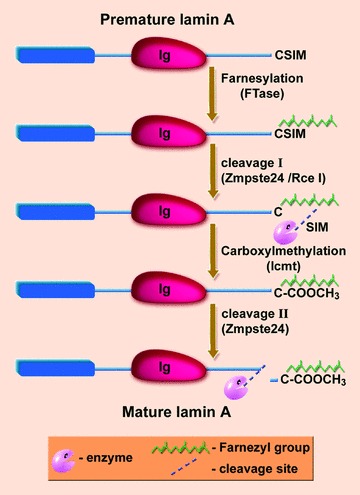
Prelamin A processing. Premature lamin A is going through four processing steps until it becomes a mature lamin A, including farnesylation of the cysteine at the carboxy terminus, cleavage of the three carboxy-terminal amino acids (aaX) by either ZMPSTE24 or RceI, carboxymethylation of the farnesylated cysteine by isoprenylcysteine methyltransferase and cleavage of the 15 terminal amino acids, including the farnesylated and carboxymethylated cysteine, by ZMPSTE24.
Lamins are divided to type A and type B
Lamins are divided into A and B type according to their expression patterns, behaviour during mitosis and biochemical characteristics. Distinct genes encode A- and B-type lamins. All metazoan cells express B-type lamins, whereas A-type lamins are expressed only in differentiated cells of higher complex organisms (see below). B-type lamins have an acidic isoelectric point, whereas A-type lamins have a neutral isoelectric point. During mitosis B-type lamins remain associated with membranes, whereas A-type lamins become solubilized [4].
Post-translational processing of lamin molecules
With the exception of the mammalian and the Drosophila lamin C, all lamins undergo a process of maturation involving the CAAX box at their carboxy terminus. At the first stage of maturation farnesyl transferase adds a farnesyl group to the cysteine. Next, either Rce1 (FACE2 in human beings) or Zmpste24 (FACE1 in human beings) cleave the last three amino acids (AAX). At the next stage the isoprenylcysteine methyltransferase methylates the farnesylated C-terminal cysteine. Although B-type lamins remain carboxy farnesylated and methylated, lamin A undergoes another cleavage event of the last 15 C-terminal amino-acids by Zmpste24, which removes the carboxy farnesylated and methylated cysteine [5] (Fig. 2). It was recently shown that blocking farnesylation of lamin A by farnesyl transferase inhibitors (FTIs) causes an alternative prenylation of geranylgeranylation by geranylgeranyl transferase [6].
Other known post-translational modifications of lamins include phosphorylation and sumoylation. Relatively little is known about the phosphorylation of lamins during interphase. The Drosophila B-type lamin (lamin Dm0) contains at least three interphase phosphorylation sites at Ser25 in the head domain at a molar ratio of 0.3 (phosphate/protein), at Thr432 or at Thr435 (TRAT sequence) in the tail domain at a molar ratio of 0.2 and at Ser595 in the tail domain at a molar ratio of 0.5 [7]. These molar ratios suggest that interphase phosphorylation of lamins is dynamic. Indeed, the phosphorylation of the TRAT sequence modulates the interaction between lamin Dm0 and the histone H2A/H2B dimer in vitro[8]. The mitotic phosphorylation of lamins was studied in more detail. Mammalian lamins contain a mitotic cyclin-dependent kinase (CDK)-1 recognition sequences flanking both sides of the rod domain. These sites are required for lamin disassembly into dimers during mitosis because mutations within these CDK1 sites inhibited lamina disassembly both in vivo[9] and in vitro[10–12]. The potential roles of other mitotic phosphorylation sites are discussed in [1]. Mammalian lamin A also undergoes sumoylation at Lys201[13]. Mutation in the Glu203, which inhibits sumoylation, causes aggregation of lamin A and increased cell death, suggesting that sumoylation regulates lamin A assembly [13].
Lamin molecules in evolution
Lamins are detected in all metazoan cells. In contrast, lamins are absent in single cell organisms or in plants. Although lamins from vertebrates and invertebrates differ significantly in their primary sequence [14], the general structural organization of lamins is evolutionary conserved with a few exceptions. Tunicate lamins are the only known lamins that have a deletion of 90 amino acids in the region of the Ig fold [15]. Caenorhabditis elegans single lamin protein (Ce-lamin) contains a Coil 2B that is shorter by 14 amino acids (two heptads), a tail domain that is shorter by ∼25 amino acids as compared to Drosophila and vertebrate lamins. It also lacks the conserved CDK1 sites [16]. Evolution trees of lamins can be found in [1, 14].
The supramolecular assembly of lamins
From lamin monomer to lamin dimer
During mitosis lamin molecules are disassembled into monomers [4]. Following mitosis they reassemble into higher-order structures. The smallest subunit detected in vitro is a dimer, where the two central α-helical rod domains within a lamin dimer form a coiled coil around each other in a parallel, unstaggered fashion [1] (Fig. 1B). The coiled-coil interactions are due to the heptad repeats; lower case letters (a through g) commonly denotes the positions within the heptad repeat. Although hydrophobic residues are usually occupying positions a and d, the e and g positions are usually being charged [2]. Electron microscope analysis showed a 52–55-nm-long rod-like shape of the lamin dimer with two globular domains representing the two tails [17].
From dimers to filaments
In the second step of assembly in vitro the lamin dimers elongate longitudinally in a polar fashion to form ∼2-nm-wide head-to-tail polymers with an axial repeat of 48–50 nm [17]. The difference between the length of the rod domain and the axial repeat represents an overlap between adjacent rod domains [18]. In the next step of assembly the head-to-tail polymers assemble laterally to form supramolecular structures. Most lamins form paracrystalline fibres with a latitudinal alternating light and dark banding pattern with 24–25 nm axial repeat, which is about half the repeating length of a lamin rod domain [1, 2]. These structures are unique to lamins, because they do not appear in an in vitro assembly of cytoplasmic IFs, which form 10-nm filaments in vitro[2]. It is noteworthy that except for the case of overexpression of lamins in insect cells, lamin paracrystalline fibres were never seen in vivo, which is likely due to the presence of lamin binding proteins in normal nuclei that are involved in the organization and regulation of lamin assembly [19].
In contrast to vertebrate and Drosophila lamins that form only paracrystalline fibres in vitro, Ce-lamin, can assemble into stable 10 nm filaments. Under different conditions containing divalent ions it can also form paracrystalline fibres [20–22]. Ciona lamin is also assembled into filaments, but with a smaller average diameter of 5.4 nm [20]. Recently, it has been shown that the IF-like filaments of the C. elegans lamin are composed of 3 and 4 tetrameric protofilaments, each of which contains two partially staggered anti-parallel head-to-tail polymers. The lamin filaments are beaded due to the paired globular tail domains, which are regularly spaced, alternating between 21 and 27 nm [18].
The roles of the different domains in the assembly of lamins
The head and tail domains of lamins play a significant role in regulating the assembly of lamins. Chicken lamins A and B2 cannot assemble into head-to-tail polymers unless they have the head domain [23]. Similarly, headless Xenopus lamin A or Drosophi lalamin Dm0 do not form head-to-tail polymers and Xenopus lamin A expressed in Sf9 insect cells can be easily extracted with physiological buffers containing Triton X-100 [19, 24]. In contrast, headless human lamin C and human lamin A lacking its 18 last amino acids form paracrystals with the same axial repeat as the wild-type protein (21 nm). Ce-lamin lacking its head domain also forms filaments and paracrystalline fibres with the same axial repeat as the wild-type protein, whereas mutations in the head domain can cause abnormal filament assembly (N.W.M. and Y.G., unpublished observation). The involvement of the head domain in lamin assembly in vivo was demonstrated by the dominant-negative effect of headless human and Xenopus lamins on the endogenous lamins. Ectopic expression of headless human lamin A in Chinese hamster ovary (CHO) cells caused aggregation of the endogenous lamins A and C, but not lamins B1 or B2 [25]. Headless human lamin A or Xenopus lamin LB3 caused aggregate formation of lamin LB3 in the nucleoplasm of nuclei assembled in vitro in Xenopus extracts [26, 27]. Taken together, the current data suggest a function for the head domain in regulating lamin filament assembly, which is evolutionarily conserved. It also suggests that the head domain interacts with specific sequences in the rod domain and interfering with these interactions can inhibit lamin polymerization.
Tailless Xenopus lamin A forms filament bundles lacking the 24-nm repeats seen in normal paracrystals [28]. Similarly, tailless Ce-lamin formed paracrystalline fibres that lost the characteristics axial repeats [18]. In contrast, tailless chicken lamin B2 enhances paracrystalline fibres assembly with normal axial repeats [23] and tailless Drosophila lamin Dm0, cannot form normal head-to-tail polymers, but surprisingly forms paracrystalline fibres without the normal axial repeats [29]. Tailless human lamin C formed 15–30 nm filaments instead of paracrystals. Taken together, these and other data [24, 25, 30] suggest that the tail domain of lamins is also involved in regulating lamin assembly and that lateral assembly of head-to-tail filaments depends in some lamins on interactions between the tail domain of one head-to-tail polymer and the rod domain of the other. Support for this conclusion comes from experiments in which the isolated tail domain of Xenopus lamin B3 inhibited the polymerization of lamin B3 in vitro and prevented the assembly of nuclei in Xenopus egg interphase extracts [31]. The Ig-fold motif in the lamin tail domain was sufficient to inhibit lamin polymerization and nuclear assembly in vitro[31].
Laminopathic mutations affect lamin filament assembly
There are an unprecedented number of disease-causing heritable mutations in the human LMNA gene (see below) [32]. Many of the mutated residues are evolutionarily conserved in Ce-lamin. A recent study has analysed the effects of 14 different disease-causing missense mutations on the assembly of Ce-lamin into 10-nm filaments or paracrystalline fibres [22]. Six of the 14 tested mutations interfered with the assembly of Ce-lamin filaments or paracrystalline fibres in vitro. The R64P or Q159K (R50P or E145K in human lamin A, causing Emery Dreifuss muscular dystrophy [EDMD] and Hutchison Gilford progeria syndrome [HGPS], respectively) mutations affected assembly of both filaments and paracrystalline fibres. The R64P mutant Ce-lamin assembled into very short (between 50 nm and several hundred nm long) 20-nm-wide filaments, suggesting that the mutation interferes with efficient assembly of head-to-tail polymers. This result also showed that lateral association of lamin dimers occurs in parallel to the elongation of the head-to-tail filament. Mutant Q159K was assembled into ‘striped’ filaments and paracrystals, due to abnormal lateral association of molecules within the filament [18]. The Y59C or D217K (Y45C or E203K in human lamin A, causing EDMD and dilated cardiomyopathy (DCM), respectively) formed wider filaments, but had no apparent effect on paracrystalline fibres assembly. Likewise, whereas none of the tested Ce-lamin tail mutations caused abnormal filament assembly, under the standard assembly conditions two of these mutations: R460W or L535P (R453W or L530P in human lamin A, respectively, both causing EDMD) affected the assembly of paracrystalline fibres, although having no apparent effect on lamin filament assembly. Taken together, these data suggest common assembly steps for lamin filament and paracrystalline fibres, which probably differ at their final stage of assembly. The observation that 8 of 14 of the missense mutations have no visible effects on the assembly suggests that defects in lamin filament assembly cannot be the only mechanism leading to laminopathic diseases (see below).
Lamin assembly in vivo
In most somatic cells the nuclear lamina appears as a thin layer between the nuclear membrane and the peripheral chromatin. In other cases, including old rat liver cells, it appears as a thick dense layer [33]. In all cases distinct filamentous structures were not visible. In CHO cells there was a complex network of 10–500-nm-wide fibres that were thicker when attached to the underlying chromatin and in the area of nuclear pore complexes (NPC) [33].
The only cell type in which distinct 10-nm lamin filaments were detected in vivo is the amphibian germinal vesicle containing lamin LB3 [34, 35]. The ability to detect 10-nm-wide filaments in this cell could be due to the ability to isolate nuclear envelope free of chro-matin and other adhering material, the reduced number and complexity of lamin binding proteins (see below) or to a unique ability of lamins to form 10-nm filaments in these cells. Electron microscopy of detergent extracted nuclear envelopes derived from Xenopus germinal vesicle showed a meshwork of filaments that were 10-nm thick [34]. A similar arrangement was detected using field emission scanning electron microscope (feSEM) and in another amphibian species, Necturus, using cryo electron microscope [1].
In a recent study, isolated and fixed nuclear envelope of the Xenopus germinal vesicle was examined by feSEM. The results of this study suggest presence of one set of parallel 10–12-nm-wide filaments with distinct irregular ∼5-nm-wide cross-connections [35]. The Xenopus germinal vesicle was further used to assemble ectopically expressed A- and B-type lamins. All these lamins were assembled on top of the existing lamin filaments. Ectopically expressed lamin B3 filaments were similar to the endogenous lamin, whereas lamin B2 assembled into filaments that were organized less precisely and lamin A formed thicker filaments that increased mechanical rigidity of the nuclear envelope [35].
Lamin-binding proteins
The many roles of lamins are mediated by interactions with numerous lamin-binding proteins both at the nuclear periphery and in the nucleoplasm (Fig. 3). There is increasingly growing number of mammalian inner nuclear membrane (INM) proteins [36], and most of these proteins probably bind lamins either directly or indirectly. For example, lamins bind in vitro to INM proteins: emerin, MAN-1, lamin B receptor – LBR, lamina-associated polypeptides-1 and -2β (LAP-1, LAP-2β), nesprin-1α, otefin, Young arrest (YA) and SUN-1. Lamins can also bind non-integral proteins including the chromatin proteins histone H2A/H2B dimers, retinoblastoma protein (pRb), barrier-to-autointegration factor (BAF), as well as LAP-2α, sterol response element binding protein (SREBP), Kruppel-like protein (MOK-2), extracellular signal-regulated kinase (ERK)-1/2, c-Fos, nuclear actin, proliferating cell nuclear antigen (PCNA) and proteins of the NPC [reviewed in 37–39].
Figure 3.
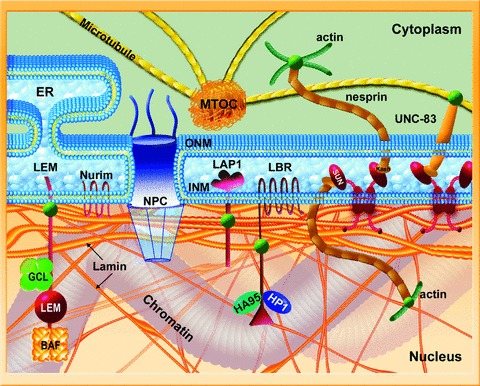
Schematic view of the nuclear envelope, lamina and chromatin. The inner nuclear membrane (INM) and the outer nuclear membranes (ONM) are joined at the nuclear pore complexes and separated by the nuclear lumen. The ONM and lumen are continuous with the endoplasmic reticulum (ER). Lamins (both A- and B-types) are shown as orange filaments; thicker at the nuclear periphery and thinner filaments in the nucleoplasm. However, the filamentous nature of the lamins, especially within the nucleus, remains hypothetical. Also shown are selected proteins of the INM including LEM-domain and SUN-domain proteins, LAP-1, Nurim and LBR (boudreaux). These proteins represent only a small fraction of proteins of the INM. Also shown few examples of non-integral proteins that interact with lamins or with their associated proteins including actin, HP1, HA95, germ cell-less and BAF. The nucleoplasmic lamins also form specific protein complexes (not shown). INM SUN-domain proteins interact with outer nuclear membrane (ONM) KASH-domain proteins, thus bridging between the nucleus and cytoplasmic structures including, actin (green), tubulin (yellow) and intermediate filament (not shown) networks and the centrosome (MTOC).
Lamins, chromatin and epigenesis
In most cells the peripheral chromatin, which includes a large fraction of the heterochromatin, is associated with the nuclear lamina [33, 40]. Electron microscopic tomography analysis showed that lamin B associates with the underlying chromatin in CHO cells [41]. In Drosophila cells the centromeres, telomeres and intercalary heterochromatin are almost always associated with the nuclear lamina [42]. The telomeres and centromeres also associate with nuclear lamina in plants, budding yeast, fission yeast and mouse meiotic cells, but not in somatic mammalian nuclei [reviewed in 14]. In addition, in both Drosophila and mammalian cells the B-type lamins interact in higher frequencies with gene-poor regions of the genome [43, 44].
Lamin binding to DNA
Lamins bind directly to nucleic acids and chromatin. Drosophila interphase lamins, but not mitotic lamins can be cross-linked to nucleic acid in vivo[45]. In vitro, lamins bind matrix attachment/ scaffold associated regions [46], single-stranded DNA [47] and centromeric and telomeric sequences [48, 49]. The binding of the Drosophila lamin Dm0 to DNA is mediated through the rod domain and requires its polymerization [50]. The significance of lamin binding to DNA is currently unclear, because regions that interact with lamins are mostly silent and therefore their compact chromatin makes the DNA less accessible.
Lamin binding to chromatin
C. elegans, Drosophila, Xenopus and mammalian lamins interact with decondensed sperm chromatin, assembled chromatin or isolated mitotic chromosomes from different species in vitro[8, 51–56], suggesting that lamin-chromatin interactions are conserved in evolution. Lamins can also bind polynucleosomes and isolated mammalian or Xenopus histones [8, 53, 57]. The major binding site of the Drosophila lamin Dm0 to mitotic chromosomes, polynucleosomes and isolated histones involves residues 425–473, which is an unstructured region located between the rod domain and the Ig fold in the tail domain. A similar region is required for the chromatin binding of human A-type lamins [57] and Xenopus lamin B2 [54]. This region binds chromatin in vitro via the histone H2A/H2B dimer [53]. The binding of lamin Dm0 to chromatin requires the conserved NLS and a sequence composed of the amino acids TRAT, which is partially phosphorylated in Drosophila interphase cells [58] on either or both threonine residues. Substituting either one of the threonine residues in the TRAT sequence with negatively charged residues decreases the binding to chromatin, suggesting that phosphorylation regulates this binding. Both Drosophila lamin Dm0 and C. elegans Ce-lamin bind directly to histone H2A in vitro and this binding requires the NLS. The amino and car-boxyl histone tails mediate this binding [8]. A weaker binding site in lamin Dm0 is located in residues 572–622 that follow the Ig fold.
The binding affinity of the human lamin A tail for both polynucleosomes and purified core histones is 0.12–0.3 μM. The binding affinity of Drosophila lamin Dm0 to the histone H2A/H2B dimer is ∼1 μM [53, 57]. The relatively low affinity of lamins binding to chromatin and the possible effect of phosphorylation on that binding suggest that the direct association of lamins with chromatin is a dynamic process. It remains to determine why heterochromatin and gene-poor regions tend to associate more frequently with lamins.
Lamins are also associate with chromatin indirectly, through their binding to different lamin-associated proteins [reviewed in 38, 59]. One of the best-studied examples is the lamins association to chromatin through BAF [reviewed in 60].
Lamins affect chromatin organization and epigenesis
Genetic analysis in C. elegans, Drosophila, mice and human cells expressing mutant lamins A and C revealed that lamins are required for normal chromatin organization. Loss of or mutations in lamins cause dissociation of heterochromatin from the nuclear periphery and its redistribution [61–64]. Mutations in the lamin A processing enzyme ZMPSTE24/FACE-1 cause similar changes in chromatin organization [65]. The redistribution of heterochromatin is accompanied with major changes in histone modifications. The best-studied model system is human fibroblasts expressing the progerin/LAΔ50 mutant isoform of lamin A (see lamin diseases section). In these cells the heterochromatin protein HP1α is reduced at least two fold and the heterochromatin marker trimethyl lysine 9 on core histone H3 (H3K9me3) is reduced or completely lost [66, 67]. In addition, the facultative heterochromatin marker trimethyl lysine 27 on core histone H3 (H3K27me3) is reduced and is lost from the inactive X chromosome, whereas there is an increase in the amounts of the constitutive heterochromatin marker tri-methyl lysine 20 on core histone H4 (H4K20me3) and up-regulation of the pericentric satellite 3 repeat [68]. These changes in chromatin organization can be reversed either following the removal of the toxic progerin isoform by morpholino anti-sense oligonucleotides [66] or by treating the cells with FTIs, which cause dissociation of progerin from the nuclear periphery [reviewed in 5].
It is currently unknown how lamins regulate the epigenetic state of chromatin. Most likely they do it through associated proteins that require lamins for their normal localization or activity and have histone acetylase, deacetylase or methylase activities. For example, LAP-2p binds and regulates the activity of histone deacetylase 3 (HDAC-3) [69] and emerin is found in protein complexes with several HDACs [70].
Lamins are involved in many nuclear functions
Lamins and their associated proteins are involved in maintaining the shape and mechanical strength of the nucleus. They are also involved in most nuclear activities, including chromatin organization, DNA replication, transcription regulation, RNA processing, linking the nucleus to all major cytoskeleton networks, apoptosis, meiosis and mitosis. Details on the roles of lamins in some of these functions are described below.
Lamins determine the shape and stiffness of the nucleus
Two of the main components that determine the shape and provide the strength to the nucleus are the nuclear lamina and chromatin. A third component is the nuclear scaffold, which most likely contains lamins. However, the specific components of the nuclear scaffold are largely unknown. The cytoplasmic filaments also help determining the shape of the nucleus [71], either directly or via their interactions with the SUN-/KASH-domain protein complexes, which bind the nuclear lamins [72].
In C. elegans down-regulation of the single lamin protein caused continuous rapid changes in nuclear shape [73]. Changes in nuclear shape that include membrane invagination and formation of nuclear blebs were also observed in Drosophila adult cells devoid of lamin Dm0[74], in mouse cells lacking lamins A and C [75, 76] and in human cells carrying disease-causing mutations in lamin A [reviewed in 77, 78].
Lamins probably provide the main mechanical support of the nucleus and changes in nuclear lamina composition have major effects on the mechanical response of both the nucleus and the cytoplasm. In vitro assembly of nuclei in Xenopus oocyte extracts depleted of most lamins resulted in fragile nuclei [79], and overex-pression of lamin A in the Xenopus germinal vesicle resulted in increased rigidity of the nuclei [35]. Studies in mouse embryonic fibroblasts (MEFs) lacking lamins A and C showed defective shape and decreased stiffness of nuclei [76, 80, 81]. MEFs lacking lamin A and expressing lamin C had only slight alterations in nuclear shape and stiffness, suggesting that lamin C can complement for lamin A role in the mechanical support of the nucleus. In contrast, while MEFs lacking lamin B1 showed alteration in nuclear shape, there was no change in nuclear stiffness [82]. The latter results suggest either an overlapping role of lamin B2 or that nuclear stiffness is determined solely by A-type lamins. Dramatic changes in nuclear shape were observed in cells overexpressing B-type lamins, which were attributed to membrane proliferation caused by the presence of the farnesyl group at the carboxy terminus [83, 84].
Cells lacking emerin or LEM2 occasionally showed changes in nuclear shape. These emerin-null cells retain their normal nuclear mechanics, whereas the mechanical properties of the LEM2 cells were not examined [85, 86]. In C. elegans, down-regulation of either Ce-emerin or LEM-2 apparently does not affect nuclear morphology. However, down-regulation of both proteins caused nuclear lobulation and membrane invagination [87]. Similar changes in nuclear shape were observed following down-regulation of BAF-1 [88], suggesting that lamin, LEM-domain proteins and BAF probably interact within the same protein complexes in vivo.
Changes in the composition of the nuclear lamina can also result in mechanically induced changes in gene expression. In response to mechanical stress, MEFs lacking lamins A and C have abnormal signalling leading to attenuate NF-kp-regulated transcription and impaired mechanically activated gene transcription [76]. Impaired cellular signalling in response to mechanical stimulation was also observed in mouse cells lacking emerin, where the expression of the NF-kβ-regulated genes was impaired in response to mechanical stimulation [85].
Lamin A is permanently farnesylated in cells either devoid of the lamin A processing enzyme Zmpste24 (FACE1 in human beings) or expressing the progerin/LAA50 lamin A isoform. The shape and the stiffness of these cells change dramatically with increased amounts of the farnesylated lamin and can be reversed when these cells are treated with either FTIs or with a combination of statins and aminobisphosphonates [5, 6]. However, FTIs had no effect of the cellular sensitivity to mechanical strain demonstrating that lamin-related effects of nuclear stiffness and mechano-sensitivity can be separated [89].
Lamins and DNA replication
There are several lines of evidence that lamins are required for DNA replication. (i) In early S phase lamin A co-localizes with sites of DNA replication [90], and in late S phase lamin B1 co-localizes with PCNA at sites of DNA synthesis [91]. (ii) MEFs that are null for lamins A and C replicate their DNA at a slower rate compared to MEFs derived from wild-type mice. Expression of lamin A in the mutant cells restore the normal rate of DNA replication [92]. (iii) Immuno-depletion of the soluble lamin LB3 from Xenopus nuclear assembly extracts results in nuclei that are not able to replicate the assembled DNA [79, 93]. Adding LB3 back to the depleted extracts restore the ability of these nuclei to replicate their DNA [94]. (iv) Expression of human lamin A or Xenopus lamin LB3 lacking their N-terminal (head) causes lamin aggregation and inhibition of the chain elongation phase of DNA replication [26, 27]. The resulting lamin aggregate contains PCNA and replication factor complex (RFC), which are required for the elongation phase of DNA replication, and did not contain XORC-2, XMCM-3 or DNA polymerase α, which are required for the initiation phase of DNA replication [27]. (E) A recent study demonstrated a potential direct role for lamins in DNA replication [95]. In nuclei assembled in vitro in Xenopus extracts lamin LB3 is closely associated with PCNA after nuclear envelope assembly. Addition of the carboxy tail domain of lamin LB3 to replicating nuclei inhibits DNA replication and causes PCNA displacement from the chromatin. Moreover, the Ig fold within the tail domain of Xenopus lamin LB3, human lamin A or human lamin B1 binds directly to PCNA in vitro, suggesting that PCNA binding to the Ig fold of lamins is required for lamin activity in DNA replication.
Lamins in transcription and splicing
In Drosophila interphase and polytene nuclei, regions of condensed chromatin with low levels of transcription are associated with the nuclear lamina [42]. In female mammalian cells the inactive X chromosome is closely associated with the nuclear lamina.
In addition, gene-poor chromosomes are peripherally located, whereas gene-rich chromosomes are more centrally located [96, 97]. By expressing Drosophila lamin Dm0 fused to the dam methylase, Pickersgill et al. have identified approximately 500 genes that were more frequently associated with lamin Dm0. These genes are clustered, transcriptionally silent and late replicating [44]. Similarly, expressing a fusion protein between the dam methylase and human lamin B1 or human emerin revealed more than 1300 large domains of 0.1–10 megabases in size that are more frequently associated with lamin B1 and emerin. These domains are gene-poor and the insulator protein CTCF separates their borders [43]. Several studies directly addressed the effects of lamins or their associated proteins on gene silencing using lacO arrays that were detected by LacI fused to either a fluorescence protein or to a myc-tag. Targeting a transcriptional activator to a nuclear periphery locus caused its migration roughly perpendicular to the nuclear envelope. The migration occurred within 1–2 hrs after transcription induction and was dependent on actin/myosin [98]. Tethering genes to the INM of mouse fibroblasts by fusing GFP-LacI to emerin lacking its LEM domain caused gene repression and accumulation of lamins A and B1 and LAP2 proteins to the chromatin domain [99]. Similarly, expressing Myc-tag LacI fused to LAP-2β caused tethering a LacO array on human chromosomes 4 and 11 to the nuclear periphery and reduction in the expression of some, but not all, genes. This repression of gene expression was dependent on HDACs activity [100]. In contrast, targeting genes to the human nuclear lamina by fusion of mCherry-LacI to lamin B1 did not affect their activity, and the kinetics of transcriptional induction at the nuclear lamina was similar to that observed at an internal nuclear region [101]. Possible explanations for the differences between these studies are the use of different promoters and cell culture systems, recruitment of different lamins (lamins B1 plus A versus lamin B1) or the recruitment of HDAC by the emerin and LAP-2β genes, which may affect gene expression regardless of lamins or nuclear position [69, 70].
Lamins are directly involved in regulating RNA polymerase II activity. Overexpression of lamins A or C in human HeLa cells causes reduction of RNA polymerase II-dependent expression in these cells [102]. Expression of human lamin A lacking their N-terminal (head) domain in BHK 21 cells or adding the mutant lamin A to an extract containing transcriptionally active Xenopus embryonic nuclei caused nucleoplasmic aggregation of A- and B-type lamins and specific inhibition of RNA polymerase II, but not RNA polymerase I or III [103].
Lamins also regulate specific transcription factors either by direct or indirect interaction through their binding partners. Lamins bind BAF both directly and through LEM domain proteins [60]. The lamin-LEM domain proteins-BAF complexes are required for nuclear assembly and chromatin organization [88]. However, recent studies suggest that BAF also functions as transcriptional repressor, where it binds specific promoters [104, 105]. The rod domain of human lamins A and C also binds directly to the transcription factor MOK-2 [106], and MOK-2 localization depends on lamins A and C [107]. Human lamin A also binds the SREBP-1 in vitro and this interaction is reduced when lamin A has a mutation in the Ig fold that causes familiar partial lipodystrophy [108]. The POU-domain protein OCT-1 is associated with the nuclear lamina and represses the collagenase gene. Interestingly, in aging cells OCT-1 dissociates from the nuclear lamina allowing the activation of the collagenase gene [109]. Another transcription factor that is confined to the nuclear lamina is insulin promoter factor-1 (IPF-1)/ pancreatic and duodenal homeobox-1 (PDX-1) [110]. At stimuli levels of glucose concentrations IPF-1/PDX-1 redistributes in the nucleoplasm. Lamin A also binds c-Fos transcription factor. The binding of Fos/Jun (AP-1) to DNA and its transcriptional activity are inhibited by its binding to lamin A in vitro. Lamin A suppresses the AP-1 complex in mammalian cells in vivo. The suppressed form of AP-1 is localized to the nuclear periphery and is misplaced in Lmna-null cells [111].
The pRb regulates both the entry into S-phase and terminal differentiation of cells. A-type lamins form protein complexes with LAP-2α, pRb and PP2A phosphatase. These complexes associate with E2F-dependent promoter sequences and regulate their transcription [112, 113]. Coil 2 in the rod domain of A-type lamins binds pocket C domain in the hypophosphorylated (repressive) form of pRb in vitro. Overexpression of lamin A mutants in cultured cells form aggregates that contain pRb, suggesting interaction between lamin protein complexes and pRb in vivo[114]. This interaction with A-type lamins stabilizes pRb, because in Lmna-null mouse cells and in liver cells of Zmpste24-null mice, pRb is rapidly degraded by the proteosomes, in a process that does not require the pRb degradation pathway of gankyrin and MDM2 [92, 115, 116]. Because lack of A-type lamins or the expression of disease-causing mutant lamin A affect the cell cycle, as well as differentiation of myocytes and adipocytes [117–119], it has been proposed that lamins A and C-LAP-2α complexes also control the balance between proliferation and differentiation of adult stem cells in tissue homeostasis and regeneration (see below) [120, 121]. Interestingly, while mouse fibroblasts derived from Lmna-null or LAP-2α-null cells showed accelerated proliferation, human dermal fibroblasts down-regulation of either lamins A and C or LAP-2α showed G1 cell cycle arrest [122]. Further studies are required to understand these differences.
Lamins also interact indirectly through lamin-associated proteins with many other transcription regulators. Some of the more studied interactions include lamin Dm0 and YA in Drosophila, which is required for the transition between meiosis and mitosis [reviewed in 15]. LAP-2p interacts with the transcription repressor germ cell-less [123] and HDAC-3 [69]. MAN-1 interacts with the receptor regulated SMADs (R-SMADs) in the transforming growth factor-β (TGF-p)/BMP pathway through the rSMAD RNA recognition motif, which inhibits rSMAD phosphorylation [reviewed in 124, 125].
The roles of lamins in RNA splicing are in debate. Using a specific lamin A antibody that preferentially binds lamin speckles, it has been shown in human and rat cells that both lamin A and lamin B colocalize with the splicing factors SC35 and U5–116 kD [126]. The size of the lamin-splicing factor speckles depended on the level of transcriptional activity and their formation occurred at anaphase [102]. In contrast, MEFs lacking lamins A and C have normal cellular distribution and morphology of splicing factor complexes and they retain their association with the nuclear matrix. In addition, the exchange rate of splicing factors between speckles and nucleoplasm remained unchanged, suggesting that the formation and maintenance of intra-nuclear splicing factor compartments is independent of lamins A and C [127]. Because lamins are thought to be a major constituent of the yet mysterious nuclear matrix, the potential roles of B-type lamins in formation and association of splicing factor complexes remain to be addressed.
Lamins and aging
In adult C. elegans the somatic cells stop dividing and the nuclear architecture in most non-neuronal cell types undergo progressive and stochastic age-dependent alterations [128]. These changes include deformation of nuclear shape, loss of peripheral heterochromatin and changes in localization of nuclear envelope proteins. At early days, nuclei appear round with nuclear envelope proteins (Ce-lamin, Ce-emerin and the NPC protein NPP-11) evenly distributed around the periphery. At later days, nuclei become convoluted and a fraction of these nuclear envelope proteins started to appear in foci. Over time there is a decrease in nuclear peripheral lamin, an increase in intra-nuclear lamin and in nuclei showing abnormal shape, extensive stretching and fragmentation (Fig. 4 shows data for Ce-lamin and Ce-emerin). Interestingly, although the classical aging pathway of the insulin/IGF-1-like/daf signalling probably does not affect the earlier changes in nuclear shape and re-distribution of nuclear lamina proteins, the later changes depend on this pathway where shortlived mutants are converted to the late phase at earlier days and long-lived mutant are converted to the late phase at later days [128]. Lamins are probably directly involved in determining lifes-pan in C. elegans, because lack of lamin activity at the adult stage, when somatic cells stop dividing, led to shortening of lifespan with the aging phenotypes appearing earlier. Interestingly, down-regulation of the two C. elegans INM LEM-domain proteins Ce-emerin and LEM-2/MAN-1 also causes reduced life span, suggesting that at least part of the effects of lamins on aging is mediated by LEM-domain proteins [128]. Because lifespan in C. elegans also depends on several other pathways [129], a goal for future studies would be to determine the epistasis relationships between the aging pathways and the nuclear lamina. The mutant fibroblasts in culture undergo age-dependent progressive changes in nuclear architecture, which are correlated with accumulation of the mutant lamin A isoform and are similar to changes observed in aging C. elegansadult cells [62].
Figure 4.
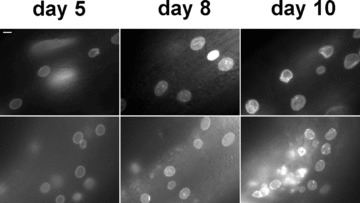
The nuclear lamina undergoes progressive changes in aging C. elegans. The nuclear lamina morphology in live C. elegans animals expressing GFP::Ce-lamin (upper panels) and Ce-emerin::GFP (lower panels) on days 5, 8 and 10 of development using a fluorescence microscope. Notice the folding and aggregations of the nuclear lamina in later days. Animals used in this experiment were grown at 20°C. Scale bar at the upper right corner is 10 μm and applies to all panels.
Another strong connection between lamins and aging was found when several premature aging diseases, including HGPS, atypical Werner syndrome and mandibuloacral dysplasia (MAD) were found to be the result of specific mutations in the LMNA gene (see below) [65].
Most HGPS cases are due to the silent mutation 1824C>T (G608G) in the LMNA gene. This mutation enhances a cryptic splice site, which results in LMNA mRNA lacking 150 bp and lamin A lacking 50 residues (progerin/LAΔ50). The missing residues include the second (more N-terminally) Zmpste24 cleavage site (Fig. 2), leaving the mutant protein permanently carboxy farnesy-lated and methylated. Progerin/LAΔ50 tends to accumulate in the nuclear periphery at later cell passages [130]. On the cellular level, the HGPS cells develop abnormal chromatin organization, a delay in nuclear assembly, binucleated cells and defects in chromosome segregation [130, 131]. Down regulating progerin with specific morpholino oligonucleotides reverses these cellular phenotypes [66].
Human beings with mutations in the ZMPSTE24 gene, as well as Zmpste24-null mice, develop early aging diseases, suggesting that the carboxy farnesylated and methylated prelamin A plays a major role in the pathogenesis of these diseases [132–134]. Based on these data, FTIs, originally developed to inhibit Ras farnesylation in cancer cells, were used to test the effects of farnesylation in HGPS or Zmpste24-null cells [115, 135]. Indeed, treating cell lines expressing progerin or lacking Zmpste24 activity with FTIs restore their shape and reverse the changes in heterochromatin markers. In addition, treating 5-week-old Zmpste24-null mice with FTIs improved their growth, body weight, life span and bone defects compared to untreated littermates [136–138]. These results have led to an ongoing clinical trial of the FTI Lonafarnib in HGPS children (see http://www.progeriaresearch.org/clinical_trial.html).
Interestingly, inhibiting farnesylation by FTIs causes geranyl-geranylation of the CAAX motif by geranylgeranyl transferase. Inhibition of both prenylations by a combination of statins and aminobisphosphonates in Zmpste24−/− mice increased median life span by 77%. Also the clinical features, including body weight and bone to tissue ratio, showed improvement [6]. In addition, down regulation of p53 targets, which are up-regulated in the mutant mice, was observed both in the liver and the heart. These data inspired another clinical trial in HGPS patients using the combination of statins and aminobisphosphonates. However, it remains to be seen whether the combination of the statin pravastatin and the aminobisphosphonate zoledronate will have similarly beneficial effects in the animal expressing progerin [6].
Recently, it has been shown that progerin is also expressed both at the mRNA and protein levels in cultures obtained from healthy human dermal fibroblasts and tends to accumulate in cells obtained from old individuals [139]. The accumulation of progerin is detected in cells with nuclear defects, similar to cells from HGPS patients or cells ectopically expressing progerin, and it is present in all binucleated cells [131]. The expression of progerin is also detected in skin biopsies from healthy individuals and the mutant protein tends to accumulates when people age [140]. Together with the results in C. elegans, it is likely that lamins play a key role in the aging process, which is conserved in evolution.
Lamins and laminopathies
Mutations in lamins and their associated proteins causing ‘laminopathies’
Most laminopathic diseases are caused by mutations in the LMNA gene including, (1) the muscle diseases autosomal dominant and recessive EDMD, limb girdle muscular dystrophy 1B, dilated cardiomyopathy 1A, (2) the adipocyte diseases autosomal dominant Dunnigan type familiar partial lipodystrophy with insulin resistance (with or without diabetes), a general lipodystrophy syndrome and type A insulin resistance syndrome without lipodystrophy, (3) the peripheral neuronal disease autosomal dominant Charcot-Mary-Tooth type 2B1 and (4) the accelerated aging diseases autosomal recessive MAD, the autosomal dominant HGPS, autosomal dominant atypical Werner syndrome and the recessive restricted dermopathy.
Mutations in the lamin A processing enzyme ZMPSTE24 also cause progeria syndrome and restrictive dermopathy. Interestingly, some patients with a single mutation in the LMNA gene suffer multiple laminopathies [reviewed in 32].
Duplication of LMNB1 gene causes adult-onset autosomal dominant leukodystrophy. The LMNB2 gene is associated with acquired partial lipodystrophy/Barraquer–Simons syndrome, because three nucleotide variants (including one in the first intron) were found in four patients, with a higher frequency than in the control population [141]. However, the evidence showing the relationship between mutations in LMNB2 and the disease are still too weak to fully establish the etiology of this disease. Mutations in lamin associated proteins also cause several heritable diseases. Loss of one copy of LBR causes Pelger-Huet anomaly and loss of both alleles of causes HEM/Greenberg dysplasia. Mutations in emerin, Nesprin-1 and Nesprin-2 cause EDMD, mutations in MAN-1 cause Buschke–Ollendorff syndrome, mutations in the LAP2 gene cause cardiomyopathy. Mutations in Nesprin-1 also cause cerebellar ataxia [reviewed in [32].
There are over 330 different disease-causing mutations in LMNA, which probably makes it the most mutated gene in the human genome. The mutations are spread throughout the gene with somewhat higher incidents in the head domain, coil 1A in the rod domain and the Ig fold in the tail domain and fewer incidents in the region between the end of the rod domain and the Ig fold, which is the region that binds to chromatin in vitro[142]. The only LMNA disease for which mutations appear to cluster in the Ig fold is partial lipodystrophy. In addition, although there are several mutations in LMNA leading to HGPS, most patients have a 1824C>T mutation (Gly608Gly) that creates an ectopic mRNA splicing site leading to the expression of progerin/LAΔ50 protein that contains eight residues of pre-lamin A and is permanently farnesylated (see section 5d).
Animal models for laminopathies
There are several animal models developed to study laminopathies. The most studied animals are mouse, C. elegans and Drosophila. Mouse with a knockout in Lmna proved to be a powerful tool in studying EDMD, cardiomyopathy and spermatogenesis [75, 135, 143]. The mouse containing the autosomal recessive mutation N195K is used as a model animal for cardiomyopathy [144] and mouse with the autosomal recessive mutation H222P is used as a model animal for both EDMD and cardiomyopathy. Several mice models were developed to study HGPS [145]. Homozygous deletion of exon 9 in Lmna causes early aging symptoms that resemble HGPS symptoms [146], and replacement of the two normal copies of Lmna with copies of the progerin isoform caused retarded post-natal growth, whereas the heterozygous mutation cause death at 6 months with osteoporosis and hair loss [147–149]. However, mouse with one copy of Lmna develops cardiomyopathy only after 12 month [150]. Similarly, while most human LMNA mutations have a dominant effect, the mouse Lmna mutations show similar phenotypes only when recessive or, in some cases, when the mice are aging. Zmpste24-null mice suffer from rib fractures, osteoporosis and muscle weakening and they die after 7 months. A gene trap in Lmnb1 results in partial or complete loss of lamin B1 activity. Mice homozygous for the deletion have respiratory failure and die [151].
Mouse models were also generated for lamin-associated proteins. Loss of the emr-1 gene resulted in apparently normal mice with minimal motor and cardiac dysfunctions that probably result from retarded muscle regeneration [152, 153]. Mutations in the mouse LBR gene result in ichthyosis [154]. The LBR-null mouse show changes in the granulocyte nuclear shape similar to changes in human granulocytes Pelger-Huet anomaly [154, 155]. A gene trapping in the MAN-1 gene causes early embryonic lethality by day 10.5 [156]. Abnormal TGF-β signalling pathway probably causes defective vascularization of the yolk sac, which is probably the cause of death. Many other mouse models for the human laminopathic disease are currently being developed around the world and should be useful for understanding the mechanisms and for developing drugs to treat these diseases.
The simple, yet evolutionary conserved, composition of the nuclear lamina in C. elegans makes this organism an attractive model for studying the basic functions of nuclear lamina genes, as well as understanding the mechanisms behind the laminopathic diseases. C. elegans has a single lamin gene that behaves both as B- and A-type lamin [16, 73]. Mutations in conserved residues of the rod and tail domains known to cause various laminopathies in human were introduced into Ce-lamin and their effects at cellular level in vivo and on lamin filament assembly in vitro were studied (see above) [22]. Transgenic animals expressing laminopathic mutations showed abnormal lamin distribution, abnormal nuclear shape or change in lamin mobility in some or all cells of the animal, similar to the phenotypes of laminopathic mutations in human cells [22]. Down-regulation or deletion mutations in emerin and LEM-2/MAN-1 homologues in C. elegans revealed overlapping functions for these LEM-domain proteins. Further analyses of these mutants revealed age-dependent muscle deterioration, similar to the EDMD disease in human (R.B. and Y.G., unpublished observations).
Drosophila is the only known invertebrate with separate genes encoding both A- and B-type lamins. Expression of lamin C constructs containing evolutionary conserved disease-causing mutations, led to phenotypes that resemble laminopathies [157]. Flies containing a P-element insertion in the Drosophila B-type lamin showed many different phenotypes including an impaired locomotion [74]. Induced expression of the Drosophila B-type lamin caused aberrant nuclear shapes and reduced the lifespan of adult flies, which correlates with an early decline of locomotion activity [158].
Molecular models for laminopathies
It is still an enigma why mutations in LMNA cause many diverse diseases. Four different, not mutually exclusive, models attempt to explain the mechanisms leading to these diseases. The first model suggests that changes in nuclear mechanics and mechano-trans-duction response contribute to the laminopathic diseases. Lamins A and C were shown to be important for the mechanical properties of the nucleus. For example mouse cells lacking lamins A and C have increased nuclear deformation, their viability under mechanical strain is reduced and their mechanical stress-dependent gene expression is impaired [76, 80]. In addition, lack of lamins A and C affects the interactions between the nucleus and the cytoskeleton networks [159]. In many laminopathic diseases cells have increased nuclear deformation. In HGPS cells the nuclear lamina has a significantly reduced ability to rearrange following mechanical stress and nuclei become progressively stiffer with increasing passage number [89, 160]. However, in contrast to the mice cells lacking lamins A and C, HGPS cells show similar resistance to acute mechanical stress [160]. Treating these cells with FTIs restored nuclear stiffness and accelerated the wound healing, but did not restore cellular sensitivity to mechanical strain [89].
The second model proposes that lamins A and C are involved in regulating cell type-specific gene expression [161]. According to this model mutations in lamins A and C cause misregulation of different tissue-specific genes, either directly or at the epigenetic level. Indeed, the activity of many different regulatory proteins depend on their interactions with lamins A and C including pRb, OCT-1, MOK-2, SREBP-1 and others [reviewed in 162, 163].
In addition, complete loss or mutations in lamins A and C or loss of C. elegans Ce-lamin cause significant changes in chromatin and heterochromatin organization. Recently, it has been shown that there are changes in the mitogen-activated protein kinase (MAPK) signalling cascade in the mouse model for cardiomyopathy in which lamins A and C have the H222P mutation [164]. In these mice ERK and Jun N-terminal kinase (JNK) are activated prior to the onset of cardiomyopathy. Ectopic expression of the mutant lamins A and C in cultured cells causes the activation of ERK and JNK signalling. Activation of ERK was also found in the heart of mice lacking emerin [164]. Systemic treatment of H222P mice with an ERK inhibitor delayed cardiomyopathy and the mutant mice were indistinguishable from wild-type mice [165].
The third model also suggests that lamins A and C are involved in regulating cell type-specific gene expression affecting adult stem cells differentiation [166]. Evidence for this model appears below in the ‘Lamins and stem cells’ section.
The last model suggests that mutations in ZMPSTE24 or in LMNA cause permanent farnesylation of lamin A that is toxic to cells (see above). The toxic lamin A causes changes in nuclear morphology, chromatin organization and gene expression. In support of this model, treating HGPS fibroblast cells or mouse models for the HGPS disease ameliorate the phenotypes (see above).
Lamins and stem cells
Unlike B-type lamins which are ubiquitously expressed in all mammalian cells and tissues throughout development, lack of lamins A and C expression is one of the defining factors of undifferentiated embryonic stem cells (ESCs) of both mouse and human origin [167]. This could provide one explanation for the diffuse and dispersed nuclear lamina structure observed in undifferentiated ESCs as compared to partially differentiated cells, which express lamins A and C [168]. Although the exact regulation of lamin A deposition in the nuclear lamina during ESC differentiation has not been studied, there is a growing body of evidence linking lamin A to somatic stem cell regulation and differentiation.
Somatic (or adult) stem cells are tissue-specific stem cells that, unlike the highly proliferative and pluripotent ESCs, are usually slowly self-renewing or quiescent. They are more restricted than ESCs in their differentiation capacity, and often give rise to specific lineages of usually only one germ layer origin. Somatic stem cells can either replenish tissues constantly, such as the situation in the gut, or can be maintained as a small slowly proliferating reservoir that, when needed, provides a source of tissue replenishment and maintenance, especially following damage. Somatic stem cells were identified in a wide and growing number of tissues, such as blood, bone, intestine, muscle, skin, brain and hair, to name a few, and they seem to be highly dependent on their niche, from which they receive signals influencing their fate [169]. Interestingly, the signalling pathways that operate in the different stem cell niches converge to four main pathways, namely Notch, Wnt, TGF-β and Sonic hedgehog [170], suggesting common mechanisms for the regulation and maintenance of adult stem cells of different origins [171].
The first hint that lamin A has a role in cellular differentiation and commitment came from work on muscle progenitor cells, demonstrating that the R453W EDMD lamin A mutation interferes with muscle progenitor differentiation [117]. Mouse C2C12 cells are capable of differentiation from myoblasts to myotubes, and expression of a R453W mutant lamin A significantly inhibited differentiation. Overexpression of wild-type lamin A also caused a partial delay in C2C12 differentiation. The effects of mutations in lamin A were also tested in the mouse 3T3-L1 cells that are capable of differentiation from fibroblasts to adipocytes [119]. Lipid accumulation was inhibited by expression of the familiar partial lipodystrophy mutations R482Q or R482W, as well as overexpression of wild-type lamin A. The role of lamin A in adipocyte differentiation was also observed in Lmna−/− fibroblasts, as these cells differentiated more readily to fat-containing cells. Therefore, lamins A and C seem to be effective inhibitors of adipocyte differentiation. Similar results were obtained from lamins A and C knockdown in human mesenchymal stem cells (MSCs). Lack of lamins A and C inhibited osteoblast differentiation and facilitated the differentiation of MSCs into adipocytes [172]. Collectively, these results highlight the connection between lamins, especially lamin A, and somatic stem cells, and demonstrate that mutations in lamin A cause somatic stem cell dysfunction. We will now turn to examine more closely the specific molecular pathways that are involved.
The Notch pathway
Notch is a highly conserved and ubiquitous cell-signalling pathway operating through four different transmembrane Notch receptors (Notch1–4). Ligand binding to the extracellular domain of Notch induces proteolytic cleavage of Notch and release of Notch intracellular domain (NICD) to exert changes in gene expression in the nucleus of Notch target genes. Conspicuous activation of the Notch signalling pathway was found in stable cell lines carrying inducible forms of progerin/LAΔ50 [173]. This effect was specific to the Notch pathway and seemed to be induced by the Notch co-activator Ski-interacting protein. Because the main tissues affected in progeria are of mesenchymal origin, the authors turned to examine human MSCs. Once again, when progerin/LAΔ50 was introduced into the MSCs, the Notch pathway was activated and the cells spontaneously differentiated along all three germ layers. When the cells were subjected to directed differentiation along the mesenchymal lineages, osteogenesis was enhanced, adipogenesis was inhibited and chondrogenesis was unperturbed. These results demonstrate the involvement of MSC regulation in the pathology of HGPS through aberrant Notch signalling.
The Wnt/β-catenin pathway
The other major pathway implicated in the stem cell-lamins A and C connection is Wnt signalling pathway. Although the Wnt pathway involves a very large number of different proteins, the canonical Wnt pathway involves binding of Wnt proteins to cell-surface receptors of the Frizzled family, causing the receptors to activate dishevelled family proteins, resulting in regulation of the concentration of β-catenin inside the nucleus. The Wnt pathway was previously found to be a master regulator of stem cell self-renewal and cancer [174] and has been studied extensively in well-characterized stem cell niches, including the intestine, blood, brain and epidermis, where it normally promotes stem cell proliferation [170]. The Wnt signalling pathway was shown to regulate hair follicle stem cells residing in the hair bulge in a mouse model of progeria [175]. Zmpste24−/−-mice, which display age-related nuclear lamina defects and progeroid-like symptoms [176], exhibited a significantly increased number of resident hair bulge stem cells and decreased proliferative potential. These cells further accumulated unprocessed prelamin A and displayed altered nuclear architecture [175]. Analysis of the signalling pathways in these cells revealed an almost complete absence of both the tran-scriptionally active form of β-catenin as well as Mitf, which is known to bind β-catenin and regulate melanocyte stem cells. Thus, the decreased epidermal stem cell proliferation observed in the Zmpste24−/−- mice is likely a result of the absence of Wnt signalling in these cells. In Zmpste24−/−- Lmna+/− mice, no accumulation of prelamin A was observed, and all of the aberrant phenotypes at the cellular and organismal levels were rescued, suggesting that the anomalous signalling pathways and stem cell dysfunction are mediated largely, if not solely, by prelamin A and/or defects in the nuclear lamina, directly linking disease genetics and phenotypes with stem cell function.
Other pathways
The TGF-β/Smad pathway is another important regulator of mesenchymal tissue homeostasis [177]. Similar to Wnt and Notch, TGF-β pathway operates through a transmembrane receptor which, when bound to extracellular TGF-β, releases intracellular Smad proteins, regulating gene expression in the nucleus. A-type lamins have been found to repress TGF-β/Smad-dependent gene expression in MEFs [113] and a decrease in the expression of TGF-β signalling in MSCs of aged mice induces depletion of osteoblasts in the bone matrix [178]. This mode of regulation resembles the situation in MSCs of Lmna-deficient mice and Lmna-knockdown human MSCs, discussed above, where differentiation was inhibited.
The Rb pathway can also cooperate with a number of tissue-specific transcription factors in order to regulate tissue-specific differentiation. The Rb/MyoD pathway has a well-established role in myogenesis of satellite myoblasts whereby the interaction of pRb with the myogenic transcription factor MyoD leads to the activation of MyoD-dependent target genes and initiation of muscle differentiation [179]. The effects of lamins A and C, which interact with pRb (see above) on muscle progenitor differentiation were verified in mice lines that are heterozygous or homozygous for a deletion in the Lmna gene. These mice displayed delayed muscle stem cell differentiation kinetics while ectopic expression of MyoD in the mutant lines partially restored the differentiation defects [118]. Similarly, two further studies have implicated a deregulated Rb/MyoD pathway as a cause of decreased differentiation potential in A-type lamin or emerin null regenerating muscle using mRNA expression profiling [152, 180].
Although the connection between lamin A and stem cells is only beginning to emerge, the examples discussed above strongly implicate lamin A and prelamin A in regulating stem cell maintenance and differentiation by influencing key signalling pathways in stem cells.
Lamins and cancer
Malignant transformation is a multi-step process of sequential alterations in the critical genes involved in cancer regulating pathways and the various mediators, which conduct the ‘crosstalk’ between them. These molecular events are accompanied, among others, by nuclear structural alterations, which differentiate cancer cells from normal cells. Alterations of the nuclear lamina are currently emerging as an additional event involved in malignant transformation. The presumed association is multi-factorial and affects tumorigenesis from its initial step to its advanced stage of metastatic spread. Lamins involvement in cancer-associated processes has been related, mainly, to their role as guardian of the nuclear architecture, their role in regulating basic nuclear activities that are implicated in tumorigenesis, their multiple interactions with major cancer gene pathways and their role in chromosomal segregation control with its resultant impact on aneuploidy.
Lamins as biomarkers for cancer
The expression of lamins in haematological malignancies has been recently reviewed [181]. Here we review the current knowledge on lamins expression patterns in non-haematological malignancies (Tables 1–4), which include, mainly, altered expression (reduced, undetectable, or elevated) and/or aberrant localization of lamins A and C. Although changes in B-type lamin expression have been observed in some cancer subtypes, one or both B-type lamins are always present in every human cell, including cancer cells. Various correlations were found between altered lamins expression and tumour subtypes and characteristics, as will be detailed below. Thus, lamins expression in tumour cells may potentially serve as a cancer-related biomarker for diagnosis, prognosis and surveillance.
Table 1.
Lung cancer: nuclear expression patterns [a–d] of lamins (positive samples/total number of tested samples)
| Tumour/method | Lamins A and C | Lamin B-type |
|---|---|---|
| Lung adenocarcinoma/IPO | 5/10 (a) | 3/10 (a) |
| 7/10 (c) | ||
| Lung squamous cell carcinoma/IPO | 8/13 (a) | 9/13 (a) |
| 1/13 (c) | 4/13 (c) | |
| Lung non-small cell carcinoma/IPO* | 0/3 (a) | 1/3 (a) |
| 1/3 (c) | 0/3 (c) | |
| 2/3 (d) | 2/3 (d) | |
| Lung small cell carcinoma/IPO | 1/15 (a) | 15/15 (a) |
| 7/15 (c) | ||
| Lung carcinoids/IPO | 2/6 (a) | 6/6 (a) |
| 2/6 (c) | ||
| Lung non-small cell carcinoma cell lines/IB | 4/4 (a) | 4/4 (a) |
| Lung small cell carcinoma cell lines/IB | 5/5 (c) | 5/5 (a) |
| Lung small cell carcinoma cell lines/IPO | 10/16 (c) | 16/16 (a) |
| 6/16 (d) | ||
| Lung adenocarcinoma cell lines/IPO | 3/4 (a) | 4/4 (a) |
| 1/4 (c) | ||
| Lung squamous cell carcinoma cell line/IPO | 1/1 (a) | 1/1 (a) |
| Lung large cell carcinoma cell line/IPO | 1/1 (a) | 1/1 (a) |
IPO – Immunoperoxidase, IB – Western/immunoblotting, (a) positive staining, (b) elevated staining/levels, (c) reduced staining/levels (d) no staining.
Lamin B1 only was evaluated.
Table 4.
Genitourinary, gynaecologic and breast cancer: nuclear expression patterns [a–d] of lamins (positive samples/total number of tested samples)
| Tumour/method | Lamin A | Lamin C | Lamins A and C | Lamin B1 | Lamin B2 |
|---|---|---|---|---|---|
| Testicular Seminoma/lPO | 2/16 (a) | nr | *1/16 (a) | 16/16 (a) | *14/15 (a) |
| 14/16 (d) | *15/16 (d) | 0/16 (d) | *1/15 (d) | ||
| ………. | ……….. | ||||
| **6/16 (a) | **16/16 (a) | ||||
| **10/16 (d) | ** 0/16 (d) | ||||
| Testicular embryonal carcinoma/IPO | 0/7 (a) | nr | *9/10 (a) | 7/7 (a) | *10/10 (a) |
| 7/7 (d) | *1/10 (d) | 0/7 (d) | *0/10 (d) | ||
| ……….. | ………… | ||||
| **8/10 (a) | **10/10 (a) | ||||
| **2/10 (d) | ** 0/10 (d) | ||||
| Testicular yolk sac tumour/IPO | 5/5 (a) | nr | * 5/5 (a) | 5/5 (a) | *6/6 (a) |
| 0/5 (d) | *0/5 (d) | 0/5 (d) | *0/6 (d) | ||
| ……….. | ……….. | ||||
| **6/6 (a) | **6/6 (a) | ||||
| **0/6 (d) | **0/6 (d) | ||||
| Testicular choriocarcinoma/lPO | 5/5 (a) | nr | *5/5 (a) | 5/5 (a) | *5/5 (a) |
| 0/5 (d) | *0/5 (d) | 0/5 (d) | *0/5 (d) | ||
| ………… | ………… | ||||
| **4/5 (a) | **5/5 (a) | ||||
| **1/5 (d) | **0/5 (d) | ||||
| Testicular teratoma/lPO | 4/4 (a) | nr | *4/5 (a) | 4/4 (a) | *5/5 (a) |
| 0/4 (d) | *1/5 (d) | 0/4 (d) | *0/5 (d) | ||
| ………… | ………. | ||||
| **4/4 (a) | **6/6 (a) | ||||
| **0/4 (d) | **0/6 (d) | ||||
| Carcinoma in situ: seminoma_ /IC | 0/4 (a) | nr | *0/6 (a) | 8/8 (a) | *7/10 (a) |
| 4/4 (d) | *6/6 (d) | 0/8 (d) | *3/10 (d) | ||
| ………… | ………… | ||||
| **2/6 (a) | **6/9 (a) | ||||
| **4/6 (d) | **3/9 (d) | ||||
| Carcinoma in situ: non-seminoma___/IC | 0/5 (a) | nr | *0/8 (a) | 6/9 (a) | *11/11 (a) |
| 5/5 (d) | *8/8 (d) | 3/9 (d) | *0/11 (d) | ||
| ………… | ………… | ||||
| **0/7 (a) | **8/9 (a) | ||||
| **7/7 (d) | **1/9 (d) | ||||
| Prostate cancer/IPO | nr | nr | 1/4 (a) | 1/4 (a) | nr |
| 3/4 (c) | 3/4 (c) | ||||
| 0/4 (d) | 0/4 (d) | ||||
| Prostate cancer/IB | nr | nr | 32/32 (c) | 37/37 (b) | nr |
| Cervical squamous cancer/IPO | nr | nr | 0/3 (a) | 1/3 (a) | nr |
| 0/3 (c) | 2/3 (c) | ||||
| 3/3 (d) | 0/3 (d) | ||||
| Uterine adenocarcinoma/IPO | nr | nr | 0/3 (a) | 1/3 (a) | nr |
| 0/3 (c) | 1/3 (c) | ||||
| 3/3 (d) | 1/3 (d) | ||||
| Ovarian cancer/PM and IB | nr | nr | 30/30 (b) | nr | nr |
| Breast adenocarcinoma/IPO | nr | nr | 0/3 (a) | 0/3 (c) | nr |
| 3/3 (c) | 3/3 (c) | ||||
| 0/3 (d) | 0/3 (d) | ||||
| NT2/D1 cell line/IB | nr | nr | present | present | nr |
| Hela cell line/IB | present | present | present | present | present |
*/** Evaluation of lamins A/C and B2 was done by two different
antibodies-yielding two results
_ Carcinoma in situ adjacent either to seminoma or __ non-seminoma
IPO– Immunoperoxidase, IB– Western/immunoblotting,
PM– Protein microarrays.NT2/D1– Testicular embryonal cell carcinoma cell line.
Hela– Cervial cancer cell line.
(a) positive staining, (b) elevated staining/levels, (c) reduced staining/levels, (d) no staining.
Altered lamin expression in cancer cells contributes, presumably, to the characteristic changes in nuclear architecture, including alterations of nuclear structure and chromatin texture, which occur in all cancer subtypes. Because lamins interact with chromatin, changes in nuclear contour might contribute to heterochromatin re-organization. Morphologically these pathognomonic changes manifest, among others, as alterations in nuclear size and shape, and in frequent appearance of irregular nuclear borders which are observed along with altered heterochromatin organization. It is of note that the morphological evaluation of tissues and cells has indeed remained the ‘gold standard’ for cancer diagnosis, providing, occasionally, even prognostic clues [182].
Lung cancer
Lamins expression has been evaluated in three major sub-types of human lung cancer: small cell lung cancer (SCLC), non-small cell (non-SCLC) squamous and adenocarcinoma (Table 1). In SCLC cell lines lamins A and C levels diminished by >80%, whereas in non-SCLC cell lines lamins A and C were readily detected. The levels of B-type lamins were roughly equal in both cell lines [183, 184]. In the latter study A-type lamins were absent or only weakly expressed in almost all SCLC cell lines, and expressed in all non-SCLC cell lines (Table 1). Similar patterns of expression were reported in SCLCs and non-SCLCs pathological sections. Frozen sections of lung cancer showed that most SCLC cells are either null or weakly expressing A-type lamins, whereas frozen sections of non-SCLC samples always expressed A-type lamins. Noteworthy, in some samples lamins A and C were detected also in the cytoplasm (Table 1). Decreased expression of B-type lamins was found in non-SCLC sections, particularly in adenocarcinoma [184] (Table 1). An additional study evaluating only three tissue samples has found reduced expression of lamins A, C and B1 in the non-SCLC tissue samples [185].
Gastrointestinal cancer
Studies in gastrointestinal neoplasms have found reduced or no expression of lamins A and C in all specimens (17 of 17) of primary colon adenocarcinomas, and in 7 of 8 primary gastric adenocarcinoma specimens [185]. Lamin B1 expression was reduced in all the 17 colonic specimens and in 6 of 8 of the gastric specimens. Aberrant cytoplasmic expression of lamins A and C occurred, occasionally, in both colonic and gastric specimens (12% and 38%, respectively). Aberrant lamin B1 expression occurred occasionally in the colonic specimens (18%) [185]. In contrast, a recent large-scale study in colorectal cancer patients showed conflicting results where lamins A and C expression was found in 463 out of 656 evaluated patients (70%). Interestingly, the expression of lamins A and C in the tumours correlated significantly with colorectal cancer related deaths [186] (Table 2). Expression of lamins A and C and B1 has been found to be reduced in most pre-malignant samples examined, such as gastric dysplasia and colonic adenoma[185].
Table 2.
Gastrointestinal cancer: nuclear expression patterns [a–d] of lamins (positive samples/total number of tested samples)
| Tumour/method | Lamins A and C | Lamin B1 |
|---|---|---|
| Oesophageal cancer: squamous/IPO | 3/6 (a) | 3/6 (a) |
| 0/6 (c) | 3/6 (c) | |
| 3/6 (d) | 0/6 (d) | |
| Oesophageal cancer: adenocarcinoma/IPO | 0/2 (a) | 1/2 (a) |
| 1/2 (c) | 0/2 (c) | |
| 1/2 (d) | 1/2 (d) | |
| Oesophageal cancer cell lines/PM | 3/3 (b) | nr |
| Gastric adenocarcinoma/IPO | 1/8 (a) | 2/8 (a) |
| 3/8 (c) | 2/8 (c) | |
| 4/8 (d) | 4/8 (d) | |
| Colonic adenocarcinoma/IPO | 0/17 (a) | 0/17 (a) |
| 3/17 (c) | 10/17(c) | |
| 14/17(d) | 7/17 (d) | |
| Colorectal cancer/IPO | 463/656 (a) | nr |
| 193/656 (d) | ||
| Pancreatic cancer/IPO | 3/3 (a) | 3/3 (a) |
| Hepatocellular cancer/IPO* | 5/5 (a) | 5/5 (a) |
IPO – Immunoperoxidase, PM – Protein microarrays.
(a) positive staining, (b) elevated staining/levels, (c) reduce staining/levels (d) no staining.
B type lamins were evaluated.
In a small cohort of patients with oesophageal squamous and adenocarcinomathere was reduced expression of both lamins A and C and B1 [185] (Table 2). Recently, a proteomic-based technology was applied for detecting malignant-related proteins in oesophageal squamous cell carcinoma (SCC) cell lines. Lamins A and C were detected among the up-regulated proteins, with more than fivefold differences between oesophageal cancer cell lines and an immortal cell line [187] (Table 2). Further studies are warranted to elucidate these findings.
Reduced lamins expression is not present in all gastrointestinal cancers. In numerous specimens of pancreaticand hepatocellular tumours, the expression of lamins A, C and B1 lamins was found to be normal [185, 188] (Table 2).
Skin cancer
Reduced or null expression of lamins A and C in basal cell carcinomas (BCC) was correlated with a rapid growth rate within the tumour, whereas absence of lamin C only was correlated with a slow growth rate within the tumour [189, 190]. In contrast, another study has found the presence of significant amounts of lamins A and C in proliferating BCC cells [191]. Methodological differences between the two studies may account for these diverse results. Lamins B1 and B2 were expressed in almost all BCCs specimens in all three studies [189] (Table 3).
Table 3.
Skin cancer: nuclear expression patterns [a–d] of lamins (positive samples/total number of tested samples)
| Tumour/method | Lamin A | Lamin C | Lamins A and C | Lamin B1 | Lamin B2 |
|---|---|---|---|---|---|
| BCC/ IF | 3/16 (a) | 7/16 (a) | 7/16 (a) | 16/16 (a) | 15/16 (a) |
| 3/16 (b) | 4/16 (b) | 7/16 (b) | 0/16 (b) | 0/16 (b) | |
| 3/16 (c) | 0/16 (c) | 1/16 (c) | 0/16 (c) | 1/16 (c) | |
| 7/16 (d) | 5/16 (d) | 1/16 (d) | 0/16 (d) | 0/16 (d) | |
| BCC/IPO | nr | nr | 0/10 (a) | 9/10 (a) | 10/10 (a) |
| 1/10 (b) | 1/10 (b) | 0/10 (b) | |||
| 8/10 (c) | 0/10 (c) | 0/10 (c) | |||
| 1/10 (d) | 0/10 (d) | 0/10 (d) | |||
| BCC/IPO | 15/15 (a) | 0/15 (a) | nr | 0/15 (a) | 15/15 (b) |
| 14/15 (b) | 14/15 (b) | ||||
| 1/15 (c) | 1/15 (c) | ||||
| 0/15 (d) | 0/15 (d) | ||||
| Bowen’s carcinoma/IPO | nr | nr | 0/6 (a) | 5/6 (a) | 3/6 (a) |
| 5/6 (b) | 1/6 (b) | 2/6 (b) | |||
| 1/6 (c) | 0/6 (c) | 1/6 (c) | |||
| 0/6 (d) | 0/6 (d) | 0/6 (d) | |||
| SCC well diff/IPO | 4/11 (a) | 1/11 (a) | 0/11 (a) | ||
| nr | nr | 7/11 (b) | 4/11 (b) | 0/11 (b) | |
| 0/11 (c) | 6/11 (c) | 7/11 (c) | |||
| 0/11 (d) | 0/11 (d) | 4/11 (d) | |||
| SCC moderate diff/IPO | 4/7 (a) | 3/7 (a) | 0/7 (a) | ||
| nr | nr | 1/7 (b) | 3/7 (b) | 5/7 (b) | |
| 2/7 (c) | 1/7 (c) | 1/7 (c) | |||
| 0/7 (d) | 0/7 (d) | 1/7 (d) | |||
| SCC poorly diff/IPO | 1/7 (a) | 4/7 (a) | 5/7 (a) | ||
| nr | nr | 2/7 (b) | 3/7 (b) | 2/7 (b) | |
| 3/7 (c) | 0/7 (c) | 0/0 (c) | |||
| 1/7 (d) | 0/7 (d) | 0/0 (d) | |||
| SCC of lips well diff/IPO | 3/5 (a) | 0/5 (a) | 0/5 (a) | ||
| nr | nr | 0/5 (b) | 1/5 (b) | 0/5 (b) | |
| 1/5 (c) | 4/5 (c) | 4/5 (c) | |||
| 1/5 (d) | 0/5 (d) | 1/5 (d) | |||
| SCC/IPO | 10/10 (a) | 10/10 (b) | nr | 10/10 (b) | 10/10 (b) |
BCC – basal cell carcinoma, SCC – squamous cell carcinoma, IF – immunofluorescence, IPO – immunoperoxidase.
(a) positive staining in all cells, (b) positive staining in most cells, (c) positive staining in part of the cells, (d) no staining.
nr – not reported.
In SCC the expression of lamins A and C was greatly reduced in some specimens of poorly and moderately differentiated SCCs [190]. In another study, lamins A and C were detected in all SCC specimens evaluated, though the stages of differentiation were not specified [191]. Lamins B1 and B2 were expressed at high or medium levels in poorly and moderately differentiated SCC and were reduced in well-differentiated SCCs [190] (Table 3).
Genitourinary cancer
Evaluation of lamins expression in testicular germ cell tumours is currently based on a single large scale study [192]. The cancers evaluated were seminomas and non-seminoma tumours including embryonal carcinoma (EC), teratoma (TE), yolk sac (YS) and choriocarcinoma(CH). In testicular germ cell tumours, expression of lamins A and C was found to be dependent on the degree of differentiation, whereas B-type lamins were always expressed. Carcinomas in situ were most often negative for lamins A and C. Differentiated non-seminomas were positive for lamins A and C, whereas EC were positive for lamin C and negative for lamin A. Seminomas were found to be negative for lamins A and C [192] (Table 4).
Evaluation of lamins expression in prostate cancer was addressed by two studies. The first study used immunohistochemistry techniques and found reduced expression of lamins A, C and B1 in three out of four patients with prostatic cancer [185]. The other study used immunoblot assays and reported 37 prostatic cancer patients with a significant increase in lamin B1 and no significant change in lamins A and C [193]. The increase in lamin B was strongly correlated with the Gleason prognostic score (Table 4).
Gynaecological cancers
In cervical and uterine cancers lamins A and C expression was not detected. Lamin B1 expression was reduced in two out of three cervical and uterine cancer specimens [185] (Table 4). High-density protein microarrays demonstrated an increased expression of lamins A and C, with no change in their cellular localization in ovarian cancer specimens. This study also found an increased expression of lamins A and C in other cancers tissues, e.g.breast and lungs (with no subtype specification), and even in a subset of healthy tissues, e.g.renal and liver specimens [194] (Table 4).
Breast cancer
Several groups evaluated lamins expression in breast cancer. An immunohistochemistry assay found reduced expression of lamins A, C and B1 in breast cancer specimens of three patients [185], while a microarray assay detected increased expression of lamins A and C in patients with breast cancer [194]. Recently, an interesting study has used immunostaining of lamin B and emerin to identify irregular folding and deep invaginations in the nuclear envelope in breast cancer tissue. An increase in the degree of folding and invagination was well correlated with an increased grade of the breast cancer [195].
Thyroid carcinoma
Papillary thyroid carcinoma (PTC) has characteristic nuclear shape irregularities appearing in conjunction with loss of heterochromatin.
Surprisingly, in a single reported study of 14 PTC patients no change was found in the expression or localization of lamins A, C and B1, or LAP2 and LBR. Similar results were found in 4 patients with follicular thyroid carcinoma. The authors suggested that changes in the nuclear morphology in PTC cells is a consequence of disrupted balance between forces exerted on the nuclear envelope from the inside (chromatin or nuclear matrix) and the outside (cytoskeleton) [196]. The complex and diverse findings presented above still support the correlation between altered lamins A and C expression and various types of human cancer. Moreover, the altered expression was found to be correlated with tumour subtype and proliferation rate, e.g.lung cancer; tumour subtype and differentiation, e.g.testicular cancer, SCC of the skin; and with an increased invasive potential of the tumour, e.g.colorectal cancer. Expression of lamin B was even correlated with the Gleason’s grading score of prostatic cancer. The medical value of these correlates has to be evaluated in further large-scale clinical studies. Nevertheless, as proliferation rate of a tumour, its differentiation status, invasive potential and grade, define, in clinical terms, its aggressiveness – altered lamins expression, correlating with these parameters, may potentially serve as an additional significant cancer biomarker.
Lamins and cancer regulating pathways
The process of tumorigenesis is initiated when a replication-competent cell acquires a mutation in a gate-keeping pathway controlled by tumour suppressor gene such as pRb or Ademomatous Polyposis Coli (APC). The nuclear lamins ‘protect’ both pathways, thus yielding an anti-cancerous effect (see above). Nuclear lamins are also involved in tumour suppressive pathways that trigger apoptosis or senescence. Thus, lamins alterations allow cancer cells to escape the normal control of cell proliferation and cell death yielding a pro-cancerous effect (Fig. 5) [197].
Figure 5.
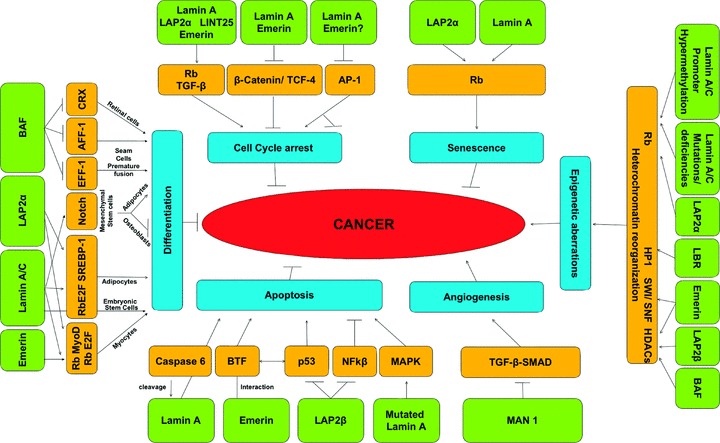
A proposed model for the involvement of the nuclear lamina in tumorigenesis. Nuclear lamins and lamin-associated proteins (green boxes) interact with major cancer-regulating pathways and their target proteins (orange boxes), mediating cell-fate decisions (blue boxes). This substantial cross-talk may yield anti-cancerous or pro-cancerous effects. The disparate net results, which have been found in various tumours, may be tissue, organ and/or tumour specific. Positive regulatory directions are denoted by arrowheads, negative regulatory by bars and ambiguous regulatory directions by straight lines.
Another level of lamins involvement in malignant transformation relates to epigenetic changes regarding DNA hypermethylation and heterochromatin reorganization. Epigenetic silencing of the lamins A and C gene by CpG island promoter hyper-methylation, has been found to be responsible for the loss of expression of A-type lamins in leukemia and lymphoma and to be associated with poor outcome in diffuse large B-cell lymphomas [198]. Lamins associated changes in pRb-dependent heterochromatin organization contribute to the permanent silencing of genes that promote proliferation, and might therefore be important for limiting the development of malignant tumours [182]. Finally, lamin A plays an intriguing role in seeding and growth of satellite cancer lesions, e.g.colon cancer. Presumably, lamin A causes a downstream up-regulation of T-plastin, which in turn leads to down-regulation of E-cadherin. Plastins are a recently described family of actin-bundling proteins, which have been implicated in invasion and metastasis [186, 199]. The complex interactions of lamins with the pRb, p53, SMAD and APC pathways (see above) and with their ‘cross-talk’ mediators (e.g. Wnt, E-cadherin) are the basis for the presumed intersections between nuclear lamina defects, cell cycle misregulation, and malignant transformation (Fig. 5). The up-regulation of p53 pathway in lamin mutant cells of HGPS patients suggests that these cells would be more cancer resistant, and promote senescence and shortened lifespan. The presence of cancer in two sporadic cases of HGPS patients reported to be suffering from osteosarcoma [200] can be explained on the basis of a rearranged p53 gene, as has been found in osteosarcoma cells [201]. Mutations in SMAD4 occur in 50% of human pancreatic cancers. SMADS are also involved in many other types of cancer and in the metastatic and angio-genetic processes [202–204].
APC is a tumour suppressor gene. In APC mutant cells, β-catenin binds to the sequence-specific DNA-binding proteins of the TCF/LEF family, thereby activating transcription of Wnt target genes involved in proliferation and invasion [205]. The repression of TCF-mediated transcription by APC is thought to be a main event in preventing tumorigenesis. Aberrant Wnt/β-catenin signalling has been found in various epithelial and haematological tumours [206]. Recently the INM lamin-binding protein, emerin, emerged as an additional negative regulator of the Wnt pathway [207]. The binding of emerin to β-catenin via an APC-like domain inhibits β-catenin activity by preventing its accumulation in the nucleus. Absence of emerin increases β-catenin accumulation in the nucleus followed by an up-regulation of its target genes. Emerin is anchored in the nucleus through interactions with lamins A and C. In cells lacking lamins A and C, overexpression of emerin does not inhibit β-catenin, indicating that this interaction is required for inhibiting β-catenin activity [207].
Lamins and cancer related aneuploidy
Present-day knowledge on the nuclear lamins sheds new light on the issue of aneuploidy and cancer. Aneuploidy, which is consistently observed in almost all cancers, is often caused by chromosomal instability, reflecting defects in mitotic segregation in cancer cells [208]. The nuclear lamins, which have recently emerged as pivotal participants in chromosomal segregation control, appear to have a crucial role in maintaining chromosomal stability, thus preventing cancer progression. There are several lines of evidence suggesting that lamins are required for normal segregation of chromosomes. (1) Assembly of BAF, LEM-domain proteins and lamins during cell division is required for proper chromosome segregation and cell cycle progression [209]. (2) Lamin A and LAP-2α prevent pRb degradation. Loss of Rb protein results in chromosomal instability by inducing defects during replication and abnormal chromosome segregation in mitosis [210]. (3) Expression of progerin/LAΔ50 leads to mitotic defects comprising delays in cytokinesis and nuclear reassembly, abnormal chromosome segregation, and binucleation [130, 131]. The presence of lamins at the centrosome during mitosis is likely to be involved in these defects [211].
Lamin and viruses
The viral cycle in the host cell includes adsorption, penetration, uncoating, viral genome replication, maturation and release of new particles from the host cell. In animal DNA viruses, transcription and translation are not coupled. Except for poxviruses, transcription occurs in the nucleus and translation in the cytoplasm. Viruses have developed different strategies for subverting, and even bypassing, host-cell nuclear transport pathways.
Herpes simplex virus (HSV) capsids are assembled in the nucleoplasm and then exit the nucleus by budding through the lamina and INM into the perinuclear space [reviewed in 212]. The HSV-1 UL34 protein targets the virus to the nuclear membrane and is essential for nuclear export of nucleocapsids. The localization of UL34 protein depends on the kinase US3 that phosphorylates both UL34 and lamins A and C. Phosphorylation of lamins by US3 alters their distribution, and allows exit of the capsids from the nucleus [213]. In vitro studies revealed that the lamins became solubilized due to the phosphorylation by US3. In addition, the UL34 protein binds specifically to lamin A and lamin B1. A recent study shows that lamin A interferes with viral infectivity and that US3 kinase activity partially inhibits this interference. Lamin B1, on the other hand, is necessary for optimal viral replication [214]. However, at least in MEFs derived from Lmna−/− mice, viral gene expression, DNA replication, and growth are reduced [215].
The human cytomegalovirus (HCMV) is a member of the herpesviridae family. Infection of human lung fibroblasts with HCMV induces mislocalization of nuclear lamins allowing virions exit from the nucleus [216]. The viral pUL50 and pUL53 proteins are required for nuclear exit. At late stages of infection both lamins A and C and lamin B show an irregular distribution at the nuclear rim, coincident with areas of pUL53 accumulation. The virions nuclear lamina localization requires co-expression of pUL53 and pUL50, which are sufficient for nuclear rim localization and lamins disorganization [216]. Another member of the herpesviridae family that its replication affects lamin A is varicella-zoster virus. During varicella-zoster virus infection the amount of lamin A mRNA decreases but the cellular content of the protein appears to remain stable at 48 hrs after infection [217].
Most retroviruses enter the nucleus only after the nuclear envelope breakdown. The retrovirus HIV-1 does not require mitosis to enter the nucleus. After more than a decade of intense research, there is still no consensus on the regulation of the entry of HIV-1 pre-integration complex into the nucleus through the NPC [218]. The exit of HIV-1 components from the nucleus to the cytoplasm involves both nuclear export signals that allow proteins to exit through the NPC and disruption of the nuclear lamina [219]. Expression of the Vpr protein suspends the cell cycle of human cells near the G2 cell-cycle checkpoint and induces local herniations at the nuclear periphery. These herniations are the result of defects in the nuclear lamina and probably contribute to the observed cell-cycle arrest. Rupture of these herniations causes mixing of nuclear and cytoplasmic components [219].
BAF is required for the biogenesis of MoMLV and HIV-1 retroviruses. At the early phases of infection, the newly transcribed dsDNA remains in the cytoplasm as a nucleoprotein pre-integration complex containing BAF. This presence of BAF blocks autointegration of the viral DNA in vitro, whereas removal of BAF from these complexes with high salt caused self-destruction of the virus by autointegration of the viral DNA [reviewed in 60]. It was also demonstrated that endogenous BAF acts as a cytosolic DNA sensor that binds cytoplasmic viral DNA and inhibits its replication. However, during vaccinia virus infection the viral B1 kinase phosphorylate BAF and therefore blocks its ability to bind to the viral genome as it is released from the core, and thus permits viral DNA replication to proceed [220].
Acknowledgments
We thank Michal Goldberg and Merav Cohen for comments on the manuscript. We also thank grants from the Israel Ministry of Health, the German-Israeli Foundation (GIF), USA-Israel Binational Science Foundation (BSF), Marie Curie (IRG), European Union FP6 (euro-laminopathies), the Israel Science Foundation (Morasha) and Muscular Dystrophy Association (MDA).
References
- 1.Stuurman N, Heins S, Aebi U. Nuclear lamins: their structure, assembly, and interactions. J Struct Biol. 1998;122:42–66. doi: 10.1006/jsbi.1998.3987. [DOI] [PubMed] [Google Scholar]
- 2.Herrmann H, Aebi U. Intermediate filaments: molecular structure, assembly mechanism, and integration into functionally distinct intracellular scaffolds. Annu Rev Biochem. 2004;74:749–89. doi: 10.1146/annurev.biochem.73.011303.073823. [DOI] [PubMed] [Google Scholar]
- 3.Krimm I, Ostlund C, Gilquin B, Couprie J, Hossenlopp P, Mornon JP, Bonne G, Courvalin JC, Worman HJ, Zinn-Justin S. The Ig-like structure of the C-terminal domain of lamin a/c, mutated in muscular dystrophies, cardiomyopathy, and partial lipodystrophy. Structure. 2002;10:811–23. doi: 10.1016/s0969-2126(02)00777-3. [DOI] [PubMed] [Google Scholar]
- 4.Gerace L, Blobel G. The nuclear envelope lamina is reversibly depolymerized during mitosis. Cell. 1980;19:277–87. doi: 10.1016/0092-8674(80)90409-2. [DOI] [PubMed] [Google Scholar]
- 5.Rusiñol AE, Sinensky MS. Farnesylated lamins, progeroid syndromes and farnesyl transferase inhibitors. J Cell Sci. 2006;119:3265–72. doi: 10.1242/jcs.03156. [DOI] [PubMed] [Google Scholar]
- 6.Varela I, Pereira S, Ugalde AP, Navarro CL, Suárez MF, Cau P, Cadiñanos J, Osorio FG, Cobo J, De Carlos F, Lèvy N, Freije JMP, López-Otín C. Combined treatment with statins and aminobisphosphonates extends longevity in a mouse model of human premature aging. Nat Med. 2008;14:767–72. doi: 10.1038/nm1786. [DOI] [PubMed] [Google Scholar]
- 7.Schneider U, Mini T, Jeno P, Fisher PA, Stuurman N. Phosphorylation of the major Drosophila lamin in vivo: site identification during both M-phase (meiosis) and interphase by electrospray ionization tandem mass spectrometry. Biochemistry. 1999;38:4620–32. doi: 10.1021/bi9827060. [DOI] [PubMed] [Google Scholar]
- 8.Mattout A, Goldberg M, Tzur Y, Margalit A, Gruenbaum Y. Specific and conserved sequences in D. melanogaster and C. elegans lamins and histone H2A mediate the attachment of lamins to chromosomes. J Cell Sci. 2007;120:77–85. doi: 10.1242/jcs.03325. [DOI] [PubMed] [Google Scholar]
- 9.Heald R, McKeon F. Mutations of phosphorylation sites in lamin A that prevent nuclear lamina disassembly in mitosis. Cell. 1990;61:579–89. doi: 10.1016/0092-8674(90)90470-y. [DOI] [PubMed] [Google Scholar]
- 10.Peter M, Heitlinger E, Haner M, Aebi U, Nigg EA. Disassembly of in vitro formed lamin head-to-tail polymers by CDC2 kinase. EMBO J. 1991;10:1535–44. doi: 10.1002/j.1460-2075.1991.tb07673.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dessev G, Iovcheva DC, Bischoff JR, Beach D, Goldman R. A complex containing p34cdc2 and cyclin B phosphorylates the nuclear lamin and disassembles nuclei of clam oocytes in vitro. J Cell Biol. 1991;112:523–33. doi: 10.1083/jcb.112.4.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nigg EA. Assembly and cell cycle dynamics of the nuclear lamina. Semin Cell Biol. 1992;3:245–53. doi: 10.1016/1043-4682(92)90026-r. [DOI] [PubMed] [Google Scholar]
- 13.Zhang YQ, Sarge KD. Sumoylation regulates lamin A function and is lost in lamin A mutants associated with familial car-diomyopathies. J Cell Biol. 2008;182:35–9. doi: 10.1083/jcb.200712124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gruenbaum Y, Goldman RD, Meyuhas R, Milles E, Margalit A, Fridkin A, Dayani Y, Prokocimer M, Enosh A. The nuclear lamina and its functions in the nucleus. Int Rev Cyt. 2003;226:1–62. doi: 10.1016/s0074-7696(03)01001-5. [DOI] [PubMed] [Google Scholar]
- 15.Melcer S, Gruenbaum Y, Krohne G. Invertebrate lamins. Exp Cell Res. 2007;313:2157–66. doi: 10.1016/j.yexcr.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 16.Riemer D, Dodemont H, Weber K. A nuclear lamin of the nematode Caenorhabditis elegans with unusual structural features; cDNA cloning and gene organization. Eur J Cell Biol. 1993;62:214–23. [PubMed] [Google Scholar]
- 17.Heitlinger E, Peter M, Haner M, Lustig A, Aebi U, Nigg EA. Expression of chicken lamin B2 in Escherichia coli: characterization of its structure, assembly, and molecular interactions. J Cell Biol. 1991;113:485–95. doi: 10.1083/jcb.113.3.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ben-Harush K, Wiesel N, Frenkiel-Krispin D, Moeller D, Soreq E, Aebi U, Herrmann H, Gruenbaum Y, Medalia O. Dissecting the supramolecular organization of Caenorhabditis elegans nuclear lamin filaments and paracrystalline fibers. J Mol Biol. 2008 doi: 10.1016/j.jmb.2008.12.024. ; in press. [DOI] [PubMed] [Google Scholar]
- 19.Klapper M, Exner K, Kempf A, Gehrig C, Stuurman N, Fisher PA, Krohne G. Assembly of A- and B-type lamins studied in vivo with the baculovirus system. J Cell Sci. 1997;110:2519–32. doi: 10.1242/jcs.110.20.2519. [DOI] [PubMed] [Google Scholar]
- 20.Karabinos A, Schunemann J, Meyer M, Aebi U, Weber K. The single nuclear lamin of Caenorhabditis elegans forms in vitro stable intermediate filaments and paracrystals with a reduced axial periodicity. J Mol Biol. 2003;325:241–7. doi: 10.1016/s0022-2836(02)01240-8. [DOI] [PubMed] [Google Scholar]
- 21.Foeger N, Wiesel N, Lotsch D, Mucke N, Kreplak L, Aebi U, Gruenbaum Y, Herrmann H. Solubility properties and specific assembly pathways of the B-type lamin from Caenorhabditis elegans. J Struc Biol. 2006;155:340–50. doi: 10.1016/j.jsb.2006.03.026. [DOI] [PubMed] [Google Scholar]
- 22.Wiesel N, Mattout A, Melcer S, Melamed-Book N, Herrmann H, Medalia O, Aebi U, Gruenbaum Y. Laminopathic mutations interfere with the assembly, localization and dynamics of nuclear lamins. Proc Natl Acad Sci USA. 2008;105:180–5. doi: 10.1073/pnas.0708974105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heitlinger E, Peter M, Lustig A, Villiger W, Nigg EA, Aebi U. The role of the head and tail domain in lamin structure and assembly: analysis of bacterially expressed chicken lamin A and truncated B2 lamins. J Struct Biol. 1992;108:74–89. doi: 10.1016/1047-8477(92)90009-y. [DOI] [PubMed] [Google Scholar]
- 24.Stuurman N, Sasse B, Fisher PA. Intermediate filament protein polymerization: molecular analysis of Drosophila nuclear lamin head-to-tail binding. J Struct Biol. 1996;117:1–15. doi: 10.1006/jsbi.1996.0064. [DOI] [PubMed] [Google Scholar]
- 25.Izumi M, Vaughan OA, Hutchison CJ, Gilbert DM. Head and/or CaaX domain deletions of lamin proteins disrupt preformed lamin A and C but not lamin B structure in mammalian cells. Mol Biol Cell. 2000;11:4323–37. doi: 10.1091/mbc.11.12.4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Spann TP, Moir RD, Goldman AE, Stick R, Goldman RD. Disruption of nuclear lamin organization alters the distribution of replication factors and inhibits DNA synthesis. J Cell Biol. 1997;136:1201–12. doi: 10.1083/jcb.136.6.1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moir RD, Spann TP, Herrmann H, Goldman RD. Disruption of nuclear lamin organization blocks the elongation phase of DNA replication. J Cell Biol. 2000;149:1179–92. doi: 10.1083/jcb.149.6.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gieffers C, Krohne G. In vitro reconstitution of recombinant lamin A and a lamin A mutant lacking the carboxy-terminal tail. Eur J Cell Biol. 1991;55:191–1999. [PubMed] [Google Scholar]
- 29.Sasse B, Aebi U, Stuurman N. A tailless Drosophila lamin Dm0 fragment reveals lateral associations of dimers. J Struct Biol. 1998;123:56–66. doi: 10.1006/jsbi.1998.4006. [DOI] [PubMed] [Google Scholar]
- 30.Moir RD, Donaldson AD, Stewart M. Expression in Escherichia coli of human lamins A and C: influence of head and tail domains on assembly properties and paracrystal formation. J Cell Sci. 1991;99:363–72. doi: 10.1242/jcs.99.2.363. [DOI] [PubMed] [Google Scholar]
- 31.Shumaker DK, Lopez-Soler RI, Adam SA, Herrmann H, Moir RD, Spann TP, Goldman RD. Functions and dysfunctions of the nuclear lamin Ig-fold domain in nuclear assembly, growth, and Emery-Dreifuss muscular dystrophy. Proc Natl Acad Sci USA. 2005;102:15494–9. doi: 10.1073/pnas.0507612102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Worman H, Bonne G. “Laminopathies”: a wide spectrum of human diseases. Exp Cell Res. 2007;313:2121–33. doi: 10.1016/j.yexcr.2007.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fawcett DW. On the occurrence of a fibrous lamina on the inner aspect of the nuclear envelope in certain cells of vertebrates. Am J Anat. 1966;119:129–45. doi: 10.1002/aja.1001190108. [DOI] [PubMed] [Google Scholar]
- 34.Aebi U, Cohn J, Buhle L, Gerace L. The nuclear lamina is a meshwork of intermediate-type filaments. Nature. 1986;323:560–4. doi: 10.1038/323560a0. [DOI] [PubMed] [Google Scholar]
- 35.Goldberg MW, Huttenlauch I, Hutchison CJ, Stick R. Filaments made from A- and B-type lamins differ in structure and organization. J Cell Sci. 2008;121:215–25. doi: 10.1242/jcs.022020. [DOI] [PubMed] [Google Scholar]
- 36.Kavanagh DM, Powell WE, Malik P, Lazou V, Schirmer EC. Organelle proteome variation among different cell types: lessons from nuclear membrane proteins. Subcell Biochem. 2007;43:51–76. doi: 10.1007/978-1-4020-5943-8_5. [DOI] [PubMed] [Google Scholar]
- 37.Mattout-Drubezki A, Gruenbaum Y. Dynamic interactions of nuclear lamina proteins with chromatin and transcriptional machinery. Cell Mol Life Sci. 2003;60:2053–63. doi: 10.1007/s00018-003-3038-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wagner N, Krohne G. LEM-Domain proteins: new insights into lamin-interacting proteins. Int Rev Cyt. 2007;261:1–46. doi: 10.1016/S0074-7696(07)61001-8. [DOI] [PubMed] [Google Scholar]
- 39.Dechat T, Pfleghaar K, Sengupta K, Shimi T, Shumaker DK, Solimando L, Goldman RD. Nuclear lamins: major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 2008;22:832–53. doi: 10.1101/gad.1652708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Franke WW. Structure, biochemistry, and function of the Nuclear envelope. Philos Trans R Soc Lond B Biol Sci. 1974;268:67–93. doi: 10.1098/rstb.1974.0016. [DOI] [PubMed] [Google Scholar]
- 41.Belmont AS, Zhai Y, Thilenius A. Lamin B distribution and association with peripheral chromatin revealed by optical sectioning and electron microscopy tomography. J Cell Biol. 1993;123:1671–85. doi: 10.1083/jcb.123.6.1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marshall WF, Sedat JW. Nuclear architecture. Results Probl Cell Differ. 1999;25:283–301. doi: 10.1007/978-3-540-69111-2_14. [DOI] [PubMed] [Google Scholar]
- 43.Guelen L, Pagie L, Brasset E, Meuleman W, Faza MB, Talhout W, Eussen BH, De Klein A, Wessels L, De Laat W, Van Steensel B. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature. 2008;453:948–51. doi: 10.1038/nature06947. [DOI] [PubMed] [Google Scholar]
- 44.Pickersgill H, Kalverda B, De Wit E, Talhout W, Fornerod M, Van Steensel B. Characterization of the Drosophila melanogaster genome at the nuclear lamina. Nat Genet. 2006;38:1005–14. doi: 10.1038/ng1852. [DOI] [PubMed] [Google Scholar]
- 45.Rzepecki R, Bogachev SS, Kokoza E, Stuurman N, Fisher PA. In vivo association of lamins with nucleic acids in Drosophila melanogaster. J Cell Sci. 1998;111:121–9. doi: 10.1242/jcs.111.1.121. [DOI] [PubMed] [Google Scholar]
- 46.Luderus ME, De Graaf A, Mattia E, Den BJ, Grande MA, De Jong L, Van Driel R. Binding of matrix attachment regions to lamin B1. Cell. 1992;70:949–59. doi: 10.1016/0092-8674(92)90245-8. [DOI] [PubMed] [Google Scholar]
- 47.Luderus ME, Den Blaauwen J, De Smit O, Compton DA, Van Driel R. Binding of matrix attachment regions to lamin polymers involves single-stranded regions and the minor groove. Mol Cell Biol. 1994;14:6297–305. doi: 10.1128/mcb.14.9.6297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Baricheva EA, Berrios M, Bogachev SS, Borisevich IV, Lapik ER, Sharakhov IV, Stuurman N, Fisher PA. DNA from Drosophila melanogaster beta-heterochromatin binds specifically to nuclear lamins in vitro and the nuclear envelope in situ. Gene. 1996;171:171–6. doi: 10.1016/0378-1119(96)00002-9. [DOI] [PubMed] [Google Scholar]
- 49.Shoeman RL, Traub P. The in vitro DNA-binding properties of purified nuclear lamin proteins and vimentin. J Biol Chem. 1990;265:9055–61. [PubMed] [Google Scholar]
- 50.Zhao K, Harel A, Stuurman N, Guedalia D, Gruenbaum Y. Binding of matrix attachment regions to nuclear lamin is mediated by the rod domain and depends on the lamin polymerization state. FEBS Lett. 1996;380:161–4. doi: 10.1016/0014-5793(96)00034-8. [DOI] [PubMed] [Google Scholar]
- 51.Burke B. On the cell-free association of lamins A and C with metaphase chromosomes. Exp Cell Res. 1990;186:169–76. doi: 10.1016/0014-4827(90)90223-w. [DOI] [PubMed] [Google Scholar]
- 52.Glass JR, Gerace L. Lamins A and C bind and assemble at the surface of mitotic chromosomes. J Cell Biol. 1990;111:1047–57. doi: 10.1083/jcb.111.3.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Goldberg M, Harel A, Brandeis M, Rechsteiner T, Richmond TJ, Weiss AM, Gruenbaum Y. The tail domain of lamin Dm0 binds histones H2A and H2B. Proc Natl Acad Sci USA. 1999;96:2852–7. doi: 10.1073/pnas.96.6.2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hoger TH, Krohne G, Kleinschmidt JA. Interaction of Xenopus lamins A and LII with chromatin in vitro mediated by a sequence element in the carboxyterminal domain. Exp Cell Res. 1991;197:280–9. doi: 10.1016/0014-4827(91)90434-v. [DOI] [PubMed] [Google Scholar]
- 55.Ulitzur N, Harel A, Feinstein N, Gruenbaum Y. Lamin activity is essential for nuclear envelope assembly in a Drosophila embryo cell-free extract. J Cell Biol. 1992;119:17–25. doi: 10.1083/jcb.119.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yuan J, Simos G, Blobel G, Georgatos SD. Binding of lamin A to polynucleosomes. J Biol Chem. 1991;266:9211–5. [PubMed] [Google Scholar]
- 57.Taniura H, Glass C, Gerace L. A chromatin binding site in the tail domain of nuclear lamins that interacts with core his-tones. J Cell Biol. 1995;131:33–44. doi: 10.1083/jcb.131.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stuurman N, Maus N, Fisher PA. Interphase phosphorylation of the Drosophila nuclear lamin: site-mapping using a monoclonal antibody. J Cell Sci. 1995;108:3137–44. doi: 10.1242/jcs.108.9.3137. [DOI] [PubMed] [Google Scholar]
- 59.Worman HJ. Components of the nuclear envelope and their role in human disease. Novartis Found Symp. 2005;264:35–42. [PubMed] [Google Scholar]
- 60.Margalit A, Brachner A, Gotzmann J, Foisner R, Y. G. Barrier-to-autointegration factor – a BAFfling little protein. Trends Cell Biol. 2007;17:202–8. doi: 10.1016/j.tcb.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 61.Cohen M, Tzur YB, Neufeld E, Feinstein N, Delannoy MR, Wilson KL, Gruenbaum Y. Transmission electron microscope studies of the nuclear envelope in Caenorhabditis elegans embryos. J Struct Biol. 2002;140:232–40. doi: 10.1016/s1047-8477(02)00516-6. [DOI] [PubMed] [Google Scholar]
- 62.Goldman RD, Shumaker DK, Erdos MR, Eriksson M, Goldman AE, Gordon LB, Gruenbaum Y, Khuon S, Mendez M, Varga R, Collins FS. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci USA. 2004;101:8963–8. doi: 10.1073/pnas.0402943101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sabatelli P, Lattanzi G, Ognibene A, Columbaro M, Capanni C, Merlini L, Maraldi NM, Squarzoni S. Nuclear alterations in autosomal-dominant Emery-Dreifuss muscular dystrophy. Muscle Nerve. 2001;24:826–9. doi: 10.1002/mus.1076. [DOI] [PubMed] [Google Scholar]
- 64.Capanni C, Cenni V, Mattioli E, Sabatelli P, Ognibene A, Columbaro M, Parnaik VK, Wehnert M, Maraldi NM, Squarzoni S, Lattanzi G. Failure of lamin A/C to functionally assemble in R482L mutated familial partial lipodystrophy fibroblasts: altered intermolecular interaction with emerin and implications for gene transcription. Exp Cell Res. 2003;291:122–34. doi: 10.1016/s0014-4827(03)00395-1. [DOI] [PubMed] [Google Scholar]
- 65.Ramírez CL, Cadiñanos J, Varela I, Freije JM, López-Otín C. Human progeroid syndromes, aging and cancer: new genetic and epigenetic insights into old questions. Cell Mol Life Sci. 2007;64:155–70. doi: 10.1007/s00018-006-6349-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Scaffidi P, Misteli T. Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome. Nat Genet. 2005;11:440–5. doi: 10.1038/nm1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Columbaro M, Capanni C, Mattioli E, Novelli G, Parnaik VK, Squarzoni S, Maraldi NM, Lattanzi G. Rescue of hete-rochromatin organization in Hutchinson-Gilford progeria by drug treatment. Cell Mol Life Sci. 2005;62:2669–78. doi: 10.1007/s00018-005-5318-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shumaker DK, Dechat T, Kohlmaier A, Adam DA, Bozovsky MR, Erdos MR, Eriksson M, Goldman AE, Khuon S, Collins FS, Jenuwein T, Goldman RE. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc Natl Acad Sci USA. 2006;103:8703–8. doi: 10.1073/pnas.0602569103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Somech R, Shaklai S, Geller O, Amariglio N, Simon AJ, Rechavi G, Gal-Yam EN. The nuclear-envelope protein and transcriptional repressor LAP2beta interacts with HDAC3 at the nuclear periphery, and induces histone H4 deacetylation. J Cell Sci. 2005;118:4017–25. doi: 10.1242/jcs.02521. [DOI] [PubMed] [Google Scholar]
- 70.Holaska JM, Wilson KL. An emerin “proteome”: purification of distinct emerincontaining complexes from HeLa cells suggests molecular basis for diverse roles including gene regulation, mRNA splicing, signaling, mechanosensing, and nuclear architecture. Biochemistry. 2007;46:8897–908. doi: 10.1021/bi602636m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Olins AL, Olins DE. Cytoskeletal influences on nuclear shape in granulocytic HL-60 cells. BMC Cell Biol. 2004;19:30. doi: 10.1186/1471-2121-5-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tzur Y, Wilson KL, Gruenbaum Y. SUN-domain proteins: ‘Velcro’ that links the nucleoskeleton to the cytoskeleton. Nat Rev Cell Mol Biol. 2006;7:782–8. doi: 10.1038/nrm2003. [DOI] [PubMed] [Google Scholar]
- 73.Liu J, Rolef-Ben Shahar T, Riemer D, Spann P, Treinin M, Weber K, Fire A, Gruenbaum Y. Essential roles for Caenorhabditis elegans lamin gene in nuclear organization, cell cycle progression, and spatial organization of nuclear pore complexes. Mol Biol Cell. 2000;11:3937–47. doi: 10.1091/mbc.11.11.3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lenz-Bohme B, Wismar J, Fuchs S, Reifegerste R, Buchner E, Betz H, Schmitt B. Insertional mutation of the Drosophila nuclear lamin dm(0) gene results in defective nuclear envelopes, clustering of nuclear pore complexes, and accumulation of annulate lamellae. J Cell Biol. 1997;137:1001–16. doi: 10.1083/jcb.137.5.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sullivan T, Escalente-Alcalde D, Bhatt H, Anver M, Naryan B, Nagashima K, Stewart CL, Burke B. Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J Cell Biol. 1999;147:913–20. doi: 10.1083/jcb.147.5.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lammerding J, Schulze PC, Takahashi T, Kozlov S, Sullivan T, Kamm RD, Stewart CL, Lee RS. Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J Clin Invest. 2004;113:370–8. doi: 10.1172/JCI19670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dahl KN, Ribeiro AJ, Lammerding J. Nuclear shape, mechanics, and mechanotransduction. Circ Res. 2008;102:1307–18. doi: 10.1161/CIRCRESAHA.108.173989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Worman HJ, Courvalin JC. How do mutations in lamins A and C cause disease. J Clin Invest. 2004;113:349–51. doi: 10.1172/JCI20832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Newport JW, Wilson KL, Dunphy WG. A lamin-independent pathway for nuclear envelope assembly. J Cell Biol. 1990;111:2247–59. doi: 10.1083/jcb.111.6.2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Broers JL, Peeters EA, Kuijpers HJ, Endert J, Bouten CV, Oomens CW, Baaijens FP, Ramaekers FC. Decreased mechanical stiffness in LMNA-/- cells is caused by defective nucleo-cytoskeletal integrity: implications for the development of laminopathies. Hum Mol Genet. 2004;13:2567–80. doi: 10.1093/hmg/ddh295. [DOI] [PubMed] [Google Scholar]
- 81.Nikolova V, Leimena C, McMahon AC, Tan JC, Chandar S, Jogia D, Kesteven SH, Michalicek J, Otway R, Verheyen F, Rainer S, Stewart CL, Martin D, Feneley MP, Fatkin D. Defects in nuclear structure and function promote dilated cardiomyopathy in lamin A/C-deficient mice. J Clin Invest. 2004;113:357–69. doi: 10.1172/JCI19448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lammerding J, Fong LG, Ji JY, Reue K, Stewart CL, Young SG, Lee RT. Lamins A and C but not lamin B1 regulate nuclear mechanics. J Biol Chem. 2006;281:25768–80. doi: 10.1074/jbc.M513511200. [DOI] [PubMed] [Google Scholar]
- 83.Ralle T, Grund C, Franke WW, Stick R. Intranuclear membrane structure formations by CaaX-containing nuclear proteins. J Cell Sci. 2004;117:6095–104. doi: 10.1242/jcs.01528. [DOI] [PubMed] [Google Scholar]
- 84.Prufert K, Vogel A, Krohne G. The lamin CxxM motif promotes nuclear membrane growth. J Cell Sci. 2004;117:6105–16. doi: 10.1242/jcs.01532. [DOI] [PubMed] [Google Scholar]
- 85.Lammerding J, Hsiao J, Schulze PC, Kozlov S, Stewart CL, Lee RT. Abnormal nuclear shape and impaired mechan-otransduction in emerin-deficient cells. J Cell Biol. 2005;170:781–91. doi: 10.1083/jcb.200502148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Brachner A, Reipert S, Foisner R, Gotzmann J. LEM2 is a novel MAN1-related inner nuclear membrane protein associated with A-type lamins. J Cell Sci. 2005;118:5797–810. doi: 10.1242/jcs.02701. [DOI] [PubMed] [Google Scholar]
- 87.Liu J, Lee KK, Segura-Totten M, Neufeld E, Wilson KL, Gruenbaum Y. MAN1 and emerin have overlapping function(s) essential for chromosome segregation and cell division in C. elegans. Proc Natl Acad Sci USA. 2003;100:4598–603. doi: 10.1073/pnas.0730821100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Margalit A, Segura-Totten M, Gruenbaum Y, Wilson KL. Barrier-to-autointegration factor is required to segregate and enclose chromosomes within the nuclear envelope and assemble the nuclear lamina. Proc Natl Acad Sci USA. 2005;102:3290–5. doi: 10.1073/pnas.0408364102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Verstraeten VL, Ji JY, Cummings KS, Lee RT, Lammerding J. Increased mechanosensitivity and nuclear stiffness in Hutchinson-Gilford progeria cells: effects of farnesyl-transferase inhibitors. Aging Cell. 2008;7:383–93. doi: 10.1111/j.1474-9726.2008.00382.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kennedy BK, Barbie DA, Classon M, Dyson N, Harlow E. Nuclear organization of DNA replication in primary mammalian cells. Genes Dev. 2000;14:2855–68. doi: 10.1101/gad.842600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Moir RD, Montag LM, Goldman RD. Dynamic properties of nuclear lamins: lamin B is associated with sites of DNA replication. J Cell Biol. 1994;125:1201–12. doi: 10.1083/jcb.125.6.1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Johnson BR, Nitta RT, Frock RL, Mounkes L, Barbie DA, Stewart CL, Harlow E, Kennedy BK. A-type lamins regulate retinoblastoma protein function by promoting subnuclear localization and preventing proteasomal degradation. Proc Natl Acad Sci USA. 2004;101:9677–82. doi: 10.1073/pnas.0403250101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Meier J, Campbell KH, Ford CC, Stick R, Hutchison CJ. The role of lamin LIII in nuclear assembly and DNA replication, in cell-free extracts of Xenopus eggs. J Cell Sci. 1991;98:271–9. doi: 10.1242/jcs.98.3.271. [DOI] [PubMed] [Google Scholar]
- 94.Goldberg M, Jenkins H, Allen T, Whitfield WG, Hutchison CJ. Xenopuslamin B3 has a direct role in the assembly of a replication competent nucleus: evidence from cell-free egg extracts. J Cell Sci. 1995;108:3451–61. doi: 10.1242/jcs.108.11.3451. [DOI] [PubMed] [Google Scholar]
- 95.Shumaker DK, Solimando L, Sengupta K, Shimi T, Adam SA, Grunwald A, Strelkov SV, Aebi U, Cardoso MC, Goldman RD. The highly conserved nuclear lamin Ig-fold binds to PCNA: its role in DNA replication. J Cell Biol. 2008;181:269–80. doi: 10.1083/jcb.200708155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Heard E, Bickmore W. The ins and outs of gene regulation and chromosome territory organisation. Curr Opin Cell Biol. 2007;19:311–6. doi: 10.1016/j.ceb.2007.04.016. [DOI] [PubMed] [Google Scholar]
- 97.Kalverda B, Röling MD, Fornerod M. Chromatin organization in relation to the nuclear periphery. FEBS Lett. 2008;582:2017–22. doi: 10.1016/j.febslet.2008.04.015. [DOI] [PubMed] [Google Scholar]
- 98.Chuang CH, AE. C, Fuchsova B, Johnson T, De Lanerolle P, Belmont AS. Long-range directional movement of an inter-phase chromosome site. Curr Biol. 2006;16:825–31. doi: 10.1016/j.cub.2006.03.059. [DOI] [PubMed] [Google Scholar]
- 99.Reddy KL, Zullo JM, Bertolino E, Singh H. Transcriptional repression mediated by repositioning of genes to the nuclear lamina. Nature. 2008;452:243–7. doi: 10.1038/nature06727. [DOI] [PubMed] [Google Scholar]
- 100.Finlan LE, Sproul D, Thomson I, Boyle S, Kerr E, Perry P, Ylstra B, Chubb JR, Bickmore WA. Recruitment to the nuclear periphery can alter expression of genes in human cells. PLoS Genet. 2008;4:e1000039. doi: 10.1371/journal.pgen.1000039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kumaran RI, Spector DL. A genetic locus targeted to the nuclear periphery in living cells maintains its transcriptional competence. J Cell Biol. 2008;180:51–65. doi: 10.1083/jcb.200706060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kumaran RI, Muralikrishna B, Parnaik VK. Lamin A/C speckles mediate spatial organization of splicing factor compartments and RNA polymerase II transcription. J Cell Biol. 2002;159:783–93. doi: 10.1083/jcb.200204149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Spann TP, Goldman AE, Wang C, Huang S, Goldman RD. Alteration of nuclear lamin organization inhibits RNA poly-merase II-dependent transcription. J Cell Biol. 2002;156:603–8. doi: 10.1083/jcb.200112047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Margalit A, Neufeld E, Feinstein N, Wilson KL, Podbilewicz B, Gruenbaum Y. Barrier-to-autointegration factor (BAF) is required for blocking premature cell fusion, vulva formation, germ cell development and survival, DTC migration and adult muscle integrity in C. elegans. J Cell Biol. 2007;178:661–73. doi: 10.1083/jcb.200704049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wang X, Xu S, Rivolta C, Li LY, Peng GH, Swain PK, Sung CH, Swaroop A, Berson EL, Dryja TP, Chen S. Barrier to autointegration factor interacts with the cone-rod homeobox and represses its transactivation function. J Biol Chem. 2002;277:43288–300. doi: 10.1074/jbc.M207952200. [DOI] [PubMed] [Google Scholar]
- 106.Dreuillet C, Tillit J, Kress M, Ernoult-Lange M. In vivo and in vitro interaction between human transcription factor MOK2 and nuclear lamin A/C. Nuc Acids Res. 2002;30:4634–42. doi: 10.1093/nar/gkf587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Dreuillet C, Harper M, Tillit J, Kress M, Ernoult-Lange M. Mislocalization of human transcription factor MOK2 in the presence of pathogenic mutations of lamin A/C. Biol Cell. 2008;100:51–61. doi: 10.1042/BC20070053. [DOI] [PubMed] [Google Scholar]
- 108.Lloyd DJ, Trembath RC, Shackleton S. A novel interaction between lamin A and SREBP1: implications for partial lipodys-trophy and other laminopathies. Hum Mol Genet. 2002;11:769–77. doi: 10.1093/hmg/11.7.769. [DOI] [PubMed] [Google Scholar]
- 109.Imai SI, Nishibayashi S, Takao K, Tomifuji M, Fujino T, Hasegawa M, Takano T. Dissociation of Oct-1 from the nuclear peripheral structure induces the cellular aging-associated collagenase gene expression. Mol Biol Cell. 1997;8:2407–19. doi: 10.1091/mbc.8.12.2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rafiq I, Kennedy HJ, Rutter GA. Glucose-dependent translocation of insulin promoter factor-1 (IPF-1) between the nuclear periphery and the nucleoplasm of single MIN6 beta-cells. J Biol Chem. 1998;273:23241–7. doi: 10.1074/jbc.273.36.23241. [DOI] [PubMed] [Google Scholar]
- 111.Ivorra C, Kubicek M, González JM, Sanz-González SM, Alvarez-Barrientos A, O’Connor JE, Burke B, Andrès V. A mechanism of AP-1 suppression through interaction of c-Fos with lamin A/C. Genes Dev. 2006;20:307–20. doi: 10.1101/gad.349506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Markiewicz E, Dechat T, Foisner R, Quinlan RA, Hutchison CJ. Lamin A/C binding protein LAP2alpha is required for nuclear anchorage of retinoblastoma protein. Mol Biol Cell. 2002;13:4401–13. doi: 10.1091/mbc.E02-07-0450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Van Berlo JH, Voncken JW, Kubben N, Broers JL, Duisters R, Van Leeuwen RE, Crijns HJ, Ramaekers FC, Hutchison CJ, Pinto YM. A-type lamins are essential for TGF-beta1 induced PP2A to dephosphorylate transcription factors. Hum Mol Genet. 2005;14:2839–49. doi: 10.1093/hmg/ddi316. [DOI] [PubMed] [Google Scholar]
- 114.Hubner S, Eam JE, Hubner A, Jans DA. Laminopathy-inducing lamin A mutants can induce redistribution of lamin binding proteins into nuclear aggregates. Exp Cell Res. 2006;312:171–83. doi: 10.1016/j.yexcr.2005.10.011. [DOI] [PubMed] [Google Scholar]
- 115.Varela I, Cadinanos J, Pendas AM, Gutierrez-Fernandez A, Folgueras AR, Sanchez LM, Zhou Z, Rodriguez FJ, Stewart CL, Vega JA, Tryggvason K, Freije JM, Lopez-Otin C. Accelerated ageing in mice deficient in Zmpste24 protease is linked to p53 signalling activation. Nature. 2005;437:564–8. doi: 10.1038/nature04019. [DOI] [PubMed] [Google Scholar]
- 116.Nitta RT, Smith CL, Kennedy BK. Evidence that proteasome-dependent degradation of the retinoblastoma protein in cells lacking A-type lamins occurs independently of gankyrin and MDM2. PLoS One. 2007;2:e963. doi: 10.1371/journal.pone.0000963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Favreau C, Higuet D, Courvalin JC, Buendia B. Expression of a mutant lamin A that causes Emery-Dreifuss muscular dystrophy inhibits in vitro differentiation of C2C12 myoblasts. Mol Biol Cell. 2004;24:1481–92. doi: 10.1128/MCB.24.4.1481-1492.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Frock RL, Kudlow BA, Evans AM, Jameson SA, Hauschka SD, Kennedy BK. Lamin A/C and emerin are critical for skeletal muscle satellite cell differentiation. Genes Dev. 2006;20:486–500. doi: 10.1101/gad.1364906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Boguslavsky RL, Stewart CL, Worman HJ. Nuclear lamin A inhibits adipocyte differentiation: implications for Dunnigan-type familial partial lipodystrophy. Hum Mol Genet. 2006;15:653–63. doi: 10.1093/hmg/ddi480. [DOI] [PubMed] [Google Scholar]
- 120.Vlcek S, Foisner R. Lamins and lamin-associated proteins in aging and disease. Curr Opin Cell Biol. 2007;19:298–304. doi: 10.1016/j.ceb.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 121.Naetar N, Korbei B, Kozlov S, Kerenyi MA, Dorner D, Kral R, Gotic I, Fuchs P, Cohen T, Bittner R, Stewart CL, Foisner R. Loss of nucleoplasmic LAP2alpha-lamin A complexes causes erythroid and epidermal progenitor hyperproliferation. Nat Cell Biol. 2008;10:1341–8. doi: 10.1038/ncb1793. [DOI] [PubMed] [Google Scholar]
- 122.Pekovic V, Harborth J, Broers JL, Ramaekers FC, Van Engelen B, Lammens M, Von Zglinicki T, Foisner R, Hutchison C, Markiewicz E. Nucleoplasmic LAP2-alpha-lamin A complexes are required to maintain a proliferative state in human fibroblasts. J Cell Biol. 2007;176:163–72. doi: 10.1083/jcb.200606139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Nili E, Cojocaru GS, Kalma Y, Ginsberg D, Copeland NG, Gilbert DJ, Jenkins NA, Berger R, Shaklai S, Amariglio N, Brok-Simoni F, Simon AJ, Rechavi G. Nuclear membrane protein, LAP2b, mediates transcriptional repression alone and together with its binding partner GCL (germ-cell-less) J Cell Sci. 2001;114:3297–307. doi: 10.1242/jcs.114.18.3297. [DOI] [PubMed] [Google Scholar]
- 124.Worman HJ. Inner nuclear membrane and regulation of Smad-mediated signaling. Bioch Biophis Acta. 2006;1761:626–31. doi: 10.1016/j.bbalip.2006.02.010. [DOI] [PubMed] [Google Scholar]
- 125.Broers JL, Ramaekers FC, Bonne G, Yaou RB, Hutchison CJ. Nuclear lamins: laminopathies and their role in premature ageing. Physiol Rev. 2006;86:967–1008. doi: 10.1152/physrev.00047.2005. [DOI] [PubMed] [Google Scholar]
- 126.Jagatheesan G, Thanumalayan S, Muralikrishna B, Rangaraj N, Karande AA, Parnaik VK. Colocalization of intranuclear lamin foci with RNA splicing factors. J Cell Sci. 1999;112:4651–61. doi: 10.1242/jcs.112.24.4651. [DOI] [PubMed] [Google Scholar]
- 127.Vecerová J, Koberna K, Malìnsky J, Soutoglou E, Sullivan T, Stewart CL, Raska I, Misteli T. Formation of nuclear splicing factor compartments is independent of lamins A/C. Mol Biol Cell. 2004;15:4904–10. doi: 10.1091/mbc.E04-07-0645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Haithcock E, Dayani Y, Neufeld E, Zahand AJ, Feinstein N, Mattout N, Gruenbaum Y, Liu J. Age-related changes of nuclear architecture in Caenorhabditis elegans. Proc Natl Acad Sci USA. 2005;102:16690–5. doi: 10.1073/pnas.0506955102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kenyon C. The plasticity of aging: insights from long-lived mutants. Cell. 2005;120:449–60. doi: 10.1016/j.cell.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 130.Dechat T, Shimi T, Adam SA, Rusinol AE, Andres DA, Spielmann HP, Sinensky MS, Goldman RD. Alterations in mitosis and cell cycle progression caused by a mutant lamin A known to accelerate human aging. Proc Natl Acad Sci USA. 2007;104:4955–60. doi: 10.1073/pnas.0700854104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Cao K, Capell BC, Erdos MR, Djabali K, Collins FS. A lamin A protein isoform overexpressed in Hutchinson-Gilford progeria syndrome interferes with mitosis in progeria and normal cells. Proc Natl Acad Sci USA. 2007;104:3939–54. doi: 10.1073/pnas.0611640104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Levy N, Lopez-Otin C, Hennekam RC. Defective prelamin A processing resulting from LMNA or ZMPSTE24 mutations as the cause of restrictive dermopathy. Arch Dermatol. 2005;14:1473–4. doi: 10.1001/archderm.141.11.1473-b. [DOI] [PubMed] [Google Scholar]
- 133.Navarro CL, Cadinanos J, Sandre-Giovannoli AD, Bernard R, Courrier S, Boccaccio I, Boyer A, Kleijer WJ, Wagner A, Giuliano F, Beemer FA, Freije JM, Cau P, Hennekam RC, Lopez-Otin C, Badens C, Levy N. Loss of ZMPSTE24 (FACE-1) causes autosomal recessive restrictive dermopathy and accumulation of Lamin A precursors. Hum Mol Genet. 2005;14:1503–13. doi: 10.1093/hmg/ddi159. [DOI] [PubMed] [Google Scholar]
- 134.Toth JI, Yang SH, Qiao X, Beigneux AP, Gelb MH, Moulson CL, Miner JH, Young SG, Fong LG. Blocking protein farnesyl-transferase improves nuclear shape in fibroblasts from humans with progeroid syndromes. Proc Natl Acad Sci USA. 2005;102:12873–8. doi: 10.1073/pnas.0505767102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Stewart CL, Kozlov S, Fong LG, Young SG. Mouse models of the laminopathies. Exp Cell Res. 2007;313:2144–56. doi: 10.1016/j.yexcr.2007.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Yang SH, Meta M, Qiao X, Frost D, Bauch J, Coffinier C, Majumdar S, Bergo MO, Young SG, Fong LG. A farnesyltransferase inhibitor improves disease phenotypes in mice with a Hutchinson-Gilford progeria syndrome mutation. J Clin Invest. 2006;116:2115–21. doi: 10.1172/JCI28968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Fong LG, Frost D, M. M, Qiao X, Yang SH, Coffinier C, Young SG. A protein farnesyltransferase inhibitor ameliorates disease in a mouse model of progeria. Science. 2006;311:1621–3. doi: 10.1126/science.1124875. [DOI] [PubMed] [Google Scholar]
- 138.Meta M, Yang SH, Bergo MO, Fong LG, Young SG. Protein farnesyltransferase inhibitors and progeria. Trends Mol Med. 2006;12:480–7. doi: 10.1016/j.molmed.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 139.Scaffidi P, Misteli T. Lamin A-dependent nuclear defects in human aging. Science. 2006;312:1059–63. doi: 10.1126/science.1127168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.McClintock D, Ratner D, Lokuge M, Owens DM, Gordon LB, Collins FS, Djabali K. The mutant form of lamin A that causes Hutchinson-Gilford progeria is a biomarker of cellular aging in human skin. PLoS One. 2007;2:e1269. doi: 10.1371/journal.pone.0001269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hegele RA, Cao H, Liu DM, Costain GA, Charlton-Menys V, Rodger NW, Durrington PN. Sequencing of the reanno-tated LMNB2 gene reveals novel mutations in patients with acquired partial lipodystrophy. Am J Hum Genet. 2006;79:383–9. doi: 10.1086/505885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Mattout A, Dechat T, Adam SA, Goldman RD, Gruenbaum Y. Nuclear lamins, diseases and aging. Curr Opin Cell Biol. 2006;18:1–7. doi: 10.1016/j.ceb.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 143.Alsheimer M, Liebe B, Sewell L, Stewart CL, Scherthan H, Benavente R. Disruption of spermatogenesis in mice lacking A-type lamins. J Cell Sci. 2004;117:1173–8. doi: 10.1242/jcs.00975. [DOI] [PubMed] [Google Scholar]
- 144.Mounkes LC, Kozlov SV, Rottman JN, Stewart CL. Expression of an LMNA-N195K variant of A-type lamins results in cardiac conduction defects and death in mice. Hum Mol Genet. 2005;14:2167–80. doi: 10.1093/hmg/ddi221. [DOI] [PubMed] [Google Scholar]
- 145.Arimura T, Helbling-Leclerc A, Massart C, Varnous S, Niel F, Lacëne E, Fromes Y, Toussaint M, Mura AM, Keller DI, Amthor H, Isnard R, Malissen M, Schwartz K, Bonne G. Mouse model carrying H222P-Lmna mutation develops muscular dystrophy and dilated cardiomy-opathy similar to human striated muscle laminopathies. Hum Mol Genet. 2005;14:155–69. doi: 10.1093/hmg/ddi017. [DOI] [PubMed] [Google Scholar]
- 146.Mounkes LC, Kozlov S, Hernandez L, Sullivan T, Stewart CL. A progeroid syndrome in mice is caused by defects in A-type lamins. Nature. 2003;423:298–301. doi: 10.1038/nature01631. [DOI] [PubMed] [Google Scholar]
- 147.Yang SH, Bergo MO, Toth JI, Qiao X, Hu Y, Sandoval S, Meta M, Bendale P, Gelb MH, Young SG, Fong LG. Blocking protein farnesyltransferase improves nuclear bleb-bing in mouse fibroblasts with a targeted Hutchinson-Gilford progeria syndrome mutation. Proc Natl Acad Sci USA. 2005;102:10291–6. doi: 10.1073/pnas.0504641102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Varga R, Eriksson M, Erdos MR, Olive M, Harten I, Kolodgie F, Capell BC, Cheng J, Faddah D, Perkins S, Avallone H, San H, Qu X, Ganesh S, Gordon LB, Virmani R, Wight TN, Nabel EG, Collins FS. Progressive vascular smooth muscle cell defects in a mouse model of Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci USA. 2006;103:3250–5. doi: 10.1073/pnas.0600012103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Sagelius H, Rosengardten Y, Hanif M, Erdos MR, Rozell B, Collins FS, Eriksson M. Targeted transgenic expression of the mutation causing Hutchinson-Gilford progeria syndrome leads to proliferative and degenerative epidermal disease. J Cell Sci. 2008;121:969–78. doi: 10.1242/jcs.022913. [DOI] [PubMed] [Google Scholar]
- 150.Wolf CM, Wang L, Alcalai R, Pizard A, Burgon PG, Ahmad F, Sherwood M, Branco DM, Wakimoto H, Fishman GI, See V, Stewart CL, Conner DA, Berul CI, Seidman CE, Seidman JG. Lamin A/C haploinsufficiency causes dilated car-diomyopathy and apoptosis-triggered cardiac conduction system disease. J Mol Cell Cardiol. 2008;44:293–303. doi: 10.1016/j.yjmcc.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Vergnes L, Pèterfy M, Bergo MO, Young SG, Reue K. Lamin B1 is required for mouse development and nuclear integrity. Proc Natl Acad Sci USA. 2004;101:10428–33. doi: 10.1073/pnas.0401424101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Melcon G, Kozlov S, Cutler DA, Sullivan T, Hernandez L, Zhao P, Mitchell S, Nader G, Bakay M, Rottman JN, Hoffman EP, Stewart CL. Loss of emerin at the nuclear envelope disrupts the Rb1/E2F and MyoD pathways during muscle regeneration. Hum Mol Genet. 2006;15:637–51. doi: 10.1093/hmg/ddi479. [DOI] [PubMed] [Google Scholar]
- 153.Ozawa R, Hayashi YK, Ogawa M, Kurokawa R, Matsumoto H, Noguchi S, Nonaka I, Nishino I. Emerin-lacking mice show minimal motor and cardiac dysfunctions with nuclear-associated vacuoles. Am J Pathol. 2006;168:907–17. doi: 10.2353/ajpath.2006.050564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Shultz LD, Lyons BL, Burzenski LM, Gott B, Samuels R, Schweitzer PA, Dreger C, Herrmann H, Kalscheuer V, Olins AL, Olins DE, Sperling K, Hoffmann K. Mutations at the mouse ichthyosis locus are within the lamin B receptor gene: a single gene model for human Pelger-Huet anomaly. Hum Mol Genet. 2003;12:61–9. doi: 10.1093/hmg/ddg003. [DOI] [PubMed] [Google Scholar]
- 155.Cohen TV, Klarmann KD, Sakchaisri K, Cooper JP, Kuhns D, Anver M, Johnson PF, Williams SC, Keller JR, Stewart CL. The lamin B receptor under transcriptional control of C/EBPepsilon is required for morphological but not functional maturation of neutrophils. Hum Mol Genet. 2008;17:2921–33. doi: 10.1093/hmg/ddn191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Cohen TV, Kosti O, Stewart CL. The nuclear envelope protein MAN1 regulates TGFbeta signaling and vasculogenesis in the embryonic yolk sac. Development. 2007;134:1385–95. doi: 10.1242/dev.02816. [DOI] [PubMed] [Google Scholar]
- 157.Schulze SR, Curio-Penny B, Li Y, Imani R, Rydberg L, Geyer PK, Wallrath LL. Molecular genetic analysis of the nested Drosophila melanogaster Lamin C gene. Genetics. 2005;171:185–96. doi: 10.1534/genetics.105.043208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Brandt A, Krohne G, Grosshans J. The farnesylated nuclear proteins KUGELKERN and LAMIN B promote aging-like phenotypes in Drosophila flies. Aging Cell. 2008;7:541–51. doi: 10.1111/j.1474-9726.2008.00406.x. [DOI] [PubMed] [Google Scholar]
- 159.Crisp M, Burke B. The nuclear envelope as an integrator of nuclear and cytoplasmic architecture. FEBS Lett. 2008;582:2023–32. doi: 10.1016/j.febslet.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 160.Dahl KN, Scaffidi P, Islam MF, Yodh AG, Wilson KL, Misteli T. Distinct structural and mechanical properties of the nuclear lamina in Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci USA. 2006;103:10271–6. doi: 10.1073/pnas.0601058103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Cohen M, Lee KK, Wilson KL, Gruenbaum Y. Transcriptional repression, apoptosis, human disease and the functional evolution of the nuclear lamina. Trends Bioc Sci. 2001;26:41–7. doi: 10.1016/s0968-0004(00)01727-8. [DOI] [PubMed] [Google Scholar]
- 162.Shaklai S, Amariglio N, Rechavi G, Simon AJ. Gene silencing at the nuclear periphery. FEBS J. 2007;274:1383–92. doi: 10.1111/j.1742-4658.2007.05697.x. [DOI] [PubMed] [Google Scholar]
- 163.Gruenbaum Y, Margalit A, Goldman RD, Shumaker DK, Wilson KL. The nuclear lamina comes of age. Nat Rev Mol Cell Biol. 2005;6:21–31. doi: 10.1038/nrm1550. [DOI] [PubMed] [Google Scholar]
- 164.Muchir A, Pavlidis P, Bonne G, Hayashi YK, Worman HJ. Activation of MAPK in hearts of EMD null mice: similarities between mouse models of X-linked and autosomal dominant Emery Dreifuss muscular dystrophy. Hum Mol Genet. 2007;16:1884–95. doi: 10.1093/hmg/ddm137. [DOI] [PubMed] [Google Scholar]
- 165.Muchir A, Shan J, Bonne G, Lehnart SE, Worman HJ. Inhibition of extracellular signal-regulated kinase signaling to prevent cardiomyopathy caused by mutation in the gene encoding A-type lamins. Hum Mol Genet. 2009;18:241–7. doi: 10.1093/hmg/ddn343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Gotzmann J, Foisner R. A-type lamin complexes and regenerative potential: a step towards understanding laminopathic diseases. Histochem Cell Biol. 2005;125:33–41. doi: 10.1007/s00418-005-0050-8. [DOI] [PubMed] [Google Scholar]
- 167.Constantinescu D, Gray HL, Sammak PJ, Schatten GP, Csoka AB. Lamin A/C expression is a marker of mouse and human embryonic stem cell differentiation. Stem Cells. 2006;24:177–85. doi: 10.1634/stemcells.2004-0159. [DOI] [PubMed] [Google Scholar]
- 168.Meshorer E, Misteli T. Chromatin in pluripotent embryonic stem cells and differentiation. Nat Rev Cell Mol Biol. 2006;7:540–6. doi: 10.1038/nrm1938. [DOI] [PubMed] [Google Scholar]
- 169.Jones DL, Wagers AJ. No place like home: anatomy and function of the stem cell niche. Nat Rev Cell Mol Biol. 2008;9:11–21. doi: 10.1038/nrm2319. [DOI] [PubMed] [Google Scholar]
- 170.Lowry WE, Richter L. Signaling in adult stem cells. Front Biosci. 2007;12:3911–27. doi: 10.2741/2360. [DOI] [PubMed] [Google Scholar]
- 171.Meshorer E, Gruenbaum Y. Gone with the Wnt/Notch: stem cells in laminopathies, progeria and aging. J Cell Biol. 2008;181:9–13. doi: 10.1083/jcb.200802155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Akter R, Rivas D, Geneau G, Drissi H, Duque G. Effect of lamin A/C knockdown on osteoblast differentiation and function. J Bone Miner Res. 2008 doi: 10.1359/jbmr.081010. ; in press. [DOI] [PubMed] [Google Scholar]
- 173.Scaffidi P, Misteli T. Lamin A-dependent misregulation of adult stem cells associated with accelerated ageing. Nat Cell Biol. 2008;10:452–9. doi: 10.1038/ncb1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature. 2005;434:843–50. doi: 10.1038/nature03319. [DOI] [PubMed] [Google Scholar]
- 175.Espada J, Varela I, Flores I, Ugalde AP, Cadiñanos J, Pendás AM, Stewart CL, Tryggvason K, Blasco MA, Freije JM, López-Otín C. Nuclear envelope defects cause stem cell dysfunction in premature-aging mice. J Cell Biol. 2008;181:27–35. doi: 10.1083/jcb.200801096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Pendas AM, Zhou Z, Cadinanos J, Freije JM, Wang J, Hultenby K, Astudillo A, Wernerson A, Rodriguez F, Tryggvason K, Lopez-Otin C. Defective prelamin A processing and muscular and adipocyte alterations in Zmpste24 metalloproteinase-deficient mice. Nat Genet. 2002;31:94–9. doi: 10.1038/ng871. [DOI] [PubMed] [Google Scholar]
- 177.Verrecchia F, Mauviel A, Farge D. Transforming growth factor-beta signaling through the Smad proteins: role in systemic sclerosis. Autoimmun Rev. 2006;5:563–9. doi: 10.1016/j.autrev.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 178.Moerman EJ, Teng K, Lipschitz DA, Lecka-Czernik B. Aging activates adipogenic and suppresses osteogenic programs in mesenchymal marrow stroma/stem cells: the role of PPAR-gamma2 transcription factor and TGF-beta/BMP signaling pathways. Aging Cell. 2004;3:379–89. doi: 10.1111/j.1474-9728.2004.00127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Novitch BG, Mulligan GJ, Jacks T, Lassar AB. Skeletal muscle cells lacking the retinoblastoma protein display defects in muscle gene expression and accumulate in S and G2 phases of the cell cycle. J Cell Biol. 1996;135:441–56. doi: 10.1083/jcb.135.2.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Bakay M, Wang Z, Melcon G, Schiltz L, Xuan J, Zhao P, Sartorelli V, Seo J, Pegoraro E, Angelini C, Shneiderman B, Escolar D, Chen YW, Winokur ST, Pachman LM, Fan C, Mandler R, Nevo Y, Gordon E, Zhu Y, Dong Y, Wang Y, Hoffman EP. Nuclear envelope dystrophies show a transcriptional fingerprint suggesting disruption of Rb-MyoD pathways in muscle regeneration. Brain. 2006;129:996–1013. doi: 10.1093/brain/awl023. [DOI] [PubMed] [Google Scholar]
- 181.Prokocimer M, Margalit A, Gruenbaum Y. The nuclear lamina and its proposed roles in tumorigenesis: projection on the hema-tologic malignancies and future targeted therapy. J Struc Biol. 2006;155:351–60. doi: 10.1016/j.jsb.2006.02.016. [DOI] [PubMed] [Google Scholar]
- 182.Zink D, Fisher AH, Nickerson JA. Nuclear structure in cancer cells. Nat Rev Cancer. 2004;4:677–87. doi: 10.1038/nrc1430. [DOI] [PubMed] [Google Scholar]
- 183.Kaufmann SH, Mabry M, Jasti R, Shaper JH. Differential expression of nuclear envelope lamins A and C in human lung cancer cell lines. Cancer Res. 1991;51:581–6. [PubMed] [Google Scholar]
- 184.Broers JL, Raymond Y, Rot MK, Kuijpers H, Wagenaar SS, Ramaekers FC. Nuclear A-type lamins are differentially expressed in human lung cancer subtypes. Am J Pathol. 1993;143:211–20. [PMC free article] [PubMed] [Google Scholar]
- 185.Moss SF, Krivosheyev V, De Souza A, Chin K, Gaetz HP, Chaudhary N, Worman HJ, Holt PR. Decreased and aberrant nuclear lamin expression in gastrointestinal tract neoplasms. Gut. 1999;45:723–9. doi: 10.1136/gut.45.5.723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Willis ND, Cox TR, Rahman-Casañs SF, Smits K, Przyborski SA, Van Den Brandt P, Van Engeland M, Weijenberg M, Wilson RG, De Bruïne A, Hutchison CJ. Lamin A/C is a risk biomarker in colorectal cance. PLoS One. 2008;3:e2988. doi: 10.1371/journal.pone.0002988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Qi YJ, He QY, Ma YF, Du YW, Liu GC, Li YJ, Tsao GS, Ngai SM, Chin JF. Proteomic identification of malignant transformation-related proteins in esophageal squamous cell carcinoma. J Cell Biochem. 2008;104:1625–35. doi: 10.1002/jcb.21727. [DOI] [PubMed] [Google Scholar]
- 188.Hytiroglou P, Choi SW, Theise ND, Chaudhary N, Worman HJ, Thung SN. The expression of nuclear lamins in human liver: an immunohistochemical study. Hum Pathol. 1993;24:169–72. doi: 10.1016/0046-8177(93)90296-s. [DOI] [PubMed] [Google Scholar]
- 189.Venables RS, McLean S, Luny D, Moteleb E, Morley S, Quinlan RA, Lane EB, CJ. H. Expression of individual lamins in basal cell carcinomas of the skin. Br J Cancer. 2001;84:512–9. doi: 10.1054/bjoc.2000.1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Oguchi M, Sagara J, Matsumoto K, Saida T, Taniguchi S. Expression of lamins depends on epidermal differentiation and transformation. Br J Dermatol. 2002;147:853–8. doi: 10.1046/j.1365-2133.2002.04948.x. [DOI] [PubMed] [Google Scholar]
- 191.Tilli CM, Ramaekers FC, Broers JL, Hutchison CJ, Neumann HA. Lamin expression in normal human skin, actinic keratosis, squamous cell carcinoma and basal cell carcinoma. Br J Dermatol. 2003;148:102–9. doi: 10.1046/j.1365-2133.2003.05026.x. [DOI] [PubMed] [Google Scholar]
- 192.Machiels BM, Ramaekers FCS, Kuijpers HJH, Groenewoud JS, Oosterhuis JW, Looijenga LHJ. Nuclear lamin expression in normal testis and testicular germ cell tumours of adolescents and adults. J Pathology. 1997;182:197–204. doi: 10.1002/(SICI)1096-9896(199706)182:2<197::AID-PATH823>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 193.Coradeghini R, Barboro P, Rubagotti A, Boccardo F, Parodi S, Carmignani G, D’Arrigo C, Patrone E, Balbi C. Differential expression of nuclear lamins in normal and cancerous prostate tissues. Oncol Rep. 2006;15:609–13. [PubMed] [Google Scholar]
- 194.Hudson ME, Pozdnyakova I, Haines K, Mor G, Snyder M. Identification of differentially expressed proteins in ovarian cancer using high-density protein microarrays. Proc Natl Acad Sci USA. 2007;104:17494–9. doi: 10.1073/pnas.0708572104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Bussolati G, Marchiò C, Gaetano L, Lupo R, Sapino A. Pleomorphism of the nuclear envelope in breast cancer: a new approach to an old problem. J Cell Mol Med. 2008;12:209–18. doi: 10.1111/j.1582-4934.2007.00176.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Fischer AH, Taysavang P, Weber CJ, Wilson KL. Nuclear envelope organization in papillary thyroid carcinoma. Histol Histopathol. 2001;16:1–14. doi: 10.14670/HH-16.1. [DOI] [PubMed] [Google Scholar]
- 197.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10:789–99. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 198.Agrelo R, Setien F, Espada J, Artiga MJ, Rodriguez M, Perez-Rosado A, Sanchez-Aguilera A, Fraga MF, Piris MA, Esteller M. Inactivation of the lamin A/C gene by CpG island promoter hypermethylation in hematologic malignancies, and its association with poor survival in nodal diffuse large B-cell lymphoma. J Clin Oncol. 2005;23:3940–7. doi: 10.1200/JCO.2005.11.650. [DOI] [PubMed] [Google Scholar]
- 199.Foran E, McWilliam P, Kelleher D, Croke DT, Long A. The leukocyte protein L-plastin induces proliferation, invasion and loss of E-cadherin expression in colon cancer cells. Int J Cancer. 2006;118:2098–104. doi: 10.1002/ijc.21593. [DOI] [PubMed] [Google Scholar]
- 200.Shalev SA, De Sandre-Giovannoli A, Shani AA, Levy N. An association of Hutchinson-Gilford progeria and malignancy. Am J Med Genet A. 2007;143:1821–6. doi: 10.1002/ajmg.a.31803. [DOI] [PubMed] [Google Scholar]
- 201.Masuda H, Miller C, Koeffler HP, Battifora H, Cline MJ. Rearrangement of the p53 gene in human osteogenic sarcomas. Proc Natl Acad Sci USA. 1987;84:7716–9. doi: 10.1073/pnas.84.21.7716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Attisano L, Wrana JL. Signal transduction by the TGF-beta superfamily. Science. 2002;296:1646–7. doi: 10.1126/science.1071809. [DOI] [PubMed] [Google Scholar]
- 203.Schmierer B, Hill CS. TGFbeta-SMAD signal transduction: molecular specificity and functional flexibility. Nat Rev Cell Mol Biol. 2007;8:970–82. doi: 10.1038/nrm2297. [DOI] [PubMed] [Google Scholar]
- 204.Ishimura A, Ng JK, Taira M, Young SG, Osada S. Man1, an inner nuclear membrane protein, regulates vascular remodeling by modulating transforming growth factor {beta} signaling. Development. 2006;133:3919–28. doi: 10.1242/dev.02538. [DOI] [PubMed] [Google Scholar]
- 205.Giles RH, Van Es JH, Clevers H. Caught up in a Wnt storm: Wnt signaling in cancer. Bioch Biophis Acta. 2003;1653:1–24. doi: 10.1016/s0304-419x(03)00005-2. [DOI] [PubMed] [Google Scholar]
- 206.Guo Z, Dose M, Kovalovsky D, Chang R, O’Neil J, Look AT, Von Boehmer H, Khazaie K, Gounari F. Beta-catenin stabilization stalls the transition from double-positive to single-positive stage and predisposes thymocytes to malignant transformation. Blood. 2007;109:5463–72. doi: 10.1182/blood-2006-11-059071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Markiewicz E, Tilgner K, Barker N, Van De Wetering M, Clevers H, Dorobek M, Hausmanowa-Petrusewicz I, Ramaekers FC, Broers JL, Blankesteijn WM, Salpingidou G, Wilson RG, Ellis JA, Hutchison CJ. The inner nuclear membrane protein emerin regulates beta-catenin activity by restricting its accumulation in the nucleus. EMBO J. 2006;25:3275–85. doi: 10.1038/sj.emboj.7601230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Rajagopalan H, Lengauer C. Aneuploidy and cancer. Nature. 2004;432:338–41. doi: 10.1038/nature03099. [DOI] [PubMed] [Google Scholar]
- 209.Margalit A, Liu J, Fridkin A, Wilson KL, Gruenbaum Y. A lamin-dependent pathway that regulates nuclear organization, cell cycle progression and germ cell development. John Wiley & Sons, Ltd; 2005. pp. 231–9. . London: . [PubMed] [Google Scholar]
- 210.Burkhart DL, Sage J. Cellular mechanisms of tumor suppression by the retinoblastoma gene. Nat Rev Cancer. 2008;8:671–82. doi: 10.1038/nrc2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Tsai MY, Wang SH, Heidinger JM, Shumaker DK, Adam SA, Goldman RD, Zheng Y. A mitotic lamin B matrix induced by RanGTP required for spindle assembly. Science. 2006;311:1887–93. doi: 10.1126/science.1122771. [DOI] [PubMed] [Google Scholar]
- 212.Stewart CL, Roux KJ, Burke B. Blurring the boundary: the nuclear envelope extends its reach. Science. 2007;318:1408–12. doi: 10.1126/science.1142034. [DOI] [PubMed] [Google Scholar]
- 213.Mou F, Forest T, Baines JD. US3 of herpes simplex virus type 1 encodes a promiscuous protein kinase that phospho-rylates and alters localization of lamin A/C in infected cells. J Virol. 2007;81:6459–70. doi: 10.1128/JVI.00380-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Mou F, Wills EG, Park R, Baines JD. Effects of Lamin A/C, B1 and viral US3 kinase activity on viral infectivity, virion egress, and targeting the herpes simplex virus UL34-encoded protein to the inner nuclear membrane. J Virol. 2008;82:8094–104. doi: 10.1128/JVI.00874-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Silva L, Cliffe A, Chang L, Knipe DM. Role for A-type lamins in herpesviral DNA targeting and heterochromatin modulation. PLoS Pathog. 2008;4:e1000071. doi: 10.1371/journal.ppat.1000071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Camozzi D, Pignatelli S, Valvo C, Lattanzi G, Capanni C, Dal Monte P, Landini MP. Remodelling of the nuclear lamina during human cytomegalovirus infection: role of the viral proteins pUL50 and pUL53. J Gen Virol. 2008;89:731–40. doi: 10.1099/vir.0.83377-0. [DOI] [PubMed] [Google Scholar]
- 217.Kuhn M, Desloges N, Rahaus M, Wolff MH. Varicella-zoster virus infection influences expression and organization of actin and alpha-tubulin but does not affect lamin A and vimentin. Intervirology. 2005;48:312–20. doi: 10.1159/000085100. [DOI] [PubMed] [Google Scholar]
- 218.Bukrinsky M. A hard way to the nucleus. Mol Med. 2004;10:1–5. [PMC free article] [PubMed] [Google Scholar]
- 219.De Noronha CM, Sherman MP, Lin HW, Cavrois MV, Moir RD, Goldman RD, Greene WC. Dynamic disruptions in nuclear envelope architecture and integrity induced by HIV-1 Vpr. Science. 2001;294:1105–8. doi: 10.1126/science.1063957. [DOI] [PubMed] [Google Scholar]
- 220.Wiebe MS, Traktman P. Poxviral B1 kinase overcomes barrier to autointegration factor, a host defense against virus replication. Cell Host Microbe. 2007;17:187–97. doi: 10.1016/j.chom.2007.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]