

Abstract
Human endothelial nitric oxide synthase (eNOS) plays a pivotal role in maintaining blood pressure homeostasis and vascular integrity. It has recently been reported that mitogen-activated protein kinases (MAPKs) are intimately implicated in expression of eNOS. However detailed mechanism mediated by them remains to be clarified. In this study, eNOS gene transactivity in human umbilical vein endothelial cells was up-regulated by stimulation of lysophosphatidylcholine (LPC). The stimulation of LPC highly activated both extracellular signal-regulated kinase 1/2 (ERK1/2) and c-Jun N-terminal kinase (JNK), with differences in the dynamic processes of activation between them. Unexpectedly, p38 MAPK could not be activated by the stimulation of LPC. The activation of JNK signalling pathway by overexpression of JNK or its upstream kinase active mutant up-regulated the transactivity of eNOS significantly, but the activation of p38 signalling pathway down-regulated it largely. The inhibition of either ERK1/2 or JNK signalling pathway by kinase-selective inhibitors could markedly block the induction of the transactivity by LPC. It was observed by electrophoretic mobility shift assay that LPC stimulated both SP1 and AP1 DNA binding activity to go up. Additionally using decoy oligonucleotides proved that SP1 was necessary for maintaining the basal or stimulated transactivity, whereas AP1 contributed mainly to the increase of the stimulated transactivity. These findings indicate that the up-regulation of the eNOS gene transactivity by LPC involves the enhancement of SP1 transcription factor by the activation of JNK and ERK1/2 signalling pathways and AP1 transcription factor by the activation of JNK signalling pathway.
Keywords: lysophosphatidylcholine, endothelial nitric oxide synthase, mitogen-activated protein kinase, transactivity, regulation
Introduction
Human endothelial nitric oxide synthase (eNOS), that is constitutively expressed primarily in endothelial cells lining blood vessels and hearts, has been known to play an important role in regulating blood pressure, vascular permeability, white blood cell adhesion and platelet aggregation, contributing to some pathological processes such as shock, hypertension, atherosclerotic heart disease, diabetes, ischaemia, vascular stenosis and occlusion [1, 2, 4, 8, 12, 14, 19, 28]. The eNOS gene knockout animals were subjected to salt-induced hypertension and ischaemia-induced tissue impairment [22]. The animals injected with a Sleeping Beauty-based transposon harboring eNOS gene showed significant reduction in pulmonary arterial pressure, attenuation of right ventricle to whole heart weight ratios and decrease in the pulmonary vessel wall thickness [26]. The eNOS-overexpressed hearts were maximally protected against infarcts and ischaemia/reperfusion injury by their elevated endogenous NO levels [9]. Local adenovirus-mediated eNOS gene transfer to the carotid artery of the stroke-prone spontaneously hypertensive animals was successful in improving endothelial function [29]. Therefore, eNOS protects against restenosis, infarction, atherosclerosis, pulmonary hypertension and ischaemia/reperfusion tissue damage, and has important therapeutic implications.
Expression of eNOS gene is up-regulated or down-regulated by a variety of exogenous and endogenous stimuli [11], at least including shear stress [40], hypoxia [17, 18, 36, 37], lysophosphatidyl-choline (LPC) [6, 42, 43], oxidized low density lipoprotein [15], histamine [23, 38], ceramide [24], flavonoid [3], resveratrol [39], transforming growth factor-β[35], tumour necrosis factor-α[20] and estrogen [27, 41]. It is, obviously, rather significant to elucidate regulation mechanism of eNOS gene expression induced by these stimuli. Little, however, is known as to how mitogen- activated protein kinase (MAPK) signalling pathways modulate eNOS gene expression even though it was reported that one of MAPK signalling pathways, i.e. phosphatidylinositol-3 kinase γ (PI-3Kγ)->Janus kinase 2(Jak2)—>mitogen-activated protein kinase/extracellular signal-regulated kinase kinase 1(MEK1)->extracellular signal-regulated kinase 1/2 (ERK1/2)->promoter selective transcription factor 1 (SP1) [6, 42], was involved in up-regulation of eNOS gene expression.
Strong evidence supports a role for LPC, which is a product of phosphatidylcholine hydrolysis by phospholipase A2 (PLA2), in the processes of atherogenesis, inflammation, and vascular wound healing [15, 21, 30]. It was demonstrated that LPC increased eNOS expression, representing a crucial mechanism by which arteries exert their plastic defence against vessel wall injury and atherogenesis, and therefore it has been used as a model system to illuminate the mechanism by which eNOS gene transcription is regulated [7, 42]. In the present study, to shed light on the regulation of human eNOS gene expression, a red fluorescent protein (RFP) reporter gene vector containing human eNOS promoter was successfully constructed and co-transfected with wild-type MAPKs and their up-stream active mutants as well as decoy oligonucleotides (ODN) into human umbilical vein endothelial cells. Using RFP as a reporter [13, 43], real time observation could be carried out to detect the activity of eNOS promoter in the cells treated with LPC. The novel findings suggest that the up-regulation of human eNOS gene transcription is implicated not only in ERK1/2, but also in JNK1/2 signalling pathway, and p38 signalling pathway exerts its effort on down-regulation of it.
Materials and methods
Construction of expression vectors
A 5′-flanking fragment of the human eNOS promoter sequence at nucleotide positions from -1600 bp to +1 bp (Gen Bank™, Accession Number: AF387340) was obtained by polymerase chain reaction with a forward primer 5′-gaagatctatctgatgctgcctgtcaccttgaccctgag-3′ (Bgl II site was underlined) and a reverse primer 5′-attaagctttgcctgctccagcagagccctg-gccttttc-3′ (Hind III site was underlined) using the genomic DNA extracted from foetus umbilical vein endothelial cells as a template. The PCR product purified by agarose gel electrophoresis was digested with Bgl II and Hind III (TaKaRa, Dalian, China) and cloned into RFP expression vector pDsRed 1–1 (Clontech, Mountain View, CA, USA). Rightness of the construct was confirmed by double restricted endonuclease digestion and DNA sequencing, and it was designated as pDseNOSRed.
Flag-tagged ERK2, JNK1 and p38(a) in pcDNA3 as well as hemagglutinin-tagged MAPKK active mutants, including MEK1(E), MKK4(E) and MKK6b(E) in pSRa or pcDNA3, were generous gifts from professor R.J. Ulevitch and Dr. J. Han in The Scripps Research Institute (La Jolla, CA, USA).
Cell culture and DNA transfection
Cultured human umbilical vein endothelial cells (HUVEC-12 cell line) were grown in a 24-well plate in DMEM containing 5% FBS. The cells were transfected with 0.5 μg of pDseNOSRed or promoterless pDsRed1–1 and 0.4 μg of MAPKK or MAPK expression vectors as indicated in the figures using LipofectAMINE reagent kit (Invitrogen, San Diego, CA, USA) following routine procedure. Then, the medium was removed and replaced with complete medium for 24 hrs. The cells were washed, incubated in medium containing 0.1% FBS for 16 hrs, and then cultured in fresh medium containing 5% FBS in the presence or absence of LPC (Sigma, St Louis, MI, USA). Selective inhibitors, including PD98059 (Sigma), SB203580 (Sigma) and curcumin (Calbiochem, Darmstadt, Germany) were added to the cells with final concentrations of 50 μMol/l, 15 μMol/l 30 μMol/l, respectively for 1.5 hrs. Then 40 μMol/l of LPC were chosen to stimulate the cells, for this concentration of LPC used had been proved to have no obvious cytotoxicity [6, 7, 43]. The eNOS promoter activity was measured at the indicated time. The transfection efficiency was normalized by an approach to co-transfect 0.2 μg of pEGFP-N1 vector as an internal control with the target constructs described above.
In the electrophoretic mobility shift assay (EMSA) experiment, HUVEC-12 cells grown in 100-mm dishes to 50% confluence were transfected with 4.0 μg of pcDNA3 or flag-tagged JNK1 in pcDNA3 using PolyFect transfection reagent kit (QIAGEN, Hilden, Germany), following the procedure from the manufacturer. The cells were gently washed by phosphate-buffered saline (PBS) 24 hrs after transfection, followed by serum starvation, drug treatment and stimulation with LPC as described above. They were harvested at the different time and the cytoplasmic protein and nuclear extracts were prepared as previously reported.
RFP reporter gene assay
The transfected cells were observed under inverted fluorescence microscope (Nikon TE300, Chiyoda-Ku, Tokyo) at each interval of 12 hrs, with wavelengths of excitation 550 nm and emission 580 nm, respectively. Red fluorescence-emitting cells in each microwell were scanned at random under the low power visual field (x100) using a high sensitivity digital camera (Penguin 150CL Pixera, Los Gatos, CA, USA) that was connected with a computer. More than 10 low power visual fields for each microwell were scanned for the avoiding the bias from RFP expression variations in the cells. Then, the optical density (OD) of red fluorescence, which represents eNOS promoter activity, was determined using the fluorescence analysis software, Image-Pro Plus (Mediacy Cybernetics, Silver Spring, MD, USA). The green fluorescence emitted by green fluorescence protein (GFP) was measured with an excitation wavelength of 488 nm 36 hrs after the transfection, by which the transfection efficiency was normalized. The same experiment was repeated more than three times.
Protein lysate and nuclear extract preparation
The harvested cells were suspended in PBS containing 0.5 mMol/l PMSF at 4°C and spun down for 15 min. at 10,000 × g. The pellet was resuspended in 400 (μl of buffer A (10 mMol/l HEPES, pH 7.9, 10 mMol/l KCl, 1.0 mMol/l DTT, 0.1 mMol/l EDTA, 0.1 mMol/l EGTA, 2.0 μg/ml aprotinin, 2.0 μg/ml leupeptin, 1.0 μg/ ml pepstatin, 1.0 mMol/l PMSF), and placed on ice for 15 min. After addition of 25 μl volume of 10% Nonidet P-40, the cells were violently rocked for 30 sec. and centrifuged at 12,000 x g, 4°C for 20 min. The supernatant was used to perform Western blot analysis. Then, the deposit was resuspended in 100 μl buffer B (20 mMol/l HEPES, pH 7.9, 400 mMol/l KCl, 1.0 mMol/l DTT, 0.1 mMol/l EDTA, 0.1 mMol/l EGTA, 2.0 μg/ml aprotinin, 2.0 μg/ml leupeptin, 1.0 μg/ ml pepstatin, 1.0 mMol/l PMSF) and rocked on ice for 45 min. The suspension mixture was centrifuged at 12,000 × g, 4°C for 20 min., and the supernatant, i.e. nuclear extract was applied to EMSA. Protein concentration was determined by Bradford assay using bovine serum albumin as a standard.
Western blot analysis
Western blot analysis was performed using PhosphoPlus p44/42 MAPK antibody kit, PhosphoPlus SAPK/JNK antibody kit and PhosphoPlus p38 MAPK antibody kit (Cell Signaling Technology, Beverly, MA, USA), following the instruction from the manufacturer. In brief, 15.0 μg of protein lysate were separated by 10% SDS-polyacrylamide gel electrophoresis, and transferred to nitrocellulose membranes (Amersham Biosciences, Piscataway, NJ, USA) using the semi-dry protein tranfer equipment, multi-phor II (Pharmacia Biotech, Piscataway, NJ, USA). The membranes were washed and incubated in blocking buffer containing 5% milk overnight at 4°C. The primary antibody incubation with gentle agitation was performed overnight at 4°C. After being washed with TBST, the membrane was incubated with horseradish peroxidase (HRP)-conjugated secondary antibody (1:2000) for 1 h at room temperature. Target proteins were detected by the enhanced chemiluminescence system, and the quantitation of protein bands was performed using densitometry software, Gel Base/Gel Plot.
Electrophoretic mobility shift assay
The double strand DNA probes of AP1 or SP1 were synthesized according to the eNOS promoter sequence in human umbilical vein endothelial cells. The AP1 probe was designed with forward sequence 5′-cctaggaaaaaat-gagtcatcct-3′ and reverse sequence 5′-aggatgactcattttcctagg-3′; the SP1 probe was designed with forward sequence 5′-gggataggggcggggcgaggg-3′ and reverse sequence 5′-ccctcgccccgcccctatccc-3′. The probes were labelled with [a-32P]dATP by terminal deoxynucleotidyl transferase (TaKaRa). The nuclear extract with 5.0 μg proteins was incubated with a cold oligonucleotide probe in a binding buffer containing 20 mMol/l Tris-HCL, pH7.5, 50 mMol/l NaCl, 0.5 mMol/l EDTA, 0.5 mMol/l DTT, 2.0 mMol/l MgCl2-, 1.5 μg of poly (dI-dC), 7.5% glycerol and 0.5 mMol/l PMSF on ice for 10 min. to confirm the specificity of binding to α-32P]dATP-labelled probes. Then, α-32P]dATP-labelled probes (10,000–25,000 cpm) were added to the nuclear extracts and incubated at room temperature for 30 min. The mixture was subjected to nondenaturing gel electrophoresis at 9 V/cm on 6% polyacrylamide gel with buffer containing 0.5×TBE and 0.5% Nonidet P-40. The gel was vacuum-dried, followed by autoradiography at -70°C overnight in a cassette with an intensifying screen.
Decoy oligonucleotides
The well-designed sense and antisense strands of AP1, SP1 and shear stress response element (SSRE) decoy oligonucleotides (ODN) as well as their mutant control decoy ODN were synthesized by TaKaRa Co. The AP1 decoy sequence was 5′-tgagtcatgagtcatgagtca-3′ with reverse sequence 5′-tgactcatgactcatgactca-3′ and its mutant control decoy sequence was 5′-tgaggaatgaggaatgaggaa-3′ with reverse sequence 5-ttcctcattcctcattcctca-3′. The SP1 decoy sequence was 5′-ggggcggggcggggcggggcggggcggggc-3′ with reverse sequence 5′-gccccgccccgccccgccccgccccgcccc-3′ and its mutant control decoy sequence was 5′-gagactgggcgagactgggcga-gactgggc-3′ with reverse sequence 5′-gcccagtctcgcccagtctcgcccagtctc-3′. The SSRE decoy sequence was 5′-ggtctcggtctcggtctc-3′ with reverse sequence 5′-gagaccgagaccgagacc-3′ and its mutant control decoy sequence was 5′-gcagtcgcagtcgcagtc-3′ with reverse sequence 5′-gactgc-gactgcgactgc-3′. Each sense and antisense oligonucleotide pair was annealed by heating to 95°C and decreasing temperature 5°C every 15 min. The reaction mixtures were held at a temperature of 4°C.
HUVEC-12 cells grown in 24-well plates were co-transfected with 0.6 μg of pDseNOSRed and 5.0 p Mol/l of ODN of AP1, SP1 or SSRE using the LipofectAMINE, in accordance with the protocol supplied by the manufacturer.
Statistical analysis
All the data were expressed as mean ± standard deviation. The software of the Statistical Package for Social Sciences (Version 10.0) was used to analyse the significance among different groups. Mean values were compared using one-way anova test. A P value <0.05 was considered statistically significant. All the results represent at least three independent experiments.
Results
Induction of human eNOS transcriptional activity by LPC
To check effect of LPC on gene expression of human eNOS, an eNOS promoter-driven RFP reporter gene expression vector was constructed. It was found that this RFP reporter system was well expressed in HUVEC-12 cells. After the stimulation of LPC (40 μMol/l), RFP expression was elevated significantly as compared with the untreated cells (P< 0.01).
Selective induction of the phosphorylation of MAPKs by LPC
After finding the induction of transactivity of eNOS in the cells by LPC, we want to elucidate intracellular signalling mechanism involved in the gene regulation. At first, we detected time-dependent effect of LPC on the activity of ERK1/2. HUVEC-12 cells were treated with 40 μM of LPC for 0 min., 15 min., 30 min., 1 hr, 2 hrs, 3 hrs, 4 hrs, 5 hrs or 6 hrs, respectively. Cell lysate was used for Western blot to detect levels of unphosphorylated and phosphory-lated ERK1/2 using specific monoclonal antibodies. It was found that the phosphorylation level of ERK1/2 began to increase after stimulation with LPC for 30 min. and reached its peak at 4 hrs, whereas the unphosphorylated level of ERK1/2 had no significant changes in the cells after the stimulation of LPC up to 6 hrs (Fig. 1A and B).
Figure 1.

Time-dependent effect of LPC stimulation on the activities of MAPKs. HUVEC-12 cells were treated with 40 μM/l of LPC at the different intervals of 0 min., 15 min., 30 min., 1 hr, 2 hrs, 3 hrs, 4 hrs, 5 hrs and 6 hrs, respectively (lanes 1–9). After harvest and lysis of the cells their cytoplasmic proteins were extracted, then Western blot was performed to detect unphosphorylated (upper panel) and phosphory-lated (lower panel) levels of ERK1/2 (A, B), JNK1/2 (C, D) and p38 (E, F), respectively. The phosphorylated levels of them were subjected to densitometric analysis. Each bar is means ± S.D. of 3 independent experiments. *P< 0.05, **P< 0.01, compared with LPC stimulation group for 0 min.
The same procedure was performed on measurement of activity of JNK1/2 using monoclonal antibodies to nonphosphorylated or phosphorylated JNK1/2. The result showed that the phosphorylation level of JNK1/2 gradually increased after LPC stimulation for 1 hr, reached the peak 3 hrs later, followed by a decrease after 4 hrs of LPC stimulation, and gradually returned to the level close to that before LPC stimulation. Like what found on ERK1/2, the nonphosphorylated protein of JNK1/2 had also no significant changes after the stimulation by LPC within the observation period (Fig. 1C and D).
For detection of the phosphorylation level on p38 MAPK, we used monoclonal antibodies specific for p38 MAPK to perform Western blot. To our surprise, the phosphorylation level of p38 MAPK was not altered by treatment of LPC in the detection period (Fig. 1E and F).
Effect of MAPK inhibitors on the phosphorylation of MAPKs induced by LPC
To further confirm the inducing action of LPC on the phosphorylation of MAPKs above, MEK1-selective inhibitor PD98059, JNK-selective inhibitor curcumin and p38-specific inhibitor SB203580 were administered to HUVEC-12 cells before treatment of LPC, respectively. The results showed that the increase of the phosphorylation level of ERK1/2 by stimulation of LPC might be abolished by PD98059 (Fig. 2A and B), and curcumin might also abrogate the induction of the JNK1 phosphorylation by LPC (Fig. 2C and D). But the phosphorylation level of p38 MAPK was not altered by treatment with LPC no matter in the presence or absence of SB203580 (Fig. 2E and F).
Figure 2.
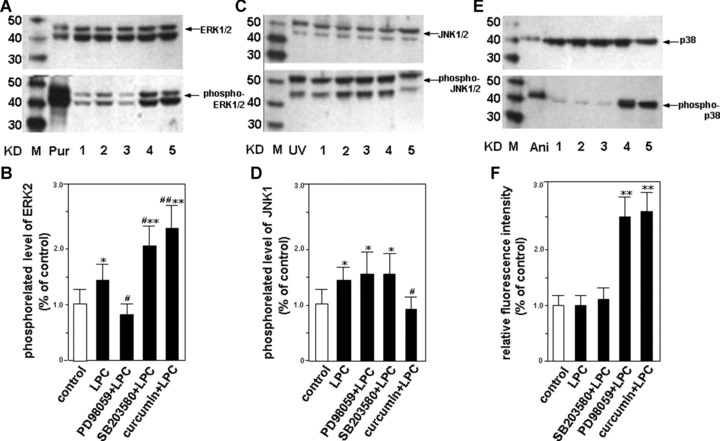
Effects of the selective inhibitors of MAPK signalling pathways on MAPK activities stimulated by LPC. HUVEC-12 cells were pretreated with selective inhibitors including PD98059 (50 μm/l), SB203580 (15 μm/l) and curcumin (30 μm/l) for 1.5 hrs, respectively, followed by stimulation with or without 40 μm/l of LPC (lanes 1–5). The cells were harvested, lysed and cytoplasmic proteins of them were extracted 2 hrs after the treatment of LPC, then Western blot was performed to detect unphosphorylated (upper panel) and phosphorylated (lower panel) levels of ERK1/2 (A, B), JNK1/2 (C, D) and p38 (E, F), respectively. The phosphorylated levels of them were subjected to densitometric analysis. Each bar is means ± S.D. of 3 independent experiments. M, molecular mass markers; Pur, purified ERK1/2 protein that bacterially expressed was phosphorylated or unphosphorylated by MEK as a positive control of ERK1/2; UV, HEK293 cells were treated with or without ultraviolet ray as a positive control of JNK1/2; Ani, C6 glioma cells were treated with or without anisomycin as a positive control of p38; control, HUVEC-12 cells were untreated with LPC as a control. *P< 0.05, **P< 0.01, compared with the control; #P< 0.05, ##P< 0.01, compared with LPC group.
Interestingly, it was found that PD98059 markedly attenuated the phosphorylation of ERK1/2 induced by LPC, but SB203580 or curcumin accelerated it and that the phosphorylation level of p38 was elevated by PD98059 or curcumin. It suggests that one of MAPK signalling pathways blocked by the corresponding inhibitor may act as a stimulating signal for other MAPK signalling pathways.
Effect of MAPK activation on human eNOS transactivity
Encouraged by the finding that LPC selectively activated different MAPKs, we want to further explore the effect of individual MAPK activation on human eNOS promoter transactivity. Expression vectors of flag-tagged ERK2, JNK1 and p38 in pcDNA3 were co-transfected with the pDseNOSRed reporter construct into HUVEC-12 cells, respectively. After measurement of the fluorescence intensity of RFP, we found that the expression of JNK1 or ERK2 significantly up-regulated human eNOS promoter transcriptional activity in the cells. In contrast, the expression of p38 markedly down-regulated the promoter transcriptional activity (Fig. 3A).
Figure 3.
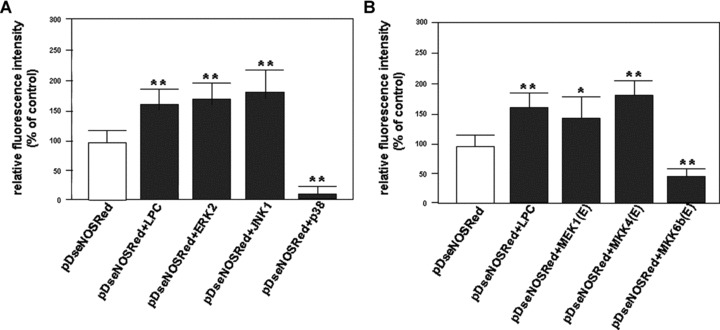
Effect of the stimulation of LPC and the activation of MAPKs on human eNOS transcriptional activity. (A) Cultured HUVEC-12 cells were co-transfected with pDseNOSRed plus pcDNA3 or ERK2 or JNK1 or p38 MAPK expression vector, respectively. (B) The cultured cells were co-transfected with pDseNOSRed plus pcDNA3 or MEK1(E) or MKK4(E) or MKK6b(E) active mutant, respectively. These cells were treated with or without stimulation of LPC (40 julMol/l) 45 hrs after the transfection. The red fluorescence-emitting cells were subjected to dynamic observation under an inverted fluorescence microscope. The fluorescence intensity of RFP expressed by the cells that represents the eNOS transactivity was quantitatively analysed using the fluorescence analysis software, Image-Pro Plus, 96 hrs after transfection. This result represents means ± S.D. of 3 independent experiments. *P< 0.05, **P< 0.01, compared with pDseNOSRed group.
In addition to overexpression of wild-type of MAPKs in the cells, the active mutants of MAPK kinases are strong activators for MAPK pathways. The eNOS RFP reporter gene construct, pDseNOSRed, was co-transfected with pcDNA3, or different active mutants of MAPK Kinases, i.e. MEK1(E), MKK4(E) and MKK6b(E), into HUVEC-12 cells. In consonance with the results of transfection of MAPKs, the co-transfection with MKK4(E) or MEK1(E) enhanced eNOS promoter activity remarkably, while MKK6b(E), an active mutant of upstream kinase of p38 MAPK, attenuated LPC-induced eNOS transactivity (Fig. 3B).
Effect of MAPK inhibitors on LPC-induced human eNOS transactivity
Specific inhibitors of MAPKs are powerful tools to explore the function of MAPK pathways. To confirm the results above, MEK1-selective inhibitor PD98059, JNK-selective inhibitor curcumin and p38-specific inhibitor SB203580 were used to determine effect of MAPK inhibition on the eNOS gene expression induced by LPC. The results showed that the human eNOS promoter activity induced by LPC was dramatically blocked by both PD98059 and curcumin. However, SB203580 lacked the ability to repress the induction of the promoter transactivity by treatment with LPC. These results showed that ERK1/2 and JNK signal pathways might be involved in the activation of the promoter transactivity induced by LPC (Fig. 4A).
Figure 4.
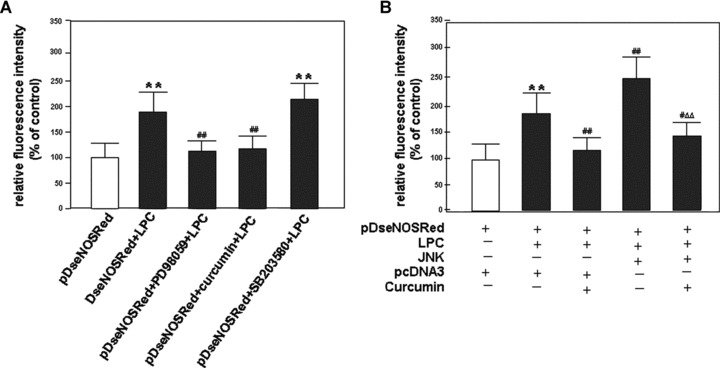
Effects of the selective inhibitors of MAPKs on human eNOS transcriptional activity induced by LPC. (A) After transfected with pDseNOSRed, HUVEC-12 cells were treated without or with PD98059 (50 μMol/l) or SB203580 (15 μMol/l) or curcumin (30 μMol/l), respectively, for 1.5 hrs before treatment of LPC. (B) After transfected with pDseNOSRed plus pcDNA3 or JNK1 expression vector, the cells were treated without or with curcumin (30 μMol/l) at the same time. 45 hrs after the transfection, the cells were stimulated without or with LPC (40 μMol/l). The treatment of each group is shown as indicated in the figure. The red fluorescence-emitting cells were subjected to dynamic observation under an inverted fluorescence microscope. The fluorescence intensity of RFP expressed by the cells that represents the eNOS transactivity was quantitatively analysed using the fluorescence analysis software, Image-Pro Plus, 96 hrs after transfection. The result represents means ± S.D. of 3 independent experiments. **P< 0.01, compared with pDseNOSRed group; #P< 0.05, ##P< 0.01, compared with pDseNOSRed plus LPC group (A) or compared with pDseNOSRed plus pcDNA3 and LPC group (B); AAP< 0.01, compared with pDseNOSRed plus JNK1 expression vector and LPC group.
The human eNOS promoter activity induced by LPC in HUVEC-12 cells that were co-transfected with the pDseNOSRed plus JNK1 expression vector was higher than that in the cells that were co-transfected with pDseNOSRed plus pcDNA3 vector. Furthermore, JNK activation-induced eNOS expression was also blocked by curcumin, strongly suggesting that JNK pathway is indeed associated with the up-regulation of the promoter transcription induced by LPC (Fig. 4B).
LPC-induced SP1 and AP1 DNA binding activities
To confirm activities of SP1 DNA binding in HUVEC-12 cells stimulated by LPC, the nuclear extracts at the different time were subjected to EMSA analysis. SP1 DNA binding activity began to go up 15 min. after treatment of LPC, reaching the peak 1 to 3 hrs after LPC treatment, followed by a gradual decrease 4 hrs after the treatment (Fig. 5A and B).
Figure 5.
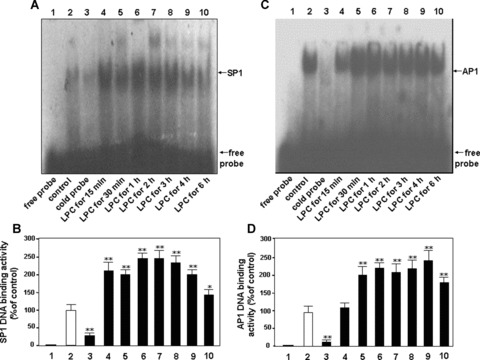
SP1 and AP1 DNA binding activities induced by LPC stimulation. After treatment of LPC, HUVEC-12 cell nuclear extracts were prepared at the different intervals for detection of SP1 (A, B) and AP1 (C, D) DNA binding activity via EMSA. The extracts were incubated with a 32P-labelled SP1 oligodeoxynucleotide probe or 32P-labelled AP1 oligodeoxynucleotide probe and subjected to nondenaturing gel electrophoresis, followed by autoradiography. Lane 1, free probe; lane 2, the cells untreated with LPC served as a control; lane 3, 100-fold molar excess of cold probe of SP1 or AP1 was used to confirm SP1- or AP1-specific DNA binding; lane 4–10, the cells were treated with 40 μMol/l of LPC for 15 min., 30 min., 1 hr, 2 hrs, 3 hrs, 4 hrs and 6 hrs, respectively. SP1 or AP1 DNA binding is indicated by the upper arrow, and free probe is indicated by the lower arrow. The result represents means ± S.D. of 3 independent experiments. *P< 0.05, **P< 0.01, compared with the control.
AP1 DNA binding activity swiftly reached the peak at 30 min. after treatment of LPC and maintained at a high level up to 4 hrs after LPC treatment, followed by a decrease 6 hrs after the treatment of LPC. The result indicates that LPC may enhance both SP1and AP1 DNA binding activities in HUVEC-12 cells (Fig. 5C and D).
Effect of MAPK inhibitors on LPC-induced SP1 and AP1 DNA binding activities
Both PD98059 and curcumin blocked SP1 DNA binding activity induced by LPC, but SB203580 failed to show any inhibitory effect on the SP1 DNA binding activity (Fig. 6A and B). These results suggested that ERK1/2 and JNK pathways were implicated in SP1 DNA binding activity induced by LPC.
Figure 6.
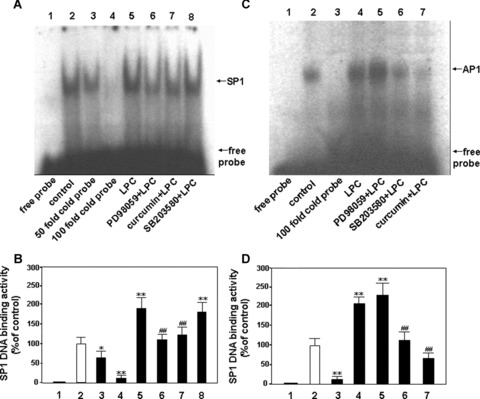
Effects of the selective inhibitors of MAPK on DNA binding activities of SP1 and AP1 in HUVEC-12 cells stimulated with LPC. The nuclear extracts from HUVEC-12 cells treated without or with 40 μMol/l of LPC for 2 hrs were used for EMSA to detect effect of the specific inhibitors of MAPKs on SP1 and AP1 DNA binding activity by stimulation of LPC. Lane 1, free probe; lane 2, treated without LPC as a control; lanes 3∼4 (A, B), 50- and 100-fold molar excess of cold probe of SP1 was used to determine SP1-specific DNA binding; lane 3 (C, D) 100-fold molar excess of cold probe of AP1 was done; lanes 5 (A, B) and 4 (C, D), treated with LPC as a stimulation group; lanes 6∼8 (A, B) and 5∼7 (C, D), pretreated with PD98059 (50 μMol/l), curcumin (30 μMol/l) and SB203580 (15 μMol/l), respectively, for 1.5 hrs before the stimulation of LPC (40 μMol/l). The data represent means ± S.D. of 3 independent experiments. *P< 0.05, **P< 0.01, compared with the control; ##P< 0.01, compared with the stimulation group.
As shown in Figure 6C and D, PD98059 could not block AP1 DNA binding activity induced by LPC, but curcumin could do it markedly, suggesting that JNK signalling pathway may involve the AP1 DNA binding activity induced by LPC.
Although SB203580 could also inhibit the AP1 DNA binding activity induced by LPC, it had no significant influence on the promoter transactivity by induction of LPC, suggesting that the reduction of the AP1 DNA binding activity by SB203580 is not associated with the enhancement of the LPC-stimulated transactivity.
Effect of MAPK inhibitors on LPC-induced SP1 and AP1 DNA binding activities in the presence of JNK1 overexpression
From the results above, it is reasonable to speculate that JNK plays a key role in the regulation of eNOS gene expression in LPC-induced physiological or pathological processes. We would like to check effect of JNK on the SP1 and AP1 DNA binding activities in HUVEC-12 cells. It was found that the overexpression of JNK1 in the cells increased the both SP1 and AP1 DNA binding activities, and also further boosted the LPC-induced SP1 and AP1 DNA binding activities (Fig. 7A and B). However, an experiment with MAPK inhibitors showed the DNA binding activities of SP1 and AP1 were not in parallel, i.e. the LPC-induced SP1 DNA binding activity was sensitive to PD98059 or curcumin, while the AP1 DNA binding activity was only inhibited by curcumin. These results further sup-ported the conclusion that JNK signal pathway was associated with the AP1 DNA binding activity induced by LPC (Fig. 7C and D).
Figure 7.
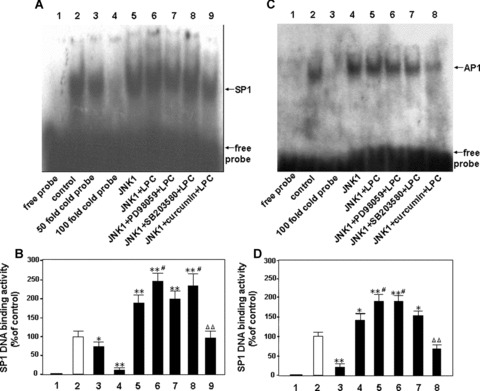
Effects of JNK signalling pathway on the DNA binding activities of SP1 and AP1 in HUVEC-12 cells by treatment of LPC. The nuclear extracts from HUVEC-12 cells treated without or with 40 μMol/l of LPC for 2 hrs were used for EMSA to detect effect of a specific inhibitor of JNK on SP1 and AP1 DNA binding activity enhanced by overexpression of JNK1 in the presence or absence of stimulation of LPC. Lane 1, free probe; lane 2, treated without LPC as a control; lanes 3∼4 (A, B), 50- and 100-fold molar excess of cold probe of SP1 was used to determine SP1-specific DNA binding; lane 3 (C, D) 100-fold molar excess of cold probe of AP1 was done; lanes 5 (A, B) and 4 (C, D), transfected alone with a JNK1 expression vector; lanes 6 (A, B) and 5 (C, D), treated with 40 μMol/l of LPC 45 hrs after tansfected with the JNK1 expression vector; lanes 7∼9 (A, B) and 6∼8 (C, D), transfected with the JNK1 expression vector, then pretreated with PD98059 (50 μMol/l), SB203580 (15 μMol/l) and curcumin (30 μMol/l) for 1.5 hrs, respectively, followed by stimulation with LPC (40 μMol/l). The data represent means ± S.D. of 3 independent experiments. *P< 0.05, **P< 0.01, compared with the control; #P< 0.05, compared with the alone transfected JNK1 group; ΔΔP< 0.01, compared with the transfected JNK1 plus LPC group.
Effect of decoy ODNs of AP1, SSRE and SP1 on human eNOS transactivity
To further analyse the response element in human eNOS promoter that involved the regulation of eNOS gene transcription, several typical decoy ODNs were used to evaluate the contribution of AP1, SSRE and SP1 in LPC-induced eNOS transcription.
It was found that SP1 decoy ODN down-regulated significantly not only the basal activity of the promoter, but also the stimulated activity of it. By contrast, the control ODN with the mutated sequence of SP1 promoted the recovery of the promoter activity inhibited by the application of SP1 decoy ODN (Fig. 8B). Although AP1 decoy ODN appeared the influence just on the stimulated activity of the promoter, the action was not as effective as the decoy ODN of wild-type of SP1 (Fig. 8A). As speculated by us, SSRE decoy ODN failed to exert any influences on the promoter activity (Fig. 8C). All these data suggested that the basal and stimulated transcriptional activity of human eNOS promoter was regulated by SP1 transcription factor, although we could not exclude the role of AP1 transcription factor in the regulation of the stimulated transactivity of it.
Figure 8.
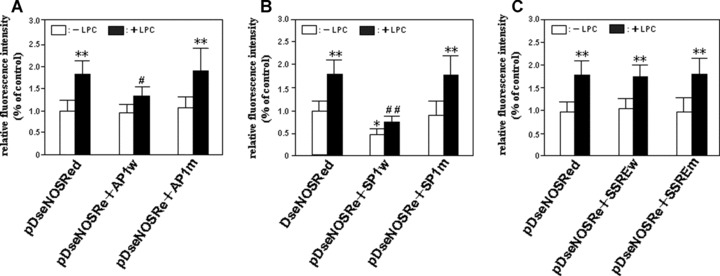
Effects of decoy ODNs of SP1, AP1 and SSRE on human eNOS transcriptional activity. The human eNOS promoter reporter gene construct, pDseNOSRed, was co-transfected with or without 5 pMol/l of wild-type ODNs of AP1, SP1 and SSRE (i.e. AP1w, SP1w and SSREw) or 5 pMol/l of mutant ODNs of them (e.g. AP1m, SP1m and SSREm), respectively, into HUVEC-12 cells to evaluate the contribution of AP1 (A), SP1 (B) and SSRE (C) in LPC-induced eNOS transcription. 45 hrs after the transfection, the cells were treated with or without 40 μMol/l of LPC. The red fluorescence-emitting cells were subjected to dynamic observation under an inverted fluorescence microscope. The fluorescence intensity of RFP expressed by the cells that represents the eNOS transactivity was quantitatively analysed using the fluorescence analysis software, Image-Pro Plus, 96 hrs after transfection. The result represents means ± S.D. of 4 independent experiments. *P< 0.05, **P< 0.01, compared with the pDseNOSRed group without the treatment of LPC; #P< 0.05, ##P< 0.01, compared with the pDseNOSRed group treated by LPC.
Discussion
Lysophosphatidylcholine (LPC), as a prominent phospholipid component of OxLDL, is responsible for various biological activities of OxLDL, such as regulation of adhesion molecules, NO, cytokines, and growth factors [25, 31, 33]. Recently, several lines of evidence showed that eNOS expression was up-regulated by LPC [32, 43, 44]. However, it was not clear how MAPK signalling transduction pathways elicit their actions on the up-regulation of eNOS induced by LPC [1, 11, 43]. In the current study, using RFP reporter gene containing human eNOS promoter we proved that eNOS gene transactivity was up-regulated by the stimulation of LPC.
Cieslik et al. showed that LPC selectively induced ERK1/2 phosphorylation in a time- and concentration-dependent manner without altering their protein levels, followed by the up-regulation of eNOS transcription activation, but that it did not induce phosphorylation of JNK or p38 MAPK [6]. Faustino’s research group also demonstrated that LPC activated ERK 1/2 MAPK pathway [10]. On the contrary, another group reported that in endothelial cells from human umbilical vein cords, LPC inhibited the hista-mine-induced arachidonic acid release, the phosphorylation of both PLA2 and ERK1/2, and led to the reduction of histamine-induced NO release through a PI3K-dependent pathway and eNOS phosphorylation [5]. In the current study, LPC stimulation resulted in the increase of phosphorylation of both ERK1/2 and JNK, but there were some differences in the dynamic processes of activation between them. However, the phosphorylation level of p38 MAPK was not altered by LPC, suggesting that ERK1/2 and JNK except p38 may involve the up-regulation of eNOS gene expression stimulated by LPC. Co-transfection experiments showed that the activation of JNK pathway by the overexpression of JNK or its upstream kinase active mutant up-regulated significantly the transactivity of eNOS promoter in HUVEC-12 cells by treatment of LPC. LPC stimulation also brought about the activation of ERK1/2 pathway in the cells, but the activation of ERK1/2 pathway alone by the overexpression of ERK1/2 or its upstream kinase active mutant was not to elicit the enough transcriptional activity. Further studies using MAPK inhibitors showed the inhibition of either ERK1/2 or JNK pathway by pretreatment of the cells with MAPK specific inhibitors, i.e. PD98059, or curcumin respectively could remarkably abrogate the induction of the transactivity in the cells stimulated by LPC. These results suggest that the induction of eNOS promoter activity by LPC depends on the activation of ERK1/2 and JNK pathways. In contrast, the activation of p38 MAPK by the overexpression of p38 highly inhibited the transcriptional activity of eNOS, while the inhibition of p38 MAPK using specific inhibitor, SB203580, would enhance the activation response of the eNOS promoter. Although it is known that both JNK and p38 MAPKs often share the identical function under stress conditions, they behaved much differently in LPC-induced gene regulation. However, there was the evidence that supported the attenuation of eNOS activity and the inhibition of eNOS phosphorylation by LPC [16]. For this reason, there appear to be different effects of LPC on distinct stages of eNOS expression.
To confirm how stimulating signals transfer from cytoplasm into nucleus through these signalling pathways, we further performed EMSA and decoy ODN trials to analyse the nuclear extracts from the cells subjected to the treatment of LPC. It was found by EMSA that LPC stimulated the elevation of the both SP1 and AP1 DNA binding activity. The SP1 DNA binding activity induced by LPC was abolished by either PD98059 or curcumin, but not SB203580. The AP1 DNA binding activity stimulated by LPC was markedly inhibited by curcumin, but not by PD98059. Overexpression of JNK1 that led to the up-regulation of LPC-stimulated eNOS transactivity enhanced the SP1 or AP1 DNA binding activity induced by LPC treatment, which was blocked by curcumin. These findings suggest that ERK1/2 signalling pathway is only responsible for the elevation of the SP1 transcription factor, and that JNK signal pathway is simultaneously associated with the elevation of the SP1 and AP1 transcription factors. Further studies with decoy oligonucleotide stratagem also confirmed that SP1 and AP1 cis-elements in the promoter of eNOS were involved in the gene regulation of eNOS. Blocking the action of the SP1 element resulted in the repression of the basal and stimulated transactivity of the promoter, while blocking the action of the AP1 element mainly inhibited the LPC-stimulated promoter transactivity. Thus, we consider that there appearss to be the cooperation between SP1 and AP1 cis-elements in eNOS promoter in the process of LPC-induced gene expression.
Interestingly, it was found that a MEK1-specific inhibitor, PD98059, markedly attenuated the phosphorylation of ERK1/2 induced by LPC, but SB203580 or curcumin accelerated it and that the phosphorylation level of p38 was elevated by PD98059 or a JNK-selective inhibitor, curcumin. What is reason about it? We consider that one of MAPK signalling pathways blocked by a corresponding inhibitor may act as a stimulating signal for other MAPK signalling pathway activation, which is likely to be a home-ostasis phenomenon of intracellular MAPK signal regulation network. Such the properties should be considered no matter in study and application concerning them.
Actually, some results showed that LPC might impair endothelium-dependent and endothelium-independent vasorelaxation [34]. Since up-regulating the expression of eNOS gene, why does LPC cause vascular impairment? It is noticed in our study that the transactivity level of eNOS induced by LPC was limited at most to two- to three-fold over the basal level of it. Consequently, this may be an adaptive mechanism that intends to counter the acute insult without producing excessive NO that may cause vascular damage, but probably inadequate to protect vascular wall from persistent, severe insults. Finally, the endothelium becomes dysfunctional and eventually, destroyed, and the blood vessels are inflicted with perpetual inflammatory and proliferative lesions leading to vascular diseases, such as atherosclerosis, restenosis and tissue infarction [42]. In my opinion, the enhancement of eNOS gene expression by the activation of ERK1/2 and JNK pathways with LPC only represents one of multiple signal regulation mechanisms for LPC to influence eNOS expression. Moreover, LPC may have different or opposite effects at different levels, including transcription, post-transcription, translation, phosphorylation and enzyme activity, on eNOS. Therefore, simultaneously utilizing multiple mechanisms to up-regulation eNOS is operative to increase vascular capacity for defending against damage.
Acknowledgments
We thank Prof. R.J. Ulevitch and Dr. J. Han at the Scripps Research Institute, La Jolla, CA 92037, USA for generously providing various MAPK and MAPKK expression constructs used in this study; and Prof. Yun-Qing. Li. at the K.K.
BRAIN Research Centre, Fourth Military Medical University, Xian 710032, China for his critical reading of this manuscript. This work was supported by the National Key Basic Research (973) Program (No. 2002CB513000) from the Ministry of Science and Technology of China, the National Natural Science Foundation of China (No. 30471635), and the Natural Science Foundation of Guangdong Province in China (No. 5006033, No. 04010451).
References
- 1.Alderton WK, Cooper CE, Knowles RG. Nitric oxide synthases: structure, function and inhibition. Biochem J. 2001;357:593–615. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Atochin DN, Wang A, Liu VW, Critchlow JD, Dantas AP, Looft-Wilson R, Murata T, Salomone S, Shin HK, Ayata C, Moskowitz MA, Michel T, Sessa WC, Huang PL. The phosphorylation state of eNOS modulates vascular reactivity and outcome of cerebral ischemia in vivo. J Clin Invest. 2007;117:1961–7. doi: 10.1172/JCI29877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bao M, Lou Y. Flavonoids from seabuck-thorn protect endothelial cells (EA.hy926) from oxidized low-density lipoprotein induced injuries via regulation of LOX-1 and eNOS expression. J Cardiovasc Pharmacol. 2006;48:834–41. doi: 10.1097/01.fjc.0000232064.64837.67. [DOI] [PubMed] [Google Scholar]
- 4.Braam B, Verhaar MC. Understanding eNOS for pharmacological modulation of endothelial function: a translational view. Curr Pharm Des. 2007;13:1727–40. doi: 10.2174/138161207780831275. [DOI] [PubMed] [Google Scholar]
- 5.Millanvoye-Van Brussel E, Topal G, Brunet A, Do Pham T, Deckert V, Rendu F, David-Dufilho M. Lysophosphatidylcholine and 7-oxocholesterol modulate Ca2+ signals and inhibit the phosphorylation of endothelial NO synthase and cytosolic phospholipase A2. Biochem J. 2004;380:533–9. doi: 10.1042/BJ20040069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cieslik K, Abrams CS, Wu KK. Up-regulation of endothelial nitric-oxide synthase promoter by the phosphatidylinositol 3-kinase gamma/Janus kinase 2/MEK-1-dependent pathway. J Biol Chem. 2001;12:1211–9. doi: 10.1074/jbc.M005305200. [DOI] [PubMed] [Google Scholar]
- 7.Cieslik K, Lee CM, Tang JL. Wu KK. Transcriptional regulation of endothelial nitric-oxide synthase by an interaction between casein kinase 2 and protein phos-phatase 2A. J Biol Chem. 1999;274:34669–75. doi: 10.1074/jbc.274.49.34669. [DOI] [PubMed] [Google Scholar]
- 8.Colombo MG, Paradossi U, Andreassi MG, Botto N, Manfredi S, Masetti S, Biagini A, Clerico A. Endothelial nitric oxide synthase gene polymorphisms and risk of coronary artery disease. Clin Chem. 2003;49:389–95. doi: 10.1373/49.3.389. [DOI] [PubMed] [Google Scholar]
- 9.Du Toit EF, Genade S, Carlini S, Moolman JA, Brunner F, Lochner A. Efficacy of ischaemic preconditioning in the eNOS overexpressed working mouse heart model. Eur J Pharmacol. 2007;556:115–20. doi: 10.1016/j.ejphar.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 10.Faustino RS, Stronger LNW, Melanie N. RanGAP-mediated nuclear protein import in vascular smooth muscle cells is augmented by lysophosphatidylcholine. Mol Pharmacol. 2007;71:438–45. doi: 10.1124/mol.105.021667. [DOI] [PubMed] [Google Scholar]
- 11.Fleming I, Busse R. Signal transduction of eNOS activation. Cardiovasc Res. 1999;43:532–41. doi: 10.1016/s0008-6363(99)00094-2. [DOI] [PubMed] [Google Scholar]
- 12.Gan L, Selin-Sjogren L, Doroudi R, Jern S. Temporal regulation of endothelial ET-1 and eNOS expression in intact human conduit vessels exposed to different intraluminal pressure levels at physiological shear stress. Cardiovas Res. 2000;48:168–77. doi: 10.1016/s0008-6363(00)00174-7. [DOI] [PubMed] [Google Scholar]
- 13.Garcia-Parajo MF, Koopman M, Van Dijk EM, Subramaniam V, Van Hulst NF. The nature of fluorescence emission in the red fluorescent protein DsRed, revealed by single-molecule detection. Proc Natl Acad Sci USA. 2001;98:14392–7. doi: 10.1073/pnas.251525598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gill RM, Braz JC, Jin N, Etgen GJ, Shen W. Restoration of impaired endothelium-dependent coronary vasodilation in failing heart: role of eNOS phosphorylation and CGMP/cGK-I signaling. Am J Physiol Heart Circ Physiol. 2007;292:H2782–90. doi: 10.1152/ajpheart.00831.2006. [DOI] [PubMed] [Google Scholar]
- 15.Gouni-Berthold I, Sachinidis A. Possible non-classic intracellular and molecular mechanisms of LDL cholesterol action contributing to the development and progression of atherosclerosis. Curr Vasc Pharmacol. 2004;2:363–70. doi: 10.2174/1570161043385466. [DOI] [PubMed] [Google Scholar]
- 16.Gousset-Dupont A, Robert V, Grynberg A. The effect of n-3 PUFA on eNOS activity and expression in Eahy 926 cells. Prostaglandins Leukot Essent Fatty Acids. 2007;76:131–9. doi: 10.1016/j.plefa.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 17.Hoffmann A, Gloe T, Pohl U. Hypoxia-induced upregulation of eNOS gene expression is redox-sensitive: a comparison between hypoxia and inhibitors of cell metabolism. J Cell Physiol. 2001;188:33–44. doi: 10.1002/jcp.1092. [DOI] [PubMed] [Google Scholar]
- 18.Jin HG, Yamashita H, Nagano Y, Fukuba H, Hiji M, Ohtsuki T, Takahashi T, Kohriyama T, Kaibuchi K, Matsumoto M. Hypoxia-induced upregulation of endothe-lial small G protein RhoA and Rho-kinase/ROCK2 inhibits eNOS expression. Neurosci Lett. 2006;408:62–7. doi: 10.1016/j.neulet.2006.08.038. [DOI] [PubMed] [Google Scholar]
- 19.Jones SP, Greer JJM, Haperen RV, Duncker DJ, Crom RD, Lefer DJ. Endothelial nitric oxide synthase overexpression attenuates congestive heart failure in mice. Proc Natl Acad Sci USA. 2003;100:4891–6. doi: 10.1073/pnas.0837428100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karantzoulis-Fegaras F, Antoniou H, Lai SL, Kulkarni G, D’Abreo C, Wong GK, Miller TL, Chan Y, Atkins J, Wang Y, Marsden PA. Characterization of the human endothelial nitric-oxide synthase promoter. J Biol Chem. 1999;274:3076–93. doi: 10.1074/jbc.274.5.3076. [DOI] [PubMed] [Google Scholar]
- 21.Kudo Y, Ootani T, Kumagai T, Fukuchi Y, Ebin K, Yokota K. A novel oxidized low-density lipoprotein-binding protein, Asphemolysin, recognizes lysophosphatidyl-choline. Biol Pharm Bull. 2002;25:787–90. doi: 10.1248/bpb.25.787. [DOI] [PubMed] [Google Scholar]
- 22.Leonard AM, Chafe LL, Montani JP, Van Vliet BN. Increased salt-sensitivity in endothelial nitric oxide synthase-knockout mice. Am J Hypertens. 2006;19:1264–9. doi: 10.1016/j.amjhyper.2006.05.025. [DOI] [PubMed] [Google Scholar]
- 23.Li H, Burkhardt C, Heinrich UR, Brausch I, Xia N, Forstermann U. Histamine upregulates gene expression of endothelial nitric oxide synthase in human vascular endothelial cells. Circulation. 2003;107:2348–54. doi: 10.1161/01.CIR.0000066697.19571.AF. [DOI] [PubMed] [Google Scholar]
- 24.Li H, Junk P, Huwiler A, Burkhardt C, Wallerath T, Pfeilschifter J, Förstermann U. Dual effect of ceramide on human endothelial cells induction of oxidative stress and transcriptional upregulation of endothelial nitric oxide synthase. Circulation. 2002;106:2250–6. doi: 10.1161/01.cir.0000035650.05921.50. [DOI] [PubMed] [Google Scholar]
- 25.Li Z, Song T, Liu GZ, Liu LY. Inhibitory effects of cariporide on LPC-induced expression of ICAM-1 and adhesion of monocytes to smooth muscle cells in vitro. Acta Pharmacol. 2006;27:1326–32. doi: 10.1111/j.1745-7254.2006.00412.x. [DOI] [PubMed] [Google Scholar]
- 26.Liu L, Liu H, Visner G, Fletcher BS. Sleeping Beauty-mediated eNOS gene therapy attenuates monocrotaline-induced pulmonary hypertension in rats. FASEB J. 2006;20:2594–6. doi: 10.1096/fj.06-6254fje. [DOI] [PubMed] [Google Scholar]
- 27.Lund CO, Mortensen A, Nilas L, Breinholt VM, Larsen JJ, Ottensen B. Estrogen and phytoestrogens: effect on eNOS expression and in vitrovasodilation in cerebral arteries in ovariectomized Watanabe heritable hyperlipidemic rabbits. Eur J Obstet Gynecol Reprod Biol. 2007;130:84–92. doi: 10.1016/j.ejogrb.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 28.Mathew R, Huang J, Gewitz MH. Pulmonary artery hypertension: caveolin-1 and eNOS interrelationship: a new perspective. Cardiol Rev. 2007;15:143–9. doi: 10.1097/01.crd.0000249381.49138.b9. [DOI] [PubMed] [Google Scholar]
- 29.Miller WH, Brosnan MJ, Graham D, Nicol CG, Morecroft I, Channon KM, Daniloy SM, Reynolds PN, Baker AH, Dominiczak AF. Targeting endothelial cells with adenovirus expressing nitric oxide synthase prevents elevation of blood pressure in stroke-prone spontaneously hypertensive rats. Mol Ther. 2005;12:321–7. doi: 10.1016/j.ymthe.2005.02.025. [DOI] [PubMed] [Google Scholar]
- 30.Murugesan G, Sandhya Rani MR, Gerber CE, Mukhopadhyay C, Ransohoff RM, Chisolm GM, Kottke-Marchant K. Lysophosphatidylcholine regulates human microvascular endothelial cell expression of chemokines. J Mol Cell Cardiol. 2003;35:1375–84. doi: 10.1016/j.yjmcc.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 31.Nakano T, Raines EW, Abraham JA, Klagsbrun M, Ross R. Lysophosphatidylcholine upregulates the level of heparin-binding epidermal growth factorlike growth factor mRNA in human monocytes. Proc Natl Acad Sci USA. 1994;91:1069–73. doi: 10.1073/pnas.91.3.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ozaki H, Ishii K, Arai H, Kume N, Kita T. Lysophosphatidylcholine activates mito-gen-activated protein kinases by a tyrosine kinase-dependent pathway in bovine aortic endothelial cells. Atherosclerosis. 1999;143:261–6. doi: 10.1016/s0021-9150(98)00297-4. [DOI] [PubMed] [Google Scholar]
- 33.Rosenblat M, Ore R, Aviram M. Lysophosphatidylcholine (LPC) attenuates macrophage-mediated oxidation of LDL. Biochem Biophys Res Commun. 2006;344:1271–7. doi: 10.1016/j.bbrc.2006.04.038. [DOI] [PubMed] [Google Scholar]
- 34.Safaya R, Chai H, Kougias P, Lin P, Lumsden A, Yao Q, Chen C. Effect of lysophosphatidylcholine on vasomotor functions of porcine coronary arteries. J Surg Res. 2005;126:182–8. doi: 10.1016/j.jss.2005.01.015. [DOI] [PubMed] [Google Scholar]
- 35.Saura M, Zaragoza C, Cao W, Bao C, Rodriguez-Puyol M, Rodriguez-Puyol D, Lowenstein CJ. Smad2 mediates transforming growth factor-beta induction of endothelial nitric oxide synthase expression. Circ Res. 2002;91:806–13. doi: 10.1161/01.res.0000040397.23817.e5. [DOI] [PubMed] [Google Scholar]
- 36.Shi Y, Baker JE, Zhang C, Tweddell JS, Su J, Pritchard KA. Chronic hypoxia increases endothelial nitric oxide synthase generation of nitric oxide by increasing heat shock protein 90 association and serine phosphorylation. Circ Res. 2002;91:300–6. doi: 10.1161/01.res.0000031799.12850.1e. [DOI] [PubMed] [Google Scholar]
- 37.Takemoto M, Sun J, Hiroki J, Shimokawa H, Liao JK. Rho-kinase mediates hpoxia-induced downregulation of endothelial nitric oxide synthase. Circulation. 2002;106:57–62. doi: 10.1161/01.cir.0000020682.73694.ab. [DOI] [PubMed] [Google Scholar]
- 38.Thors B, Halldorsson H, Clarke GD, Thorgeirsson G. Inhibition of Akt phosphorylation by thrombin, histamine and lysophosphatidylcholine in endothelial cells Differential role of protein kinase C. Atherosclerosis. 2003;168:245–53. doi: 10.1016/s0021-9150(03)00127-8. [DOI] [PubMed] [Google Scholar]
- 39.Wallerath T, Deckert G, Ternes T, Anderson H, Li H, Witte K, Forstermann U. Resveratrol, a polyphenolic phytoalexin present in red wine, enhances expression and activity of endothelial nitric oxide synthase. Circulation. 2002;106:1652–8. doi: 10.1161/01.cir.0000029925.18593.5c. [DOI] [PubMed] [Google Scholar]
- 40.Wedgwood S, Mitchell CJ, Fineman JR, Black SM. Developmental differences in the shear stress-induced expression of endothelial NO synthase: changing role of AP-1. Am J Physiol Lung Cell Mol Physiol. 2003;284:L650–2. doi: 10.1152/ajplung.00252.2002. [DOI] [PubMed] [Google Scholar]
- 41.Weiner CP, Lizasoain I, Baylis SA, Knowles RG, Charles IG, Moncada S. Induction of calcium-dependent nitric oxide synthases by sex hormones. Proc Natl Acad Sci USA. 1994;91:5212–6. doi: 10.1073/pnas.91.11.5212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu KK. Regulation of endothelial nitric oxide synthase activity and gene expression. Ann NY Acad Sci. 2002;962:122–30. doi: 10.1111/j.1749-6632.2002.tb04062.x. [DOI] [PubMed] [Google Scholar]
- 43.Xing FY, Jiang Y, Liu J, Zhao KS, Mo YY, Qin QH, Wang JZ, Ouyang JM, Zeng YY. Role of AP1 element in the activation of human eNOS promoter by lysophosphatidylcholine. J Cell Biochem. 2006;98:872–84. doi: 10.1002/jcb.20739. [DOI] [PubMed] [Google Scholar]
- 44.Yamakawa T, Tanaka S, Yamakawa Y, Kamei J, Numaguchi K, Motley ED, Inagami T, Eguchi S. Lysophos-phatidylcholine activates extracellular signal-regulated kinases 1/2 through reactive oxygen species in rat vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2002;22:752–8. doi: 10.1161/01.atv.0000015903.02749.71. [DOI] [PubMed] [Google Scholar]