

Abstract
Hystidyl-proline [cyclo(His-Pro)] is an endogenous cyclic dipeptide produced by the cleavage of thyrotropin releasing hormone. Previous studies have shown that cyclo(His-Pro) protects against oxidative stress, although the underlying mechanism has remained elusive. Here, we addressed this issue and found that cyclo(His-Pro) triggered nuclear accumulation of NF-E2-related factor-2 (Nrf2), a transcription factor that up-regulates antioxidant-/electrophile-responsive element (ARE-EpRE)-related genes, in PC12 cells. Cyclo(His-Pro) attenuated reactive oxygen species production, and prevented glutathione depletion caused by glutamate, rotenone, paraquat and β-amyloid treatment. Moreover, real-time PCR analyses revealed that cyclo(His-Pro) induced the expression of a number of ARE-related genes and protected cells against hydrogen peroxide-mediated apoptotic death. Furthermore, these effects were abolished by RNA interference-mediated Nrf2 knockdown. Finally, pharmacological inhibition of p-38 MAPK partially prevented both cyclo(His-Pro)-mediated Nrf2 activation and cellular protection. These results suggest that the signalling mechanism responsible for the cytoprotective actions of cyclo(His-Pro) would involve p-38 MAPK activation leading to Nrf2-mediated up-regulation of antioxidant cellular defence.
Keywords: PC12, Nrf2 silencing, rotenone, paraquat, β-amyloid, glutamate, hydrogen peroxide
Introduction
Thyrotropin-releasing hormone (TRH) is a tripeptide expressed in the central nervous system, where it acts as a hypothalamic neuroendocrine signal eliciting a number of behavioural responses [1]. Upon release, TRH is rapidly subjected to cleavage on its pyroglutamyl residue, both in the blood and in the central nervous system, producing the cyclic dipeptide histidyl-proline [cyclo(His-Pro)]. Thus, cyclo(His-Pro) is ubiquitously distributed, although it is mainly found in gut and brain [2]. Since cyclo(His-Pro) does not undergo further metabolic changes, its half-life is higher than that of TRH, and it diffuses across the blood brain barrier [3].
Cyclo(His-Pro) – and certain newly synthesized derivatives – have been shown to prevent apoptotic death induced by free radicals or calcium mobilization, suggesting its possible usefulness for the treatment of cellular/neuronal death [1, 4, 5]. However, the molecular mechanism underlying these protective actions of cyclo(His-Pro) is unknown.
Cells respond to environmental injury by expressing genes protecting them against oxidants and electrophilic compounds-mediated stress [6]. The induction of these genes are largely mediated by Nrf2, a ubiquitously expressed member of the bZIP transcription factor family [7–9]. Nrf2 is normally sequestered in the cytoplasm bound to the repressor protein Keap1 [9, 10] but, upon the stress signal, it is dissociated from Keap1 and translocated to the nucleus [11]. In the nucleus, Nrf2 forms a heterodimer with members of the small Maf family [12] and recognizes – and binds to – the antioxidant response element (ARE) promoter sequences, hence promoting the transcriptional activation of antioxidant genes [13, 14].
Here, we aimed to investigate whether the cytoprotective role of cyclo(His-Pro) against a wide range of toxic insults is mediated by an improvement of the cellular antioxidant defence. We found evidence consistent with the notion that cyclo(His-Pro) promotes cyto-protection through a mechanism involving Nrf2 activation, leading to ARE-dependent gene expression and reduced oxidative stress.
Materials and methods
Materials
Cyclic peptide-(His-Pro) (Fig. 1) and other analogues were synthesized at Dipartimento di Scienze del Farmaco, Università degli Studi G. D’Annunzio, Chieti, Italy [15]. All the reagents, unless otherwise stated, were from Sigma Aldrich (St. Luis, MO, USA). RPMI-1640 medium, foetal bovine serum (FBS), horse serum (HS), trypsin, streptomycin/penicillin and glutamine were from Life Technologies (GibcoBRL, Gaithersburg, MD, USA).
Figure 1.
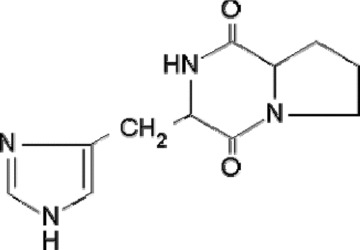
Chemical structure of cyclo(His-Pro).
Cell culture
PC12 cells, purchased from ATCC (Manassas, VA, USA), were routinely cultured in RPMI 1640 supplemented with 10% heat-inactivated horse serum and 5% heat-inactivated foetal bovine serum, 4 mM glutamine, 50 U/ml penicillin and 50 μg/ml streptomycin at 37°C in a humidified atmosphere of 5% CO2. The medium was changed every other day, and cells were plated at an appropriate density according to each experimental scale. After 24 hrs subculture, cells were switched to 0.5% FBS medium, incubated for 24 hrs with cyclic peptide-(His-Pro), exposed for 30 min. to H2O2, washed and grown for additional 24 hrs. p-38 MAP kinase (p-38 MAPK) inhibitor (10 μM SB203580, Tocris Bioscience, Bristol, UK) was added 2 hrs prior to cyclo(His-Pro) treatment.
For other treatments, cells were incubated for 24 hrs with 10 mM glutamate, 100 μM rotenone, 100 julM paraquat and 25 μM β-amyloid 25–35, either in the absence or in the presence of cyclo(His-Pro).
Measurement of cell viability
Cell viability was determined by the conventional MTT (3-[4, 5-dimethylthiazol-2-yl]-2, 5-dephenyl tetrazolium bromide) reduction assay. The dark blue formazan crystals formed in intact cells were solubilized with lysis buffer (10% sodium dodecylsulfate, 0.01 M HCl) and the absorbance at 550 nm was measured with a microplate reader (Seac, Florence, Italy). Results were expressed as the percentages of reduced MTT, assuming the absorbance of control cells as 100%.
Hoechst 33258 staining
PC12 cells were grown on glass coverslips in the presence or in the absence of 50 μM cyclo(His-Pro) for 24 hrs and then treated with 100 μM H2O2. After fixation, cells were stained with Hoechst 33258 (5 μg/ml in PBS) for 5 min. at room temperature and then examined with a Nikon Eclipse Te2000-S fluorescent microscope (Nikon Intrnational SpA, Calenzano, Italy).
Nrf2 siRNA knockdown
PC12 cells (0.2 × 106 cells/well) were seeded in antibiotic-free medium for 24 hrs prior to transfection. DharmaFECT™ 1 transfection reagent (Dharmacon RNA Technologies, Lafayette, CO, USA) was used to transfect cells with 100 nM siRNA pool against rat Nrf-2 (Dharmacon RNA Technologies) for 48 hrs according to manufacturer’s protocol with a transfection efficiency of >80%. Control siRNA with no known mammalian homology (siCONTROL Non-Targeting siRNA #1; Dharmacon) and non-transfected cells were used as negative controls.
Real-time PCR
Total RNA was isolated with TRIZOL Reagent (Invitrogen Ltd, Paisley, UK) according to the manufacturer’s instructions and cDNA was synthesized using iScript cDNA synthesis kit (Bio-Rad Lab, Hercules, CA, USA). Realtime PCR was performed using the iCycler iQ detection system (Bio-Rad) and SYBR Green chemistry. Rat primers, obtained from Invitrogen (Invitrogen Ltd), are listed in Table 1. SYBR Green RT-PCR amplifications were carried out in a 96-well plate in a 25-μl reaction volume that contained 12.5 μl of 2x iQ™ SYBR ® Green SuperMix (Bio-Rad), 400 nM forward and reverse primers, and 5 to 40 ng of cDNA. In each assay, no-template controls were included and each sample was run in triplicates. The thermal profile consisted of incubation at 95°C 3 min., followed by 40 cycles of denaturation for 10 sec. at 95°C and an annealing/extension step of 30 sec. at 60°C. Mean of Ct values of the stimulated sample was compared to the untreated control sample. ΔCt is the difference in Ct values derived from the target gene (in each assayed sample) and ß-actin, while ΔΔCt represents the difference between the paired samples. The n-fold differential ratio was expressed as 2ΔΔCt .
Table 1.
List of primers
| Accession number | Gene name | Gene symbol | Primer sequences (F: forward; R: reverse) |
|---|---|---|---|
| EF156276 | β-Actin | β-Actin | F. GGCCAACCGTGAAAAGATGAC |
| R. ACACAGCCTGGATGGCTACGT | |||
| NM_12815 | Glutamate cysteine ligase catalytic subunit | GCLC | F. GGGAAGGAAGGCGTGTTTCCT |
| R. GTCGACTTCCATGTTTTCAAGGT | |||
| NM_17305 | Glutamate cysteine ligase modifier subunit | GCLM | F. CCAGGAGTGGGTGCCACTGT |
| R. TTTGACTTGATGATTCCTCTGCT | |||
| NM_030826 | Glutathione peroxidase | Gpx | F. CAGTTCGGACATCAGGAGAAT |
| R. AGAGCGGGTGAGCCTTCT | |||
| MM_53906 | Glutathione reductase | GR | F GGGCAAAGAAGATTCCAGGTT |
| R. GGACGGCTTCATCTTCAGTGA | |||
| NM_17013 | Glutathione-S-transferase-2 | GSTA-2 | F GCCAAGACTACCTTGTAGGT |
| R. GTCAGCCTGTTCCCTACAAG | |||
| X07467. | Glucose-6-phosphate dehydrogenase | G6PDH | F GCCTTCTACCCGAAGACACCTT |
| R. CTGTTTGCGGATGTCATCCA | |||
| BC083542 | NAD(P)H:quinone oxidoreductase 1 | NQO1 | F GCCTTTGTTCCACAAGGATAGG |
| R. GCCCCTAATCTGACCTCGTT | |||
| NM_053800 | Thioredoxin | Trx-1 | F CATGCCGACCTTCCAGTTC |
| R. GGCTTCGAGCTTTTCCTTGT | |||
| NM_52799 | Neuronal nitric oxide synthase | nNOS | F ACGGACCCGACCTCAGAGAC |
| R. AGAGACGAGGCCGAACACTG | |||
| NM_012611 | Inducible nitric oxide synthase | iNOS | F GTCACCTATCGCACCCGAGATC |
| R. GCCACTGACACTCCGCACAAAG | |||
| NM_31789 | NF-E2-related factor-2 | Nrf2 | F GTTGAGAGCTCAGTCTTCAC |
| R. CAGAGAGCTATCGAGTGACT |
Accession number and gene symbol are indicated for each primer pair.
Western blot analysis
PC12 cells were washed with Tris-buffered saline on ice, harvested using a cell scraper, and lysed in 20 mM Tris buffer (pH 7.0) containing 25 mM (β-glycerophosphate, 2 mM EGTA, 1% Triton X-100, 1 mM vanadate, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 10 μg/ml pepstatin A, 1 mM PMSF, and 2 mM dithiothreitol (DTT). Lysates, homogenized twice in an ultrasonicator (10 sec), were incubated on ice for 30 min., centrifuged at 15,000 xg for 30 min. and the supernatants were denatured by boiling. Proteins (40 μg) were loaded on 12% SDS-PAGE, separated electrophoretically, and transferred to a PVDF membrane (Millipore Corp., Bedford, MA, USA). After blocking with non-fatty milk, the membrane was incubated with the primary antibody anti-phospho-p38 MAPK (1:1000, Cell Signaling Technology, Inc., Beverly, MA, USA), anti-p38 MAPK (1:200, Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA). After secondary incubation in horseradish peroxidase-conjugated anti-rabbit IgG antibody (1:2500) (Amersham Bioscience, Little Chalfont, UK), the immunocomplexes were visualized with an enhanced chemiluminescence kit (ECL, Pierce). Bands were analysed with the Bandscan software.
Preparation of nuclear extract and Western blot for Nrf2
PC12 cells were washed with Tris-buffered saline on ice, harvested using a cell scraper, and lysed in 10 mM HEPES, pH 7.9, containing 10 mM KCl, 0.1 mM EGTA, 0.1 mM EDTA, 0.5% Nonidet P-40, 1 mM vanadate, 0.5 mM PMSF, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 10 μg/ml pepstatin A, and 1 mM DTT (Buffer A). PC12 cells were then incubated on ice for 15 min. and centrifuged at 14,000 xg for 3 min. at 4°C. The supernatant (cytosolic extract) was collected and stored at -80°C until use. The resulting nuclear pellet, washed twice in ice-cold Buffer A, was resuspended in ice-cold Buffer B (20 mM HEPES, pH 7.9, containing 20% glycerol, 420 mM NaCl, 1 mM EGTA, 1 mM EDTA, 1 mM DTT, 0.5 mM PMSF, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 10 μg/ml pepstatin A) for 30 min. at 0°C on a rotating wheel. The nuclear extract was finally centrifuged at 14,000 xg for 10 min. Cytosolic and nuclear extracts (40 μg) were loaded on 12% SDS-PAGE and Nrf2 levels determined by Western blotting using Nrf2(C-20) antibody (1:200; Santa Cruz Biotech). α-tubulin (B-7) and Lamin B(H-90) antibodies (1:100, Santa Cruz Biotech) were used as marker proteins for cytosolic and nuclear extracts. After secondary incubation in horseradish peroxidase-conjugated anti-rabbit IgG antibody (1:5000) (Amersham Bioscience, Little Chalfont, UK), the immunocomplexes were visualized with an enhanced chemiluminescence kit (ECL, Pierce). Bands were analysed with the Bandscan software.
Glutathione determination
The concentration of glutathione (GSH) was determined in whole cell lysate after perchloric acid precipitation using the dithionitrobenzoic acid (DTNB) method at 412 nm (molar extinction coefficient 13,6 mmoP1 cm−1), as described in [16]. GSH concentrations (obtained in μM) were expressed as percentage of control.
Measurement of intracellular fluorescence
The DCFH-DA method was used to detect the levels of intracellular reactive oxygen species. DCFH-DA diffuses into cells, where it is hydrolysed by intracellular esterase to polar 2’,7’-dichlorodihydrofluorescein. This non-fluorescent fluorescein analogue gets trapped inside the cells and is oxidized by intracellular oxidants to a highly fluorescent, 2’,7’-dichlorofluo-rescein and fluorescence intensity is proportional to the amount of oxidant species produced by the cells. Cells (10×10)3 were treated as described and DCFH-DA (100 μM) was added into the medium for a further 30 min. at 37°C. The fluorescence of 2’,7’-dichlorofluorescein was detected at 485 nm excitation and at 535 nm emission, using a Titertek Fluoroscan II (Flow Laboratories, McLean, VA, USA). Results, expressed as percentage of the control DCF fluorescence, were normalized to cell viability.
Measurement of NO production
Nitric oxide (NO) production was determined indirectly through the measurement of nitrite, a stable metabolite of nitric oxide, by the Griess reaction. This assay relies on a simple colorimetric reaction between nitrite and Griess reagent [0.1% N-(naphthyl)ethylenediamine dihydrochloride, 1% sulfanil-amide in 5% phosphoric acid] to produce a pink azo product. After treating with 100 μM H2O2, cells were washed and grown in 0.5% FBS medium for 24 hrs, then a 50 μl aliquot of medium was mixed with an equal volume of Griess reagent and incubated for 20 min. at room temperature. The absorbance was read at 550 nm using a microtitre plate reader and results, expressed as percentage of the control, were normalized to cell viability.
Measurement of intracellular calcium level
Intracellular calcium was measured by the fluorescence ratio of the calcium indicator dye Fura-2 AM. Briefly, after a 24-hrs incubation with cyclo(His-Pro), the medium was removed and cells washed with Krebs-Ringer-HEPES (KRH) buffer (1.5 mM CaCl2, 131 mM NaCl, 1.3 mM MgSO2, 5 mM KCl, 0.4 mM KH2PO4, 6 mM glucose, 20 mM HEPES, pH 7.4). They were then loaded with 3 μM Fura-2 AM at 37°C for 30 min. After removal of Fura-2 AM, cells (5 × 106) were incubated for an additional 30 min. in KRH buffer and subsequently exposed to 100 μM H2O2 for 30 min. Fluorescence intensity was then recorded using a Perkin-Elmer LS50B fluorescence spectrofluorimeter (Wellesley, MA, USA). There was no significant change in the viability of the cells after removal of the medium and during calcium measurement in KRH. Average calcium levels were expressed as a ratio of the fluorescence emissions (510 nm) obtained using two different excitation wavelengths (340 and 380 nm). The fluorescence ratio (F340/F380) was calculated as an indicator of intracellular Ca2+.
Protein determination
Protein concentration was measured by BioRad Assay kit (Bio-Rad Lab).
Statistical analysis
All results were confirmed in at least three separate experiments and expressed as mean ± S.D. Data were analysed for statistical significance by Student’s t-test. P-values <0.05 were considered significant.
Results
Cyclo(His-Pro) protects PC12 cells against H2O2-induced apoptotic death, oxidative/ nitrosative stress and calcium accumulation
In a previous study, we reported that cyclo(His-Pro) can prevent PC12 cells from serum deprivation-mediated apoptotic death [5]. To investigate whether such cytoprotection could be ascribed an antioxidant effect, we first assessed the effect of cyclic dipeptide on H2O2-mediated PC12 cell injury. Cytotoxicity induced by H2O2 was initially measured by determining MTT reduction in cells incubated with increasing concentrations (50–500 μM) of H2O2. As shown in Figure 2A, treatment of PC12 cells with cyclo(His-Pro) abolished H2O2-mediated cytotoxicity, an effect that was dose-dependent (Fig. 2B). Analogues of cyclo(His-Pro), i.e. cyclo-(His-His), cyclo-(Leu-Pro) and cyclo-(Gly-His) showed no cytoprotection (data not shown). Morphological analysis of PC12 cells treated with 100 μM H2O2 exhibited the typical phenotypic changes due to apoptotic cell death, i.e. cell shrinkage and membrane bleb-bing (not shown). Furthermore, quantification of cells exhibiting fragmented DNA revealed that cyclo(His-Pro) fully prevented apoptotic cell death (Fig. 2C). Treatment of PC12 cells with cyclo(His-Pro) after H2O2- injury resulted in cytoprotection, at least up to 12 hrs (Fig. 2D). Next, we assessed whether cyclo(His-Pro) modifed cellular redox status. Thus, H2O2 induced a significant increase in ROS and NO production, as well as a marked decrease in intracellular GSH concentrations (Fig. 2E), an effect that was fully prevented by cyclo(His-Pro) (50 μM). Since oxidative stress is accompanied by the occurrence of large increases in intracellular calcium [17–19], we next determined intracellular calcium levels by fluorescence using Fura-2 AM. As shown in Figure 2E, H2O2 (100 μM) increased the fluorescence ratio (F340/F380) from 100% in controls to 248 ± 20% in H2O2-exposed cells, whereas pre-treatment with cyclo(His-Pro) (50 μM), led F340/F380 ratio to decrease to 158 ± 18%. As negative control, we assessed the F340/F380 ratio in calcium-free Krebs’ solution, resulting in no alterations under either condition (Fig. 2E).
Figure 2.
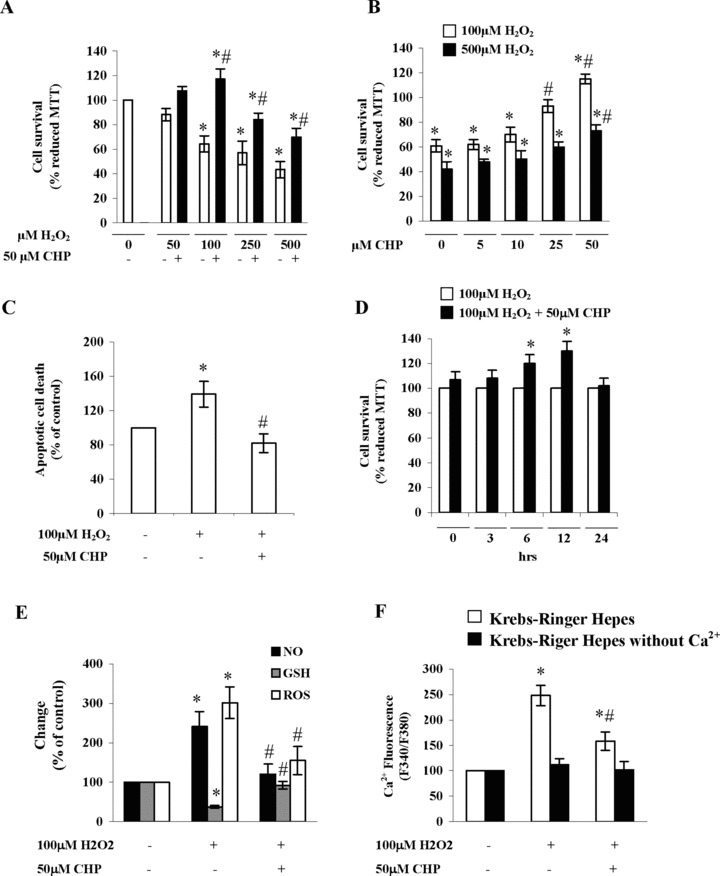
Cyclo(His-Pro) protects PC12 cells against H2O2-induced apoptotic death, oxidative/nitrosative stress and calcium accumulation. (A) PC12 cells were incubated with 50 μM cyclo(His-Pro) for 24 hrs, then H2O2 was added at the indicated concentrations and the analyses performed as described. Reduced MTT data in control cells (absorbance at 550 nm = 1.66 ± 0.2) were considered as 100%. Results are expressed as the mean ± S.D. (n= 8). *P< 0.05 versus control; #P< 0.05 versus H2O2-treated cells. (B) PC12 cells were incubated with increasing cyclo(His-Pro) concentrations for 24 hrs, then 100 μM or 500 μM H2O2 were added and the analyses performed as described. Results are expressed as the mean ± S.D. (n= 8). *P< 0.05 versus control cells (100 μM H2O2); #P< 0.05 versus control cells (500 μM H2O2). (C) PC12 cells were incubated with 50 μM cyclo(His-Pro) for 24 hrs, then 100 (jlM H2O2 was added and condensed and/fragmented nuclei were determined by Hoechst staining and expressed as percentages of control (no treatment). Results are given as the mean ± S.D. (n= 8). *P< 0.05 versus control; #P< 0.05 versus H2O2-treated cells. (D). PC12 cells were treated with 100 (jlM H2O2, then 50 μM cyclo(His-Pro) was added after the indicated time intervals. Reduced MTT data in H2O2-treated cells were considered as 100%. Results are expressed as the mean ± S.D. (n= 5). *P< 0.05 versus H2O2-treated cells. (E) Cells were incubated with 50 μM cyclo(His-Pro) for 24 hrs, then 100 μM of H2O2 was added and the analyses performed as described. Values (mean ± S.D., n = 7), given as percentages of control (no treatment; considered as 100%), were: NO (absorbance values at 550 nm): 0.8 ± 0.02; glutathione (GSH, in μM): 4.5 ± 0.1; ROS (DCF fluorescence): 0.9 ± 0.2; *P< 0.05 versus control; #P< 0.05 versus H2O2-treated cells. (F) Cells were incubated for 24 hrs either in the absence or in the presence of 50 |jJM cyclo(His-Pro), loaded with 3 μM Fura-2/AM in Krebs-Ringer HEPES (KRH), and incubated with 100 μM H2O2. Intracellular Ca2+ was measured by Fura-2/AM F340/F380 fluorescence ratio. Data are presented as mean ± S.D., (n= 5). *P< 0.05 versus control; #P< 0.05 versus H2O2-treated cells.
Effects of cyclo(His-Pro) treatment on Nrf2 activation and oxidative/nitrosative stress-related gene expression
To investigate the molecular mechanism of the protection exerted by cyclo(His-Pro), we focused on Nrf2, a transcription factor responsible for ARE-mediated gene expression. As shown in Figure 3A, H2O2 triggered a slight, not significant increase in nuclear levels of Nrf2 protein, whereas cyclo(His-Pro) promoted a significant increase in Nrf2. Cyclo(His-Pro) alone had no effect when compared with PC12 cells maintained in standard conditions (data not shown). Real-time PCR analyses showed that the treatment of the cells with cyclo(His-Pro) enhanced mRNA expression of Nrf2-regulated genes (Fig. 3B).
Figure 3.
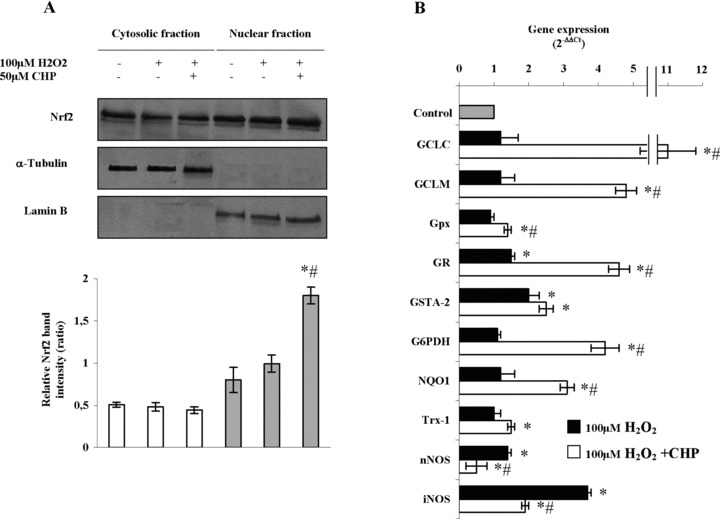
Cyclo(His-Pro) triggers nuclear translocation of Nrf2 leading to changes in the antioxidant gene expression pattern in PC12 cells. Cells were incubated with 50 μM cyclo(His-Pro) for 24 hrs prior to treatment with 100 μM H2O2. (A) Nrf2 protein in cytosol (white bars) and nuclear (grey bars) fractions. The nuclear and cytosolic extracts containing 40 |ag of proteins were subjected to Western blotting analysis with the indicated antibodies. Anti-α-tubulin and anti-lamin B antibodies were used as markers for the cytosolic and nuclear extracts, respectively. Quantification of the scanned band intensities revealed a ∼2-fold increase in Nrf2 intensity in the nuclear fraction (lower panel). (B) Real-time PCR analyses of the samples revealing changes in the expression of several genes, whose values were normalized to β-actin expression, and presented as 2−ΔΔ. Relative mRNA abundance of each gene in untreated cells was assumed to be 1.0 (control). Results are given as mean ± S.D. values for n= 3 independent experiments. *P< 0.05 versus control; #P< 0.05 versus H2O2-treated cells.
Expression of genes involved in glutathione synthesis, i.e. GCLC and GCLM, was significantly increased by cyclo(His-Pro) treatment prior to H2O2 insult (Fig. 3B). GSTA-2 and GR expression were moderately increased by H2O2, whereas Gpx was unaffected. However, pre-treatment with the cyclic dipeptide significantly increased GR and GSTA-2 and, at a lesser extent, Gpx (Fig. 3B). Other genes involved in the redox potential maintenance, i.e. Trx-1, NQO1 and G6PDH, were up-regulated by cyclo(His-Pro) (Fig. 3B). We found that H2O2 increased nNOS expression moderately, whereas iNOS expression was strongly enhanced; however, pre-treatment with cyclo(His-Pro) fully prevented the expression of both NOS isoforms (Fig. 3B). On the whole, these findings suggest a marked effect of cyclo(His-Pro) on the expression of genes involved in oxidative stress cellular response.
Effects of Nrf2 siRNA on cyclo(His-Pro)-induced Nrf2 activation oxidative/nitrosative stress-related gene expression
To provide more direct evidence for the role of Nrf2 in cyclo(His-Pro)-induced regulation of the redox status, we used the small interfering RNA (siRNA) strategy to inhibit the endogenous expression of Nrf2 in PC12 cells. The transfection of PC12 cells with an siRNA specific for Nrf2 significantly reduced Nrf2 mRNA (Fig. 4A) and protein (Fig. 4B) abundances, whereas the scrambled control siRNA had no effect (Fig. 4A and B). Interestingly, the decrease in Nrf2 mRNA and protein levels caused by Nrf2 siRNA treatment was partially, but still significantly counteracted by cyclo(His-Pro) (Fig. 4A and B). Next, we aimed to investigate further the implication of Nrf2 in cyclo(His-Pro)-triggered oxidative/nitrosative stress-related gene expression. To perform this, Nrf2 was silenced in PC12 cells previously treated with cyclo(His-Pro) plus H2O2, i.e.the experimental condition found to significantly alter oxidative/ nitrosative gene expression (see Fig. 3B). As shown in Figure 4C, real-time PCR analyses revealed that Nrf2 knockdown (Nrf2 siRNA) abolished the increase in the mRNA levels of every analysed gene, except NOS isoforms. Thus, Nrf2 silencing increased nNOS, whereas it did not alter iNOS mRNA levels. To confirm the functional effects of these observations, we next analysed cell survival and the concentrations of ROS, NO and GSH. As shown in Figure 4D, Nrf2 knockdown reduced cell viability (MTT analyses), increased ROS and NO concentrations and decreased GSH concentrations in cyclo(His-Pro) plus H2O2-treated PC12 cells.
Figure 4.
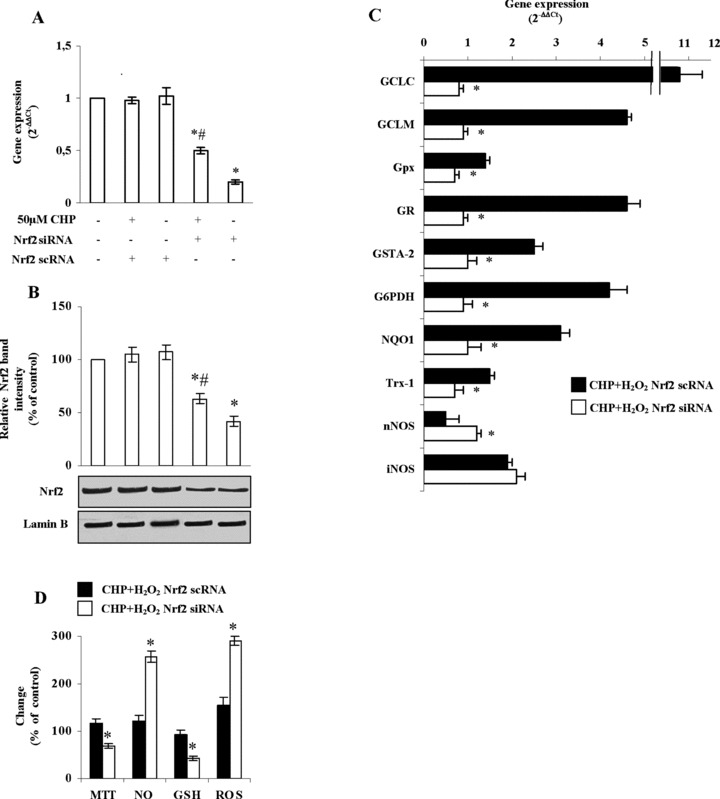
Knockdown of Nrf2 by RNA interference prevents cyclo(His-Pro)-mediated regulation of antioxidant gene expression in PC12 cells. Cells were transfected with 100 nM siRNA or 100 nM scRNA (scrambled RNA), as described in Experimental, followed by incubation with 50 μM cyclo(His-Pro) for 24 hrs. (A) Nrf2 mRNA abundance, as revealed by real-time PCR. (B) Nrf2 in nuclear extracts analysed by Western blotting, revealing a ∼50% decrease in Nrf2 protein content. Results are given as mean ± S.D. values for n = 3 independent experiments. *P< 0.05 versus control. #P < 0.05 versus siRNA values. (C) Real-time PCR analyses showing expression of Nrf2 downstream genes. Changes in gene expression were normalized to p-actin expression and presented as 2−ΔΔCT. Relative mRNA abundance of each gene in untreated cells was assumed to be 1.0 (control). (D) Cell viability (MTT reduction), ROS, NO and glutathione (GSH) were assessed in Nrf2-silenced cells treated with 50 μM cyclo(His-Pro) for 24 hrs prior to H2O2 treatment. Control values (100%) are indicated in the Figure 2 legend. Results are given as mean ± S.D. values for n = 3 independent experiments. *P<0.05 versus CHP+H2O2- Nrf2scRNA.
Role of p-38 MAPK on cyclo(His-Pro)-induced Nrf2 activation and ARE-dependent gene expression
It is proposed that the phosphorylation of Nrf2 may play a role in the transcriptional activation of genes through ARE [20–24]. Moreover, we have previously shown that cyclo(His-Pro) promotes p-38 MAPK phosphorylation in serum-starved PC12, and that inhibition of p-38 MAPK prevents cyclo(His-Pro)-mediated cytoprotection [5]. Now, we aimed to confirm whether cyclo(His-Pro)-triggered p-38 MAPK phosphorylation and cytoprotection were related phenomena. To perform this, we first analysed total and phosphorylated p-38 MAPK protein abundances by Western blotting in PC12 cells previously incubated in the absence or in the presence of SB203580 (10 μM), a p38 MAPK inhibitor. As shown in Figure 5A, H2O2 slightly, but not significantly, increased the relative abundance of phospho-p-38 MAPK versus total-p-38 MAPK. Phosphorylation of p-38 MAPK was, however, significantly increased by cyclo(His-Pro), an effect that was abolished by p-38 MAPK inhibition (Fig. 5A). We next assessed whether p-38 MAPK is a factor involved in cyclo(His-Pro)-induced Nrf2 activation. As shown in Figure 5B, p-38 MAPK inhibition significantly decreased Nrf2 protein abundance in the nucleus, as well as the mRNA levels of every analysed gene, except NOS isoforms (Fig. 5C). Thus, p-38 MAPK inhibition increased the expression of both nNOS and iNOS (Fig. 5C). The relative changes in gene expression under all the experimental conditions used are summarized in Table 2. To confirm the functional relevance of these results, we analysed cell survival and the concentrations of ROS, NO and GSH. As shown in Figure 5D, p-38 MAPK inhibition reduced cell viability (MTT analyses), increased ROS and NO concentrations and decreased GSH concentrations in cyclo(His-Pro) plus H2O2-treated PC12 cells.
Figure 5.
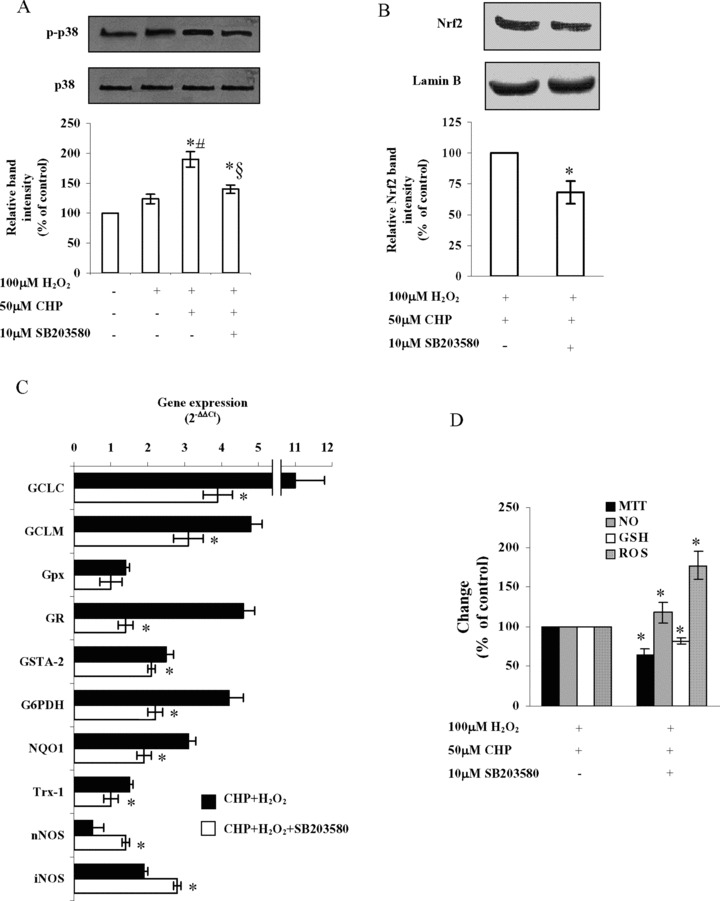
p-38 MAPK mediates cyclo(His-Pro)-induced Nrf2 translocation and antioxidant gene expression in PC12 cells. Cells were pre-incubated in the presence of 10 μM of SB203580, a well-known compound proved to be selective for p-38 MAPK at such concentration. After 2 hrs, 50 μM of cyclo(His-Pro) was added, and cells were incubated for further 24 hrs followed by 100 μM H2O2 treatment. (A) Involvement of p-38 MAPK in cyto-protection by cyclo(His-Pro), as assumed by Western blotting. Results are given as mean ± S.D. values for n = 3 independent experiments. *P< 0.05 versus control; #P< 0.05 versus H2O2-treated cells; §P< 0.05 versus CHP-treated cells. (B) Nrf2 protein is decreased in the nuclear extracts of cells treated with the p-38 MAPK inhibitor, as assessed by Western blotting. (C) Real-time PCR analyses of the samples revealing changes in the expression of Nrf2 downstream genes, whose values were normalized to (J-actin expression, and presented as 2−AACt. Relative mRNA abundance of each gene in untreated cells was assumed to be 1.0 (control). (D) Cell viability (MTT reduction), ROS, NO and glutathione (GSH) were assessed in the SB203580-treated cells incubated with 50 μM cyclo(His-Pro) for 24 hrs prior to H2O2 treatment. Control values (100%) are indicated in the Figure 2 legend. Results are given as mean ± S.D. values for n = 3 independent experiments. *P < 0.05 versus CHP+H2O2-treated cells.
Table 2.
Summary of fold changes of the investigated genes
| Treatment | ||||
|---|---|---|---|---|
| H202 | + | + | + | + |
| CHP | - | + | + | + |
| Nrf2 siRNA | - | - | + | - |
| SB203580 | - | - | - | + |
| Gene | Fold change | |||
|---|---|---|---|---|
| GCLC | 1.2 ± 0.5 | 11 ± 1.2 | 0.8 ± 0.1 | 3.9 ± 0.4 |
| GCLM | 1.2 ± 0.4 | 4.8 ± 0.3 | 1.0 ± 0.2 | 3.1 ± 0.4 |
| Gpx | 0.9 ± 0.1 | 1.4 ± 0.1 | 0.7 ± 0.1 | 1.0 ± 0.3 |
| GR | 1.5 ± 0.1 | 4.6 ± 0.3 | 0.9 ± 0.1 | 1.4 ± 0.2 |
| GSTA-2 | 2.0 ± 0.3 | 2.5 ± 0.2 | 1.0 ± 0.2 | 2.1 ± 0.4 |
| G6PDH | 1.1 ± 0.1 | 4.2 ± 0.4 | 0.9 ± 0.2 | 2.2 ± 0.2 |
| NQ01 | 1.2 ± 0.4 | 3.1 ± 0.2 | 1.0 ± 0.3 | 1.9 ± 0.2 |
| Trx-1 | 1.0 ± 0.2 | 1.5 ± 0.1 | 0.7 ± 0.2 | 1.0 ± 0.2 |
| nNOS | 1.4 ± 0.1 | 0.5 ± 0.3 | 1.2 ± 0.1 | 1.4 ± 0.1 |
| iMOS | 3.7 ± 0.1 | 1.9 ± 0.1 | 2.1 ± 0.2 | 2.8 ± 0.1 |
Effects of cyclo(His-Pro) treatment on glutamate-, rotenone-, paraquat- and (β-amyloid injured PC12 cells
Glutamate, rotenone, paraquat and β-amyloid are neurotoxic compounds that trigger cell death through several mechanisms that involve oxidative stress [25–28]. Therefore, we decided to investigate whether cyclo(His-Pro) could exert protective effects against these toxic insults and to verify the molecular mechanism underlying the protection in PC12 cells. Under these experimental conditions, maximal protection was achieved with 100 μM cyclo(His-Pro) (data not shown). Incubation of PC12 cells with glutamate decreased both cell survival and GSH concentrations, whereas it increased by twofold the production of NO and ROS (Fig. 6A). Under these conditions, cyclo(His-Pro) fully prevented the increase in NO and ROS, and partially prevented cell death and GSH depletion (Fig. 5A). Rotenone (Fig. 6B) and paraquat (Fig. 6C) decreased cell viability and GSH levels to similar extent values, but the effects on NO and ROS generation were notably different. Thus, rotenone markedly increased ROS production, whilst paraquat increased NO levels (Fig. 6B and C). All effects caused by both toxins (rotenone and paraquat) were fully counteracted by cyclo(His-Pro) (Fig. 6B and C). β-amyloid reduced by ∼50% cell viability and GSH levels, and doubled NO and ROS production (Fig. 6D); cyclo(His-Pro) fully prevented the increase in NO and ROS, and partially prevented cell death and GSH depletion (Fig. 6D).
Figure 6.
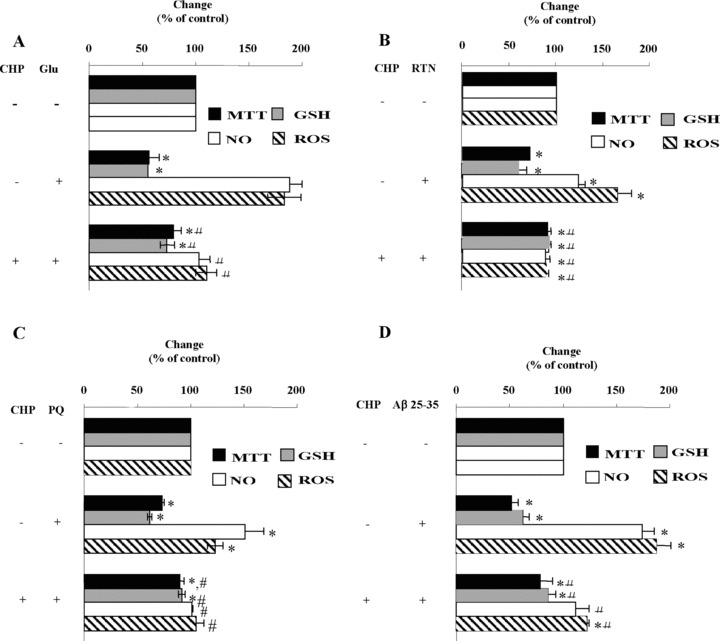
Cyclo(His-Pro) attenuates ROS production and prevents glutathione depletion caused by glutamate, rotenone, paraquat and p-amyloid in PC12 cells. Cells were incubated for 24 hrs with 10 mM glutamate (A), 100 μM rotenone (B), 100 μM paraquat (C) or 25 μM β-amyloid 25–35 (D), either in the absence or in the presence of 100 μM cyclo(His-Pro). After 24 hrs, cells were harvested and used to assess cell viability, GSH concentration, NO and ROS production. Control values (100%; mean ± S.D., n = 3) are indicated in the Figure 2 legend. *P < 0.05 versus control; #P < 0.05 versus injured cells.
Discussion
Here we show that the cyclic dipeptide cyclo(His-Pro) provides cytoprotection in PC12 cells through a mechanism involving nuclear accumulation and activation of Nrf2. This confirms and provides a mechanistic explanation for the previous observations reporting protection by cyclo(His-Pro) both in cultured cells and in vivo[1, 4, 5]. Hydrogen peroxide-mediated ROS and NO generation and glutathione depletion leading to apoptotic cell death were also abolished by treatment with cyclo(His-Pro), suggesting that the dipeptide may act as an endogenous antioxidant. Here, we also show that cyclo(His-Pro) prevented H2O2-induced rise in intracellular calcium. Whether the ability of the dipeptide to chelate calcium, previously reported [29], is responsible for the reduction in H2O2-induced calcium accumulation and cell protection against oxidative stress remains to be elucidated.
The maintenance of cellular redox status is mainly due to the interplay between the thioredoxin and the glutathione systems. Thus, whilst glutathione maintains a low redox potential and high free thiol levels, thioredoxin regulates the redox status of protein thiols involved in signal transduction and gene regulation [30]. Our results showing up-regulation of genes related to both redox systems (glutathione-synthesizing/regenerating enzymes and thioredoxin-1 isoform) highlight the biological relevance of cyclo(His-Pro) to act as an inducing agent after interaction with H2O2. In fact, disruption of glutathione metabolism has been implied in aging and certain neurodegenerative disorders, in which glutathione deficiency is associated with mitochondrial damage and increased ROS [25, 31]. NAD(P)H:quinone oxidoreductase 1 (NQO1) inhibition by NO has been shown to be synergistic with glutathione deficiency [32] and the activity of α-ketoglutarate dehydrogenase, the enzyme responsible for NADH supply to complex I, is inhibited by H2O2[33, 34]. In our hands, cyclo(His-Pro) counterbalanced the H2O2-mediated increase in NO production (along with the expression of both NOS isoforms), as well as prevented glutathione deficiency and complex I repression. These results suggest that the use of cyclo(His-Pro) may be of interest as a potential therapeutic strategy in oxidative stress-based diseases.
Because of the wide-range induction of antioxidant genes caused by cyclo(His-Pro) treatment, it appears that multiple factors may be responsible for the cytoprotective role of the dipeptide. Thus, RNAi-mediated Nrf2 knockdown counteracted cytoprotection by cyclo(His-Pro), strongly suggesting the involvement of multiple antioxidant genes. In this context, to counteract oxidative insults, cells induce the Phase I and Phase II enzymes, i.e. the series of gene products that reduce reactive electrophiles and detoxify carcinogens [14]. Phase II enzymes are transcriptionally regulated in a coordinated fashion through the cis-acting enhancer sequence ARE [7, 14, 21]. Here, we show that cyclo(His-Pro) triggers the expression of a panel of antioxidant enzymes through Nrf2, a result that is compatible with the notion that Nrf2-ARE interaction is involved in the dipeptide effect. In fact, Nrf2 silencing increased nNOS, whereas it did not alter iNOS mRNA levels, a result that is in agreement with the fact that NO levels do not change in Nrf2−/– fibroblasts treated with inhibitors of cellular inflammatory processes [35]. The mechanism whereby cyclo(His-Pro) activates Nrf2 is intriguing. Nrf2 is a transcription factor that binds to Keap1, forming a complex that remains sequestered in the cytosol under non-oxidizing conditions [11, 21, 36]. Following an oxidative insult, Nrf2 dissociates from the Keap1-Nrf2 complex and translocates to the nucleus, binding to ARE, thereby potentiating the cellular response against electrophile/oxidative stress [9]. Direct modification of critical cysteine residues on Keap1 and phosphorylation of the Nrf2-Keap1 complex by mitogen-activated protein kinase(s) have been implied in the mechanism for the heterodimer disruption and increased Nrf2 stabilization [10, 11, 14, 21, 22, 24, 37]. Here, we show that pharmacological inhibition of p-38 MAPK significantly prevented Nrf2 nuclear translocation and attenuated cyclo (His-Pro) cytoprotection under redox oxidizing conditions. Although further experiments would be required to confirm this pathway, these results are compatible with the notion that both redox changes and MAPK-mediated phosphorylation would be required for Nrf2 nuclear translocation and/or stabilization.
Taken together, our results suggest that the dipeptide cyclo(His-Pro) protects PC12 cells against oxidative insults through the activation of Nrf2-mediated detoxifying/antioxidants gene expression. These findings provide the first evidence of a molecular mechanism responsible for the cytoprotective effects of cyclo(His-Pro) against oxidative stress-based pathologies.
Acknowledgments
The authors thank Dr. M. Kerrigan (Cantab, MA) for helpful linguistic suggestions. This study was supported by grants from Murst-Italia and by Fondazione Cassa di Risparmio di Perugia, Italia (Scientific and Technological Research, Grant n. 2006.0216.020). J.P.B. is funded by MEC (SAF2007–61492 and CONSOLIDER) and JCyL (SA066A07 and Red Terapia Celular-ISCIII).
References
- 1.Prakash KR, Tang Y, Kozikowski AP, Flippen-Anderson JL, Knoblach SM, Faden AI. Synthesis and biological activity of novel neuroprotective diketopiperazines. Bioorg Med Chem. 2002;10:3043–8. doi: 10.1016/s0968-0896(02)00132-3. [DOI] [PubMed] [Google Scholar]
- 2.Prasad C. Bioactive cyclic dipeptides. Peptides. 1995;16:151–64. doi: 10.1016/0196-9781(94)00017-z. [DOI] [PubMed] [Google Scholar]
- 3.Jaspan JB, Banks WA, Kastin AJ. Study of passage of peptides across the blood-brain barrier: biological effects of cyclo(His-Pro) after intravenous and oral administration. Ann N Y Acad Sci. 1994;739:101–7. doi: 10.1111/j.1749-6632.1994.tb19811.x. [DOI] [PubMed] [Google Scholar]
- 4.Faden AI, Movsesyan VA, Knoblach SM, Ahmed F, Cernak I. Neuroprotective effects of novel small peptides in vitro and after brain injury. Neuropharmacology. 2005;49:410–24. doi: 10.1016/j.neuropharm.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 5.Minelli A, Bellezza I, Grottelli S, Pinnen F, Brunetti L, Vacca M. Phosphoproteomic analysis of the effect of cyclo-[His-Pro] dipeptide on PC12 cells. Peptides. 2006;27:105–13. doi: 10.1016/j.peptides.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 6.Holtzclaw WD, Dinkova-Kostova AT, Talalay P. Protection against electrophile and oxidative stress by induction of phase 2 genes: the quest for the elusive sensor that responds to inducers. Adv Enzyme Regul. 2004;44:335–67. doi: 10.1016/j.advenzreg.2003.11.013. [DOI] [PubMed] [Google Scholar]
- 7.Ishii T, Itoh K, Takahashi S, Sato H, Yanagawa T, Katoh Y, Bannai S, Yamamoto M. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J Biol Chem. 2000;275:16023–9. doi: 10.1074/jbc.275.21.16023. [DOI] [PubMed] [Google Scholar]
- 8.Rushmore TH, Morton MR, Pickett CB. The antioxidant responsive element. Activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. J Biol Chem. 1991;266:11632–9. [PubMed] [Google Scholar]
- 9.Wakabayashi N, Dinkova-Kostova AT, Holtzclaw WD, Kang MI, Kobayashi A, Yamamoto M, Kensler TW, Talalay P. Protection against electrophile and oxidant stress by induction of the phase 2 response: fate of cysteines of the Keap1 sensor modified by inducers. Proc Natl Acad Sci USA. 2004;101:2040–5. doi: 10.1073/pnas.0307301101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Padmanabhan B, Tong KI, Ohta T, Nakamura Y, Scharlock M, Ohtsuji M, Kang MI, Kobayashi A, Yokoyama S, Yamamoto M. Structural basis for defects of Keap1 activity provoked by its point mutations in lung cancer. Mol Cell. 2006;21:689–700. doi: 10.1016/j.molcel.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 11.Velichkova M, Hasson T. Keap1 regulates the oxidation-sensitive shuttling of Nrf2 into and out of the nucleus via a Crm1-dependent nuclear export mechanism. Mol Cell Biol. 2005;25:4501–13. doi: 10.1128/MCB.25.11.4501-4513.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Itoh K, Igarashi K, Hayashi N, Nishizawa M, Yamamoto M. Cloning and characterization of a novel erythroid cell-derived CNC family transcription factor heterodimerizing with the small Maf family proteins. Mol Cell Bio. 1995;15:4184–93. doi: 10.1128/mcb.15.8.4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jaiswal AK. Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radic Biol Med. 2004;36:1199–207. doi: 10.1016/j.freeradbiomed.2004.02.074. [DOI] [PubMed] [Google Scholar]
- 14.Kobayashi A, Ohta T, Yamamoto M. Unique function of the Nrf2-Keap1 pathway in the inducible expression of antioxidant and detoxifying enzymes. Methods Enzymol. 2004;378:273–86. doi: 10.1016/S0076-6879(04)78021-0. [DOI] [PubMed] [Google Scholar]
- 15.Kukla MJ, Breslin HJ, Bowden CR. Synthesis, characterization, and anorectic testing of the four stereoisomers of cyclo(histidylproline) J Med Chem. 1985;28:1745–7. doi: 10.1021/jm00149a035. [DOI] [PubMed] [Google Scholar]
- 16.Tietze F. Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione: applications to mammalian blood and other tissues. Anal Biochem. 1969;27:502–22. doi: 10.1016/0003-2697(69)90064-5. [DOI] [PubMed] [Google Scholar]
- 17.Alberdi E, Sanchez-Gomez MV, Matute C. Calcium and glial cell death. Cell Calcium. 2005;38:417–25. doi: 10.1016/j.ceca.2005.06.020. [DOI] [PubMed] [Google Scholar]
- 18.Thompson RJ, Zhou N, MacVicar BA. Ischemia opens neuronal gap junction hemichannels. Science. 2006;312:924–7. doi: 10.1126/science.1126241. [DOI] [PubMed] [Google Scholar]
- 19.Zoccarato F, Toscano P, Alexandre A. Dopamine-derived dopaminochrome promotes H(2)O(2) release at mitochondrial complex I: stimulation by rotenone, control by Ca(2+), and relevance to Parkinson disease. J Biol Chem. 2005;280:15587–94. doi: 10.1074/jbc.M500657200. [DOI] [PubMed] [Google Scholar]
- 20.Chan K, Han XD, Kan YW. An important function of Nrf2 in combating oxidative stress: detoxification of acetaminophen. Proc Natl Acad Sci USA. 2001;98:4611–6. doi: 10.1073/pnas.081082098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nguyen T, Sherratt PJ, Pickett CB. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu Rev Pharmacol Toxicol. 2003;43:233–60. doi: 10.1146/annurev.pharmtox.43.100901.140229. [DOI] [PubMed] [Google Scholar]
- 22.Sherratt PJ, Huang HC, Nguyen T, Pickett CB. Role of protein phosphorylation in the regulation of NF-E2-related factor 2 activity. Methods Enzymol. 2004;378:286–301. doi: 10.1016/S0076-6879(04)78022-2. [DOI] [PubMed] [Google Scholar]
- 23.Yamamoto N, Sawada H, Izumi Y, Kume T, Katsuki H, Shimohama S, Akaike A. Proteasome inhibition induces glutathione synthesis and protects cells from oxidative stress: relevance to Parkinson disease. J Biol Chem. 2007;282:4364–72. doi: 10.1074/jbc.M603712200. [DOI] [PubMed] [Google Scholar]
- 24.Yuan X, Xu C, Pan Z, Keum YS, Kim JH, Shen G, Yu S, Oo KT, Ma J, Kong AN. Butylated hydroxyanisole regulates ARE-mediated gene expression via Nrf2 coupled with ERK and JNK signaling pathway in HepG2 cells. Mol Carcinog. 2006;45:841–50. doi: 10.1002/mc.20234. [DOI] [PubMed] [Google Scholar]
- 25.Hsu M, Srinivas B, Kumar J, Subramanian R, Andersen J. Glutathione depletion resulting in selective mitochon-drial complex I inhibition in dopaminergic cells is via an NO-mediated pathway not involving peroxynitrite: implications for Parkinson’s disease. J Neurochem. 2005;92:1091–103. doi: 10.1111/j.1471-4159.2004.02929.x. [DOI] [PubMed] [Google Scholar]
- 26.Indo HP, Davidson M, Yen HC, Suenaga S, Tomita K, Nishii T, Higuchi M, Koga Y, Ozawa T, Majima HJ. Evidence of ROS generation by mitochondria in cells with impaired electron transport chain and mitochondrial DNA damage. Mitochondrion. 2007;7:106–18. doi: 10.1016/j.mito.2006.11.026. [DOI] [PubMed] [Google Scholar]
- 27.Parihar MS, Brewer GJ. Simultaneous age-related depolarization of mitochondrial membrane potential and increased mitochondrial reactive oxygen species production correlate with age-related glutamate excitotoxicity in rat hippocampal neurons. J Neurosci Res. 2007;85:1018–32. doi: 10.1002/jnr.21218. [DOI] [PubMed] [Google Scholar]
- 28.Smith DG, Cappai R, Barnham KJ. The redox chemistry of the Alzheimer’s disease amyloid beta peptide. Biochim Biophys Acta. 2007;1768:1976–90. doi: 10.1016/j.bbamem.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 29.Gockel P, Vogler R, Gelinsky M, Meissner A, Albrich H, Vahrenkamp H. Zinc com-plexation of cyclic dipeptides containing cysteine and/or histidine. Inorganica Chimica Acta. 2001;323:16–22. [Google Scholar]
- 30.Arner ES, Holmgren A. Physiological functions of thioredoxin and thioredoxin reductase. Eur J Biochem. 2000;267:6102–9. doi: 10.1046/j.1432-1327.2000.01701.x. [DOI] [PubMed] [Google Scholar]
- 31.Zhu Y, Carvey PM, Ling Z. Age-related changes in glutathione and glutathione-related enzymes in rat brain. Brain Res. 2006;1090:35–44. doi: 10.1016/j.brainres.2006.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Borutaite V, Brown GC. S-nitrosothiol inhibition of mitochondrial complex I causes reversible increase in mitochondrial hydrogen peroxide production. Biochim Biophys Acta. 2006;1757:562–6. doi: 10.1016/j.bbabio.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 33.Dhakshinamoorthy S, Jain AK, Bloom DA, Jaiswal AK. Bach1 competes with Nrf2 leading to negative regulation of the antioxidant response element (ARE)-mediated NAD(P)H: quinone oxidoreductase 1 gene expression and induction in response to antioxidants. J Biol Chem. 2005;280:16891–900. doi: 10.1074/jbc.M500166200. [DOI] [PubMed] [Google Scholar]
- 34.Sheline CT, Wei L. Free radical-mediated neurotoxicity may be caused by inhibition of mitochondrial dehydrogenases in vitro and in vivo. Neuroscience. 2006;140:235–46. doi: 10.1016/j.neuroscience.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 35.Dinkova-Kostova AT, Liby KT, Stephenson KK, Holtzclaw WD, Gao X, Suh N, Williams C, Risingsong R, Honda T, Gribble GW, Sporn MB, Talalay P. Extremely potent triterpenoid inducers of the phase 2 response: correlations of protection against oxidant and inflammatory stress. Proc Natl Acad Sci USA. 2005;102:4584–9. doi: 10.1073/pnas.0500815102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nguyen T, Sherratt PJ, Nioi P, Yang CS, Pickett CB. Nrf2 controls constitutive and inducible expression of ARE-driven genes through a dynamic pathway involving nucleocytoplasmic shuttling by Keap1. J Biol Chem. 2005;280:32485–92. doi: 10.1074/jbc.M503074200. [DOI] [PubMed] [Google Scholar]
- 37.Nguyen T, Sherratt PJ, Huang HC, Yang CS, Pickett CB. Increased protein stability as a mechanism that enhances Nrf2-mediated transcriptional activation of the antioxidant response element. Degradation of Nrf2 by the 26 S protea-some. J Biol Chem. 2003;278:4536–41. doi: 10.1074/jbc.M207293200. [DOI] [PubMed] [Google Scholar]