

Abstract
Accumulating evidence suggest that alterations in energy metabolism are among the earliest events that occur in the Alzheimer disease (AD) affected brain. Energy consumption is drastically decreased in the AD-affected regions of cerebral cortex and hippocampus pointing towards compromised mitochondrial function of neurons within specific brain regions. This is accompanied by an elevated production of reactive oxygen species contributing to increased rates of neuronal loss in the AD-affected brain regions. In this review, we will discuss the role of mitochondrial function and dysfunction in AD. We will focus on the consequences of amyloid precursor protein and amyloid-β peptide accumulation in mitochondria and their involvement in AD pathogenesis.
Keywords: Alzheimer disease, mitochondria, oxidative stress, amyloid precursor protein, amyloid-β peptide
Introduction
Age and AD related mitochondrial changes in brain and peripheral tissues
Effect of APP accumulation on mitochondrial function
Mitochondria as a target and mediator of Aβ toxicity
Conclusions
Introduction
Alzheimer disease (AD) is a devastating neurodegenerative disease characterized by a progressive decline in memory and cognitive functions such as language and perception [1]. It affects mostly elderly people and appears both sporadically (accounting for about 95% of all cases) and in familiar autosomal dominant (FAD) form [2, 3]. AD is characterized by two brain lesions, intraneuronal fibrillary tangles and extracellular amyloid plaques [4, 5]. Neurofibrillary tangles are composed of aggregated hyperphosphorylated τ protein. The role of aggregated τ in neurodegeneration is still controversial, as evidence points to either a toxic or protective role in the disease [6–8]. The major constituent of the amyloid plaques is a hydrophobic 39–43 amino acid peptide named amyloid-β peptide (Aβ), a cleavage product of the much larger transmembrane protein, amyloid precursor protein (APP) encoded by a single gene on chromosome 21 [9]. Aβ is generated from the C-terminal end of APP by the sequential action of β- and γ-secretases [10]. Amyloid formation is generally associated with the clinical manifestations of AD and the Aβ cascade hypothesis is the main pathogenic model of AD [11]. Almost all gene mutations linked to the early-onset FAD cases are associated with deregulated metabolism of APP resulting in enhancement of production of Aβ or increase of Aβ42/40 ratio [12]. Although the aetiology of sporadic AD is largely unknown, accumulating data suggest that mitochondrial dysfunction and oxidative stress occur in brain as well as in peripheral tissues of AD patients [13]. Mitochondria produce most of the cell energy, they are key regulators of cell survival and death, have a central role in ageing and have recently been found to harbour specific proteins implicated in AD. A better understanding of how mitochondrial functions change in aging and neurodegenerative diseases could reveal novel mitochondria-directed approaches for therapy of AD. Here we review and discuss the role of mitochondrial changes during aging and AD progression as well as recent findings connecting the amyloid cascade hypothesis and mitochondria.
Age and AD related mitochondrial changes in brain and peripheral tissues
Today the free radical as well as the mitochondrial decline theories gains most in popularity among the different theories of aging. According to the free radical theory of aging, proposed by Harman [14], reactive oxygen species (ROS) produced in the body as a by-product of oxidative metabolism initiate changes associated with aging. Free radicals such as hydroxyl radical (OH.), superoxide anion radical (O2.-), hydroperoxyl radical (HO2.) or nitric oxide radical (NO.) can indiscriminately react with a wide variety of organic substrates causing peroxidation of lipids, cross-linking and modifications of proteins and mutations in DNA [15]. Cells have a variety of defence mechanisms to protect themselves against the harmful effects of ROS. Still, over time ROS-induced damage interfere with cell communication, disturb DNA, RNA and protein synthesis, decrease energy levels and generally impede vital chemical processes. Most of the cellular ATP is produced in mitochondria during respiration in a process called oxidative phosphorylation. This process encompasses electron transfer between protein complexes of respiratory chain in the inner mitochondrial membrane, vectorial transfer of protons into intermembrane space and ATP synthesis by ATP synthase upon re-entry of protons into the matrix [16]. During respiration a small portion of electrons passing through the electron transport chain, mostly at complex I and complex III, react with molecular oxygen and generate ROS through enzymatic and non-enzymatic reactions [17, 18]. The constant ROS production inside mitochondria subjects them to substantial free radical damage. Because the mitochondrion contains its own intron-less and histone-less ∼16-kb circular DNA, a central role for mtDNA mutations caused by free radicals in aging has been postulated [19, 20]. In fact, mtDNA mutations have been shown to accumulate with aging in different tissues of various species [21–23]. As an extension and refinement of the free radical theory of aging the mitochondrial ‘vicious cycle’ theory was put forward [24]. This theory postulates that mtDNA mutations, which accumulate progressively during life, as a side effect of respiration, are directly responsible for a measurable deficiency in cellular oxidative phosphorylation activity leading to an enhanced ROS production. Trifunovic and colleagues have developed a ‘mtDNA mutator’ homozygote mouse expressing a proofreading-deficient form of the DNA polymerase (Polg) [25]. These mice have widespread tissue distribution of mutated mtDNA and a remarkable phenotype with several examples of premature aging. Interestingly, the increased levels of mtDNA mutations were not associated with increased ROS production [26] and this animal model has thus questioned the mitochondrial ‘vicious cycle’ theory. In a similar animal model developed by another group, the heterozygote Polg+/D257A mice were also shown to have strikingly high mtDNA mutation frequency albeit lower than PolgD257A/D257A mice [27] despite the fact that they lack the accelerating aging features of homozygous mice. It was found that there is a threshold of mtDNA mutations to reach the phenotypic expression, which differs in various tissues and ability of cells to repair large mtDNA deletions would determine tissue tolerance [28]. Despite the missing backward link between increased rate of mtDNA mutations and increased ROS production in the mtDNA mutator mice, another mouse model still points towards age-associated oxidative stress correlated with increased rate of mtDNA mutations [29]. In this model, mice expressing catalase targeted to mitochondria but not to peroxisomes or nuclei have extended lifespan by approximately 20%. Their results suggest that scavenging harmful ROS in mitochondria protect mtDNA and prolong lifespan [29].
A large number of reports support the fact that decreased mitochondrial functions are associated with aging. In general mitochondria isolated from old animals show lower respiration rates due to decreased activity of electron transfer chain enzymes [30, 31]. Respiratory chain complexes I and IV show significantly decreased enzymatic activities in mitochondria isolated from different tissues derived from old animals, whereas complexes II and III are less affected [32–36]. One plausible explanation for this phenomena is the large proportion of mtDNA encoded subunits of complex I (seven subunits) and complex IV (three subunits) in comparison to only one subunit of complex III and no nuclear encoded subunits in complex II. Time course analysis of mitochondrial DNA gene expression in C57BL6 mouse brain reveal increased expression of mitochondrial encoded genes of the respiratory chain at 12 and 18 months of age as compared to 2-month-old controls [37]. These data suggest age dependent compensatory mechanism for mitochondrial respiratory chain dysfunction.
Other mitochondrial enzymes such as adenine nucleotide translocase [38], nitric oxide synthase [39] and carnitine acyltransferase [40] show 30–60% decreased activities in senescent brain. The observed decrease of electron transfer in aged brain mitochondria is paralleled with the development of mitochondria with increased size [41] and higher proportion of depolarized non-functional mitochondria [42]. In summary, physiological aging is associated with general decline in mitochondrial functions.
Numerous reports show abnormal mitochondrial dysfunction associated with AD pathology. For example, a decline in cerebral glucose metabolism occurs before pathology and symptoms manifest, continues as symptoms progress and is more severe than in normal aging [43]. Studies of neural cells devoid in mtDNA and fused with platelets that lack intact nucleus (cybrid cells) provide important evidence for mitochondrial dysfunction in AD. In such cells that differ only in their mitochondrial content cybrids obtained from AD patients show decreased activities of mitochondrial enzymes and increased oxidative stress and, remarkably, elevated extra and intracellular levels of Aβ[41–45]. The cybrid model implicates mtDNA as important factor in AD pathogenesis and the mtDNA mutation rate is increased in AD as compared to age-matched controls. However, no consistent abnormalities in mtDNA associated with AD were reported [44], suggesting random mtDNA damage due to elevated ROS production.
Mitochondrial dysfunction of APP transgenic mice was demonstrated by an increased expression of genes related to mitochondrial energy metabolism as well as apoptosis [45]. These data implicate mitochondrial stress as common denominator for AD and aging [37].
Among the most consistent findings in mitochondrial AD associated defects are decreased activities of α-ketoglutarate dehydrogenase [46, 47], pyruvate dehydrogenase [48] and cytochrome c oxidase (complex IV) of electron transfer chain [49, 50]. Interestingly, the activities of all of these enzymes are inhibited by Aβ[51] providing a possible link between amyloid cascade and mitochondrial dysfunction in AD. Crouch et al. showed that low molecular weight Aβ1–42 oligomers inhibits cytochrome c oxidase in a copper-dependent manner [52]. Maximal cytochrome c oxidase inhibition was reached with Aβ solutions aged for up to 6 hrs. These Aβ1–42 preparations consisted of around 10% toxic oligomeric species (∼17 μM). The remaining 90% are monomeric Aβ, high molecular weight oligomers, protofibrils and fibrils and regarded as relatively non-toxic. It remains to be determined whether concentrations of low molecular weight Aβ1–42 oligomers required to inhibit complex IV are present in AD brain mitochondria.
Effect of APP accumulation on mitochondrial functions
Recently it was shown that mitochondria isolated from affected AD brain regions contain substantial amounts of APP as compared to age-matched controls [53]. APP forms stable translocation intermediate complexes with translocase of the outer membrane (TOM) in the AD mitochondria (Fig. 1). APP also links TOM and translocase of the inner membrane (TIM) together. Mitochondrial accumulation of APP correlates with decreased ability of mitochondria to import nuclear encoded proteins and with decreased activity of cytochrome c oxidase in AD-affected brain regions [53]. It was suggested that APP has a typical endoplasmic reticulum (ER) signal peptide followed by a cryptic mitochondrial targeting signal, whereas a more C-terminally located domain enriched in acidic amino acids acts as a mitochondrial translocation arrest sequence [54]. Expression of full length APP, in contrast to APP lacking the acidic domain, impedes mitochondrial functions suggesting that APP exert its toxicity mainly via formation of protein translocation intermediates [54]. In yeast in vivo mitochondrial targeting of a chimeric mitochondrial precursor resulted in irreversible accumulation of precursor protein in mitochondrial translocation contact sites, progressive zippering of the outer and the inner membranes, cell growth arrest and cell death [55, 56]. Interestingly, transgenic mice overexpressing APP show accumulation of APP in brain mitochondria in contrast to the non-transgenic age-matched animals [54]. In 2008, Hauptmann and colleagues reported early mitochondrial dysfunction associated with increased ROS production, decreased mitochondrial membrane potential, ATP level and complex IV activity in double Swedish and London mutant APP transgenic mice [57]. In this animal model a several fold increased expression of APP in the brain was detected. At present it is not clear whether the observed mitochondrial toxicity is due to APP or Aβ accumulation.
Fig 1.
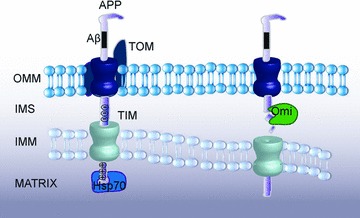
APP is localized to mitochondria. APP N-terminal contains a mitochondrial targeting sequence (positively charged aa 40, 44 and 51) and is incompletely imported into mitochondria due to an acidic stop translocation sequence (aa 220–290). Subsequently APP may either be degraded by proteases such as Omi in the intermembrane space or form supercomplexes with the TOM and TIM complexes whereby blocking the import machinery.
A combination of several factors may cause abnormal accumulation of APP in the mitochondrial translocation channels during AD progression. APP is normally targeted to ER on its way to the plasma membrane and the cryptic mitochondrial targeting signal is not exposed. Recently, an inducible cytosolic endoprotease that specifically activates mitochondrial cryptic signals by removal of the preceding ER signal peptide was characterized [58]. Upon activation of this protease several proteins including CYP1A1, glucocorticoid receptor, retinoid X receptor and p53 underwent cleavage-dependent mitochondrial import. Although in vitro mitochondrial import of APP does not require removal of ER signal peptide [54] it cannot be excluded that such processing occurs in vivo.
Post-translational modifications represent another mechanism that can control protein targeting to mitochondria. For example, post-translational N-terminal myristoylation of pro-apoptotic protein BID is required for targeting its proteolytic fragment to mitochondria during apoptosis [59]. Phosphorylation also can regulate the movement of proteins from cytosol to mitochondria [60] or promote mitochondrial localization of ER-targeted proteins [61]. Enhanced mitochondrial targeting was shown to be mediated through increased binding of client phosphoproteins to cytosolic chaperones [60, 62, 63]. Selective mitochondrial relocalization of proteins can also be achieved without their post-translational modification. Conformational change in the pro-apoptotio protein BAX structure as the result of cellular stress promotes its rapid translocation into mitochondria [64, 65]. Moreover, in response to decreased mitochondrial membrane potential parkin is re-localized from cytosol to mitochondria, which results in elimination of mitochondria by autophagosomes [66]. All together the changes in intracellular homeostasis during the initial course of AD can potentially increase mitochondrial APP targeting, resulting in mitochondrial dysfunction and acceleration of disease progression. Complete impairment of protein import into mitochondria will eventually result in collapse of mitochondrial function and ultimately to neuronal cell death. Therefore it can be assumed that during the AD progression a gradual accumulation of APP in the mitochondrial import channels is a dynamic and regulated process.
Several distinct mechanisms are known for mitochondrial protein turnover: autophagic degradation of entire organelles [67], the proteolysis of proteins within the matrix or intermembrane space [68], and proteasome-dependent outer mitochondrial membrane-associated degradation [69]. Careful balance between mitochondrial fission and fusion ensures the mitochondrial quality; however, mitochondria lacking electrochemical potential for extended periods will be selectively cleared through steady-state autophagy, or mitophagy [66, 70]. The destruction of whole organelle will eliminate non-functional mitochondria as well as prevent uncontrolled release of mitochondrial intermembrane space proapoptotic proteins into cytosol. Interestingly, abnormal mitochondrial morphology, dynamics and distribution have been observed both in fibroblasts from sporadic AD patients as well as in mice overexpressing APP [71, 72]. Furthermore, overexpression of APP in neuroblastoma cell lines displayed fragmentation and abnormal distribution of mitochondria caused by an imbalance of the mitochondrial fission and fusion system, leading to mitochondrial and neuronal dysfunction [72]. This indicates that the balance between mitochondrial fission and fusion can be affected by mitochondrial accumulation of APP or Aβ, or that a change in the fission and fusion balance towards increased fission during AD progression could result in mitochondrial APP and Aβ accumulation.
Mitochondrial protein turnover is a balance between protein synthesis inside the organelle, import of nuclear encoded proteins from cytosol and protein degradation by mitochondrial proteases. Several proteases have been identified and extensively studied in mitochondria. For example, the mitochondrial protease HtrA2/Omi has been studied in connection to APP turnover [73, 74]. HtrA2/Omi was able to cleave APP in vitro, inside mitochondria in vivo[73] as well as during ER-associated APP degradation [74]. HtrA2/Omi is a serine protease located in the intermembrane space of mitochondria. During cell death HtrA2/Omi translocates from intermembrane space to cytosol and enhances apoptosis via cleavage of inhibitory apoptosis proteins and subsequent activation of caspases [75, 76]. However, animal studies using mice with a targeted deletion of the HtrA2/Omi gene [77] as well as motor neuron degeneration 2 mice (Mnd2) [78] expressing homozygous Ser276Cys mutation in the HtrA2/Omi protease domain that greatly reduces its catalytic activity, did not exhibit decreased apoptosis. On the contrary HtrA2/Omi-deficient mice exhibit neurodegenerative phenotype, failed to gain weight and die prematurely clearly demonstrating the essential protective role of HtrA2/Omi in vivo[79]. Recent data indicate that HtrA2/Omi can play a role in the mitochondrial quality control response monitoring and controlling protein folding in the mitochondrial intermembrane space [80, 81]. HtrA2/Omi protease activity is regulated through serine phosphorylation having stimulating (Ser142) [82] or inhibitory (Ser212) [83] effect. HtrA2/Omi protease activity in mitochondria is also regulated by protein-protein interactions via the PDZ domain of HtrA2/Omi [79]. Such interactions can either promote or inhibit apoptosis probably depending on the function of HtrA2/Omi-interacting partner protein [82, 84–86]. Interestingly HtrA2/Omi interacts with presenilin-1, a catalytic subunit of the γ-secretase that cleaves APP to produce Aβ peptide [86].
It remains to be tested if the inhibition of the APP accumulation in mitochondrial import channels or increased proteolysis of APP would protect cells from mitochondria-mediated injury and potentially slow down AD progression.
Mitochondria as a target and mediator of Aβ toxicity
We and others have shown that Aβ is accumulating in mitochondria from post-mortem AD brain, living patients with cortical plaques and TgAPP mice [87–90]. In TgAPP mice mitochondrial Aβ accumulation occurs prior to plaque formation, indicating that this is an early event in the pathogenesis [87]. Even though both APP [54] and γ-secretase complexes [91] have been detected in mitochondria it is not likely that Aβ is produced locally in mitochondria. The reason is that the import of APP is arrested due to an acidic domain at amino acids 220–290 leaving the Aβ-region outside the membrane. Since γ-secretase cleaves its substrates by regulated intramembrane proteolysis such localization of APP excludes it as a γ-secretase substrate in mitochondria. Therefore, Aβ found in AD mitochondria must have been taken up. We decided to investigate these uptake mechanisms using isolated mitochondria treated with Aβ in the absence or presence of antibodies or inhibitors directed to various mitochondrial translocases and pores [90]. The uptake of Aβ was not affected by the presence of antibodies directed towards the voltage-dependent anion channel nor in the presence of cyclosporine A which is an inhibitor of the mitochondrial permeability transition pore (mPTP). Interestingly, import of both Aβ1–40 and Aβ1–42 was prevented when import competent mitochondria were pre-incubated with antibodies directed towards proteins of the TOM complex, i.e. Tom20, Tom40, Tom70 (Fig. 2). Aβ import was not affected by the addition of valinomycin, an ionophore which cause depolarization of the mitochondrial inner membrane, indicating that the Aβ import was not dependent on the mitochondrial membrane potential. After import Aβ was mostly localized to mitochondrial cristae and associated with the inner membrane fraction. It was earlier reported that Aβ co-localizes with the mitochondrial matrix protein Hsp60 in mouse and human samples [87]. One explanation to this discrepancy might be that in our in vitro assay we studied Aβ localization after 30 min. of import, whereas Caspersen et al. report data from post-mortem AD brains and 8-month-old transgenic APP mice. However, our data from brain biopsies obtained from people, who display Aβ aggregates in the neuropil, show Aβ immuno-gold labelling in association with mitochondrial inner membranes [90]. Still we cannot exclude that Aβ can be released or escapes from the membrane and also localizes to the matrix.
Fig 2.
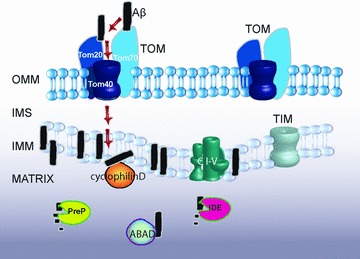
Aβ1–40 and Aβ1–42 are taken up by mitochondria through the TOM machinery. After import most of the Aβ will reside in the inner mitochondrial membrane, where it possibly can inhibit the respiration chain (CI-V) resulting in an increased production of ROS. However, a fraction will also reach the matrix where it either can be degraded by proteases like PreP and IDE or interact with proteins such as CypD and ABAD.
Aβ-binding alcohol dehydrogenase (ABAD) was identified as an Aβ-binding protein in a yeast two-hybrid screen [88]. ABAD is localized to the mitochondrial matrix and has an essential physiological role in mitochondria (Fig. 2). ABAD-Aβ complexes were detected in AD brain and in Tg mutantAPP/ABAD (Tg mAPP/ABAD) mice. Cortical neurons cultured from Tg mAPP/ABAD mice show increased production of ROS and decreased mitochondrial membrane potential, ATP levels and activity of respiratory chain complex IV. Consistently, these neurons displayed DNA-fragmentation and caspase-3 activity resulting in cell death by day 5–6 in culture [92]. ABAD uses NAD+ and/or NADH as its cofactor and catalyses the reversible oxidation and/or reduction of alcohol group in its substrates [93]. The crystal structure of ABAD-Aβ complexes has been determined showing that the NAD+ binding pocket is distorted, hindering NAD+ from binding to ABAD in the presence of Aβ[88, 93]. Thus Aβ blocks ABAD activity causing mitochondrial dysfunction and ultimately cell death. Two stretches of ABAD residues in the LD loop region (amino acids 95–113) have been shown to be important for Aβ binding. Cell permeable peptides (amino acids 92–120, ABAD-DP) protected cells from cytochrome c release, DNA fragmentation and attenuated cell death in neuronal cultures. ABAD-DP also blocked the production of ROS both in cultured neurons and in mouse brain tissue. Small-molecule inhibitors of the ABAD-Aβ interaction belonging to a class of benzothiazole ureas have been identified [94] and ABAD emerges as a new drug target for AD. Aβ has also been shown to specifically interact with cyclophilin D (CypD), a mitochondrial matrix protein that associates with the inner membrane during opening of the mPTP [95]. Cortical mitochondria from CypD-deficient mice are resistant to Aβ- and calcium-induced mitochondrial swelling and permeability transition. Moreover, Tg mAPP/CypD-null mice had improved learning and memory and synaptic function both in 12- and 24-month-old animals [95, 96].
Recently, we have identified an additional Aβ target in mitochondria, namely a novel zinc-metalloendopeptidase called PresequenceProtease, PreP that has been shown to be the protease responsible for degradation of Aβ in mitochondria [97]. PreP was originally found and characterized in Arabidopsis thaliana[98] as a protease degrading targeting peptides that are cleaved off in mitochondria after completed protein import as well as other unstructured peptides up to 65 amino acid residues in length, but not small proteins [99, 100]. Crystal structure of AtPreP has been solved and refined at 2.1Å[101]. The structure revealed a unique totally enclosed large peptidasome cavity and a novel catalytic mechanism involving hinge bending motions of the enzyme in response to peptide binding.
A human homologue of PreP, hPreP, originally identified as hMP1 [102] is a 1037 amino acids protein (AAH05025) that belongs to M16C pitrilysin oligopeptidase family. Its gene is located on chromosome 10. The protein has a predicted mitochondrial targeting peptide of 29 amino acids and its mitochondrial localization has been confirmed by proteomic studies of human mitochondria [103]. We have showed that the recombinant hPreP completely degraded both Aβ40 and Aβ42 as well as Aβ Arctic protein (42, E22G) at unique cleavage sites including several sites in a very hydrophobic C-terminal Aβ29–42 segment that is prone to aggregation. Interestingly, PreP is an organellar functional analogue of the human insulin degrading enzyme (IDE), implicated in AD as it cleaves Aβ before insoluble amyloid fibres are formed [104–106]. IDE is also located on chromosome 10 (10q) and genetic association between single nucleotide polymorphisms (SNPs) in IDE and late onset of AD has been reported [107]. In contrast to IDE, hPreP does not cleave insulin despite the fact that the overall 3D structure of IDE is highly similar to PreP [108]. However, there exist differences that may explain varying cleavage specificity [109]. 3D structural model of hPreP generated from the 3D structure of AtPreP [97, 101] revealed the presence of two cysteine residues in close proximity to each other. These residues form a disulphide bridge under oxidizing conditions locking the enzyme in a closed conformation and hindering substrate binding. This finding might be of physiological importance as it implies a possible inhibition of hPreP under conditions of elevated ROS production in mitochondria. An increased ROS production has been reported upon accumulation of Aβ in mitochondria [88]. Therefore, the degradation of the mitochondrial Aβ by hPreP may potentially be of importance in the pathology of AD. Furthermore, we make the assumption that hPreP may prevent Aβ–ABAD and Aβ–CypD interactions by clearance of the Aβ peptides that may provide a new strategy against AD.
One important question is how Aβ can reach the mitochondrial surface. Aβ is generated in the lumen of the endoplasmatic reticulum/intermediate compartment, trans-Golgi network and endosomal/lysosomal pathway as well as released from the plasma membrane [110]. Intracellular Aβ42 has been shown to accumulate in intracellular multivesicular bodies [111] and it is possible that Aβ leaking from these vesicles could reach the mitochondria. Moreover, in our confocal microscopy analysis fluorescent Aβ1–40 applied extracellulary is taken up by the cells and later partly localized to mitochondria [90]. Accordingly, Saavedra et al. have recently shown that Aβ1–42 is internalized by primary neurons in the absence of apolipoprotein E [112]. These data suggest that secreted Aβ can be re-internalized into cells either itself or through some kind of vesicular transport and come in contact with mitochondria. These mechanisms require further investigation.
Conclusions
Mitochondria have a central role in AD pathogenesis and maintenance of mitochondrial functions arises as a novel approach for AD treatment. As we have discussed here, both APP and Aβ accumulate in AD mitochondria leading to impairment of mitochondrial functions and cell toxicity. We have recently shown that Aβ is imported into mitochondria through the TOM complex and that PreP can degrade Aβ inside mitochondria. Other studies have shown that Aβ causes toxicity inside mitochondria by interactions with, e.g. ABAD or CypD or by inhibiting complex IV activity in the respiratory chain. Accumulation of APP in the TOM complex prevents import of other proteins and thus it impairs both mitochondrial and cellular functions. Avoiding APP targeting to mitochondria or clearance of the import pore by, e.g. HtrA2/Omi would abolish such toxicity. Taken all together, prevention of mitochondrial Aβ and APP uptake, enhancing Aβ degradation while inside mitochondria and prevention of Aβ-protein interactions are all worthwhile novel strategies for AD-drug development.
Acknowledgments
This work was supported by Dainippon Sumitomo Pharma Co., Ltd. (Osaka, Japan), Gamla Tjänarinnor Foundation, Gun och Bertil Stohnes Foundation, The Foundation for Geriatric Diseases at Karolinska Institutet, Wallenbergs Foundation and the Swedish Research Council (to EG).
References
- 1.Selkoe DJ. Translating cell biology into therapeutic advances in Alzheimer’s disease. Nature. 1999;399:A23–31. doi: 10.1038/399a023. [DOI] [PubMed] [Google Scholar]
- 2.Harman D. Alzheimer’s disease pathogenesis: role of aging. Ann N Y Acad Sci. 2006;1067:454–60. doi: 10.1196/annals.1354.065. [DOI] [PubMed] [Google Scholar]
- 3.Campion D, Dumanchin C, Hannequin D, et al. Early-onset autosomal dominant Alzheimer disease: prevalence, genetic heterogeneity, and mutation spectrum. Am J Hum Genet. 1999;65:664–70. doi: 10.1086/302553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Katzman R, Saitoh T. Advances in Alzheimer’s disease. FASEB J. 1991;5:278–86. [PubMed] [Google Scholar]
- 5.Katzman R, Jackson JE. Alzheimer disease: basic and clinical advances. J Am Geriatr Soc. 1991;39:516–25. doi: 10.1111/j.1532-5415.1991.tb02500.x. [DOI] [PubMed] [Google Scholar]
- 6.Drewes G. Marking tau for tangles and toxicity. Trends Biochem Sci. 2004;29:548–55. doi: 10.1016/j.tibs.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 7.Bretteville A, Planel E. Tau aggregates: toxic, inert, or protective species. J Alzheimers Dis. 2008;14:431–6. doi: 10.3233/jad-2008-14411. [DOI] [PubMed] [Google Scholar]
- 8.Congdon EE, Duff KE. Is tau aggregation toxic or protective. J Alzheimers Dis. 2008;14:453–7. doi: 10.3233/jad-2008-14415. [DOI] [PubMed] [Google Scholar]
- 9.Kang J, Lemaire HG, Unterbeck A, et al. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987;325:733–6. doi: 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- 10.Sisodia SS, St George-Hyslop PH. Gamma-Secretase, Notch, Abeta and Alzheimer’s disease: where do the presenilins fit in. Nat Rev Neurosci. 2002;3:281–90. doi: 10.1038/nrn785. [DOI] [PubMed] [Google Scholar]
- 11.Mudher A, Lovestone S. Alzheimer’s disease-do tauists and baptists finally shake hands. Trends Neurosci. 2002;25:22–6. doi: 10.1016/s0166-2236(00)02031-2. [DOI] [PubMed] [Google Scholar]
- 12.Crentsil V. The pharmacogenomics of Alzheimer’s disease. Ageing Res Rev. 2004;3:153–69. doi: 10.1016/j.arr.2003.07.003. [DOI] [PubMed] [Google Scholar]
- 13.Fukui H, Moraes CT. The mitochondrial impairment, oxidative stress and neurodegeneration connection: reality or just an attractive hypothesis. Trends Neurosci. 2008;31:251–6. doi: 10.1016/j.tins.2008.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 15.Barja G. Free radicals and aging. Trends Neurosci. 2004;27:595–600. doi: 10.1016/j.tins.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 16.Mitchell P, Moyle J. Chemiosmotic hypothesis of oxidative phosphorylation. Nature. 1967;213:137–9. doi: 10.1038/213137a0. [DOI] [PubMed] [Google Scholar]
- 17.St-Pierre J, Buckingham JA, Roebuck SJ, et al. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J Biol Chem. 2002;277:44784–90. doi: 10.1074/jbc.M207217200. [DOI] [PubMed] [Google Scholar]
- 18.Tahara EB, Navarete FD, Kowaltowski AJ. Tissue-, substrate-, and site-specific characteristics of mitochondrial reactive oxygen species generation. Free Radic Biol Med. 2009;46:1283–97. doi: 10.1016/j.freeradbiomed.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 19.Harman D. The biologic clock: the mitochondria. J Am Geriatr Soc. 1972;20:145–7. doi: 10.1111/j.1532-5415.1972.tb00787.x. [DOI] [PubMed] [Google Scholar]
- 20.Fleming JE, Miquel J, Cottrell SF, et al. Is cell aging caused by respiration-dependent injury to the mitochondrial genome. Gerontology. 1982;28:44–53. doi: 10.1159/000212510. [DOI] [PubMed] [Google Scholar]
- 21.Corral-Debrinski M, Horton T, Lott MT, et al. Mitochondrial DNA deletions in human brain: regional variability and increase with advanced age. Nat Genet. 1992;2:324–9. doi: 10.1038/ng1292-324. [DOI] [PubMed] [Google Scholar]
- 22.Khaidakov M, Heflich RH, Manjanatha MG, et al. Accumulation of point mutations in mitochondrial DNA of aging mice. Mutat Res. 2003;526:1–7. doi: 10.1016/s0027-5107(03)00010-1. [DOI] [PubMed] [Google Scholar]
- 23.Wang Y, Michikawa Y, Mallidis C, et al. Muscle-specific mutations accumulate with aging in critical human mtDNA control sites for replication. Proc Natl Acad Sci USA. 2001;98:4022–7. doi: 10.1073/pnas.061013598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alexeyev MF, Ledoux SP, Wilson GL. Mitochondrial DNA and aging. Clin Sci. 2004;107:355–64. doi: 10.1042/CS20040148. [DOI] [PubMed] [Google Scholar]
- 25.Trifunovic A, Wredenberg A, Falkenberg M, et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–23. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- 26.Trifunovic A, Hansson A, Wredenberg A, et al. Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. Proc Natl Acad Sci USA. 2005;102:17993–8. doi: 10.1073/pnas.0508886102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vermulst M, Bielas JH, Kujoth GC, et al. Mitochondrial point mutations do not limit the natural lifespan of mice. Nat Genet. 2007;39:540–3. doi: 10.1038/ng1988. [DOI] [PubMed] [Google Scholar]
- 28.Vermulst M, Wanagat J, Kujoth GC, et al. DNA deletions and clonal mutations drive premature aging in mitochondrial mutator mice. Nat Genet. 2008;40:392–4. doi: 10.1038/ng.95. [DOI] [PubMed] [Google Scholar]
- 29.Schriner SE, Linford NJ, Martin GM, et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. 2005;308:1909–11. doi: 10.1126/science.1106653. [DOI] [PubMed] [Google Scholar]
- 30.Huang JH, Hood DA. Age-associated mitochondrial dysfunction in skeletal muscle: Contributing factors and suggestions for long-term interventions. IUBMB Life. 2009;61:201–14. doi: 10.1002/iub.164. [DOI] [PubMed] [Google Scholar]
- 31.Leuner K, Hauptmann S, Abdel-Kader R, et al. Mitochondrial dysfunction: the first domino in brain aging and Alzheimer’s disease. Antioxid Redox Signal. 2007;9:1659–75. doi: 10.1089/ars.2007.1763. [DOI] [PubMed] [Google Scholar]
- 32.Benzi G, Pastoris O, Marzatico F, et al. The mitochondrial electron transfer alteration as a factor involved in the brain aging. Neurobiol Aging. 1992;13:361–8. doi: 10.1016/0197-4580(92)90109-b. [DOI] [PubMed] [Google Scholar]
- 33.Navarro A, Boveris A. Rat brain and liver mitochondria develop oxidative stress and lose enzymatic activities on aging. Am J Physiol Regul Integr Comp Physiol. 2004;287:R1244–9. doi: 10.1152/ajpregu.00226.2004. [DOI] [PubMed] [Google Scholar]
- 34.Lenaz G, Bovina C, Castelluccio C, et al. Mitochondrial complex I defects in aging. Mol Cell Biochem. 1997;174:329–33. [PubMed] [Google Scholar]
- 35.Martinez M, Ferrandiz ML, De Juan E, et al. Age-related changes in glutathione and lipid peroxide content in mouse synaptic mitochondria: relationship to cytochrome c oxidase decline. Neurosci Lett. 1994;170:121–4. doi: 10.1016/0304-3940(94)90254-2. [DOI] [PubMed] [Google Scholar]
- 36.Navarro A, Boveris A. Brain mitochondrial dysfunction in aging: conditions that improve survival, neurological performance and mitochondrial function. Front Biosci. 2007;12:1154–63. doi: 10.2741/2133. [DOI] [PubMed] [Google Scholar]
- 37.Manczak M, Jung Y, Park BS, et al. Time-course of mitochondrial gene expressions in mice brains: implications for mitochondrial dysfunction, oxidative damage, and cytochrome c in aging. J Neurochem. 2005;92:494–504. doi: 10.1111/j.1471-4159.2004.02884.x. [DOI] [PubMed] [Google Scholar]
- 38.Yan LJ, Sohal RS. Mitochondrial adenine nucleotide translocase is modified oxidatively during aging. Proc Natl Acad Sci USA. 1998;95:12896–901. doi: 10.1073/pnas.95.22.12896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Navarro A, Gomez C, Sanchez-Pino MJ, et al. Vitamin E at high doses improves survival, neurological performance, and brain mitochondrial function in aging male mice. Am J Physiol Regul Integr Comp Physiol. 2005;289:R1392–9. doi: 10.1152/ajpregu.00834.2004. [DOI] [PubMed] [Google Scholar]
- 40.Liu J, Killilea DW, Ames BN. Age-associated mitochondrial oxidative decay: improvement of carnitine acetyltransferase substrate-binding affinity and activity in brain by feeding old rats acetyl-L- carnitine and/or R-alpha -lipoic acid. Proc Natl Acad Sci USA. 2002;99:1876–81. doi: 10.1073/pnas.261709098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sastre J, Millan A, Garcia de la Asuncion J, et al. A Ginkgo biloba extract (EGb 761) prevents mitochondrial aging by protecting against oxidative stress. Free Radic Biol Med. 1998;24:298–304. doi: 10.1016/s0891-5849(97)00228-1. [DOI] [PubMed] [Google Scholar]
- 42.Terman A, Gustafsson B, Brunk UT. The lysosomal-mitochondrial axis theory of postmitotic aging and cell death. Chem Biol Interact. 2006;163:29–37. doi: 10.1016/j.cbi.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 43.Giannakopoulos P, Hof PR, Michel JP, et al. Cerebral cortex pathology in aging and Alzheimer’s disease: a quantitative survey of large hospital-based geriatric and psychiatric cohorts. Brain Res Brain Res Rev. 1997;25:217–45. doi: 10.1016/s0165-0173(97)00023-4. [DOI] [PubMed] [Google Scholar]
- 44.Onyango I, Khan S, Miller B, et al. Mitochondrial genomic contribution to mitochondrial dysfunction in Alzheimer’s disease. J Alzheimers Dis. 2006;9:183–93. doi: 10.3233/jad-2006-9210. [DOI] [PubMed] [Google Scholar]
- 45.Reddy PH, McWeeney S, Park BS, et al. Gene expression profiles of transcripts in amyloid precursor protein transgenic mice: up-regulation of mitochondrial metabolism and apoptotic genes is an early cellular change in Alzheimer’s disease. Hum Mol Genet. 2004;13:1225–40. doi: 10.1093/hmg/ddh140. [DOI] [PubMed] [Google Scholar]
- 46.Gibson GE, Haroutunian V, Zhang H, et al. Mitochondrial damage in Alzheimer’s disease varies with apolipoprotein E genotype. Ann Neurol. 2000;48:297–303. [PubMed] [Google Scholar]
- 47.Bubber P, Haroutunian V, Fisch G, et al. Mitochondrial abnormalities in Alzheimer brain: mechanistic implications. Ann Neurol. 2005;57:695–703. doi: 10.1002/ana.20474. [DOI] [PubMed] [Google Scholar]
- 48.Sorbi S, Bird ED, Blass JP. Decreased pyruvate dehydrogenase complex activity in Huntington and Alzheimer brain. Ann Neurol. 1983;13:72–8. doi: 10.1002/ana.410130116. [DOI] [PubMed] [Google Scholar]
- 49.Kish SJ, Bergeron C, Rajput A, et al. Brain cytochrome oxidase in Alzheimer’s disease. J Neurochem. 1992;59:776–9. doi: 10.1111/j.1471-4159.1992.tb09439.x. [DOI] [PubMed] [Google Scholar]
- 50.Cardoso SM, Proenca MT, Santos S, et al. Cytochrome c oxidase is decreased in Alzheimer’s disease platelets. Neurobiol Aging. 2004;25:105–10. doi: 10.1016/s0197-4580(03)00033-2. [DOI] [PubMed] [Google Scholar]
- 51.Casley CS, Canevari L, Land JM, et al. Beta-amyloid inhibits integrated mitochondrial respiration and key enzyme activities. J Neurochem. 2002;80:91–100. doi: 10.1046/j.0022-3042.2001.00681.x. [DOI] [PubMed] [Google Scholar]
- 52.Crouch PJ, Blake R, Duce JA, et al. Copper-dependent inhibition of human cytochrome c oxidase by a dimeric conformer of amyloid-beta1–42. J Neurosci. 2005;25:672–9. doi: 10.1523/JNEUROSCI.4276-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Devi L, Prabhu BM, Galati DF, et al. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J Neurosci. 2006;26:9057–68. doi: 10.1523/JNEUROSCI.1469-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Anandatheerthavarada HK, Biswas G, Robin MA, et al. Mitochondrial targeting and a novel transmembrane arrest of Alzheimer’s amyloid precursor protein impairs mitochondrial function in neuronal cells. J Cell Biol. 2003;161:41–54. doi: 10.1083/jcb.200207030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schulke N, Sepuri NB, Pain D. In vivo zippering of inner and outer mitochondrial membranes by a stable translocation intermediate. Proc Natl Acad Sci USA. 1997;94:7314–9. doi: 10.1073/pnas.94.14.7314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schulke N, Sepuri NB, Gordon DM, et al. A multisubunit complex of outer and inner mitochondrial membrane protein translocases stabilized in vivo by translocation intermediates. J Biol Chem. 1999;274:22847–54. doi: 10.1074/jbc.274.32.22847. [DOI] [PubMed] [Google Scholar]
- 57.Hauptmann S, Scherping I, Drose S, et al. Mitochondrial dysfunction: An early event in Alzheimer pathology accumulates with age in AD transgenic mice. Neurobiol Aging. 2009;30:1574–86. doi: 10.1016/j.neurobiolaging.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 58.Boopathi E, Srinivasan S, Fang JK, et al. Bimodal protein targeting through activation of cryptic mitochondrial targeting signals by an inducible cytosolic endoprotease. Mol Cell. 2008;32:32–42. doi: 10.1016/j.molcel.2008.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zha J, Weiler S, Oh KJ, et al. Posttranslational N-myristoylation of BID as a molecular switch for targeting mitochondria and apoptosis. Science. 2000;290:1761–5. doi: 10.1126/science.290.5497.1761. [DOI] [PubMed] [Google Scholar]
- 60.Zha J, Harada H, Yang E, et al. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14–3-3 not BCL-X(L) Cell. 1996;87:619–28. doi: 10.1016/s0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]
- 61.Anandatheerthavarada HK, Biswas G, Mullick J, et al. Dual targeting of cytochrome P4502B1 to endoplasmic reticulum and mitochondria involves a novel signal activation by cyclic AMP-dependent phosphorylation at ser128. EMBO J. 1999;18:5494–504. doi: 10.1093/emboj/18.20.5494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Robin MA, Anandatheerthavarada HK, Biswas G, et al. Bimodal targeting of microsomal CYP2E1 to mitochondria through activation of an N-terminal chimeric signal by cAMP-mediated phosphorylation. J Biol Chem. 2002;277:40583–93. doi: 10.1074/jbc.M203292200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Anandatheerthavarada HK, Sepuri NB, Biswas G, et al. An unusual TOM20/ TOM22 bypass mechanism for the mitochondrial targeting of cytochrome P450 proteins containing N-terminal chimeric signals. J Biol Chem. 2008;283:19769–80. doi: 10.1074/jbc.M801464200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 64.Suzuki M, Youle RJ, Tjandra N. Structure of Bax: coregulation of dimer formation and intracellular localization. Cell. 2000;103:645–54. doi: 10.1016/s0092-8674(00)00167-7. [DOI] [PubMed] [Google Scholar]
- 65.Nechushtan A, Smith CL, Hsu YT, et al. Conformation of the Bax C-terminus regulates subcellular location and cell death. EMBO J. 1999;18:2330–41. doi: 10.1093/emboj/18.9.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Narendra D, Tanaka A, Suen DF, et al. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mijaljica D, Prescott M, Devenish RJ. Different fates of mitochondria: alternative ways for degradation. Autophagy. 2007;3:4–9. doi: 10.4161/auto.3011. [DOI] [PubMed] [Google Scholar]
- 68.Escobar-Henriques M, Langer T. Mitochondrial shaping cuts. Biochim Biophys Acta. 2006;1763:422–9. doi: 10.1016/j.bbamcr.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 69.Neutzner A, Youle RJ, Karbowski M. Outer mitochondrial membrane protein degradation by the proteasome. Novartis Found Symp. 2007;287:4–14. ; discussion -20. [PubMed] [Google Scholar]
- 70.Twig G, Elorza A, Molina AJ, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–46. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang X, Su B, Fujioka H, et al. Dynamin-like protein 1 reduction underlies mitochondrial morphology and distribution abnormalities in fibroblasts from sporadic Alzheimer’s disease patients. Am J Pathol. 2008;173:470–82. doi: 10.2353/ajpath.2008.071208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang X, Su B, Siedlak SL, et al. Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc Natl Acad Sci USA. 2008;105:19318–23. doi: 10.1073/pnas.0804871105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Park HJ, Kim SS, Seong YM, et al. Beta-amyloid precursor protein is a direct cleavage target of HtrA2 serine protease. Implications for the physiological function of HtrA2 in the mitochondria. J Biol Chem. 2006;281:34277–87. doi: 10.1074/jbc.M603443200. [DOI] [PubMed] [Google Scholar]
- 74.Huttunen HJ, Guenette SY, Peach C, et al. HtrA2 regulates beta-amyloid precursor protein (APP) metabolism through endoplasmic reticulum-associated degradation. J Biol Chem. 2007;282:28285–95. doi: 10.1074/jbc.M702951200. [DOI] [PubMed] [Google Scholar]
- 75.Vaux DL, Silke J. Mammalian mitochondrial IAP binding proteins. Biochem Biophys Res Commun. 2003;304:499–504. doi: 10.1016/s0006-291x(03)00622-3. [DOI] [PubMed] [Google Scholar]
- 76.Challa M, Malladi S, Pellock BJ, et al. Drosophila Omi, a mitochondrial-localized IAP antagonist and proapoptotic serine protease. EMBO J. 2007;26:3144–56. doi: 10.1038/sj.emboj.7601745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Martins LM, Morrison A, Klupsch K, et al. Neuroprotective role of the Reaper-related serine protease HtrA2/Omi revealed by targeted deletion in mice. Mol Cell Biol. 2004;24:9848–62. doi: 10.1128/MCB.24.22.9848-9862.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jones JM, Datta P, Srinivasula SM, et al. Loss of Omi mitochondrial protease activity causes the neuromuscular disorder of mnd2 mutant mice. Nature. 2003;425:721–7. doi: 10.1038/nature02052. [DOI] [PubMed] [Google Scholar]
- 79.Vande Walle L, Lamkanfi M, Vandenabeele P. The mitochondrial serine protease HtrA2/Omi: an overview. Cell Death Differ. 2008;15:453–60. doi: 10.1038/sj.cdd.4402291. [DOI] [PubMed] [Google Scholar]
- 80.Radke S, Chander H, Schafer P, et al. Mitochondrial protein quality control by the proteasome involves ubiquitination and the protease Omi. J Biol Chem. 2008;283:12681–5. doi: 10.1074/jbc.C800036200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Moisoi N, Klupsch K, Fedele V, et al. Mitochondrial dysfunction triggered by loss of HtrA2 results in the activation of a brain-specific transcriptional stress response. Cell Death Differ. 2009;16:449–64. doi: 10.1038/cdd.2008.166. [DOI] [PubMed] [Google Scholar]
- 82.Plun-Favreau H, Klupsch K, Moisoi N, et al. The mitochondrial protease HtrA2 is regulated by Parkinson’s disease-associated kinase PINK1. Nat Cell Biol. 2007;9:1243–52. doi: 10.1038/ncb1644. [DOI] [PubMed] [Google Scholar]
- 83.Yang L, Sun M, Sun XM, et al. Akt attenuation of the serine protease activity of HtrA2/Omi through phosphorylation of serine 212. J Biol Chem. 2007;282:10981–7. doi: 10.1074/jbc.M700445200. [DOI] [PubMed] [Google Scholar]
- 84.Ma X, Kalakonda S, Srinivasula SM, et al. GRIM-19 associates with the serine protease HtrA2 for promoting cell death. Oncogene. 2007;26:4842–9. doi: 10.1038/sj.onc.1210287. [DOI] [PubMed] [Google Scholar]
- 85.Chao JR, Parganas E, Boyd K, et al. Hax1-mediated processing of HtrA2 by Parl allows survival of lymphocytes and neurons. Nature. 2008;452:98–102. doi: 10.1038/nature06604. [DOI] [PubMed] [Google Scholar]
- 86.Gupta S, Singh R, Datta P, et al. The C-terminal tail of presenilin regulates Omi/HtrA2 protease activity. J Biol Chem. 2004;279:45844–54. doi: 10.1074/jbc.M404940200. [DOI] [PubMed] [Google Scholar]
- 87.Caspersen C, Wang N, Yao J, et al. Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J. 2005;19:2040–1. doi: 10.1096/fj.05-3735fje. [DOI] [PubMed] [Google Scholar]
- 88.Lustbader JW, Cirilli M, Lin C, et al. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease. Science. 2004;304:448–52. doi: 10.1126/science.1091230. [DOI] [PubMed] [Google Scholar]
- 89.Manczak M, Anekonda TS, Henson E, et al. Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet. 2006;15:1437–49. doi: 10.1093/hmg/ddl066. [DOI] [PubMed] [Google Scholar]
- 90.Hansson Petersen CA, Alikhani N, Behbahani H, et al. The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc Natl Acad Sci USA. 2008;105:13145–50. doi: 10.1073/pnas.0806192105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hansson CA, Frykman S, Farmery MR, et al. Nicastrin, presenilin, APH-1, and PEN-2 form active gamma-secretase complexes in mitochondria. J Biol Chem. 2004;279:51654–60. doi: 10.1074/jbc.M404500200. [DOI] [PubMed] [Google Scholar]
- 92.Takuma K, Yao J, Huang J, et al. ABAD enhances Abeta-induced cell stress via mitochondrial dysfunction. FASEB J. 2005;19:597–8. doi: 10.1096/fj.04-2582fje. [DOI] [PubMed] [Google Scholar]
- 93.Yan Y, Liu Y, Sorci M, et al. Surface plasmon resonance and nuclear magnetic resonance studies of ABAD-Abeta interaction. Biochemistry. 2007;46:1724–31. doi: 10.1021/bi061314n. [DOI] [PubMed] [Google Scholar]
- 94.Xie Y, Deng S, Chen Z, et al. Identification of small-molecule inhibitors of the Abeta-ABAD interaction. Bioorg Med Chem Lett. 2006;16:4657–60. doi: 10.1016/j.bmcl.2006.05.099. [DOI] [PubMed] [Google Scholar]
- 95.Du H, Guo L, Fang F, et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat Med. 2008;14:1097–105. doi: 10.1038/nm.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Du H, Guo L, Zhang W, et al. Cyclophilin D deficiency improves mitochondrial function and learning/memory in aging Alzheimer disease mouse model. Neurobiol Aging. doi: 10.1016/j.neurobiolaging.2009.03.003. doi: 10.1016/jneurobiologing.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Falkevall A, Alikhani N, Bhushan S, et al. Degradation of the amyloid beta-protein by the novel mitochondrial peptidasome, PreP. J Biol Chem. 2006;281:29096–104. doi: 10.1074/jbc.M602532200. [DOI] [PubMed] [Google Scholar]
- 98.Stahl A, Moberg P, Ytterberg J, et al. Isolation and identification of a novel mitochondrial metalloprotease (PreP) that degrades targeting presequences in plants. J Biol Chem. 2002;277:41931–9. doi: 10.1074/jbc.M205500200. [DOI] [PubMed] [Google Scholar]
- 99.Moberg P, Stahl A, Bhushan S, et al. Characterization of a novel zinc metalloprotease involved in degrading targeting peptides in mitochondria and chloroplasts. Plant J. 2003;36:616–28. doi: 10.1046/j.1365-313x.2003.01904.x. [DOI] [PubMed] [Google Scholar]
- 100.Stahl A, Nilsson S, Lundberg P, et al. Two novel targeting peptide degrading proteases, PrePs, in mitochondria and chloroplasts, so similar and still different. J Mol Biol. 2005;349:847–60. doi: 10.1016/j.jmb.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 101.Johnson KA, Bhushan S, Stahl A, et al. The closed structure of presequence protease PreP forms a unique 10,000 Angstroms3 chamber for proteolysis. EMBO J. 2006;25:1977–86. doi: 10.1038/sj.emboj.7601080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mzhavia N, Berman YL, Qian Y, et al. Cloning, expression, and characterization of human metalloprotease 1: a novel member of the pitrilysin family of metalloendoproteases. DNA Cell Biol. 1999;18:369–80. doi: 10.1089/104454999315268. [DOI] [PubMed] [Google Scholar]
- 103.Taylor SW, Fahy E, Zhang B, et al. Characterization of the human heart mitochondrial proteome. Nat Biotechnol. 2003;21:281–6. doi: 10.1038/nbt793. [DOI] [PubMed] [Google Scholar]
- 104.Kurochkin IV. Insulin-degrading enzyme: embarking on amyloid destruction. Trends Biochem Sci. 2001;26:421–5. doi: 10.1016/s0968-0004(01)01876-x. [DOI] [PubMed] [Google Scholar]
- 105.Selkoe DJ. Clearing the brain’s amyloid cobwebs. Neuron. 2001;32:177–80. doi: 10.1016/s0896-6273(01)00475-5. [DOI] [PubMed] [Google Scholar]
- 106.Tanzi RE, Moir RD, Wagner SL. Clearance of Alzheimer’s Abeta peptide: the many roads to perdition. Neuron. 2004;43:605–8. doi: 10.1016/j.neuron.2004.08.024. [DOI] [PubMed] [Google Scholar]
- 107.Bertram L, Blacker D, Mullin K, et al. Evidence for genetic linkage of Alzheimer’s disease to chromosome 10q. Science. 2000;290:2302–3. doi: 10.1126/science.290.5500.2302. [DOI] [PubMed] [Google Scholar]
- 108.Shen Y, Joachimiak A, Rosner MR, et al. Structures of human insulin-degrading enzyme reveal a new substrate recognition mechanism. Nature. 2006;443:870–4. doi: 10.1038/nature05143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Im H, Manolopoulou M, Malito E, et al. Structure of substrate-free human insulin-degrading enzyme (IDE) and biophysical analysis of ATP-induced conformational switch of IDE. J Biol Chem. 2007;282:25453–63. doi: 10.1074/jbc.M701590200. [DOI] [PubMed] [Google Scholar]
- 110.Gouras GK, Almeida CG, Takahashi RH. Intraneuronal Abeta accumulation and origin of plaques in Alzheimer’s disease. Neurobiol Aging. 2005;26:1235–44. doi: 10.1016/j.neurobiolaging.2005.05.022. [DOI] [PubMed] [Google Scholar]
- 111.Gouras GK, Tsai J, Naslund J, et al. Intraneuronal Abeta42 accumulation in human brain. Am J Pathol. 2000;156:15–20. doi: 10.1016/s0002-9440(10)64700-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Saavedra L, Mohamed A, Ma V, et al. Internalization of beta-amyloid peptide by primary neurons in the absence of apolipoprotein E. J Biol Chem. 2007;282:35722–32. doi: 10.1074/jbc.M701823200. [DOI] [PubMed] [Google Scholar]