

Abstract
SIRT1, a nicotinamide adenine dinucleotide (NAD+)-dependent histone/protein deacetylase, has been extensively studied recently for its critical role in the regulation of physiology, calorie restriction and aging. Studies on laboratory mice showed that expression of SIRT1 can be induced by starvation in a p53-dependent manner and requires the p53-binding sites present in the Sirt1 promoter. However, it remains to be determined whether these findings based on rodents apply to human beings. In this paper, we characterized a putative p53-binding element in the human SIRT1 promoter that might be required for the up-regulation of SIRT1 in response to nutritional stress. The p53-binding site in the promoter of human SIRT1 is more deviant from the consensus sequence than the corresponding sequence in the mouse Sirt1. There is a C to A change at the second half site in human SIRT1, thus disrupting the core-binding element CWWG in the canonical RRRCWWGYYY. To test whether such sequence change would affect its binding with p53 and the SIRT1 expression under stress, we studied various human cell lines with different p53 status and cells with ectopic expression of functionally distinct p53. We found that serum withdrawal also up-regulates human SIRT1 gene expression in a p53-dependent manner and that the p53-binding element in SIRT1 is required for the up-regulation. Thus, the mechanism responsible for the regulation of SIRT1 expression by p53 is conserved between mice and human beings.
Keywords: SIRT1, nutritional stress, p53, gene expression, promoter
Introduction
The silent information regulator 2 (Sir2) family of genes encodes a group of nicotinamide adenine dinucleotide (NAD+)-dependent histone/protein deacetylases (sirtuins) that are ubiquitously distributed [1, 2]. Sir2, the founding member of the family, was first characterized in yeast, and is a heterostructural component of silent chromatin that is required for transcriptional silencing of the silent mating-type loci, telomeres and rDNA repeats [3–5]. Besides gene silencing, Sir2 proteins were also implicated in the regulation of many biological processes such as cell cycle regulation [6], fatty acid metabolism [7] and lifespan extension [5, 8, 9]. Overexpression of Sir2 extends the lifespan of budding yeast, while its knockout shortens the lifespan by about 50%[8, 10]. A similar paradigm applies to C. elegans in which the dosage of Sir2.1 gene is positively correlated to lifespan [9]. SIRT1, the closest mammalian homologue of the yeast Sir2, has been extensively studied recently [11]. It regulates cell cycle progression, apoptosis, and other metabolic processes by interacting with a number of molecules, including the forkhead transcription factor, the tumour suppressor p53, DNA repair protein Ku70 and PPAR-γ[12–17]. It was found that overexpression of SIRT1 can elicit beneficial phenotypes resembling calorie restriction [18]. Nutrient withdrawal concomitantly augments the expression of the mouse Sirt1 and activates the forkhead transcription factor Foxo3a in rodent cells [19]. Moreover, starvation-induced increase in Sirt1 expression requires Foxo3a, and stimulation of Sirt1 expression by Foxo3a was mediated through physical interaction between Foxo3a and p53, which binds to two p53-binding sites present in the mouse Sirt1 promoter [19].
The p53 plays a pivotal role in the maintenance of cellular homeostasis. It binds to DNA in a sequence-specific fashion. The consensus DNA-binding sequence for p53 consists of two decameric motifs or half-sites of the general form RRRCWWGYYY(R = A, G; W = A, T; Y = C, T) separated by 0–13 base pairs [20]. Upon binding to DNA targets containing two half-site motifs, p53 functions as tetramers to activate or repress transactivation [21].
A sequence analysis indicated that the p53-binding sites in the promoter of human SIRT1 is more deviant from the consensus sequence than those in the mouse Sirt1. There is a C to A change at the second half site in human SIRT1. With the concern that such a deviation might affect the binding of the DNA sequence to p53, and consequently the transcriptional regulation of SIRT1 by p53 under stress, we determined whether nutritional withdrawal would up-regulate human SIRT1 expression in a p53-dependent manner as in the mouse. Using various human cell lines with different p53 status, we were able to confirm that serum withdrawal also up-regulates human SIRT1 gene expression in a p53-dependent manner.
Materials and methods
Chemicals and reagents
Anti-p53(DO-1)sc-126, anti-SIRT1(B-7) sc-74465 and anti-β-actin(AC-15)sc-69879 were all obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Lipofectamine 2000 and RNA isolation reagent TRIZOL were from Invitrogen (Carlsbad, CA, USA). Dual luciferase reporter assay kit and AMV Reverse Transcriptase were from Promega (Madison, WI, USA). Dulbecco’s modified Eagle’s medium, McCoy’5A medium, RPMI1640 and foetal bovine serum were obtained from Gibco (Carlsbad, CA, USA). All restriction enzymes and T4 DNA ligase were from New England Biolabs (Beverly, MA, USA). Electrophoretic mobility shift assay was performed with the Roche (Basel, Switzerland) DIG Gel shift kit, second generation. Other chemicals were from Sigma (St. Louis, MO, USA) unless otherwise noted.
Cell culture
The human osteosarcoma U2OS and Saos-2 cells were cultured in McCoy’5A medium supplemented with 15% foetal bovine serum and 100 U/ml penicillin-streptomycin. The human hepatoma cell lines HepG2 and Hep3B cells as well as HeLa cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% foetal bovine serum and antibiotics. HEK293 cells were grown in RPMI1640 contained 10% foetal bovine serum. Cells were incubated in the humidified incubator equilibrated with 5% CO2 at 37°C and passed using standard cell culture techniques.
RNA isolation and real-time PCR analysis
Total RNA was extracted using TRIzol reagents per the protocol provided by the supplier (Invitrogen). The first-strand cDNA was synthesized from 3 μg of total RNA by AMV reverse transcriptase (Promega). SIRT1 mRNAs were measured by TaqMan PCR assay in a 10-μl reaction volume on 96-well plates using the Applied Biosystems 7500 Real Time PCR System. The primers and TaqMan probes were designed with Primer Express 2.0 software (Applied Biosystems, Foster City, CA, USA). Transcript levels were normalized to GAPDH levels. The sequence of the primers were: human SIRT1 sense: 5′-AGAGCCTCACATGCAAGCTCTAG-3′ and antisense: 5′-GCCAATCATAAGATGTTGCTGAAC-3′, probe: 5′-(FAM)ACTGGACTCCAAGGCCACGGATAGGT(TAMRA) -3′ and human GAPDH sense: 5′-CCAGGTTGGTCTCCTCTGACTT-3 and antisense: 5′-GTTGCTGTAGCCAAATTCGTTGT-3′; probe: 5′-(FAM)AACAGCGACACCCACTCCTCCACC (TAMRA)-3′. Cycle conditions for PCRs were at 50°C for 2 min., at 95°C for 10 min. and 40 cycles each at 95°C for 15 sec. and 60°C for 1 min.
Western blot analysis
Cells were harvested 16 hrs after serum starvation, and lysed with passive lysis buffer (Promega). The protein concentration was determined by BCA protein assay method (Pierce, Rockford, IL, USA) using bovine serum albumin as a standard. Samples containing equal amounts of protein (30 μg) were subjected to 10% SDS-PAGE and transferred to a polyvinylidene difluoride membrane (0.2 μm; GE Healthcare, Little Chalfont, Buckinghamshire, UK). After blocking with 5% skim milk, blots were probed with anti-p53 antibody, anti-SIRT1 antibody or anti-β-actin (Santa Cruz Biotechnology at 1:500 dilution in 1% skimmed milk in TBS-T) at 4°C overnight. After washing with TBS-T, the membranes were treated with horseradish peroxidase-conjugated secondary antibody (1:10,000) for 1 hr and visualized by ECL PLUS (GE Healthcare) as specified by the manufacturer.
Generation of the SIRT1 reporter constructs
The human SIRT1 fragments with or without the putative p53 response elements were generated by PCR using a common reverse primer R-54 (5′- CCCAAGCTTTCTTCCAACTGCCTCTCTG-3′) and the following sense primers that are flanked by Xho I and Hind III restriction sites (restriction sites are underlined): for construct P-158 which contains p53 response elements: 5′-CCGCTCGAGAGACGCAACAGCCTCCGCCC-3′; for construct P-111 (without p53 response elements): 5′-CCGCTCGAGGGCCCGCGTGGGTGGCGGGAG-3′; for construct P-158mut which contains two point mutations in the core sequence of p53 response element): 5′-AGACGCAACAGCCTCCGCCCGCCAAGAGACCCGTAGTG-3′. PCRs were performed with the following cycle conditions: one cycle 94°C for 4 min.; 35 cycles 94°C for 40 sec., 58°C for 40 sec., 72°C for 60 sec.; one cycle 72°C for 10 min. The amplicon was gel-purified and subcloned into the promoterless luciferase reporter plasmid pGL3-Basic (Promega). The sequences of all constructs were confirmed by restriction digestion and direct sequencing. The mouse Sirt1 fragment was as described [19], except that it was named P-163 instead of P-202.
Transient transfection and luciferase assay
Transient transfections were performed with Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer’s instructions. Briefly, cells were seeded in culture medium without antibiotics at a density of 6 × 104 cells/well in a 24-well plate for 24 hrs before transfection. For each well, 0.2 μg of the SIRT1 reporter constructs (deletion and mutated constructs) were cotransfected with 0.02 μg of pRL-TK vector that provides constitutive expression of Renilla luciferase serving as an internal control to normalize transfection efficiencies. Sixteen hours after transfection, cells were harvested, and luciferase activity was measured using Perkin-Elmer 1420 multilabel counter (Waltham, MA, USA). All transfection experiments were carried out in triplicate.
Electrophoretic mobility shift assay (EMSA)
Nuclear extract was prepared from U2OS cells as described previously [22]. The oligonucleotides corresponding to the putative p53-binding site found in the human SIRT1 promoter and its complementary strand were annealed and end-labelled with digoxin using T4 polynucleotide kinase. The sequence of wild-type and mutant p53 response element (sense strand) are 5′-CCACGTGACCCGTAGTGTTGT-3′ and 5′-CCAAGAGACCCGTAGTGTTGT-3′, respectively. EMSA was performed according to the manufacturer’s instructions. Briefly, 2 μl of 0.4 ng/μl labelled probes were incubated with or without nuclear extract in gel shift binding buffer at room temperature for 20 min. For the competition assay, nuclear extracts were incubated with 100-fold amount of unlabelled competitor oligonucleotides for 10 min. prior to adding labelled wild-type probe. Supershift experiments were performed with a specific p53 antibody. The samples were resolved by 6% non-denaturing polyacrylamide gel, and electrophoretically transferred to a hybond-N+ nylon membrane (Amersham, Little Chalfont, Buckinghamshire, UK). The nylon membranes were then blocked with the blocking buffer supplied in the kit, incubated with the primary antibody for 1 hr at room temperature, followed by CSPD-conjugated secondary antibody, and visualized by ECL PLUS (GE Healthcare).
Statistical analysis
The statistical significance of differences between experimental groups was calculated using Student’s test. Groups were considered statistically different if P < 0.05.
Results
Up-regulation of human SIRT1 gene expression in response to serum withdrawal was dependent on p53
A previous study showed that expression of the mouse Sirt1 could be induced by acute nutrient withdrawal, and the induction was mediated via two p53-binding sites present in the Sirt1 promoter in a p53-dependent manner [19]. We therefore reasoned that SIRT1 induction by serum deprivation in human cell lines may similarly be regulated in a p53-dependent manner. To determine whether p53 affected the SIRT1 induction in human cells subjected to serum withdrawal, we examined the SIRT1 levels in six cell lines of different p53 status, four cell lines with wild-type p53 (HEK293, HeLa, HepG2, and U2OS) and two p53-null cell lines (Saos-2 and Hep3B), under normal growth condition or under serum starvation. The mRNA and protein levels of SIRT1 were measured by real-time PCR and Western blot, respectively. As shown in Fig. 1A–C, serum withdrawal resulted in a significant induction of SIRT1 at both mRNA and protein levels in the p53-functional, but not p53-null, cells, suggesting that SIRT1 induction by serum withdrawal could be p53 dependent. In consistence with a requirement of p53 function for SIRT1 induction, the p53 level was concomitantly increased 16 hrs after serum withdrawal (Fig. 1C).
Fig 1.
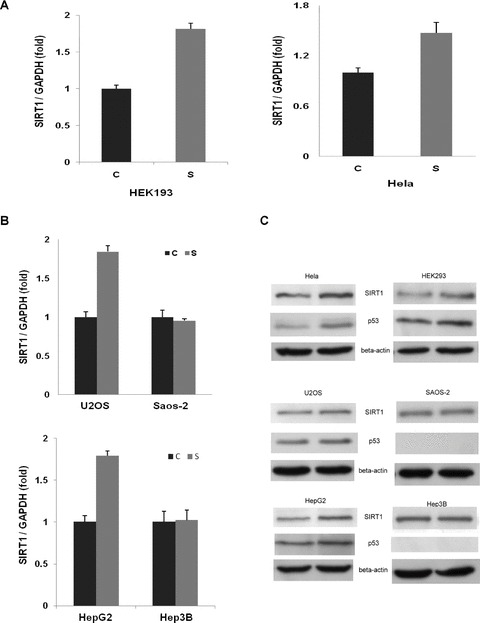
Induction of SIRT1 expression by serum starvation in human cell lines is dependent on p53. The cells were grown under normal nutrients (C) or under serum starvation (S) for 16 hrs. Real-time PCR analysis and Western blot were performed as described in the Material and Methods. Significant induction of SIRT1 expression was observed in p53-expressing cells (HEK293, HeLa, U2OS and HepG2), but not p53-null cells (Saos-2 and Hep3B). GAPDH RNA and β-actin protein were used as normalizing control for RNA and protein quantification assay, respectively. (A) and (B), mRNA levels quantified with real-time PCR. (C) Protein levels determined by Western blot.
The p53 response element in the promoter of human SIRT1 gene was required for the induction of the SIRT1 expression by serum withdrawal
The p53 target genes typically contain p53 consensus binding sites in their promoters or other regulatory regions. Mouse Sirt1 gene contains two p53-binding sites in its promoter [19]. However, an analysis using software MatInspector indicated that the p53 response element in human SIRT1 promoter differs from that in the mouse Sirt1 promoter at one position (Fig. 2A). There is a C to A substitution in the second half site. This represents a further deviation from the consensus binding site than the mouse Sirt1 promoter and might impair its binding with p53 and, consequently lead to reduced induction of SIRT1 expression in response to nutritional stress in human cells. To examine this, we first tested if there is a functional difference in the promoters of mouse Sirt1 and human SIRT1. As shown in Fig. 2B, the luciferase reporter containing the human SIRT1 promoter showed about half of the activity of the reporter containing the mouse Sirt1 promoter, under normal growth condition and under nutritional stress. Although human SIRT1 promoter had a lower reporter activity than the mouse counterpart, it was still more than 40 times higher than the promoterless pGL3-basic control. This result indicated that the human SIRT1 promoter may possess a similar function as the one in the mouse, although the p53 responsive element it contains is more deviant from the consensus sequence.
Fig 2.
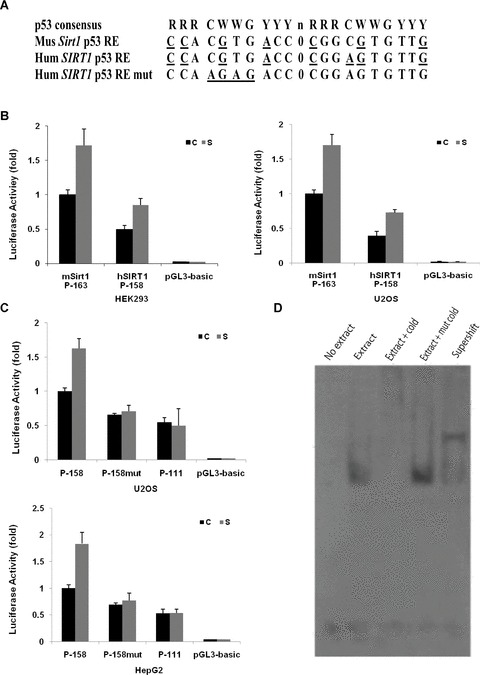
A putative p53 response element present in the SIRT1 promoter contributes to the induction of human SIRT1 expression by serum withdrawal. (A) Schematic representation of the putative p53 response elements in the promoter sequence of the SIRT1 gene, where R is purine, Y is pyrimidine, W is A or T, and n is a spacer of 0–13 nt between the two half sites. The deviations from consensus were underlined. Seven and eight deviations exist in mouse and human SIRT1 promoter, respectively. Two point mutations (bold) were introduced to change the core sequence from CGTG to AGAG. (B) Functional comparison of the mouse Sirt1 and human SIRT1 promoters in luciferase reporter assay. Same amount of luciferase constructs containing mouse Sirt1 or human SIRT1 were transfected into HEK293 and U2OS cells and cells were maintained in normal nutrients (C) or under serum starvation (S). Cells were harvested 16 hrs after treatments and luciferase activities were determined. The activity of the construct containing mouse Sirt1 promoter under normal nutrients was arbitrarily set as 100%, and the relative luciferase activity under normal or starved condition was calculated accordingly. Each bar represents the value of mean ± S.E.M. (C) Deletion or mutation of p53 response element in SIRT1 promoter abolishes the expression of luciferase stimulated by serum starvation. Constructs with native (P-158) or mutant p53 response element (P-158mut) or without p53 response element (P-111) were transfected into U2OS and HepG2 cells, and the relative luciferase activity under either normal nutrients (C) or serum-starved condition (S) was determined. The activity of construct P-158 under normal nutrient was arbitrarily set to 100%, and the relative luciferase activity under normal or serum-starved condition was calculated accordingly. Each bar represents the value of mean ± S.E.M. (D) Physical binding of p53 response element with p53. The protein-DNA complexes were resolved by native polyacrylamide gel electrophoresis. The retarded mobility of the oligonucleotide was observed in the presence of nuclear extract, lane 2. The formation of DNA-protein complex was competitively inhibited by 100-fold molar excess of unlabelled (cold) wild-type oligonucleotides, lane 3, but not by the same amount of cold mutant oligonucleotides. The retarded mobility of the probe in the presence of p53 protein was supershifted with p53 antibody, lane 5.
We next focused on the characterization of promoter activity of human SIRT1. Two luciferase reporters driven by the promoter with (P-158) or without (P-111) p53 response element were constructed and transfected into two p53-expressing cell lines, U2OS and HepG2 cells, and luciferase activity was measured after serum deprivation. Consistent with the above findings, the stimulatory effect of serum withdrawal was observed for the construct containing p53 response element, but not in P-111 construct in which the p53 response element was deleted, indicating that the p53 response element is essential for the induction of SIRT1 promoter activity in response to serum deprivation (Fig. 2C). To further confirm this observation, two point mutations were introduced into P-158 construct to generate a p53 mutant construct, P-158mut, that have the core of p53 consensus sequence changed from CGTG to AGTG. The mutation of p53 response element abolished the stimulatory effect of serum withdrawal (Fig. 2C). Notably, although the absolute luciferase activities of the P-158mut and P-111 were lower when compared to P-158, decreased to about 40% and 45%, respectively; these two reporters still showed distinct luciferase activities, nearly 30 times of the background activity of the promoterless reporter pGL3-basic.
To determine whether endogenous p53 is capable of binding specifically to this p53 response element in the human SIRT1 promoter, we performed a gel shift assay using a digoxin labelled 21bp oligonucleotide and nuclear protein from U2OS cells. The protein-DNA complexes were resolved by native polyacrylamide gel electrophoresis. The retarded mobility of the oligonucleotide was observed in the presence of nuclear extract (Fig. 2D, lane 2). The formation of DNA-protein complex was competitively inhibited by 100-fold molar excess of unlabelled (cold) wild-type oligonucleotides (Fig. 2D, lane 3), but not by the same amount of (cold) mutant oligonucleotides (Fig. 2D, lane 4). The retarded mobility of the probe in the presence of p53 protein was supershifted with p53 antibody (Fig. 2D, lane 5). These results show that endogenous p53 binds specifically to the 21 base pairs of the p53 response element in the human SIRT1 promoter.
Up-regulation of SIRT1 in response to serum withdrawal requires p53 protein
If up-regulation of the human SIRT1 expression is p53 dependent, one would predict that this stimulatory effect by serum withdrawal would be abolished in p53-deficient cells. Towards this end, Saos-2 cells and Hep3B cells, which lack endogenous p53 protein, were transfected with luciferase reporter constructs containing SIRT1 promoter. As shown in Fig. 3A, luciferase activities of the reporters driven by the SIRT1 promoter were not significantly increased by serum deprivation in the absence of p53 protein, although the p53 response element was present.
Fig 3.
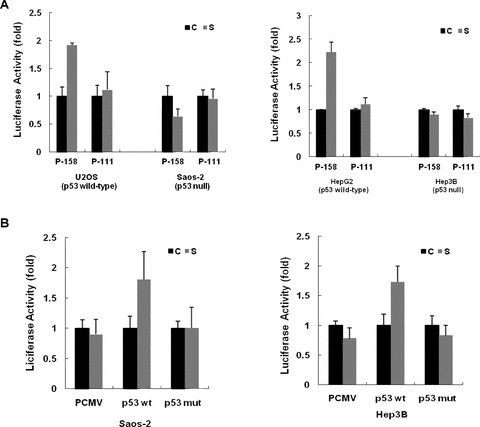
p53 protein is required for the induction of human SIRT1 promoter activity by serum withdrawal. (A) Constructs with (P-158) or without p53 response element (P-111) were transfected into either p53-positive cells (U2OS and HepG2) or p53-null cells (Saos-2 and Hep3B), and the relative luciferase activity was determined. The activity under normal nutrient (C) was arbitrarily set to 100%, and the relative luciferase activity under starved condition (S) was calculated accordingly. Each bar represent the value of mean ± S.E.M. (B) Saos-2 and Hep3B cells were cotransfected with the P-158 construct along with the expression plasmids encoding either wild-type p53 (p53 wt) or a truncation mutant (p53mut), and the luciferase activity was determined. The activity under normal nutrient (C) was arbitrarily set to 100%, and the relative luciferase activity under starved condition (S) was calculated accordingly. Each bar represents the value of mean ± S.E.M.
To further address the impact of p53 expression on up-regulation of human SIRT1 promoter, Saos-2 and Hep3B cells were cotransfected with the P-158 construct and the expression vectors encoding either wild-type p53 or a truncation mutant. The introduction of the wild-type p53 led to a significant induction of SIRT1 upon serum withdrawal. In contrast, cotransfection of a vector expressing p53 truncation mutation was unable to restore the stimulatory effect of serum withdrawal in p53-deficient cells (Fig. 3B).
Discussion
Collectively, our results demonstrated that up-regulation of human SIRT1 expression in response to serum withdrawal is p53 dependent. This conclusion was supported by several lines of evidence. First, serum withdrawal resulted in a significant induction of SIRT1 at the mRNA and at the protein level in p53-expressing cells, but not p53-null cells. Second, deletion or mutation of the p53 response element in the SIRT1 promoter abolished the stimulatory effect of serum deprivation. Third, the SIRT1 promoter was unable to drive the luciferase reporter in response to serum starvation in p53-null cells. Fourth, cotransfection assay indicated that re-introduction of wild-type p53, but not truncated (mutant) p53, could restore the stimulatory effect of serum withdrawal in p53-deficient cells. These findings indicated that the up-regulation of human SIRT1 expression under the nutrient stress is mediated in a p53-dependent manner.
Sirtuins are up-regulated by various biological stresses including caloric restriction. Caloric restriction has been shown to extend lifespan and prevent numerous diseases associated with aging in mammals [10, 17, 23–26]. The induction of SIRT1 expression under various stress conditions probably acts as a protective adaptation response. Nemoto et al. reported that acute nutrient withdrawal could simultaneously augment the expression of the Sirt1 and activate the Foxo3a, a forkhead transcription factor, in rodent cells. Knockdown of Foxo3a expression inhibited the starvation-induced increase in Sirt1 expression. Furthermore, they found that stimulation of Sirt1 transcription by Foxo3a was mediated via a p53-binding site (two half sites) in the mouse Sirt1 promoter, and there was a nutrient-sensitive physical interaction between Foxo3a and p53. Sirt1 expression was not induced in starved p53-deficient mice [19]. These results suggested that in mammalian cells, p53, Foxo3a, and SIRT1, which are all implicated in aging, constitute a nutrient-sensing pathway. Consistent with these observations, we found that serum withdrawal also up-regulates human SIRT1 gene expression in a p53-dependent manner and the p53-binding element in SIRT1 is required for the up-regulation.
The p53 protein is estimated to regulate several hundred target genes that are involved in pathways like apoptosis, DNA damage repair and cell growth arrest [27]. According to in vitro experiments, the p53 protein binds specifically to a palindromic consensus sequence, RRRCWWGYYY(R = A, G; W = A, T; Y = C, T) separated by 0–13 base pairs [20], with nearly all response elements containing at least one mismatch; in vivo results have suggested that the spacer region may be much smaller [27]. It was suggested that the ‘rules of engagement’ for p53 response elements may differ depending on the activated pathway, particularly in the regulation of apoptosis and cell-cycle checkpoints [28]. Although some p53-binding sites match the consensus sequence quite well, others can be consensus poor and yet they are all essential, and efficient, in the transactivating process [29]. Although p53-binding sites in human SIRT1 promoter differ from those in mouse Sirt1 promoter, our experiments indicated that this response element is essential and sufficient to mediate starvation-induced increase in SIRT1 expression. Interestingly, a comparison of mouse Sirt1 and human SIRT1 promoters in their ability to drive luciferase reporter revealed that the fragment in human SIRT1 promoter was about half as efficient as the one in mouse Sirt1 promoter. Probably, the sequence variation in p53-binding element in human SIRT1 promoter, which is more deviant from the consensus p53-binding sequence, rendered a lower p53-binding capacity and led to a lower transcriptional activity. Nevertheless, the p53-binding element in human SIRT1 promoter is functional in response to nutrient stress. This finding suggests that the mechanism responsible for p53-mediated regulation of SIRT1 expression is conserved between mice and human beings.
It should be noted that although the up-regulation of SIRT1 expression stimulated by serum starvation requires p53 and Foxo3a, SIRT1 also down-regulates p53 and Foxo3a proteins [16, 30, 31]. SIRT1 has been demonstrated to bind and deacetylate p53 in vitro and in vivo and to attenuate its ability to transactivate its downstream target genes, such as p21 for cell-cycle arrest and Bax for apoptosis [16, 30]. Overexpression of sirtuins has been shown to inhibit p53-dependent apoptosis in response to DNA damage and oxidative stress [16]. SIRT1 interacts with Foxo3a in a similar manner as it does with p53. In response to serum withdrawal, Foxo proteins move from cytoplasm to the nucleus, where they function as transcription factors to activate or repress a suite of genes, including SIRT1. On the other hand, SIRT1 deacetylates and represses the activity of Foxo3a [31], paralleling its effect on p53. Thus, under nutritional stress, p53 and Foxo proteins promote SIRT1 expression, which serves as a protective mechanism against excessive apoptosis. Thanks to deacetylation activity of SIRT1, p53 and Foxo proteins are only kept at appropriate levels. Therefore, SIRT1 interacts with p53 and Foxo in a feedback loop. Such feedback loops are critical in the maintenance of homeostasis.
It remains to be determined how serum withdrawal activates p53. A recent study showed that glucose deprivation can activate AMPK, which in turn activates p53, via phosphorylation on serine 15 and induces cell cycle arrest [32]. However, it is unknown if changes of SIRT1 expression are involved in this process.
In summary, our results demonstrate that the up-regulation of human SIRT1 expression in response to serum withdrawal is p53 dependent. The mechanism responsible for the regulation of SIRT1 expression is conserved between mice and human beings. These findings should be helpful in understanding other factors involved in the regulation of SIRT1 gene expression in human beings.
Acknowledgments
This work was supported by National Basic Research Program of China (973 Program); grant number 2007CB512001.
References
- 1.Blander G, Guarente L. The Sir2 family of protein deacetylases. Annu Rev Biochem. 2004;73:417–35. doi: 10.1146/annurev.biochem.73.011303.073651. [DOI] [PubMed] [Google Scholar]
- 2.Gray SG, Ekstrom TJ. The human histone deacetylase family. Exp Cell Res. 2001;262:75–83. doi: 10.1006/excr.2000.5080. [DOI] [PubMed] [Google Scholar]
- 3.Aparicio OM, Billington BL, Gottschling DE. Modifiers of position effect are shared between telomeric and silent mating-type loci in S. cerevisiae. Cell. 1991;66:1279–87. doi: 10.1016/0092-8674(91)90049-5. [DOI] [PubMed] [Google Scholar]
- 4.Rine J, Herskowitz I. Four genes responsible for a position effect on expression from HML and HMR in Saccharomyces cerevisiae. Genetics. 1987;116:9–22. doi: 10.1093/genetics/116.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sinclair DA, Guarente L. Extrachromosomal rDNA circles – a cause of aging in yeast. Cell. 1997;91:1033–42. doi: 10.1016/s0092-8674(00)80493-6. [DOI] [PubMed] [Google Scholar]
- 6.Dryden SC, Nahhas FA, Nowak JE, et al. Role for human SIRT2 NAD-dependent deacetylase activity in control of mitotic exit in the cell cycle. Mol Cell Biol. 2003;23:3173–85. doi: 10.1128/MCB.23.9.3173-3185.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Starai VJ, Celic I, Cole RN, et al. Sir2-dependent activation of acetyl-CoA synthetase by deacetylation of active lysine. Science. 2002;298:2390–2. doi: 10.1126/science.1077650. [DOI] [PubMed] [Google Scholar]
- 8.Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999;13:2570–80. doi: 10.1101/gad.13.19.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tissenbaum HA, Guarente L. Increased dosage of a Sir-2 gene extends lifespan in Caenorhabditis elegans. Nature. 2001;410:227–30. doi: 10.1038/35065638. [DOI] [PubMed] [Google Scholar]
- 10.Lin SJ, Defossez PA, Guarente L. Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science. 2000;289:2126–8. doi: 10.1126/science.289.5487.2126. [DOI] [PubMed] [Google Scholar]
- 11.North BJ, Verdin E. Sirtuins: Sir2-related NAD-dependent protein deacetylases. Genome Biol. 2004;5:224.1–12. doi: 10.1186/gb-2004-5-5-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Greer EL, Brunet A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene. 2005;24:7410–25. doi: 10.1038/sj.onc.1209086. [DOI] [PubMed] [Google Scholar]
- 13.Motta MC, Divecha N, Lemieux M, et al. Mammalian SIRT1 represses forkhead transcription factors. Cell. 2004;116:551–63. doi: 10.1016/s0092-8674(04)00126-6. [DOI] [PubMed] [Google Scholar]
- 14.Picard F, Kurtev M, Chung N, et al. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature. 2004;429:771–6. doi: 10.1038/nature02583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith J. Human Sir2 and the ‘silencing’ of p53 activity. Trends Cell Biol. 2002;12:404–6. doi: 10.1016/s0962-8924(02)02342-5. [DOI] [PubMed] [Google Scholar]
- 16.Vaziri H, Dessain SK, Ng Eaton E, et al. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–59. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- 17.Brunet A, Sweeney LB, Sturgill JF, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–5. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 18.Bordone L, Cohen D, Robinson A, et al. SIRT1 transgenic mice show phenotypes resembling calorie restriction. Aging Cell. 2007;6:759–67. doi: 10.1111/j.1474-9726.2007.00335.x. [DOI] [PubMed] [Google Scholar]
- 19.Nemoto S, Fergusson MM, Finkel T. Nutrient availability regulates SIRT1 through a forkhead-dependent pathway. Science. 2004;306:2105–8. doi: 10.1126/science.1101731. [DOI] [PubMed] [Google Scholar]
- 20.El-Deiry WS, Kern SE, Pietenpol JA, et al. Definition of a consensus binding site for p53. Nat Genet. 1992;1:45–9. doi: 10.1038/ng0492-45. [DOI] [PubMed] [Google Scholar]
- 21.McLure KG, Lee PW. How p53 binds DNA as a tetramer. EMBO J. 1998;17:3342–50. doi: 10.1093/emboj/17.12.3342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dhar SK, Xu Y, Chen Y, et al. Specificity protein 1-dependent p53-mediated suppression of human manganese superoxide dismutase gene expression. J Biol Chem. 2006;281:21698–709. doi: 10.1074/jbc.M601083200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen D, Steele AD, Lindquist S, et al. Increase in activity during calorie restriction requires Sirt1. Science. 2005;310:1641. doi: 10.1126/science.1118357. [DOI] [PubMed] [Google Scholar]
- 24.Cohen HY, Miller C, Bitterman KJ, et al. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science. 2004;305:390–2. doi: 10.1126/science.1099196. [DOI] [PubMed] [Google Scholar]
- 25.Kim D, Nguyen MD, Dobbin MM, et al. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer’s disease and amyotrophic lateral sclerosis. EMBO J. 2007;26:3169–79. doi: 10.1038/sj.emboj.7601758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Viswanathan M, Kim SK, Berdichevsky A, et al. A role for SIR-2.1 regulation of ER stress response genes in determining C. elegans life span. Dev Cell. 2005;9:605–15. doi: 10.1016/j.devcel.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 27.Wei CL, Wu Q, Vega VB, et al. A global map of p53 transcription-factor binding sites in the human genome. Cell. 2006;124:207–19. doi: 10.1016/j.cell.2005.10.043. [DOI] [PubMed] [Google Scholar]
- 28.Qian H, Wang T, Naumovski L, et al. Groups of p53 target genes involved in specific p53 downstream effects cluster into different classes of DNA binding sites. Oncogene. 2002;21:7901–11. doi: 10.1038/sj.onc.1205974. [DOI] [PubMed] [Google Scholar]
- 29.Contente A, Dittmer A, Koch MC, et al. A polymorphic microsatellite that mediates induction of PIG3 by p53. Nat Genet. 2002;30:315–20. doi: 10.1038/ng836. [DOI] [PubMed] [Google Scholar]
- 30.Luo J, Nikolaev AY, Imai S, et al. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107:137–48. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- 31.Motta MC, Divecha N, Lemieux M, et al. Mammalian SIRT1 represses forkhead transcription factors. Cell. 2004;116:551–63. doi: 10.1016/s0092-8674(04)00126-6. [DOI] [PubMed] [Google Scholar]
- 32.Jones RG, Plas DR, Kubek S, et al. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell. 2005;18:283–93. doi: 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]