

Abstract
Taxol is a powerful chemotherapeutic agent that binds to microtubules to prevent tumour cell division. However, a traditional high dose of taxol may also induce apoptosis in normal cells. The anti-apoptotic molecule Bcl-2 is up-regulated in tumour cells to prevent apoptosis. We designed this study to determine whether use of a low dose of taxol and anti-apoptotic Bcl-2 gene silencing would effectively induce apoptosis in human glioblastoma U251MG cells and also inhibit invasion, angiogenesis and intracranial as well as subcutaneous tumour growth. We treated the cells with either 100 nM taxol or transfected with a plasmid vector expressing Bcl-2 siRNA or both agents together for 72 h. Knockdown of Bcl-2 potentiated efficacy of taxol for cell death. Fluorescence-activated cell sorting analysis, double immunofluorescent staining and TUNEL assay demonstrated apoptosis in about 70% of the cells after treatment with the combination of taxol and Bcl-2 siRNA. In vitro Matrigel invasion assay demonstrated dramatic decrease in glioblastoma cell invasion and in vivo angiogenesis assay showed complete inhibition of neovascularization in athymic nude mice after treatment with the combination. Further, treatment with the combination of taxol and Bcl-2 siRNA caused suppression of intracranial tumour growth and subcutaneous solid tumour development. In conclusion, our results indicate that the combination of taxol and Bcl-2 siRNA effectively induces apoptosis and inhibits glioblastoma cell invasion, angiogenesis and intracranial as well as subcutaneous tumour growth. Therefore, the combination of a low dose of taxol and Bcl-2 siRNA is a promising therapeutic strategy for controlling the aggressive growth of human glioblastoma.
Keywords: angiogenesis, apoptosis, Bcl-2 siRNA, cell invasion, glioblastoma, taxol
Introduction
Glioblastomas are the most malignant and common primary brain tumours in adults and these are associated with a dismal prognosis [1]. Glioblastomas comprise 23% of primary brain tumours in the United States [2]. Since brain tumour cells often infiltrate deep into the normal brain tissue, complete surgical removal of a brain tumour is almost impossible, keeping the high possibility of recurrence of the tumour.
Cell invasion, angiogenesis and tumour growth are complex mechanisms that involve a variety of biochemical and cellular processes [3]. The degree of a primary brain tumour growth is directly correlated with its invasive potency and angiogenesis [4]. Inhibition of these processes may not only suppress tumour growth but also improve the prognosis of aggressive brain tumours. Thus, exploring the innovative procedures to intervene the processes of cell invasion and angiogenesis would arrest the aggressive growth of human glioblastomas.
Dysregulation of apoptotic mechanisms plays an important role in the pathogenesis and progression of various cancers and determines the responses of tumours to therapeutic interventions [5]. Highly invasive cancer cells are protected from apoptosis by up-regulation of various anti-apoptotic molecules such as Bcl-2 [6]. Furthermore, Bcl-2 can protect cancer cells from taxol-mediated apoptosis by inducing multi-nucleation [7]. Significant knockdown of expression of Bcl-2 could effectively induce apoptosis in glioblastoma cells and also inhibit cell invasion, angiogenesis and tumour growth.
Taxol is a powerful anti-cancer drug that strongly binds to the β-subunit of tubulin and promotes the formation of highly stable microtubules that resist depolymerization, thus preventing active tumour cell division and arresting the cells at the G2/M phase of the cell cycle [8, 9]. Taxol induces apoptosis in cancer cells through activation of caspase pathways [6, 10]. Although anti-cancer agents including taxol have improved therapeutic responses in some advanced glioblastomas, long-term prognosis for glioblastoma patients remains unsatisfactory. Moreover, taxol at a high dose inhibits normal cell division and causes undesirable side effects. So, we contemplated that the use of a low dose of taxol and simultaneous down-regulation of Bcl-2 expression could effectively induce apoptosis in glioblastoma cells, prevent cell invasion and inhibit both angiogenesis and tumour growth.
The expression of small interfering RNA (siRNA) through mammalian vector is a powerful tool to knockdown mRNA expression of a particular gene and thereby the protein level of the targeted gene [11]. Up-regulation of the anti-apoptotic Bcl-2 molecule in tumour cells prevents cell death due to inhibition of mitochondrial release of cytochrome c into the cytosol and thereby exerts a survival advantage to tumour cells in response to various apoptotic stimuli [12, 13]. Introduction of Bcl-2 siRNA into tumour cells through a plasmid vector could degrade Bcl-2 mRNA and thus down-regulate the cognate protein level. Therefore, treatment with the combination of a low dose of taxol and a plasmid vector carrying the Bcl-2 siRNA cDNA could be an innovative therapeutic strategy to inhibit cell division and induce apoptosis in glioblastoma cells
This investigation was designed to use a low dose of taxol and knockdown the expression of Bcl-2 using a gene specific siRNA for efficiently induction of apoptosis in highly invasive human glioblastoma cells and also inhibition of cell invasion, in vivo angiogenesis and both intracranial and subcutaneous tumourigenesis in athymic nude mice.
Materials and methods
Cell culture conditions
Human glioblastoma U138MG cell line was purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). Another human glioblastoma U251MG cell line, one of the most invasive and virulent among human gliobalstoma cell lines, was procured from the National Cancer Institute (NCI, Frederick, MD, USA). The U138MG and U251MG cells were propagated in DMEM and RPMI 1640 (Mediatech, Herndon, VA, USA), respectively, supplemented with 10% foetal bovine serum (FBS) and antibiotics in a fully humidified incubator containing 5% CO2 at 37°C. Taxol (also known as paclitaxel, 6 mg/ml) was procured from Bristol-Myers Squibb (Princeton, NJ, USA) and diluted to a 1-mM solution in dimethylsulfoxide and protected from light. Taxol was further diluted in serum-free media and used in the cell culture and animal experiments.
Construction of the Bcl-2 siRNA cDNA expression vector
The Bcl-2 siRNA cDNA was constructed into a mammalian expression vector, pRNAT-CMV3.2/Neo (GenScript, Piscataway, NJ, USA), between the BamHI and XhoI sites. We prepared three Bcl-2 siRNA sequences and the most effective one was selected for this investigation on the basis of percent knockdown of Bcl-2 expression both at the mRNA and protein levels. The selected Bcl-2 siRNA cDNA sequence began at nucleotide 974 (BC027258): 5′-GGA TGC CTT TGT GGA ACT GTA TT-3′ (sense) and 3′-TAC AGT TCC ACA AAG GCA TCC-5′ (antisense). The scrambled siRNA sequences were 5′-GTG TTA AGG TGC TGT TCG ACA TT-3′ (sense) and 3′-TGT CGA ACA GCA CCT TAA CAC-5′ (antisense). The loop selected was 5′-TTG ATA TCC G-3′. The linear siRNA construct – with the sense and antisense strands, loop, termination signal and BamHI and XhoI restriction sites – was annealed with the complimentary strand and ligated into the siRNA expression vector (pRNAT-CMV3.2/Neo) between the BamHI and XhoI sites. In this vector, the powerful cytomegalovirus (CMV) promoter drives the expression of siRNA and the SV40 promoter drives the expression of the neomycin resistance gene. This expression vector also carries the coral green fluorescence protein (cGFP) for tracking of transfection efficiency in cell cultures. The siRNA sequence was confirmed by DNA sequencing using the pRNA reverse sequencing primer: 5′-TAG AAG GCA CAGTCG AGG-3′. The forward sequencing primer used for CMV was 5′-GTA CGG TGG GAG GTC TAT AT-3′. The plasmid vector carrying the Bcl-2 siRNA cDNA was transformed into the JM109 competent cells (Promega, Madison, WI, USA) and the positive colonies were screened using Qiagen miniprep plasmid DNA purification system (Qiagen, Valencia, CA, USA). The highly expressing colony was selected and propagated in LB broth containing neomycin. The plasmid vector expressing Bcl-2 siRNA was purified using maxiprep (Qiagen) and used for both cell culture and animal experiments.
Treatment of U138MG and U251MG cells with taxol and Bcl-2 siRNA cDNA plasmid vector
Cells in culture were treated with a final concentration of 100 nM taxol or transfected with mammalian expression vector carrying the Bcl-2 siRNA cDNA or both agents together in culture medium containing 1% FBS. The concentration of 100 nM taxol was selected on the basis of our previous study [6]. The cells were transfected using 3-μl Fugene HD (Roche Diagnostics, IN, USA) and 1-μg plasmid DNA vector. In a six-well plate, 2 μg DNA was used/well. The transfection efficiency was monitored through the expression of cGFP using a fluorescent microscope (Olympus IX71, Olympus, Tokyo, Japan). Also, the cells were transfected with the same vector carrying the scrambled siRNA cDNA sequence. After 24 hrs, the medium was replaced with regular serum medium and the cultures were incubated for another 48 hrs. After treatments with taxol or/and Bcl-2 siRNA, we determined changes in cell viability and proliferation using the 3-(4, 5-dimethylthiazolyl-2)-2, 5-diphenyltetrazolium bromide (MTT) assay [14].
RT-PCR to examine Bcl-2 mRNA
The reverse transcription-polymerase chain reaction (RT-PCR) experiments were carried out to monitor the knockdown of Bcl-2 mRNA after treatment with the cognate siRNA. Total RNA was isolated from the cells using Aurum kit (Bio-Rad, Hercules, CA, USA). We used the following primer sequences for PCR amplifications of Bcl-2 (BC027258, forward: 5′-TGT GGA TGA CTG AGT ACC-3′ and reverse 5′- AGG AGA AAT CAA ACA GAG G-3′) and GAPDH (NM_002046, forward: 5′-CCA CCC ATG GCA AAT TCC-3′ and reverse: 5′-CAG GAG GCA TTG CTG ATG AT-3′) genes. The primers were transcribed using a single-step RT-PCR kit (Invitrogen, Carlsbad, CA, USA) on a PCR cycler (Eppendorf, Westbury, NY, USA) with annealing temperature at 58°C. The RT-PCR products were resolved by eletrophoresis on 1.5% agarose gels containing ethidium bromide (1 μg/ml) and visualized using a UV chamber (Alpha Innotech, San Leandro, CA, USA). Expression of GAPDH was used as an internal standard.
Western blotting for Bcl-2 protein
Following treatments, the cells were washed with ice-cold phosphate-buffered saline (PBS, pH 7.4) and scraped with the freshly prepared R buffer containing protease inhibitors. Cells were centrifuged at 12,000 rpm for 10 min. at 4°C. The supernatant was discarded and the cell pellet was suspended in 100 to 200 μl of RIPA buffer depending on the size of the pellet and then sonicated using a sonicator (Kontes, Vineland, NJ, USA). Cell lysates were centrifuged at 14,000 rpm for 10 min. at 4°C and the supernatants were collected. Protein concentration in each supernatant was determined and all the samples were stored at –20°C until used. Protein samples (25 μg each) were resolved on the SDS-polyacrylamide gels and then electroblotted to nitrocellulose membranes. After blocking the non-specific sites with 5% non-fat milk, the membranes were incubated overnight at 4°C on a rocker with the monoclonal Bcl-2 primary IgG antibody (Santa Cruz Biotechnology, CA, USA). The membranes were washed and incubated with horseradish peroxidase conjugated secondary IgG antibody (Biomeda, Foster City, CA, USA) at room temperature for 2 hrs. The membranes were washed again, treated with chemiluminescence reagent (Amersham, Buckinghamshire, UK), exposed to Kodak autoradiography film, and developed. The membranes were re-probed with a mouse monoclonal GAPDH primary IgG antibody (Novus Biologicals, Littleton, CO, USA) for GAPDH content to demonstrate that the same amounts of protein were loaded in all lanes.
FACS analysis for determining DNA fragmentation
Fluorescence-activated cell sorting (FACS) analysis was performed for determining DNA fragmentation and the percentage of apoptotic/dead cells after treatment with taxol or Bcl-2 siRNA or both agents together. Untreated and treated cells were harvested using TrypLE (Invitrogen, Carlsbad, CA, USA) and washed once with PBS. The cells were then dispersed in 1 ml propidium iodide (50 μg/ml) (Biosure, Grass Valley, CA, USA) with gentle vortex and incubated for 30 min. in the dark at 4°C. The cells were sorted on a FACS machine (FACSCalibur, BD, Franklin Lakes, NJ, USA) and analysed for population of apoptotic/dead cells and DNA fragmentation based on the red fluorescence at 488 nm. Data were represented as FACS dot plot graphs and FACS histograms.
Immunofluorescent stainings for active fragments of caspase-9 and caspase-3
We cultured the U251MG cells on the four-well chamber slides (Lab-Tek, Rochester, NY, USA) at a density of 1 × 104 cells per well. After 24 hrs, the cells were treated with 100 nM taxol or transfected with Bcl-2 siRNA vector or both together. The growth of cultures was terminated at 72 hrs after the start of treatment and the cells were fixed in 95% cold-ethanol for 15 min. Then, cells were washed twice with PBS and blocked with 2% donkey and 2% goat serum (50:50) for 1 hr. The cells were washed and treated with goat polyclonal active caspase-9 primary IgG antibody (Santa Cruz Biotechnology) and rabbit monoclonal active caspase-3 primary IgG antibody (Cell Signaling, Danvers, MA, USA) simultaneously and incubated overnight at 4°C. The cells were then washed and incubated with fluorescein isothiocyanate (FITC) conjugated donkey anti-goat secondary IgG antibody (MP Biomedicals, Solon, OH, USA) and Texas red conjugated goat anti-rabbit secondary IgG antibody (Biomeda) at room temperature for 1 hr. The cells were counterstained with Hoechst 33342 (Invitrogen) and mounted with Vectashield (Vector, Burlingame, CA, USA). Then, cells were examined under a fluorescence microscope (Olympus IX71) and photographed electronically. The pictures were merged electronically along with the nuclear counterstaining using the Spot advanced software (Meyer Instruments, Houston, TX, USA).
TUNEL assay
The recombinant terminal deoxynucleotidyl transferase (rTdT) mediated-dUTP Nick End Labelling (TUNEL) assay is a widely used technique for the detection of apoptotic cells within a cell population. We cultured the U251MG cells in chamber slides and treated as described above. The TUNEL staining was performed with a fluorometric TUNEL assay kit (Promega). Briefly, the fixed cells were washed in PBS and permeabilized with 0.2% Triton X-100 for 5 min. The cells were washed, equilibrated with the equilibration buffer (supplied by Promega) and incubated with 50 μl of a cocktail of rTdT and fluorescein-12-dUTP for 1 hr at 37°C in a humidified chamber. The reaction was terminated and the slides were washed thrice in PBS and mounted with an anti-fading agent (Biomeda). Slides were dried in the dark, examined under a fluorescence microscope (Olympus) and then photographed. Apoptotic cells were quantitatively evaluated using the Image-Pro plus software (Media Cybernetics, Silver Spring, MD, USA).
In vitro matrigel invasion assay
In vitro Matrigel invasion assay [15] was performed to assess the effect of taxol and Bcl-2 siRNA on invasive property of the human glioblastoma U138MG and U251MG cells. Transwell inserts (12 well, 12 mm with 12.0-μm pore size, Corning) were coated with 200 μl of the Matrigel (1.0 mg/ml in ice-cold serum-free medium) solution (BD Biosciences, San Jose, CA, USA) and allowed to dry at 37°C. The control and treated cells were detached using trypsin solution and 200 μl of cell suspension (2 × 105 cells) from each sample was added to the transwells in triplicate. After 48 hrs incubation at 37°C in a CO2 incubator, the Matrigel membranes were collected and stained with HEMA stain (Fisher Scientific, Pittsburg, PA, USA). The number of cells that migrated to the undersurface of the membrane were examined under a microscope, photographed and counted in 10 randomly selected microscopic fields.
In vivo angiogenesis (dorsal skinfold) assay
Anti-angiogenic effect of taxol and Bcl-2 siRNA in U251MG cells was examined using the in vivo angiogenesis (dorsal skinfold) assay [16]. The diffusion chamber rings (Millipore, Bedford, MA, USA) were prepared with Millipore membrane filters (0.45 μm) on both sides. The chambers were sterilized by UV irradiation and injected with 200 μl of cell suspension (2 × 105 cells) after treatment with either taxol or Bcl-2 siRNA or both agents together for 72 hrs. The opening of the chamber was subsequently sealed with sterile bone wax and the chambers were surgically implanted under the dorsal skin of athymic nude mice (Charles River Laboratories, MA, USA). After 10 days, the implanted chambers were removed surgically and the superficial fascia exposed to the chamber was harvested. The formation of new vasculature (neovascularization) in zigzag pattern was observed using a stereomicroscope (Olympus SZX12, Olympus) equipped with a Spot RT Slider digital camera (Meyer Instruments) and photographed. The tumour-induced neovasculature was measured and quantitated with the help of an ocular micrometer. This and all other animal experiments were performed in compliance with our Institutional Animal Care and Use Committee (IACUC) guidelines and the Guide for the Care and Use of Laboratory Animals of the US Department of Health and Human Services (National Institutes of Health, Bethesda, MD, USA).
Bioluminescence imaging of intracerebral and subcutaneous tumours in nude mice
We have selected the U251MG cells for further in vivo studies because it is one of the most invasive and virulent cell line among all human glioblastoma cell lines. The U251MG cells were stably transfected with a plasmid vector (phCMV-FSR, Genlantis, San Diego, CA, USA) carrying luciferase gene and propagated in media containing G-418 (500 μg/ml). The stably transfected U251MG cells were treated with either 100 nM taxol or transfected with Bcl-2 siRNA or both agents together for 72 hrs. The cells were harvested and 1 × 106 cells suspended in 10 μl of serum-free medium were injected into the cerebrum of athymic nude mice (Charles River Laboratories) with the help of a stereotaxic apparatus (Stoelting, Wood Dale, IL, USA). Similarly, 2 × 106 cells suspended in 100 μl of serum-free medium were injected subcutaneously to different groups of nude mice. Afterwards, the mice were injected intraperitoneally with either taxol (50 μg/injection/mouse) or Bcl-2 siRNA plasmid vector (50 μg DNA/injection/mouse) or both agents together on alternate days for 20 days. Mice with implantation of the cells transfected with the scrambled siRNA vector also received similar treatments. On days 7, 14 and 21, the mice were injected with 100 μl (50 mg/ml) of luciferin (Genlantis). After 10 min. of luciferin injection, the mice were visualized for bioluminescence from luciferase activity using the Xenogen IVIS-200 imaging system (Xenogen, Hopkinton, MA, USA).
Examination of subcutaneous tumourigenesis and solid tumour development in nude mice
We examined the effect of taxol and Bcl-2 siRNA on subcutaneous tumourigenesis and solid tumour development in athymic nude mice. The U251MG cells in culture were treated with 100 nM taxol or transfected with Bcl-2 siRNA or both agents together for 72 hrs. The control and treated cells were detached using trypsin solution and 1 × 107 cells suspended in 100 μl of serum-free medium were mixed with 100 μl of high concentration of the Matrigel (BD Biosciences). Then, 100 μl of this cell suspension (5 × 106 cells) in Matrigel was injected subcutaneously into athymic nude mice. We used six mice in each treatment group to determine the significance of treatments. The animals were left for tumour development for 2 weeks without any treatment. Afterwards, the mice were injected intraperitoneally with taxol (50 μg/injection/mouse) or Bcl-2 siRNA plasmid vector (50 μg DNA/injection/mouse) or both agents together on alternate days for 4 weeks. Mice implanted with the cells transfected with the scrambled siRNA vector also received similar treatments. Tumour volume was measured beginning from third week using a digital vernier caliper. Tumour volume was calculated using the formula ([smallest diameter2× widest diameter]/2) on the basis a previous report [17] and the growth curves were plotted. The animals were killed at the end of the sixth week, solid tumours were surgically removed, photographed and the tumour weights were recorded.
Statistical analysis
The arithmetic mean and standard deviation (S.D.) were calculated for all quantitative experiments. The results were statistically evaluated using a one-way ANOVA. The least significant difference method was used to compare mean values for the control samples with those of taxol or Bcl-2 siRNA plasmid vector treated samples. Taxol or Bcl-2 siRNA alone mean values were also compared with the combination treatment values. The data were presented as mean ± S.D. of independent experiments (n≥ 3). The difference between two values was considered statistically significant at P < 0.05. Pearson and Lee’s correlation coefficient was used to study the correlation between the quantitative data from FACS analysis and TUNEL assay.
Results
Down-regulation of Bcl-2 at mRNA and protein levels
Remarkable knockdown of Bcl-2 at mRNA and protein levels occurred in both U138MG and U251MG cells after transfection with the plasmid vector expressing Bcl-2 siRNA. The data were presented only for U251MG cells (Fig. 1). We found no appreciable alterations in Bcl-2 mRNA (Fig. 1A) and Bcl-2 protein (Fig. 1B) levels after transfection with a scrambled siRNA vector or treatment with taxol alone. Treatment with Bcl-2 siRNA alone or combination of taxol and Bcl-2 siRNA resulted in approximately 80% knockdown of Bcl-2 mRNA (Fig. 1A). Bcl-2 siRNA alone resulted in almost 60% down-regulation of cognate protein and the combination of taxol and Bcl-2 siRNA resulted in about 70% down-regulation of Bcl-2 (Fig. 1B). Expression of GAPDH was used as an internal control to demonstrate equal loading of both RNA and protein samples.
Fig 1.
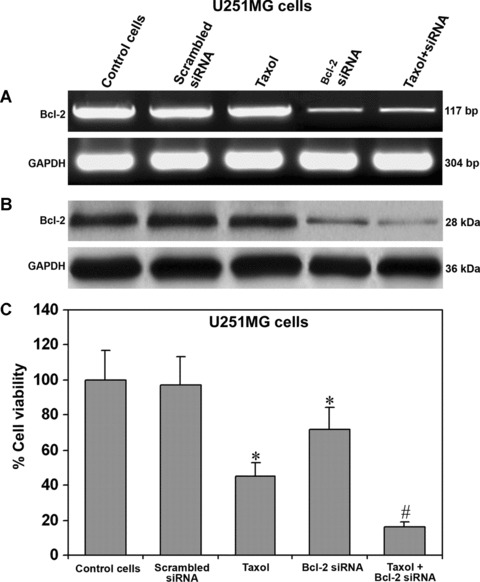
Expression of Bcl-2 at mRNA and protein levels in U251MG cells. Treatment (72 hrs) of cells: control, 100 nM taxol, transfection with a plasmid vector carrying the scrambled siRNA cDNA, transfection with a plasmid vector carrying the Bcl-2 siRNA cDNA and taxol + Bcl-2 siRNA. In determination of both RNA and protein levels, GAPDH was used as an internal control. (A) Semi-quantitative RT-PCR for examination of Bcl-2 mRNA. (B) Western blotting for examination of Bcl-2 protein. (C) MTT assay for determination of percent changes in cell viability. For MTT assay, the cells were treated for 48 hrs. Data are representative of six independent experiments in duplicate (*P < 0.001 when compared with the control mean values and #P < 0.001 when compared with taxol or Bcl-2 siRNA mean values).
Effect of taxol and Bcl-2 siRNA on cell viability and proliferation
The MTT assay is widely used for characteristic measurements for cell viability and proliferation. We observed a significant decrease in the percentage of cell viability in both U138MG and U251MG cells after treatment with taxol or/and Bcl-2 siRNA. The data were presented only for U251MG cells (Fig. 1C). Significant decrease in cell viability occurred due to treatment with taxol or Bcl-2 siRNA. The decrease in cell viability was most remarkable and synergistic after treatment with both agents together (Fig. 1C).
Increase in apoptosis as indicated by FACS analysis
FACS analysis is a useful tool for detection of apoptotic cells and DNA fragmentation. So, we performed FACS analysis for determining apoptosis and DNA fragmentation in U251MG cells after treatment with taxol or Bcl-2 siRNA or both agents together (Fig. 2). An increase in cell population in the M1 area indicated apoptotic cells, as shown in the FACS dot plot graphs (Fig. 2A). A significant increase (P < 0.001) in cell population in the M1 area occurred due to treatment with either taxol or Bcl-2 siRNA, compared with the scrambled siRNA transfected cells. The cell population in the M1 area was doubled after the treatment with combination of taxol and Bcl-2 siRNA. Further, we monitored DNA fragmentation in the cells after the treatments, as shown in the FACS histograms (Fig. 2B). The M1 area represented the population of apoptotic or dead cells with DNA fragmentation and the M2 area denoted the population of healthy cells. The apoptotic DNA fragmentation was remarkably increased after the treatment with combination of taxol and Bcl-2 siRNA (Fig. 2B). Concomitantly, the population of healthy cells was decreased. We presented the quantitative FACS data for percentage of live and apoptotic cells based on DNA fragmentation (Fig. 2C). Quantitation of data in the M1 area for DNA fragmentation demonstrated 28%, 30% and 65.5% apoptotic cells after treatment with taxol, Bcl-2 siRNA and combination of both, respectively (Fig. 2C). Results indicated that the treatment with combination of taxol and Bcl-2 siRNA caused more apoptosis in U251MG cells than either treatment alone. The untreated controls were not shown.
Fig 2.
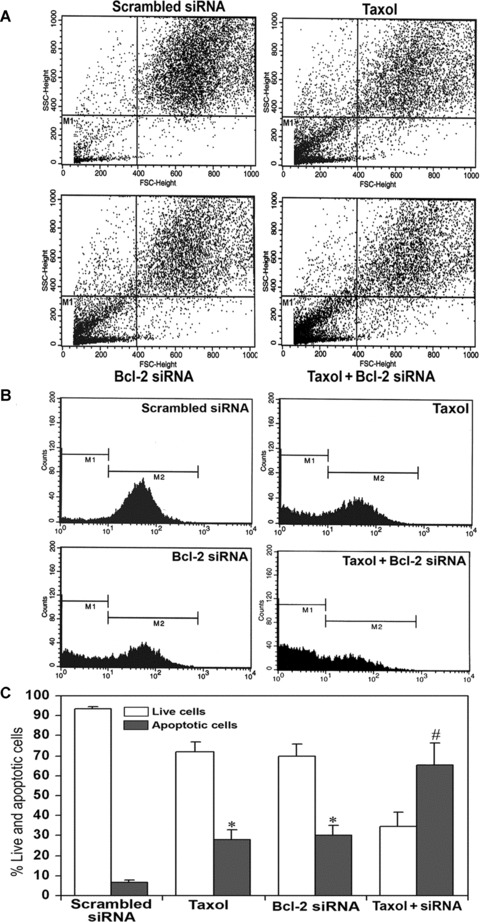
FACS analysis for detection of apoptotic cells and DNA fragmentation. Treatment (72 hrs) of cells: transfection with a plasmid vector expressing scrambled siRNA (treated control), 100 nM taxol, transfection with a plasmid vector expressing Bcl-2 siRNA and taxol + Bcl-2 siRNA. (A) FACS dot plots of U251MG cells. Before FACS analysis, the cells were treated with 50 μg/ml propidium iodide for 30 min. at 4°C in dark. Population in the M1 area represents the apoptotic/dead cells. (B) FACS histograms of U251MG cells. The prominent increase in population of cells in the sub-G1 phase (M1) indicated increase in apoptosis after treatment with taxol or Bcl-2 siRNA or both. (C) Quantitative presentation of DNA fragmentation data from FACS analysis to indicate percent changes in live and apoptotic cells. Data are representative of four independent experiments (*P < 0.001 when compared with scrambled siRNA treatment mean values and #P < 0.001 when compared with taxol or Bcl-2 siRNA treatment mean values).
Increased expression of active fragments of caspase-9 and caspase-3
Double immunofluorescent staining was employed to examine the active fragments (cleaved subunits) of caspase-9 and caspase-3 (Fig. 3). We used specific antibodies for the active fragments to avoid cross-reaction with the parent molecules. Immunofluorescent stainings demonstrated remarkable increases in the active fragments of caspase-9 and caspase-3 after treatments with taxol, Bcl-2 siRNA and both agents together (Fig. 3A). The stainings were highly conspicuous after treatment of cells with combination of taxol and Bcl-2 siRNA. Counterstaining of the nucleus with Hoechst 33342 revealed nuclear fragmentation in cells treated with taxol, Bcl-2 siRNA and combination of both. Merged microphotographs clearly demonstrated simultaneous stainings of active fragments of caspase-9 and caspase-3 as well as of disintegrating nucleus in the apoptotic cells (shown with arrows). In both untreated control (not shown) and scrambled Bcl-2 siRNA transfected cells, the stainings for active fragments of caspase-9 and caspase-3 were feeble. The scrambled Bcl-2 siRNA treatment was considered as the treated control.
Fig 3.
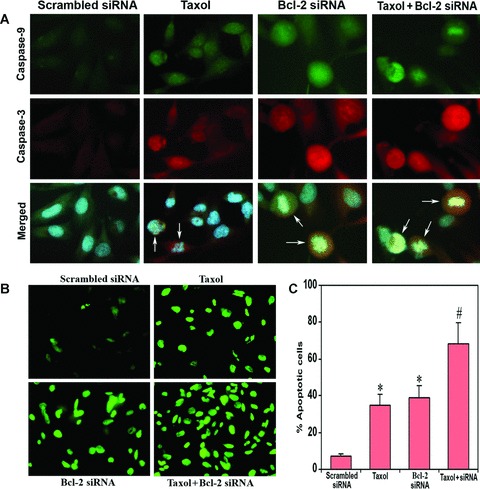
In situ stainings for biochemical markers of apoptosis in U251MG cells. Treatment (72 hrs) of cells: transfection with a plasmid vector expressing scrambled siRNA (treated control), 100 nM taxol, transfection with a plasmid vector expressing Bcl-2 siRNA and taxol + Bcl-2 siRNA. (A) Double immunofluorescent stainings to examine active fragments of caspase-9 and caspase-3. The cells were incubated overnight at 4°C with specific antibodies for active fragments of caspase-9 and caspase-3, washed and then treated with FITC conjugated and Texas red conjugated secondary antibodies at room temperature for 1 hr. Hoechst 33342 was used to counterstain the nucleus. Merged microphotographs demonstrated simultaneous expression of active fragments of caspase-9 and caspase-3 as well as disintegration of nucleus in the apoptotic cells (shown with arrows). (B) Fluorescent TUNEL assay for detection of apoptotic cells. Treatment with combination of taxol and Bcl-2 siRNA resulted in more apoptotic cell death than either treatment alone. (C) Quantitation of TUNEL-positive cells. Data are representative of four independent experiments (*P < 0.001 when compared with the scrambled siRNA treatment mean values and #P < 0.001 when compared with taxol or Bcl-2 siRNA treatment mean values).
Increased apoptosis as indicated by TUNEL assay
The major hallmark of apoptosis is nuclear DNA fragmentation. We also used TUNEL assay to determine apoptotic DNA fragmentation and found increase in number of TUNEL-positive cells after treatment of cells with taxol or Bcl-2 siRNA (Fig. 3B). Treatment with combination of both agents resulted in doubling the number of apoptotic cells, compared with treatment with either agent alone. The TUNEL staining was absent in untreated control (not shown) and scrambled Bcl-2 siRNA transfected cells. Quantitative evaluation of the TUNEL-positive cells using the Image-Pro Plus software revealed 35%, 39% and 68% apoptotic cells after treatment with taxol, Bcl-2 siRNA and combination of both, respectively (Fig. 3C). The mean values of the combination treatment were significantly different (P < 0.001) from the individual mean values of taxol or Bcl-2 siRNA treatment. The quantitative data of TUNEL staining correlated positively (r= 0.998) with the FACS data for apoptosis. The scrambled Bcl-2 siRNA treatment was considered as the treated control.
Marked reduction in tumour cell invasion
We examined the effects of taxol, Bcl-2 siRNA and combination of both agents on the invasive properties of both U138MG and U251MG cells using the Matrigel invasion assay (Fig. 4). The staining of invading cells through the polycarbonate membranes demonstrated that the invasion of cells was very low after treatment with taxol or Bcl-2 siRNA, compared with invasion of untreated control cells (not shown) or scrambled siRNA vector transfected cells (Fig. 4A and C). Taxol was more effective than Bcl-2 siRNA to prevent cell invasion through the Matrigel. The treatment with combination of taxol and Bcl-2 siRNA resulted in a remarkable decrease in the number of invading cells. Quantitation of the invading cells using Image-Pro Plus software showed that the U138MG cells after treatment with taxol, Bcl-2 siRNA and both agents together resulted in only 31%, 57% and 12% invasion, respectively, compared with cells after transfection with scrambled siRNA (Fig. 4B). In U251MG cells, we observed only 35%, 49% and 10% invasion after treatment with taxol, Bcl-2 siRNA and both agents together, respectively (Fig. 4D). The scrambled siRNA treatments were considered as treated controls for both cell lines.
Fig 4.
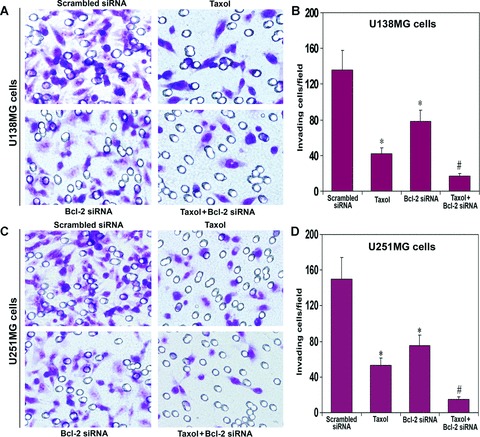
In vitro Matrigel invasion assay using U138MG and U251MG cells. Treatment (72 hrs) of cells: transfection with a plasmid vector expressing scrambled siRNA (treated control), 100 nM taxol, transfection with a plasmid vector expressing Bcl-2 siRNA and taxol + Bcl-2 siRNA. Invasion assays were carried out in 12-well transwell inserts of polycarbonate filters. After 48 hrs incubation at 37°C in a CO2 incubator, the membranes were collected and stained with HEMA. The number of cells that migrated to the undersurface of the membrane were examined under a microscope, counted and photographed. (A) The changes in capability of invasion of U138MG cells after the treatments. (B) Quantitative evaluation of invading U138MG cells. (C) The changes in capability of invasion of U251MG cells after the treatments. (D) Quantitative evaluation of invading U251MG cells. The quantitative data are presented as mean ± S.D. of cells from 10 randomly selected microscopic fields from three independent wells (*P < 0.001 when compared with the scrambled siRNA treatment mean values and #P < 0.001 when compared with taxol or Bcl-2 siRNA treatment mean values).
Inhibition of in vivo angiogenesis
We examined the effect of taxol, Bcl-2 siRNA and both agents together on in vivo angiogenesis in terms of neovascularization in the dorsal skin of nude mice (Fig. 5). The implantation of diffusion chambers containing U251MG untreated cells (not shown) and scrambled siRNA vector transfected cells (treated control) maintained the formation of well-developed blood microvessels as indicated by the thin and curved structures arising from the pre-existing vessels in a zigzag manner (Fig. 5A). The formation of similar structures was partially inhibited in cells treated with taxol or Bcl-2 siRNA but completely inhibited in cells treated with both agents together (Fig. 5A). Quantitative measurements of the tumour-induced neovasculature revealed inhibition of 75% and 70% neuvasculature in cells treated with taxol alone and Bcl-2 siRNA alone, respectively, while inhibition of 100% neovasculature in cells treated with both agents together (Fig. 5B). These results established that combination of taxol and Bcl-2 siRNA could completely inhibit in vivo angiogenesis.
Fig 5.
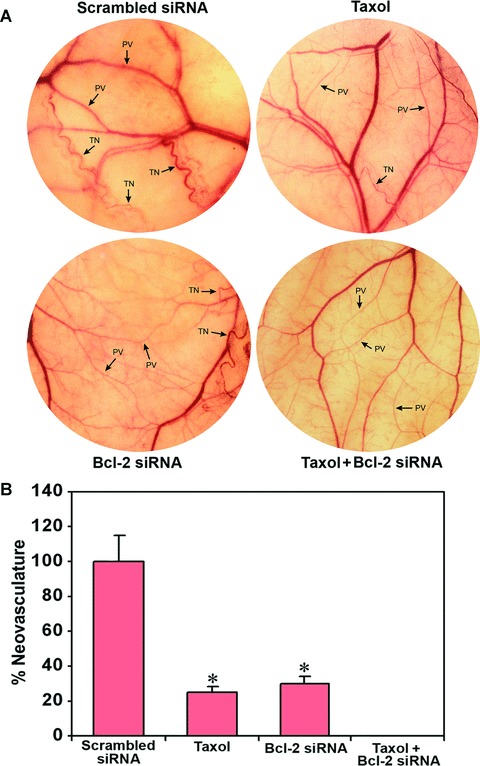
In vivo angiogenesis (dorsal skinfold) assay. Treatment (72 hrs) of cells: transfection with a plasmid vector expressing scrambled siRNA (treated control), 100 nM taxol, transfection with a plasmid vector expressing Bcl-2 siRNA and taxol + Bcl-2 siRNA. The U251MG cells (2 × 105) from each treatment were suspended in 200 μl of serum-free medium and injected into a diffusion chamber. Then, diffusion chambers were surgically implanted under the dorsal skin of nude mice and left for 10 days. (A) In vivo angiogenesis in terms of development of neovasculature. Strong development of tumour-induced neovasculature (TN) with curved thin structures (as indicated by TN arrows) arising from pre-existing vasculature (PV) with relatively straight structures (as indicated by PV arrows) was observed in nude mice with U251MG cells untreated (not shown) and transfected with scrambled siRNA vector. The formation of such neovasculature was considerably reduced in nude mice with U251MG cells treated with taxol or Bcl-2 siRNA and completely inhibited in nude mice with U251MG cells treated with combination of taxol and Bcl-2 siRNA. (B) Quantitative presentation of neovasculature to indicate the extent of in vivo angiogenesis. The measurement of the TN was performed with the help of an ocular micrometer. Values are presented as mean ± S.D. of TN in six animals from each treatment group (*P <0.001 when compared with the scrambled siRNA treatment mean values).
Inhibition of intracranial tumour growth in nude mice
We injected cell suspensions of U251MG untreated control cells, cells transfected with scrambled Bcl-2 siRNA (treated control) and cells treated with either taxol or Bcl-2 siRNA or both agents together, into the cerebrum of nude mice and allowed them to grow for 3 weeks (Fig. 6). All animals were scanned using bioluminescence imaging to examine the changes in tumour growth on days 7, 14 and 21 (Fig. 6). The implantations of untreated control cells and cells transfected with scrambled siRNA resulted in similar rate of cell multiplication and tumourigenesis as indicated by the large bioluminescence image produced by the tumour cells carrying a luciferase gene (Fig. 6). The production of such bioluminescence image was partially inhibited in tumour cells treated with taxol or transfected with Bcl-2 siRNA but remarkably reduced in tumour cells treated with both agents together. The extent of inhibition of intracranial tumour growth in nude mice gradually decreased with prolongation of treatment with the combination of taxol and Bcl-2 siRNA.
Fig 6.
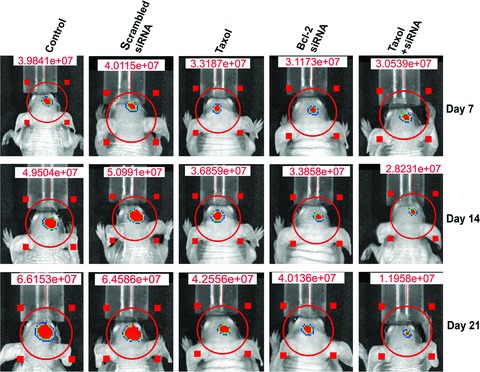
Longitudinal bioluminescence imaging of intracranial tumour growth in nude mice. The U251MG cells (1 × 106) stably transfected with luciferase gene were used for treatments and implantations in nude mice. Treatment (72 hrs) of U251MG cells: control (untreated control), transfection with a plasmid vector expressing scrambled siRNA (treated control), 100 nM taxol, transfection with a plasmid vector expressing Bcl-2 siRNA and taxol + Bcl-2 siRNA. The cells from each treatment were harvested and 1 × 106 cells were suspended in 10 μl of serum-free medium and injected into the cerebrum of nude mice with the help of a stereotaxic apparatus. On day 3 onwards, the mice were injected intraperitoneally with nothing (control) or scrambled siRNA vector (treated control) or taxol or Bcl-2 siRNA vector or taxol + Bcl-2 siRNA vector for 20 days on alternate days. On days 7, 14 and 21, the mice were injected with luciferin for visualization of bioluminescence from luciferase activity to detect the extent of intracranial tumour growth in nude mice. The data are representative of six mice in each treatment group.
Combination of taxol and Bcl-2 siRNA inhibited subcutaneous tumourigenesis and solid tumour development in nude mice
Further, we examined the effects of taxol or/and Bcl-2 siRNA on U251MG cells for capability of subcutaneous tumourigenesis and solid tumour development in nude mice (Fig. 7). The capability of U251MG cells for subcutaneous tumourigenesis was remarkably inhibited after treatment with combination of taxol and Bcl-2 siRNA (Fig. 7A). The mice with control and scrambled siRNA treated U251MG cells produced almost similar well-developed solid tumours in a 6-week period (Fig 7B). During the same period, there was remarkable reduction in solid tumour growth in the animals that received taxol or Bcl-2 siRNA treatment. The treatment with combination of taxol and Bcl-2 siRNA resulted in the most remarkable reduction in solid tumour growth (Fig 7B). The longitudinal measurements of solid tumour volume revealed steady state growth in animals with control and scrambled siRNA vector transfected U251MG cells (Fig. 7C). The tumour growth curve switched significantly downhill after treatment with taxol or Bcl-2 siRNA and it did not rise and remained almost straight after treatment with combination of taxol and Bcl-2 siRNA (Fig. 7C). Moreover, measurements of tumour weight demonstrated 59% and 61% reduction in tumour weight after treatments with taxol alone and Bcl-2 siRNA alone, respectively (Fig. 7D). Most importantly, treatment with combination of taxol and Bcl-2 siRNA resulted in 94% reduction in tumour weight (Fig. 7D). These results confirmed that combination of taxol and Bcl-2 siRNA was the most effective treatment for inhibition of subcutaneous tumourigenesis and solid tumour development in nude mice.
Fig 7.
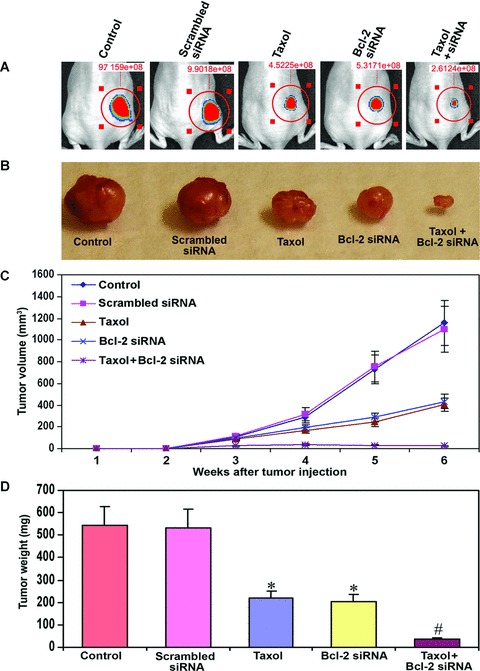
Examination of subcutaneous tumourigenesis and solid tumour development in nude mice. Treatment (72 hrs) of U251MG cells: control (untreated control), transfection with a plasmid vector expressing scrambled siRNA (treated control), 100 nM taxol, transfection with a plasmid vector expressing Bcl-2 siRNA and taxol + Bcl-2 siRNA. (A) Inhibition of subcutaneous tumourigenesis in nude mice. The U251MG cells (carrying the luciferase gene) were treated as mentioned above and the, injected under the dorsal of skin of nude mice. Beginning from day 3, the mice received intraperitoneal injection of nothing (control) or scrambled siRNA vector or taxol or Bcl-2 siRNA vector or taxol + Bcl-2 siRNA vector on alternate days for 20 days. On day 21, the mice were injected with luciferin and visualized for luciferase activity. The data are representative of six mice in each treatment group. (B) Inhibition of solid tumour development in the subcutaneous tissue of nude mice. The U251MG cells (not carrying the luciferase gene) were treated as mentioned above, harvested and suspended in an equal volume of the Matrigel, and 100 μl of this suspension (5 × 106 cells) was injected subcutaneously in nude mice. The animals were left for 2 weeks without any treatment. Afterwards, the mice received intraperitoneal injection of nothing (control) or scrambled siRNA vector or taxol or Bcl-2 siRNA vector or taxol + Bcl-2 siRNA vector on alternate days for 4 weeks. At the end of the sixth week, the tumours were surgically removed, weighed and photographed. (C) Longitudinal measurement of tumour volume in nude mice using a digital vernier caliper. The data are presented as mean ± S.D. of six animals in each treatment group. (D) Measurement of tumour weight following the treatments. The data are presented as mean ± S.D. of six animals in each treatment group (*P < 0.001 when compared with the scrambled siRNA treatment mean values and #P < 0.001 when compared with taxol or Bcl-2 siRNA treatment mean values).
Discussion
Taxol is one of the most powerful anti-cancer drugs that strongly bind to β-tubulin to prevent tumour cell division and induce cell death [18, 19]. In this study, we presented data demonstrating that combination of a low dose of taxol and Bcl-2 siRNA most effectively induces apoptosis in the most aggressive human glioblastoma cells. We further demonstrated that treatment with combination of taxol and Bcl-2 siRNA most dramatically decreased tumour cell invasion, completely prevented in vivo angiogenesis, and efficiently reduced intracranial and subcutaneous tumourigenesis. To the best of our knowledge, this is the first report to show that combination of taxol and Bcl-2 siRNA is a highly effective treatment to induce apoptosis in aggressive human glioblastoma cells and inhibit cell invasion, in vivo angiogenesis and tumour growth both intracranially and subcutaneously in nude mice. Thus, combination of a low dose taxol and Bcl-2 siRNA should be a promising tool for the treatment of malignant brain tumours.
We previously employed various chemotherapeutic strategies to induce apoptosis in human glioblastoma cells both in vitro and in vivo[20, 21]. In the current investigation, we used an innovative strategy to combine a powerful chemotherapy with a gene therapy for controlling the aggressive growth of human glioblastoma. The siRNAs using direct oligonucleotides and small hairpin RNAs (shRNAs) through plasmid vectors are being widely employed for induction of apoptosis in various cancers including glioblastomas. Indeed, synthetic siRNA molecules targeting Bcl-2 in malignant melanoma cells significantly down-regulated Bcl-2 expression at mRNA and protein levels, induced apoptotic cell death and inhibited cell growth [22]. Introduction of Bcl-2 siRNA into human breast cancer MCF-7 cells increased apoptosis and decreased cell proliferation [23]. Here, we observed 70% knockdown of Bcl-2 expression at mRNA and protein levels in human glioblastoma U251MG cells after introduction of a plasmid vector carrying Bcl-2 siRNA cDNA that transcribed cognate siRNA. Subsequently, we observed that treatment with combination of taxol and Bcl-2 siRNA most effectively increased apoptosis in glioblastoma cells, as demonstrated from FACS analysis, double immunofluoresecent stainings of active fragments of caspase-9 and caspase-3, disintegration of nuclei and TUNEL assay.
The primary cause of therapeutic failure in treatment of glioblastoma is invasion, which is the ability of cells to penetrate surrounding tissue as a consequence of degradation of extracellular matrix. Invasion of tumour cells into the Matrigel is widely used to characterize the involvement of extracellular matrix receptors and matrix degrading enzymes, which play prominent roles in tumour progression. Our in vitro Matrigel invasion assay demonstrated highly appreciable impairment of the invasive capability of both human glioblastoma U138MG and U251MG cells after treatment with combination of taxol and Bcl-2 siRNA. It has previously been reported that taxol treatment significantly decreased in vitro cell migration and invasion in malignant ovarian cancer cells [24]. The decrease in cell invasion after treatment of cells with combination of taxol and Bcl-2 siRNA potentiated their growth arrest and apoptosis.
The ability of glioblastoma cells to induce neovascularization plays an important role in tumour invasion and metastasis. Various factors that regulate angiogenesis have been identified [25]. During tumourigenesis, angiogenesis depends on the production of a variety of pro-angiogenic molecules including vascular endothelial growth factor (VEGF) by tumour cells [26]. It is now known that VEGF plays a key role in angiogenesis and tumour cell metastasis [27, 28]. Up-regulation of VEGF in various tumours, especially in malignant brain tumours [26] potentiate the proliferation of microvascular endothelial cells, which are the building blocks of neovascularization. Angiogenesis is a major rate-limiting event for the invasion of glioblatomas, as the presence of blood vessels not only sustains tumour growth but also facilitates tumour cell penetration deep inside normal brain tissue. Thus, development of appropriate therapeutic strategies for blockage of neovascularization is an important milestone in cancer research. Taxol treatment down-regulates VEGF and inhibits in vitro angiogenesis in human ovarian carcinoma cells [29]. In this study, we observed complete inhibition of neovascularization capability of U251MG cells under the dorsal of skin of nude mice after treatment with combination of taxol and Bcl-2 siRNA. This result indicated that glioblastoma cells treated with the combination of taxol and Bcl-2 siRNA failed to secrete angiogenic factors. Silencing of various anti-apoptotic molecules is a novel strategy to enhance the efficacy of chemotherapy. Treatment with combination of Bcl-2 siRNA and a low dose of cisplatin resulted in massive induction of apoptotic death with almost complete suppression of cell growth in malignant melanoma [22]. Silencing of Bcl-2 and Bcl-xL genes in conjunction with chemotherapy provided a potential therapeutic strategy against human hepatoblastoma [30]. Furthermore, down-regulation of Bcl-2 and XIAP using gene silencing technology in human breast cancer MCF-7 cells has been shown to enhance the effects of etoposide and doxorubicin for increasing apoptosis and reducing cell growth [23].
Gene silencing through intraperitoneal injection of plasmid vector carrying the siRNA cDNA is an effective method [11]. Recently, it has also been demonstrated that intraperitoneal injection of a hairpin RNA-expressing plasmid vector targeting urokinase-type plasminogen activator receptor can retard angiogenesis and inhibit intracranial tumour growth in nude mice [31]. In addition, intraperitoneal injections of DY547 and rhodamine-labelled siRNA showed strong signals in the liver, spleen and bone marrow [32]. In the present study, we have observed inhibition of both intracranial and subcutaneous tumourigenesis as well as remarkable decrease (94%) in the solid tumour development in nude mice after treatment with combination of taxol and Bcl-2 siRNA.
In conclusion, our results demonstrate that a low dose of taxol in combination with a plasmid vector expressing Bcl-2 siRNA effectively induces apoptosis in glioblastoma cells, markedly decreases tumour cell invasion, completely inhibits in vivo angiogenesis, and remarkably suppresses both intracranial and subcutaneous tumour growth in nude mice. Taken together, our study indicates that the combination of taxol and Bcl-2 siRNA offers a novel and potential therapeutic strategy for controlling the aggressive growth of human glioblastoma.
Acknowledgments
This investigation was supported in part by the R01 grants (CA-91460 and NS-57811 to S.K.R.) from the National Institutes of Health.
References
- 1.Pulkkanen KJ, Yla-Herttuala S. Gene therapy for malignant glioma: current clinical status. Mol Ther. 2005;12:585–98. doi: 10.1016/j.ymthe.2005.07.357. [DOI] [PubMed] [Google Scholar]
- 2.Donaldson SS, Laningham F, Fisher PG. Advances toward an understanding of brainstem gliomas. J Clin Oncol. 2006;24:1266–72. doi: 10.1200/JCO.2005.04.6599. [DOI] [PubMed] [Google Scholar]
- 3.Hood JD, Cheresh DA. Role of integrins in cell invasion and migration. Nat Rev Cancer. 2002;2:91–100. doi: 10.1038/nrc727. [DOI] [PubMed] [Google Scholar]
- 4.Zagzag D, Friedlander DR, Margolis B, et al. Molecular events implicated in brain tumor angiogenesis and invasion. Pediatr Neurosurg. 2000;33:49–55. doi: 10.1159/000028975. [DOI] [PubMed] [Google Scholar]
- 5.Kitada S, Pedersen IM, Schimmer AD, et al. Dysregulation of apoptosis genes in hematopoietic malignancies. Oncogene. 2002;21:3459–74. doi: 10.1038/sj.onc.1205327. [DOI] [PubMed] [Google Scholar]
- 6.George J, Banik NL, Ray SK. Bcl-2 siRNA augments taxol mediated apoptotic death in human glioblastoma U138MG and U251MG cells. Neurochem Res. 2009;34:66–78. doi: 10.1007/s11064-008-9659-z. [DOI] [PubMed] [Google Scholar]
- 7.Nuydens R, Dispersyn G, Van Den Kieboom G, et al. Bcl-2 protects neuronal cells against taxol-induced apoptosis by inducing multi-nucleation. Apoptosis. 2000;5:335–43. doi: 10.1023/a:1009683425260. [DOI] [PubMed] [Google Scholar]
- 8.Ling X, Bernacki RJ, Brattain MG, et al. Induction of survivin expression by taxol (paclitaxel) is an early event, which is independent of taxol-mediated G2/M arrest. J Biol Chem. 2004;279:15196–203. doi: 10.1074/jbc.M310947200. [DOI] [PubMed] [Google Scholar]
- 9.Ganesh T, Yang C, Norris A, et al. Evaluation of the tubulin-bound paclitaxel conformation: synthesis, biology, and SAR studies of C-4 to C-3′ bridged paclitaxel analogues. J Med Chem. 2007;50:713–25. doi: 10.1021/jm061071x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huisman C, Ferreira CG, Broker LE, et al. Paclitaxel triggers cell death primarily via caspase-independent routes in the non-small cell lung cancer cell line NCI-H460. Clin Cancer Res. 2002;8:596–606. [PubMed] [Google Scholar]
- 11.George J, Tsutsumi M. siRNA-mediated knockdown of connective tissue growth factor prevents N-nitrosodimethylamine-induced hepatic fibrosis in rats. Gene Ther. 2007;14:790–803. doi: 10.1038/sj.gt.3302929. [DOI] [PubMed] [Google Scholar]
- 12.Metrailler-Ruchonnet I, Pagano A, Carnesecchi S, et al. Bcl-2 protects against hyperoxia-induced apoptosis through inhibition of the mitochondria-dependent pathway. Free Radic Biol Med. 2007;42:1062–74. doi: 10.1016/j.freeradbiomed.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 13.Murphy KM, Ranganathan V, Farnsworth ML, et al. Bcl-2 inhibits Bax translocation from cytosol to mitochondria during drug-induced apoptosis of human tumor cells. Cell Death Differ. 2000;7:102–111. doi: 10.1038/sj.cdd.4400597. [DOI] [PubMed] [Google Scholar]
- 14.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 15.Tolboom TC, Huizinga TW. In vitro matrigel fibroblast invasion assay. Methods Mol Med. 2007;135:413–22. doi: 10.1007/978-1-59745-401-8_27. [DOI] [PubMed] [Google Scholar]
- 16.Sckell A, Leunig M. Dorsal skinfold chamber preparation in mice – studying angiogenesis by intravital microscopy. In: Murray JC, editor. Methods in molecular medicine – angiogenesis protocols. Vol. 46. Totowa, New Jersey: Humana Press; 2001. pp. 95–105. [DOI] [PubMed] [Google Scholar]
- 17.Wachsberger PR, Burd R, Marero N, et al. Effect of the tumor vascular-damaging agent, ZD6126, on the radioresponse of U87 glioblastoma. Clin Cancer Res. 2005;11:835–42. [PubMed] [Google Scholar]
- 18.Díaz JF, Strobe R, Engelborghs Y, et al. Molecular recognition of taxol by microtubules. Kinetics and thermodynamics of binding of fluorescent taxol derivatives to an exposed site. J Biol Chem. 2000;275:26265–76. doi: 10.1074/jbc.M003120200. [DOI] [PubMed] [Google Scholar]
- 19.Ganesh T, Guza RC, Bane S, et al. The bioactive Taxol conformation on β-tubulin: experimental evidence from highly active constrained analogs. Proc Natl Acad Sci USA. 2004;101:10006–11. doi: 10.1073/pnas.0403459101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.George J, Gondi CS, Dinh DH, et al. Restoration of tissue factor pathway inhibitor-2 in a human glioblastoma cell line triggers caspase-mediated pathway and apoptosis. Clin Cancer Res. 2007;13:3507–17. doi: 10.1158/1078-0432.CCR-06-3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karmakar S, Banik NL, Ray SK. Combination of all-trans retinoic acid and paclitaxel-induced differentiation and apoptosis in human glioblastoma U87MG xenografts in nude mice. Cancer. 2008;112:596–607. doi: 10.1002/cncr.23223. [DOI] [PubMed] [Google Scholar]
- 22.Wacheck V, Losert D, Günsberg P, et al. Small interfering RNA targeting bcl-2 sensitizes malignant melanoma. Oligonucleotides. 2003;13:393–400. doi: 10.1089/154545703322617078. [DOI] [PubMed] [Google Scholar]
- 23.Lima RT, Martins LM, Guimarães JE, et al. Specific downregulation of bcl-2 and XIAP by RNAi enhances the effects of chemotherapeutic agents in MCF-7 human breast cancer cells. Cancer Gene Ther. 2004;11:309–16. doi: 10.1038/sj.cgt.7700706. [DOI] [PubMed] [Google Scholar]
- 24.Westerlund A, Hujanen E, Höyhtyä M, et al. Ovarian cancer cell invasion is inhibited by paclitaxel. Clin Exp Metastasis. 1997;15:318–28. doi: 10.1023/a:1018481617275. [DOI] [PubMed] [Google Scholar]
- 25.Pandya NM, Dhalla NS, Santani DD. Angiogenesis – a new target for future therapy. Vascul Pharmacol. 2006;44:265–74. doi: 10.1016/j.vph.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 26.Machein MR, Plate KH. VEGF in brain tumors. J Neurooncol. 2000;50:109–20. doi: 10.1023/a:1006416003964. [DOI] [PubMed] [Google Scholar]
- 27.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–76. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 28.Goh PP, Sze DM, Roufogalis BD. Molecular and cellular regulators of cancer angiogenesis. Curr Cancer Drug Targets. 2007;7:743–58. doi: 10.2174/156800907783220462. [DOI] [PubMed] [Google Scholar]
- 29.Hata K, Osaki M, Dhar DK, et al. Evaluation of the antiangiogenic effect of taxol in a human epithelial ovarian carcinoma cell line. Cancer Chemother Pharmacol. 2004;53:68–74. doi: 10.1007/s00280-003-0693-x. [DOI] [PubMed] [Google Scholar]
- 30.Lei XY, Zhong M, Feng LF, et al. siRNA-mediated Bcl-2 and Bcl-xL gene silencing sensitizes human hepatoblastoma cells to chemotherapeutic drugs. Clin Exp Pharmacol Physiol. 2007;34:450–6. doi: 10.1111/j.1440-1681.2007.04593.x. [DOI] [PubMed] [Google Scholar]
- 31.Gondi CS, Lakka SS, Dinh DH, et al. Intraperitoneal injection of a hairpin RNA-expressing plasmid targeting urokinase-type plasminogen activator (uPA) receptor and uPA retards angiogenesis and inhibits intracranial tumor growth in nude mice. Clin Cancer Res. 2007;13:4051–60. doi: 10.1158/1078-0432.CCR-06-3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Larson SD, Jackson LN, Chen LA, et al. Effectiveness of siRNA uptake in target tissues by various delivery methods. Surgery. 2007;142:262–9. doi: 10.1016/j.surg.2007.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]