

Abstract
Primary and metastatic neoplasms of the liver account for more than a million deaths per year worldwide. Despite decades of research, effective novel therapies for these cancers are urgently needed. Oncolytic virotherapeutics represent a novel class of pharmacophore that holds promise for the treatment of hepatic neoplasms. Cancer-specific replication is followed by oncolysis, virus spreading and infection of adjacent cancer cells. This process is then repeated. Virotherapeutics target multiple genetic pathways involved in carcino-genesis, and demonstrate activity against apoptosis-resistant tumour cells. This platform can also exploit the advantage of multiple intrinsic anti-cancer therapeutic mechanisms, combining direct viral oncolysis with therapeutic transgene expression. Recent advances in pre-clinical and clinical studies are revealing the potential of this unique therapeutic class, in particular for liver cancers. This review summarizes the available data on applying oncolytic virotherapeutics to hepatic neoplasms to date, and discusses the challenges and future directions for virotherapy.
Keywords: oncolytic virus, liver tumour, hepatic neoplasm, hepatocellular carcinoma, gene therapy, clinical trial
Introduction
Pre-clinical studies
Clinical studies
Challenges for the use of virotherapy agents in liver tumour treatment
Future directions
Introduction
Both primary hepatocellular carcinomas (HCC) and cancers metastasized to the liver are notoriously aggressive. Most patients with unresec tumours do not respond to existing systemic therapies [1]. Recent advance include the approval of multikinase inhibitor sorafenib (Nexavar; Bayer, Morristown, NJ, USA) by the Food and Drug Administration for advanced HCC. Survival of these patients is improved for approximately 2 months [2]. Mortality of colorectal cancers (CRC) largely reflects the occurrence and progression of liver metastases, and treatment for metastatic CRC is nearly always palliative [3]. Bevacizumab (Avastin; Genentech, San Francisco, CA, USA) is recently approved for metastatic CRC, and improved the survival by approximately 4 months when added to standard chemotherapy regimens [4]. These are important advancements for the treatment of these diseases. Some of these advanced primary and secondary liver cancers may also be addressed by locoregional therapies such as radiofrequency ablation and transarterial chemoembolization (TACE) or radioembolization, but only a minority of patients are eligible for these treatment options.
Therefore, novel therapies that act through mechanisms other than those of traditional therapies are urgently needed. In addition, patients will benefit from new therapies that not only prolong survival but also induce significantly higher response rates.
Viral oncolysis has been a recognized phenomenon in human beings for over a century, and engineered cancer-selective viruses have been tested for over a decade [5]. The safety of this therapeutic platform has been consistent, and antitumoural efficacy has been demonstrated in various tumour types [5, 6]. Pre-clinical and clinical studies have identified HCC and other liver tumours as appropriate targets for oncolytic virotherapy. The underlying molecular pathology of these tumours renders them susceptible to hosting viral replication. These tumours are amenable to multiple routes of administration, including direct intratumoural (IT), intraarterial, intraportal, intrabiliary and intravenous (IV) [7]. This review summarizes laboratory and clinical studies in targeting these tumours with oncolytic viruses, and discusses unique challenges and opportunities for this field.
Pre-clinical studies
Both broad-spectrum and tumour type-specific oncolytic viruses have been tested in pre-clinical liver tumour models. The first engineered oncolytic viruses were designed to replicate in and destroy multiple tumour types based on common molecular pathways/mechanisms. As shown in Table 1, these viruses were also tested in liver tumour models.
Table 1.
Pre-clinical studies of the oncolytic virotherapy in liver tumours
| Product name | Virus Species | Genetic deletions in virus/ genetic targets in cancer | Transgene expression | Animal model tested | Dose/administration route | References |
|---|---|---|---|---|---|---|
| hrR3 | HSV-1 | Deletion in ICP6/ RR complementation by cancer cells | None | MC26 murine CRC liver metastases model; MC26 subcutaneous model (pre-immunized) | 5 × 107 pfu / intrasplenic; 1 × 108 pfu / intravenous | [8] |
| rVSV-GFP | VSV | GFP expression/ Inherent tumour selective (IFN-resistance in tumours) | None | MCA-RH777 rat orthotopic HCC model (solitary nodule) | 1 × 108 pfu / intratumoural | [9] |
| 1.3 × 107 pfu / single hepatic arterial infusion (HAI) | [10] | |||||
| rVSV-NDV/F (L289A) | VSV | GFP-expression/ Inherent tumour selective (IFN-resistance in tumours) | fusogenic protein (from Newcastle disease virus) | MCA-RH777 rat orthotopic HCC model (multifocal) | 1 × 107 pfu / 3 HAI | [11] |
| rVSV-F | VSV | None/ Inherent tumour selective (IFN-resistance in tumours) | fusogenic protein (from Newcastle disease virus) | MCA-RH777 rat orthotopic HCC model (multifocal) | 4 × 106 to 4 × 107 pfu / 3 HAI; IFN-α66 IU/Kg | [12] |
| ZD55-Smac, ZD55-TRAIL | Adenovirus | E1B-55K-/E3B-deletion/ p53 pathway abnormality | Smac and TRAIL | BEL7404 (HCC) mouse xenograft model (subcutaneous) | 2 × 109 pfu / intratumoural injection | [13] |
| MV-Edm-CEA, MV-Edm-hNIS | Measles | None/ Inherent tumour selective (IFN-resistance in tumours) | CEA or NIS | HuH7, Hep3B mouse xenograft models (subcutaneous) | 2 × 106 pfu / 5 intratumoural injections | [14] |
| JX-594 | Vaccinia | TK deletion/ High cellular TK drives replication | GM-CSF | VX2 rabbit HCC model; carcinogen-induced rabbit HCC model | 1 × 108 to 1 x 109 pfu / intratumoural and intravenous injections | [15] |
| CV890 | Adenovirus | AFP promoter-driven E1A/ AFP-secreting cells | - | Hep3B mouse xenograft model (subcutaneous) | 1 × 1011 vp / intravenous injection | [19] |
Several oncolytic Herpes simplex virus (HSV) vectors have been tested for liver tumours. Oncolytic HSV hrR3 has a deletion in ICP6 (ribonucleotide reductase; RR), and therefore its replication is restricted to cells with high cellular RR activities. Cancer cells have high cellular RR and therefore support the replication of hrR3. hrR3 replication is highly tumour selective; viral burst titres in CRC cells are up to three logarithmic orders greater than that in normal hepatocytes. IV administration of hrR3 was able to suppress tumour growth in a CRC diffuse liver metastasis model, even when animals were pre-immunized [8].
Ebert et al. tested vesicular stomatitis virus (VSV) expressing green fluorescent protein (GFP) for the treatment of HCC. rVSV-GFP replicates in and destroys exclusively HCC cells but not in benign human or rat hepatocytes. In vivo, a single IT administration of 1 × 10 plaque-forming units (pfu) of rVSV-GFP into an orthotopic solitary tumour was able to slow tumour growth and prolong survival in an immunocompetent rat tumour model [9]. rVSV-GFP was also tested in a multifocal HCC animal model using hepatic arterial infusion (HAI) [10]. The feasibility of re-dosing VSV via HAI was addressed in a separate study. Repeated HAI did not increase liver toxicity, but resulted in enhanced antitumoural efficacy [11]. Prophylactic treatment with interferon (IFN)-α increased the maximal tolerated dose (MTD) for twofold without affecting IT VSV replication [12].
Pei et al. showed that HCC cells express high levels of inhibitor of apoptosis proteins, and are resistant to tumour necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL)-mediated killing. E1B-55K-deleted oncolytic adenovirus expressing second mitochondria-derived activator of caspases (Smac) or TRAIL showed partial antitumoural efficacy in the BEL7404 xenograft tumour model [13]. This study demonstrated that engineering an oncolytic adenovirus with apoptosis-inducing mechanism(s) can greatly enhance efficacy. However, the potential negative impact of enhanced apoptosis on viral replication was not addressed.
Blechacz et al. described the use of oncolytic measles viruses (MV) for the treatment of HCC [14]. The mechanism of cancer selectivity of the Edmonston strain-based measles virus (MV-Edm) vectors is based on its receptor CD46, which is expressed exclusively on most tumour cells. MV-Edm vectors engineered to express transgenes that allow in vivo monitoring (e.g. soluble CEA) were evaluated for the treatment of HCC. Recombinant MV-Edm vectors effectively infected HCC cell lines, resulting in syn-cytium formation that led to cell death. In vivo, MV-Edm vectors were not toxic in susceptible animals, and IT and IV administrations of MV-Edm vectors were able to induce antitumoural efficacy in HCC tumour models [14].
JX-594 is a thymidine kinase-deleted, granulocyte macrophage-colony stimulating factor (GM-CSF)-expressing oncolytic poxvirus. IV delivery of JX-594 was well tolerated and showed efficacy against primary liver tumour, and successfully prevented lung metastases. JX-594 also showed efficacy in a carcinogen-induced tumour model [15]. JX-594 has been tested in human melanoma patients, and safety and antitumoural efficacy has been demonstrated [16]. Based on these data, a phase I clinical trial testing JX-594 in patients with liver tumours was carried out [17, 18].
Tumour type-specific oncolytic viruses were developed by limiting the capability for viral replication to certain host tumour types. This was achieved by engineering the promoters of the viruses to restrict viral replication specifically to cells that express/secrete certain proteins. For instance, engineering an AFP promoter into the virus would limit the viral replication, but not infection, to those AFP overexpressing cells (i.e. HCC tumour cells). Replication of AFP-driven oncolytic adenovirus CV890 in vitro was limited to AFP-secreting HCC cell lines, and not in non-AFP-secreting HCC cells and benign cells. Interestingly, the level of AFP secretion did not seem to affect the level of replication [19]. In vivo, CV890 showed antitumoural efficacy, and doxorubicin synergized with CV890 in killing AFP-secreting HCC cells in vitro and tumours in vivo[19]. Application of these viruses, however, is limited to tumours that activate the transcription of the promoter-containing virus genome. Before clinical benefit can be assessed, the efficiency of promoter-activated transcription needs to be determined, as patients often experience fluctuating AFP levels over time, and antitumoural efficacy is dependent on efficient replication in tumour cells.
One critical issue when considering these pre-clinical models is how relevant they are to clinical practice. The use of orthotopic and spontaneous models provide superior anatomical and physiological correlation. In addition, the use of immunocompetent animals enhances our understanding of the potentially beneficial interaction between these viruses and the host immune system [9, 15, 20]. Therefore, spontaneous, orthotopic, immunocompetent models, although rare, are considered optimal for their close relevance to human cancers. One also needs to consider the susceptibility of the animal species to the virus species tested. Certain human virus species do not replicate well in animals, and hence toxicity might be different from that of infecting susceptible hosts. Using murine viruses in murine models could provide substantially different information regarding safety and biodistribution.
Clinical studies
Since the first report of an engineered oncolytic virus, liver cancers have been targets in clinical studies (Table 2). Liver tumour-targeted trials of oncolytic adenovirus Onyx-015, for example, were some of the first performed. Onyx-015 (aka dl1520) is an E1B-55K-/E3B-deleted adenovirus [21]. IT administration of Onyx-015 for liver metastases was followed by studies using HAI [7, 22, 23]. Similar results were reported in pilot studies testing multiple administration routes [24, 25]. These studies were designed to determine the safety and MTD of Onyx-015 viadifferent administration routes. Biological end-points (e.g. viral replication, cytokine induction, etc.) were also analysed. Overall, Onyx-015 was safe when administered intratumourally, intraperitoneally, intraarterially or intravenously at doses up to 3 × 1011 pfu [7, 22, 24–27]. Subsequently, Onyx-015 was administered via HAI into patients with liver-predominant gastrointestinal cancers receiving 5-FU. Onyx-015 was well tolerated at doses ranging up to 2 × 1012 viral particles (vp), with flu-like symptoms the most common adverse event (AE). No dose-limiting toxicity (DLT) or MTD was reached, and viral replication and objective response were noted in selected cases [26]. In a phase II study, Onyx-015 replication after repeated HAI was shown despite the development of high titres of neutralizing antibodies. Combination with chemotherapy resulted in antitumoural efficacy in several chemotherapy-resistant patients [27]. These pioneering studies demonstrated the feasibility and safety of oncolytic virus by HAI, including in combination with chemotherapy.
Table 2.
Clinical experience of virotherapy in liver cancers
| Virus | Route/ phase | Cancer type/patient number | Doses/schedule (vp: viral particles; pfu: plaque-forming units; other therapies in italics) | AE (G3/G4 episodes; DLT; most freq. AE) | Antitumoural response† | PD | Viral end-points: gene expression, replication, shedding, and pharma-cokinetics | Immune response | References |
|---|---|---|---|---|---|---|---|---|---|
| Ad-d/1520 (Onyx-015; AE1B-55K, AE3B) | I (IT) | GI liver mets/ 19 | 2 × 109 to 2 × 1012 vp/ days 1, 29, 57 | No DLT; fever, asthenia, chills | n.a. | n.a. | n.a. | n.a. | [7] |
| I (HAI) | CRC liver mets/11 | 2 × 108 to 2 × 1012 vp/ days 1, 8, 22, 50, 78; 5-FU 425 mg/m2 iv days 22, 50, 78, Leucovorin 20 mg/m2 iv days 22,50, 78 | No DLT; 30 G3/G4 AE; fever, chills, transaminitis | n.a. (2 PR at high doses) | n.a. | Q-PCR + (blood, d 4) >2 × 1011 vp; 1-5 × 106 genome/ml (d 4) | All Ab (50%+ at baseline) | [26] | |
| II (HAI) | CRC liver mets/27 | 2 × 1012 vp/ days 1, 8, 22, 50, 78; 5-FU 425 mg/m2 iv days 22,50, 78, Leucovorin 20 mg/m2 iv days 22,50, 78 | 27 G3/G4 AE; fever, chills, ALP ↑ | 3/27 (11%) chemo-refractory: 2/24 (8%) | 11/27 (41%) | 6/8 Q-PCR + (blood); 5/7 viremia (genome copies) on d 4 | All Ab (50%+ at baseline); TNF, IFN-γ, IL-1, IL-6, IL-10 induction | [27] | |
| I/ II (IT, HAI, IV) | Liver/ 16 | 1.5 × 108- 1.5 × 1010 vp7days 1,2,15,16,29,30 (IT); days 1-5 (HAI, IV); 5-FU 300 mg/m2 qd x 3m, oxaleplatin 85 mg/m2 q3w (extra-hepatic cases) | No DLT; fever, chills | 0 (3/6 CEA ↓ >50%) | 1/7 (14%) | ISH, EM, HE + (Bx) | n.a. | [24] | |
| II (IV then IT) | HCC/ 5 | 1.5 × 109 vpVday 1 (IV); days 2, 15, 16, 29, 30 (IT) | 3 G3/G4 AE; fever, chills | 1/5 (1/5 AFP ↓) | 4/5 (80%) | HE, EM + (Bx); serum PCR + (disappeared after 4 hrs) | All Ab (100%+ at baseline) | [25] | |
| I/ II (IT, IP) | Hepatobiliary / 19 | 3 × 108 to 5 × 108 vp* (IT) up to 1.5 × 109vp* total; 5 × 108 vp* (IP) for ascites | 6 G3/G4 AE; fever, myalgia, abd pain | 1/19 (8 others have AFP↓ > 50%) | 6/19 (32%) | 0 CPE (urine); 2/2 bile stent PCR +; 4/4 ascites PCR + (d 1-9) | All Ab (100%+ at baseline) | [22] | |
| HSV-NV 1020 (Δ1 copy ICP34.5, ΔUL24, ΔUL56) | I (HAI) | CRC liver mets/ 12 | 3 × 106 to 1 × 108 pfu | No DLT; fever, nausea, headache | Reduced CEA in some patients | n.a. | 1 PCR + (serum, saliva) | n.a. | [29] |
| II (HAI) | CRC liver mets/ 21 | 1 × 108 pfu x 4 | No significant toxicity | 0 (single agent); 3/11 PR (after C/T) | 11/21 | n.a. | n.a. | [30] | |
| VV-JX-594 (ATK, GM-CSF-express-ing) | I (IT) | Primary and secondary liver tumours | 1 × 108 to 3 × 109 pfu | Fever, chills; DLT: transient hyper-bilirubinemia | 70% (Choi); 30% (RECIST) | 1/10 (10%) | All Q-PCR+ (serum) | All ↑ Ab (21%+ at baseline); TNF, IFN-γ induction | [17], [18] |
Abbreviations: AE: adverse effect; DLT: dose-limiting toxicity; HCC: hepatocellular carcinoma; CRC: colorectal carcinoma; IT: intratumoural; IP: intraperitoneal; IV: intravenous; HAI: hepatic arterial infusion; EM: electron microscopy; EUS: endoscopic ultrasound; R/T: radiotherapy; C/T: chemotherapy; n.a.: non-available; ND: not done; bx: biopsy; IHC: immunohistochemistry; PCR: polychain reaction; PD: progressive disease; PR: partial response; Q-PCR: quantitative PCR; ISH: in situ hybridization; ALP: alkine phosphate; vp: viral particle and pfu: plaque-forming unit.
Estimated based on particle-to-pfu ratio of 20.
Antitumoural response = complete remission + PR.
HSV mutant NV1020 is a clonal derivative of R7020, which was constructed as a vaccine for HSV, contains deletions in thymidine kinase locus and also across the joint region of long and short components of the HSV genome [28]. Safety of NV1020 in CRC liver metastases through single HAI was tested in a phase I trial [29]. Treatment was well tolerated, with flu-like symptoms being the most common AE. No DLT was noted up to 1 × 108 pfu. However, it is unclear why no further dose escalation was performed. Interestingly, no significant viral replication or induction of inflammatory cytokines was noted after treatment. A phase II study examining four weekly HAI administrations of NV1020 prior to second-line chemotherapy in CRC liver metastases has been recently completed. Patients with measurable liver metastases from relapsing CRC received NV1020 (1 × 108 pfu) by weekly HAI (x4) as single agent, followed by two additional cycles of chemotherapy. Twenty-one patients were treated, among which over 40% showed stable disease, and three patients showed partial regression after chemotherapy [30].
More recently, a clinical trial was conducted using oncolytic vaccinia virus JX-594 (TK-deleted/GM-CSF-expressing Wyeth strain vaccinia virus) in patients with liver tumours [17, 18]. The primary end-point is determination of MTD/MFD and safety. JX-594 was administered via direct IT injection. JX-594 was well tolerated at the MTD (3 × 109 pfu), with transient flu-like symptoms the most common AE. Transient asymptomatic hyperbilirubinemia at the highest dose level (3×109 pfu) was dose-limiting. Despite IT administration, JX-594 genomes were detected in peripheral venous circulation as soon as 15 min. after injection. Secondary and tertiary waves of JX-594 in the blood following replication were also detected (Fig. 1A). Interestingly, despite systemic exposure, there was no significant toxicity to liver or other organs (Fig. 2B). Viral replication was detected in distant tumour sites following JX-594 viremia. Antitumoural efficacy by response evaluation criteria in solid tumours (RECIST) criteria was demonstrated in 30% of evaluable patients, and in 80% when using the Choi criteria [17, 31]. Significant (>50%) reduction of serum tumour markers was also noted in several patients. Furthermore, the trial demonstrated the feasibility of re-dosing patients in the presence of neutralizing antibodies. Patients who developed new tumours after completing the initial JX-594 treatment course were given JX-594 into the new tumours. Despite the presence of high titres of neutralizing antibodies, these tumours responded to JX-594 administration similarly to the original tumours (Fig. 2A). This is consistent with previous reports that the presence of circulating neutralizing antibodies does not preclude the antitumoural efficacy of locally delivered oncolytic virus [5, 7]. Phase II study with IT JX-594 in HCC is underway.
Figure 1.
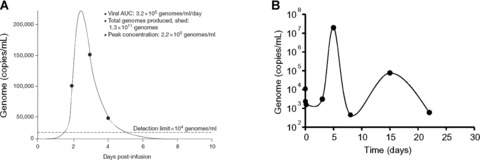
(A) Systemic JX-594 genome levels after intratumoural administration. Reproduced from [17]. (B) Mathematical modelling of oncolytic virus Onyx-015 replication after hepatic arterial infusion. Reproduced from [27].
Figure 2.
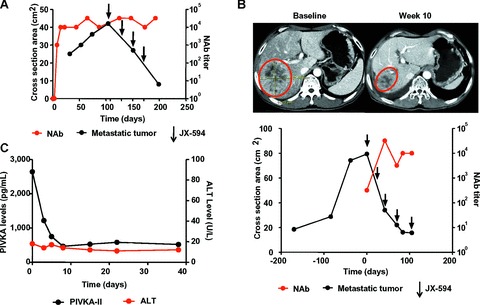
JX-594 induces antitumoural efficacy without hepatotoxicity in the presence of neutralizing antibodies. Representative data from one patient who underwent long-term JX-594 treatment and developed an extrahepatic lesion that received four cycles of treatment. (A) Objective tumour response (after four cycles) of metastatic tumour in neck, injected after induction of high titre neutralizing antibodies to JX-594. Black circles: tumour measurement; red circles: neutralizing antibody titres; arrows: JX-594 treatment. Reproduced from (18). (B) AFP and ALT levels throughout the study. The last four JX-594 treatments were given to the neck tumour. Black circles: AFP levels; red circles: ALT levels.
Challenges for the use of virotherapy agents in liver tumour treatment
For liver tumours, one of the biggest challenges is proving whether local control and/or cure of intrahepatic disease will result in clinical benefit, as measured by overall survival and quality of life. Although survival benefit has not been demonstrated in all studies, TACE is widely used in unresec HCC, and is being validated for other liver-predominant tumours [32]. HAI increases overall survival and progression-free survival in patients with post-resection CRC liver metastases [33]. For HCCs, an additional challenge is the high prevalence of underlying liver cirrhosis and dysfunction. HCC most often arises from cirrhotic livers, and severe liver cirrhosis limits the tolerance for collateral hepatocel-lular damage. Certain virus species are hepatotropic. For example, the liver has been shown to be a critical organ for adenovirus tropism, and therefore patients with poor hepatic reserve may be especially susceptible to adenovirus-related liver toxicity after receiving intrahepatic or systemic virotherapy. Dosing and toxicity monitoring will require special attention in these patients.
In addition, our previous clinical experiences have shown that effective locoregional therapy frequently results in transient tumour swelling prior to tumour shrinkage [17, 27, 34]. Possible mechanisms include edema formation (e.g.due to vascular leakage) and cellular and/or cytokine-mediated acute local inflammation. The mechanism of action for this phenomenon needs further exploration. The frequency and clinical relevance of the transient swelling will vary by tumour type and location. Obstruction of the biliary system, inferior vena cava, and other vessels has been observed when the treated tumour was adjacent to these structures.
Response and efficacy assessment also need refinement. Recent studies on multikinase inhibitors (e.g.sorafenib, sunitinib, etc.) have shown that tumour response and clinical benefit are reflected by changes in tumour character (e.g.density) rather than by changes in size only [31]. While tumour sizes may remain unchanged, tumour necrosis may be induced by these agents, with objective clinical benefit [31]. These studies showed that new imaging criteria (e.g.Choi criteria) increases the prediction value of clinical benefit when compared to standard RECIST criteria [35]. For example, Choi criteria, currently applied to response assessment in gastrointestinal stromal tumours only, are being actively evaluated in several HCC trials with small molecules.
Similar phenomena have been noted for stereotactic radiotherapy, TACE, radioembolization, and ablative techniques such as radiofrequency ablation. Oncolytic virotherapy is another platform that will require modification of the paradigm of how to measure efficacy. Tumour necrosis is indeed a major mechanism of effective virotherapy. Therefore, response may be explained by changes in functional imaging such as fluorodeoxyglucose-positron emission tomgraphy (FDG-PET), and changes in vascularity as shown by perfusion computed tomography (CT) [19] or dynamic contrast-enhanced magnetic resonance imaging (DCE-MRI). These technologies may prove to be superior to traditional WHO or RECIST criteria when assessing efficacy in liver tumours.
Future directions
Oncolytic virotherapy holds promise for the treatment of liver tumours. For this therapeutic platform to be successful, however, several clinical development strategy issues need to be refined. The optimal routes of administration will need to be determined, and will vary according to viral species, cancer types and stages. Local and/or locoregional administration (IT, HAI, portal venous, intrabiliary, retrograde hepatic venous) can each deliver high titre of viruses. Direct IT injection into tumours may theoretically maximize tumour targeting and subsequent viral replication. The spectrum of AEs in human beings has been relatively mild and accep to date. Efficacy has not been jeopardized by the development of neutralizing antibodies. However, the IT route may be limited by the feasibility of injecting multiple tumours or micrometastases, and the anatomical characteristics of some tumours represent higher risk of injections (e.g.close proximity to diaphragm, major vessels or biliary structures). HAI, either through transfemoral catheterization or via an implanted pump, can deliver the viruses directly to the tumours, can be repeated, can be combined with TACE or other embolization techniques, and could be better tolerated than systemic delivery. Intratumoural viral replication and persistence and systemic viremia have been demonstrated with both IT and intraarterial administration routes [6, 17, 27] (Fig. 1). Systemic delivery, on the other hand, could theoretically deliver the viruses to all tumours irrespective of location including outside of the liver, but the quantity of virus that reaches each tumour would be severely limited by pulmonary, vascular, and non-specific uptake and adhesion. The efficacy could also be severely limited by circulating neutralizing antibodies. In addition, toxicities tend to be more profound than with local administrations, and may be more likely to involve organs other than the targeted liver.
When evaluating the efficacy and clinical response to viral oncolysis, the appropriate metrics need to be established. Local tumour control in the liver has been demonstrated to prolong survival and/or to improve the quality of life. Chemoembolization has proven to be beneficial in certain unresec HCC patients, and systemic chemotherapy prolongs disease-free survival in CRC patients with liver metastases [36–38]. Therefore, local and locoregional administration of oncolytic viruses should be compared with TACE, and systemic virotherapy should be compared with sorafenib (in HCC) [1, 38]. In contradistinction to TACE and other embolic techniques, though, oncolytic viruses can enter the systemic circulation within minutes despite intraparenchy-mal administration. This is likely due to leaky tumour vascula-ture. Replication and generations of viral shedding may enhance the distribution and duration of viral treatment. Antitumoural immunity may also be induced with IT injection. Thus, unlike the situation with TACE, evaluation of untreated distant tumours needs to be included when evaluating the efficacy of oncolytic viral treatments.
Given the feasibility of re-dosing patients locally in the presence of high level neutralizing antibodies, it is possible that local and systemic administration routes could complement each other. For instance, IT and/or intraarterial administrations can be used to target tumours that are amenable to treatment, followed by systemic administration to target micrometastases.
Another important direction of investigation is to study potential interactions between oncolytic viruses and other molecular therapeutics. This is important as several oncolytic viruses target specific cancer-associated signalling pathways (e.g.poxvirus for EGF pathway), which are also targets of those molecular therapeutics. Small molecule-based drugs are being approved for multiple cancer types, which will affect the clinical development strategies of oncolytic viruses. It might be necessary to compare virotherapeutics to small molecules, or to explore the effect of combining these agents. As virus replication is dependent on the activation of those pathways in cancer cells, blocking those pathways with small molecules could have deleterious effects on viral replication. Furthermore, systemic circulation of oncolytic viruses relies at least in part on leaky tumour vasculature. Anti-angiogenic agents are known to ‘normalize’ tumour vasculature [39], and therefore it is possible that systemic virus exposure will be affected when anti-angiogenic agents are administered prior to or concurrent with oncolytic viruses. Pre-clinical studies should therefore focus on different dosing schema to optimize the effects of combining different agents.
In addition to the antitumoural effect, it will be crucial to determine what impact oncolytic virotherapy has on underlying hepatitis in HCC patients. Three HCC patients treated with oncolytic vaccinia virus JX-594 via IT injection were chronically infected with hepatitis B virus (HBV) and had been treated with antiviral medications prior to study enrolment. After JX-594 treatment, all three patients experienced sustained reduction in HBV genome levels, with decreases ranging from 51% to 90% (Fig. 3) [18]. Possible mechanisms include induction of antiviral cytokines, many of which have been known to have anti-HBV effects (e.g. TNF-α, IFN-γ) [18]. Whether this phenomenon is applicable to other HBV- or hepatitis C virus (HCV)-associated HCCs is yet to be determined. In addition, most patients with chronic HBV infection are also on chronic antiviral medications, and the impact of concurrent anti-hepatitis medications on oncolytic viruses needs to be studied. Testing virotherapeutics in HBV animal models [40] will also provide more insights.
Figure 3.

Long-term inhibition of HBV replication in a representative HCC patient after treatment with oncolytic poxvirus JX-594.
Finally, how liver cirrhosis affects the biology of oncolytic viruses and vice versa need to be studied. Pre-clinical models suggest that biodistribution of systemically administered viruses could be different in the presence of cirrhosis. Adenovirus, for example, has been tested in mice with cirrhotic livers, and instead of liver, the predominant organ in which the virus accumulates immediately after IV delivery is lung [41]. There are little data on other virus species, but these interactions may prove to be complex and will warrant further study.
In summary, oncolytic virotherapeutics are a novel and promising treatment platform for HCC and other liver tumours. This new therapy may complement the mainstream regimens based upon surgery, ablation, chemotherapy, targeted therapies, radiation and embolization. More translational research and clinical trials that address the critical issues outlined will be key to the success of this therapeutic class.
References
- 1.El-Serag HB, Marrero JA, Rudolph L, Reddy KR. Diagnosis and treatment of hepatocellular carcinoma. Gastroenterology. 2008;134:1752–63. doi: 10.1053/j.gastro.2008.02.090. [DOI] [PubMed] [Google Scholar]
- 2.Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–90. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 3.Saad ED, Hoff PM. Chemotherapy of metastatic colorectal cancer. Curr Treat Options Gastroenterol. 2005;8:239–47. doi: 10.1007/s11938-005-0016-x. [DOI] [PubMed] [Google Scholar]
- 4.Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic col-orectal cancer. N Engl J Med. 2004;350:2335–42. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- 5.Liu TC, Galanis E, Kirn D. Clinical trial results with oncolytic virotherapy: a century of promise, a decade of progress. Nat Clin Pract Oncol. 2007;4:101–17. doi: 10.1038/ncponc0736. [DOI] [PubMed] [Google Scholar]
- 6.Liu TC, Kirn D. Systemic efficacy with oncolytic virus therapeutics: clinical proof-of-concept and future directions. Cancer Res. 2007;67:429–32. doi: 10.1158/0008-5472.CAN-06-2871. [DOI] [PubMed] [Google Scholar]
- 7.Kirn D. Clinical research results with dl1520 (Onyx-015), a replication-selective adenovirus for the treatment of cancer: what have we learned? Gene Ther. 2001;8:89–98. doi: 10.1038/sj.gt.3301377. [DOI] [PubMed] [Google Scholar]
- 8.Yoon SS, Nakamura H, Carroll NM, et al. An oncolytic herpes simplex virus type 1 selectively destroys diffuse liver metas-tases from colon carcinoma. FASEB J. 2000;14:301–11. [PubMed] [Google Scholar]
- 9.Ebert O, Shinozaki K, Huang TG, et al. Oncolytic vesicular stomatitis virus for treatment of orthotopic hepatocellular carcinoma in immune-competent rats. Cancer Res. 2003;63:3605–11. [PubMed] [Google Scholar]
- 10.Shinozaki K, Ebert O, Kournioti C, et al. Oncolysis of multifocal hepatocellular carcinoma in the rat liver by hepatic artery infusion of vesicular stomatitis virus. Mol Ther. 2004;9:368–76. doi: 10.1016/j.ymthe.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 11.Shinozaki K, Ebert O, Woo SL. Eradication of advanced hepatocellular carcinoma in rats via repeated hepatic arterial infusions of recombinant VSV. Hepatology. 2005;41:196–203. doi: 10.1002/hep.20536. [DOI] [PubMed] [Google Scholar]
- 12.Shinozaki K, Ebert O, Suriawinata A, et al. Prophylactic alpha interferon treatment increases the therapeutic index of oncolytic vesicular stomatitis virus virotherapy for advanced hepatocellular carcinoma in immune-competent rats. J Virol. 2005;79:13705–13. doi: 10.1128/JVI.79.21.13705-13713.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pei Z, Chu L, Zou W, et al. An oncolytic adenoviral vector of Smac increases antitumor activity of TRAIL against HCC in human cells and in mice. Hepatology. 2004;39:1371–81. doi: 10.1002/hep.20203. [DOI] [PubMed] [Google Scholar]
- 14.Blechacz B, Splinter PL, Greiner S, et al. Engineered measles virus as a novel oncolytic viral therapy system for hepato-cellular carcinoma. Hepatology. 2006;44:1465–77. doi: 10.1002/hep.21437. [DOI] [PubMed] [Google Scholar]
- 15.Kim JH, Oh JY, Park BH, et al. Systemic armed oncolytic and immunologic therapy for cancer with JX-594, a targeted poxvirus expressing GM-CSF. Mol Ther. 2006;14:361–70. doi: 10.1016/j.ymthe.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 16.Mastrangelo MJ, Maguire HC, Jr, Eisenlohr LC, et al. Intratumoral recombi-nant GM-CSF-encoding virus as gene therapy in patients with cutaneous melanoma. Cancer Gene Ther. 1999;6:409–22. doi: 10.1038/sj.cgt.7700066. [DOI] [PubMed] [Google Scholar]
- 17.Park BH, Hwang TH, Liu TC, et al. Use of a targeted oncolytic poxvirus, JX-594, in patients with refractory primary or metastatic liver cancer: a phase I trial. The Lancet Oncology. 2008;9:533–42. doi: 10.1016/S1470-2045(08)70107-4. [DOI] [PubMed] [Google Scholar]
- 18.Liu TC, Hwang T, Park BH, et al. The targeted oncolytic poxvirus JX-594 demonstrates antitumoral, antivascular, and anti-HBV activities in patients with hepatocellular carcinoma. Mol Ther. 2008;16:1637–42. doi: 10.1038/mt.2008.143. [DOI] [PubMed] [Google Scholar]
- 19.Li Y, Yu DC, Chen Y, et al. A hepatocellular carcinoma-specific adenovirus variant, CV890, eliminates distant human liver tumors in combination with doxorubicin. Cancer Res. 2001;61:6428–36. [PubMed] [Google Scholar]
- 20.Huang TG, Ebert O, Shinozaki K, et al. Oncolysis of hepatic metastasis of colorectal cancer by recombinant vesicular stomatitis virus in immunecompetent mice. Mol Ther. 2003;8:434–40. doi: 10.1016/s1525-0016(03)00204-1. [DOI] [PubMed] [Google Scholar]
- 21.Bischoff JR, Kirn DH, Williams A, et al. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science. 1996;274:373–6. doi: 10.1126/science.274.5286.373. [DOI] [PubMed] [Google Scholar]
- 22.Makower D, Rozenblit A, Kaufman H, et al. Phase II clinical trial of intralesional administration of the oncolytic aden-ovirus ONYX-015 in patients with hepatobiliary tumors with correlative p53 studies. Clin Cancer Res. 2003;9:693–702. [PubMed] [Google Scholar]
- 23.Hamid O, Varterasian ML, Wadler S, et al. Phase II trial of intravenous CI-1042 in patients with metastatic colorec-tal cancer. J Clin Oncol. 2003;21:1498–504. doi: 10.1200/JCO.2003.09.114. [DOI] [PubMed] [Google Scholar]
- 24.Habib NA, Sarraf CE, Mitry RR, et al. E1B-deleted adenovirus (dl1520) gene therapy for patients with primary and secondary liver tumors. Hum Gene Ther. 2001;12:219–26. doi: 10.1089/10430340150218369. [DOI] [PubMed] [Google Scholar]
- 25.Habib N, Salama H, Abd El Latif Abu Median A, et al. Clinical trial of E1B-deleted adenovirus (dl1520) gene therapy for hepatocellular carcinoma. Cancer Gene Ther. 2002;9:254–9. doi: 10.1038/sj.cgt.7700431. [DOI] [PubMed] [Google Scholar]
- 26.Reid T, Galanis E, Abbruzzese J, et al. Intra-arterial administration of a replication-selective adenovirus (dl1520) in patients with colorectal carcinoma metastatic to the liver: a phase I trial. Gene Ther. 2001;8:1618–26. doi: 10.1038/sj.gt.3301512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reid T, Galanis E, Abbruzzese J, et al. Hepatic arterial infusion of a replication-selective oncolytic adenovirus (dl1520): phase II viral, immunologic, and clinical endpoints. Cancer Res. 2002;62:6070–9. [PubMed] [Google Scholar]
- 28.Delman KA, Bennett JJ, Zager JS, et al. Effects of preexisting immunity on the response to herpes simplex-based oncolytic viral therapy. Hum Gene Ther. 2000;11:2465–72. doi: 10.1089/10430340050207957. [DOI] [PubMed] [Google Scholar]
- 29.Kemeny N, Brown K, Covey A, et al. Phase I, open-label, doseescalating study of a genetically engineered herpes simplex virus, NV1020, in subjects with metastatic colorectal carcinoma to the liver. Hum Gene Ther. 2006;17:1214–24. doi: 10.1089/hum.2006.17.1214. [DOI] [PubMed] [Google Scholar]
- 30.Nemunaitis J, Chari RS, Gambhir SS, et al. Phase II assessment of NV1020, a gene modified oncolytic herpes simplex viral therapeutic in advanced colorectal cancer (CRC) metastatic to liver. J Clin Oncol. 2008;26:15S. [Google Scholar]
- 31.Choi H, Charnsangavej C, Faria SC, et al. Correlation of computed tomography and positron emission tomography in patients with metastatic gastrointestinal stromal tumor treated at a single institution with imatinib mesylate: proposal of new computed tomography response criteria. J Clin Oncol. 2007;25:1753–9. doi: 10.1200/JCO.2006.07.3049. [DOI] [PubMed] [Google Scholar]
- 32.Ruan DT, Warren RS. Liver-directed therapies in colorectal cancer. Semin Oncol. 2005;32:85–94. doi: 10.1053/j.seminoncol.2004.09.025. [DOI] [PubMed] [Google Scholar]
- 33.Elias D, Goere D, Boige V, et al. Outcome of posthepatectomy-missing colorectal liver metastases after complete response to chemotherapy: impact of adjuvant intra-arterial hepatic oxaliplatin. Ann Surg Oncol. 2007;14:3188–94. doi: 10.1245/s10434-007-9482-9. [DOI] [PubMed] [Google Scholar]
- 34.Sze DY, Freeman SM, Slonim SM, et al. Dr. Gary J. Becker Young Investigator Award: intraarterial adenovirus for metastatic gastrointestinal cancer: activity, radiographic response, and survival. J Vasc Interv Radiol. 2003;14:279–90. doi: 10.1097/01.rvi.0000058422.01661.1e. [DOI] [PubMed] [Google Scholar]
- 35.Benjamin RS, Choi H, Macapinlac HA, et al. We should desist using RECIST, at least in GIST. J Clin Oncol. 2007;25:1760–4. doi: 10.1200/JCO.2006.07.3411. [DOI] [PubMed] [Google Scholar]
- 36.Llovet JM, Bruix J. Novel advancements in the management of hepatocellular carci-noma in 2008. J Hepatol. 2008;48:S20–37. doi: 10.1016/j.jhep.2008.01.022. [DOI] [PubMed] [Google Scholar]
- 37.Lee JJ, Chu E. An update on treatment advances for the first-line therapy of metastatic colorectal cancer. Cancer J. 2007;13:276–81. doi: 10.1097/PPO.0b013e3181570062. [DOI] [PubMed] [Google Scholar]
- 38.Llovet JM, Di Bisceglie AM, Bruix J, et al. Design and endpoints of clinical trials in hepatocellular carcinoma. J Natl Cancer Inst. 2008;100:698–711. doi: 10.1093/jnci/djn134. [DOI] [PubMed] [Google Scholar]
- 39.Jain RK, Duda DG, Clark JW, Loeffler JS. Lessons from phase III clinical trials on anti-VEGF therapy for cancer. Nat Clin Pract Oncol. 2006;3:24–40. doi: 10.1038/ncponc0403. [DOI] [PubMed] [Google Scholar]
- 40.Huang LR, Wu HL, Chen PJ, Chen DS. An immunocompetent mouse model for the tolerance of human chronic hepatitis B virus infection. Proc Natl Acad Sci USA. 2006;103:17862–7. doi: 10.1073/pnas.0608578103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smith JS, Tian J, Lozier JN, Byrnes AP. Severe pulmonary pathology after intravenous administration of vectors in cirrhotic rats. Mol Ther. 2004;9:932–41. doi: 10.1016/j.ymthe.2004.03.010. [DOI] [PubMed] [Google Scholar]