

Abstract
The longest open reading frame of PKHD1 (polycystic kidney and hepatic disease 1), the autosomal recessive polycystic kidney disease (ARPKD) gene, encodes a single-pass, integral membrane protein named polyductin or fibrocystin. A fusion protein comprising its intracellular C-terminus, FP2, was previously used to raise a polyclonal antiserum shown to detect polyductin in several human tissues, including liver. In the current study, we aimed to investigate by immunohistochemistry the detailed polyductin localization pattern in normal (ductal plate [DP], remodelling ductal plate [RDP], remodelled bile ducts) and abnormal development of the primitive intrahepatic biliary system, known as ductal plate malformation (DPM). This work also included the characterization of polyductin expression profile in various histological forms of neonatal and infantile cholestasis, and in cholangiocellular carcinoma (CCC) and hepatocellular carcinoma (HCC). We detected polyductin expression in the intrahepatic biliary system during the DP and the RDP stages as well as in DPM. No specific staining was found at the stage of remodelled bile ducts. Polyductin was also detected in liver biopsies with neonatal cholestasis, including mainly biliary atresia and neonatal hepatitis with ductular reaction as well as congenital hepatic fibrosis. In addition, polyductin was present in CCC, whereas it was absent in HCC. Polyductin was also co-localized in some DP cells together with oval stem cell markers. These results represent the first systematic study of polyductin expression in human pathologies associated with abnormal development of intrahepatic biliary tree, and support the following conclusions: (i) polyductin expression mirrors developmental properties of the primitive intrahepatic biliary system; (ii) polyductin is re-expressed in pathological conditions associated with DPM and (iii) polyductin might be a potential marker to distinguish CCC from HCC.
Keywords: polyductin, fibrocystin, bile duct, ductal plate, hepatocellular carcinoma, cholangiocellular carcinoma, ARPKD, PKHD1 gene
Introduction
Autosomal recessive polycystic kidney disease (ARPKD) is a significant cause of polycystic kidney disease in paediatric patients, with an estimated incidence of 1:20,000 live births [1]. ARPKD affects both kidneys and biliary tract and is associated with a variable profile of phenotypes. Patho-anatomical findings related to this disease include fusiform dilatation of renal collecting ducts and biliary dysgenesis associated with portal tract fibrosis. Mutations in the PKHD1 (polycystic kidney and hepatic disease 1) gene are responsible for all typical forms of ARPKD [2, 3]. Mutations in this gene can also be found in patients with primary diagnosis of congenital hepatic fibrosis (CHF) [4]. The PKHD1 gene is associated with a complex splicing pattern and its longest open reading frame product, polyductin, also known as fibrocystin, is a single transmembrane domain glycoprotein with a molecular weight of more than 440 kD in its unglycosylated form [5].
A fusion protein containing the polyductin’s intracellular carboxy-terminus, FP2, was previously used as a target to generate a polyclonal antibody that was applied to study the polyductin expression profile in human tissues [6]. The anti-FP2 purified antiserum stains the renal collecting ducts and thick ascending limbs of Henle in humans and the branching ureteric bud in mouse. Specific staining was also detected in murine intrahepatic and extrahepatic biliary system [7, 8]. The absence of positive signal in human adult hepatocytes might be a clue to speculate that FP2-positive foetal hepatoblasts could undergo transformation into other cell types and thus participate in biliary tract formation. We hypothesized that the polyductin expression profile might be abnormal in liver disorders associated with biliary tract anomalies. Because ductal plate malformation (DPM) is linked to an abnormal development of the primitive intrahepatic biliary system, its study should contribute to a better understanding of the maturation of these structures [9, 10]. Although controversially discussed, biliary disorders with or without metabolic diseases may show an increased risk to develop cancer (more commonly cholangiocellular carcinoma (CCC) than hepatocellular carcinoma (HCC)) [11]. CCC can arise in solitary cysts of the liver and has been reported in CHF, Caroli’s disease, von Meyenburg complexes and polycystic liver disease. Other reported associations of CCC with biliary disorders include choledochal cyst, primary sclerosing cholangitis, and biliary atresia (BA). The cancer may arise either in the cyst wall itself, or in remnant tissue or undilated parts of the extrahepatic or intrahepatic biliary system [12].
During liver development, liver progenitors differentiate into hepatocytes or biliary cells and data seem favour the notion that the segregation between hepatocytes and biliary cells is dependent on a gradient of Activin/TGFbeta signalling, and an aberrant and/or excessive differentiation by activin/TGFbeta signalling is at the basis of the abnormality of development of the intrahepatic biliary system [13, 14]. Lack of remodelling of the primitive biliary system results in the persistence of an excess of embryonic bile duct structures. This abnormality has been termed the DPM [15].
In the current work, we investigated its expression pattern in foetal, neonatal, infantile disease states and adult hepatobiliary carcinomas. Our findings suggest that polyductin may be involved in the origin of the intrahepatic biliary system and that specific insults may lead to its increased expression during different phases of organogenesis. This proposed model might provide consequential information to better understand biliary dysgenesis.
Materials and methods
Definitions
The intrahepatic bile ducts develop out of primitive hepatic epithelial cell sheets and are mostly determined by the progressive development and branching of the portal vein with its surrounding mesenchyme [16].
The primitive hepatic epithelial cells that are in direct contact with the mesenchyme around the portal vein transform into bile duct type cells, first in one and later in two layers. The development of the intrahepatic biliary system comprises three stages: ductal plate (DP), remodelling ductal plate (RDP) and remodelled bile ducts (RBD). First, a cleft occurs at ∼9th–12th gestational week between the two layers of bile duct type epithelial cells (DP stage). Second, biliary cells migrate from the DP into the mesenchyme whereas biliary structures are still partially in contact with the DP, at ∼13th–17th gestational week (RDP stage). Third, remodelled bile ducts are now centrally located in the portal tracts and have no contact with the DP, at ∼18th–40th gestational week (RBD). However, because the intrahepatic bile duct development proceeds from the hilum to peripheral portions of the liver, two or more of these stages may be present in the same liver specimen, independently of the gestational age [17, 18].
Morphologically, DPM is manifested by persistence or incomplete disappearance of the DP and excessive ramification of the leading portal vein and is frequently seen in Meckel syndrome [19–21].
Several pathological conditions associated with liver disease at neonatal and infantile age were examined, including mostly BA, neonatal hepatitis (NH) and paucity of intrahepatic bile ducts. BA consists of a partial or complete obliteration of mainly extrahepatic bile ducts and covers up to 30% of the neonatal cholestasis cases [22–24]. In BA, the extrahepatic tract shows often a shrunken or missing gallbladder, whereas intrahepatic bile ducts may form irregular, distorted tubular structures with portal fibrosis. NH is an intrahepatic cholestatic non-obstructive reaction of viral, metabolic or idiopathic nature, characterized by cholestasis, lobular inflammation, focal fibrosis and hepatocytes with giant cell changes [25, 26]. Paucity of the intrahepatic bile ducts (PIBD) is observed in diseases with loss of interlobular bile ducts [27]. Diagnosis is based on the number of the interlobular bile ducts, defined as round duct with a well-developed lumen and an arteriole near the centre of the portal tract. Liver biopsies with at least six portal tracts are associated with a normal development of the intrahepatic biliary system if small portal tracts contain 0.9 to 1.8 interlobular bile ducts per tract. PIBD is defined by a BD/PT ratio below 0.5, whereas ratios between 0.6 and 0.9 may be considered suspicious for paucity or show development retardation of the intrahepatic biliary system. PIBD is frequently accompanied by cholestasis and portal fibrosis and comprises two clinical forms: syndromic (or Alagille syndrome) and non-syndromic [28]. The non-syndromic form can be observed in association with several neonatal and infantile diseases.
Subjects
Formalin-fixed and paraffin-embedded human liver tissues from 11 foetuses were retrieved from the files of the Institute of Pathology, University of Heidelberg, Germany. Seven samples were from normal foetuses (21.3 ± 4.6 weeks of gestational age, range: 15–29 weeks) and four from foetuses with Meckel syndrome (20.8 ± 1.8 weeks of gestational age, range: 19–23 weeks). The control specimens came either from spontaneous abortions (placental or amniotic cause of death: premature rupture of the membranes, placenta praevia, amnionitis, or acute placenta dysfunction) or elective terminations of pregnancy (e.g. hysterectomy in a mother suffering from mammary carcinoma). These foetuses were checked for the absence of external and/or internal malformations, including normalcy of the extrahepatic and intrahepatic biliary. Gestational age was confirmed by somatometric measurements [29]. To avoid tissue autolysis artifacts autopsies were often performed as rapid procedures (<6 hrs) or at latest within 36 hrs following parental consent. We carried out a preliminary assessment of staining variation with postmortem tissue arising from autopsies performed between 4 and 36 hrs after death. These preliminary studies did not identify any staining variation in this time range (data not shown). Chromosomal studies, when performed by amniocentesis or autopsy, revealed numerically and structurally normal karyotypes. In the foetuses without karyotype analysis, aneuploidy phenotypes were carefully excluded according online dysmorphology databases. Foetuses from pregnant women with history of cigarette smoking, carbohydrate intolerance or drug addiction (confounders) were not included in the study.
Liver tissue was also collected from 60 newborns and infants with age less than 1 year undergoing liver biopsy for abnormal liver function parameters (Table 1). In addition, liver tissue with normal bile duct development from a newborn (cause of death: pneumonia) and an infant (sudden infantile death syndrome) were also evaluated as postnatal controls. These cases were the only two liver specimens obtained at autopsy in this group.
Table 1.
Age at time of the liver biopsy of 60 patients with neonatal and infantile liver disease
| Disease | Number of cases | Age at time of liver biopsy (days) (Mean +/− S.D.) |
|---|---|---|
| PIBD | 16 | 137.87 +/− 147.93 (S.E.M.: 38.19 days) |
| BA | 10 | 87.00 +/− 61.49 (S.E.M.: 20.5 days) |
| NH | 9 | 109.38 +/− 93.08 (S.E.M.: 32.91 days) |
| NC-NOS | 5 | 70.60 +− 67.11 (S.E.M.: 30.01 days) |
| Gly-I | 3 | 295 +/− 191.23 (S.E.M.: 110.41 days) |
| CHF | 2 | 227.5 +/− 74.25 |
| CVD-HI | 2 | 294.5 +/− 70.00 |
| DILD | 2 | 292.0 +/− 42.43 |
| FFAOD | 2 | 70.00 +/− 42.46 |
| CC-LD | 1 | 174 |
| APO-LD | 1 | 266 |
| PDHP-LD | 1 | 343 |
| CMV-H | 1 | 88 |
| WFASS | 1 | 418 |
| CID-NOS | 1 | 106 |
| NBLD-NOS | 1 | 144 |
| NEC-LD | 1 | 140 |
| Sepsis | 1 | 12 |
APO-LD (annular pancreas obstruction-associated liver disease), BA (biliary atresia), CC-LD (choledocal cyst-associated liver disease), CID NOS (congenital immunodeficiency, not otherwise specified), CHF (congenital hepatic fibrosis), CMV-H (cytomegalovirus hepatitis), CVD-HI (cardiovascular disease with hepatic involvement), DILD (drug-induced liver disease), FFAOD (fetal fatty acid oxidation disorder associated with maternal HELLP syndrome (hemolysis, elevated liver enzymes, low platelets), GlyI (glycogenosis type I), NC NOS (neonatal cholestasis not otherwise specified), NH (neonatal hepatitis), PDHP-LD (liver disease associated with deficiency of pyruvate dehydrogenase phosphate), PIBD (paucity of intrahepatic bile ducts), NBLD NOS (nonbiliary liver disease not otherwise specified), NEC-LD (hepatic involvement associated with necrotizing enterocolitis), and WFASS (Wissler-Fanconi allergic subsepsis syndrome). S.D.: standard deviation, S.E.M.: standard error of the mean.
Thirty-nine adult liver samples were analysed, including HCC (n= 16), CCC (n= 23). All specimens included in this study were from liver resection or liver explants (no adult autopsy material). We used liver tissue without neoplastic change close to malignancy as normal control liver tissue. HCC and CCC were reviewed and classified into well, moderately, or poorly differentiated according to conventional WHO criteria, evaluated by two pathologists (HFO, CS). HCC was associated with chronic hepatitis B virus (HBV), hepatitis C virus (HCV) and long-term alcoholism-related liver cirrhosis. Thorotrast exposure was found in eight CCC patients.
To better address specific pathogenesis issues of FP2 staining, we also added the liver tissue of an infant affected with alpha-1-antitrypsin deficiency (Pi ZZ), the liver tissue of an infant with progressive familiar intrahepatic cholestasis, a juvenile liver case of Caroli’s disease as well as an adult case of Caroli’s disease at a cirrhotic stage, and two cases of acute and chronic liver rejection.
Tissue collection (Germany) was always performed in accordance to German federal laws and foetal tissue was obtained from legal abortion material only. This study is related to an extensive DPM study of the liver, carried out primarily on foetal and infantile material at the University of Heidelberg and originally approved by the Ethics Committee of the University of Heidelberg (Resolution 186/98). This study was continuously reviewed and approved in Germany (Heidelberg Ethics Committee), United Kingdom (Bristol Ethics Committee), and Austria (Innsbruck, Departmental Authority as restrospective study) during the period 2002–2007.
Immunohistochemistry
All tissue specimens were routinely fixed in formalin and embedded in paraffin wax. Three-μm thick tissue sections were mounted onto glass slides pretreated with 2% 3-aminopropyltriethoxysilane (APES, Sigma, Steinheim, Germany) and dried at 37°C overnight. The previously characterized immuno-purified anti-FP2 antiserum was used as described by Menezes et al.[6]. In addition, several antibodies were used to differentiate CCC from HCC (Table 2). The intensity of the FP2-staining was evaluated in pre-formed ductular structures, neo-ductuli, and hepatocytes as none, mild, moderate and intense. When present, we regarded the faint diffuse staining of the hepatocytes as ‘baseline expression’[30]. Immunolabelling was considered antibody-specific according to two criteria: normal serum producing no consistent immunostaining of any cells and signal intensity diminished as the dilution of antibody was increased. The slides were evaluated independently by two authors (LD, CS) without knowledge of the gestational age of the cases and aetiology. A negative control slide was included in each immunostaining, by replacing the primary antibody with normal serum. To validate the staining pattern, fresh collected and rapid formalin-fixed liver specimens were compared with archived liver specimens. The staining pattern did not change between fresh and stored specimens. Moreover, staining was repeated and blindly evaluated for consistency (data not shown).
Table 2.
Antibodies used for immunohistochemical labeling to differentiate cholangiocellular carcinoma (CCC) from hepatocellular carcinoma (HCC) (other than anti-FP2)
| Antibody | Source | Clone | Dilution | Cellular location |
|---|---|---|---|---|
| Anti-CK 7 | Dako Corp., Carpinteria, CA | OV-TL12/30 | 1:100 | Cytoplasmic |
| Anti-CK 19 | Dako Corp., Carpinteria, CA | RCK108 | 1:100 | Cytoplasmic |
| Anti-CEA | Dako Corp., Carpinteria, CA | Polyclonal | 1:400 | Cytoplasmic vs. pericanalicular |
| Hep Par 1 | Dako Corp., Carpinteria, CA | OCH 1E5.2.10 | 1:100 | Cytoplasmic, granular |
CK7: Cytokeratin 7; CK19: Cytokeratin 19, CEA: Carcinoembryonic antigen, Hep Par 1: Hepatocyte Paraffin 1. Selected cases were also stained with alpha-fetoprotein and CD10 (data not shown).
Double immunostaining was also performed to identify FP2 staining in oval cells. The first staining was performed for FP2 by the avidin-biotinylated peroxidase complex method followed by chromogenic reaction with diaminobenzidine (brown). The second staining was for epithelial cell adhesion molecule (Ep-CAM) (1:75), cytokeratin 19 (CK-19) (1:100) and mucin-1 (MUC-1) (1:100) by the alkaline phosphatase anti-alkaline phosphatase method (Dako Corp. Carpinteria, CA, USA), followed by chromogenic reaction with Vector blue (Vector Laboratories, Burlingame, CA, USA). As controls for the specificity of double immunostaining, the specific antibodies were replaced by normal serum.
Statistical analysis
The Fisher’s exact test was used to compare anti-FP2 staining between CCC and HCC (GraphPad 2.01, San Diego, CA, USA). All P-values were two-sided. Values of less than 0.05 were considered to indicate statistical significance. Intra- and interobserver differences were less than 5% and non-concordant cases were re-evaluated simultaneously by both observers (LD, CS).
Results
Foetal liver
Liver tissue from seven foetuses without intra- and extrahepatic biliary system abnormalities was initially examined. In the DP stage (Fig. 1A), FP2 staining was diffusely more intense in the hepatocytes close to the portal vein in most cases (six out of seven), whereas a faint staining was detected in one case. A faint staining was present in almost all hepatocytes at this stage. In RDP stage, FP2 staining was slightly more variable. The remodelling biliary structures (Fig. 1B) showed an intense staining in one liver sample, a weak labelling signal in four liver samples, and no staining in two liver samples. A faint staining was also present in almost all hepatocytes at this stage. Remodelled biliary structures of the portal tracts at RBD stage (Fig. 1C) remained unstained in all cases and a very faint staining was detected in the hepatocytes of these liver samples. Liver tissue from four foetuses affected with Meckel syndrome (Fig. 1D) was then investigated and showed intense labelling of the abnormally remodelling biliary structures as seen in DP stage during the normal development of the intrahepatic biliary system. A faint staining was also present in almost all hepatocytes of these liver samples.
Figure 1.
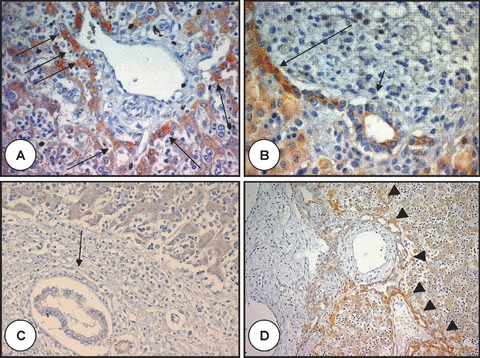
Polyductin expression in foetal liver without and with ductal plate malformation. In (A), there is a portal tract at the stage of ductal plate with intense staining (arrows) of the hepatocytes at the limiting plate (anti-FP2, ×400); (B) shows a remodelling ductal plate structure with a gradient of staining intensity: the intensity is greater close to the periportal hepatocytes (long arrow) than close to the duct or in the remodelling bile duct (short arrow) (anti-FP2, ×400); (C) shows no staining of the remodelled bile duct (arrow) (anti-FP2, ×200). A baseline staining is present in the hepatocytes; (D) shows a liver of foetus with Meckel syndrome and ductal plate malformation with intense staining of the abnormal biliary structures (arrowheads) close to the periportal hepatocytes (anti-FP2, ×200).
Neonatal, infantile and juvenile liver
None or very faint FP2 staining was noted in the hepatocytes or biliary structures of unaffected liver tissue and FP2 immunostained slides did not show any signal with regard to the canals of Hering. Figure 2A shows liver tissue with a baseline staining. In five out of ten cases with BA without DPM at early and at advanced stage with ductular proliferation (Fig. 2B and C), there was a faint staining of the portal ductal structures (early stage, Fig. 2B) and moderate FP2 staining of the biliary proliferations of the portal tracts (advanced stage, Fig. 2C). In three additional cases of BA with DPM at early (Fig. 2D) and at advanced stage (Fig. 2E) with ductular proliferation, FP2 staining of the biliary structures was strong. In two cases of BA without DPM, no staining was detected. NH showed a moderate to strong staining of the liver tissue in six out of nine cases (Fig. 2F and G), whereas a mild staining was found in three cases. In particular, pseudorosettes and neoductuli showed mostly an intense FP2 staining, whereas giant cells showed mostly very faint staining. Liver tissue from two patients with CHF (Fig. 2H) showed a moderate diffuse staining of the ductular structures, whilst the hepatocytes showed a mild diffuse staining. In liver biopsies with paucity of the intrahepatic bile ducts (PIBD) (Fig. 3A), intense, mild and no FP2 staining were found in two, nine and five cases, respectively. Finally, a mild staining was found in single cases of sepsis, choledocal cyst-associated liver disease (CC-LD), annular pancreas obstruction-associated liver disease (APO-LD), cardiovascular disease with hepatic involvement (CVD-HI), FFAOD (foetal fatty acid oxidation disorder associated with maternal HELLP syndrome), drug-induced liver disease (DILD), non-biliary liver disease (NBLD NOS), and liver disease associated with necrotizing enterocolitis (NEC-LD) as well as in two cases of neonatal cholestasis not otherwise specified (NC NOS). An additional case of an infant with alpha-1-antitrypsin deficiency (Fig. 3B) was added to the study and showed more intense FP2 staining at the periportal area. The liver tissue of progressive familiar intrahepatic cholestasis (Fig. 3C) showed an intense staining of the biliary structures and periportal hepatocytes. In Caroli’s disease (Fig. 3D), there were intraluminal bulbar protrusions of the ductal wall with moderate to intense staining of the biliary structures. The other cases of Table 1 did not show any staining. No age-dependent differences were found within each group.
Figure 2.
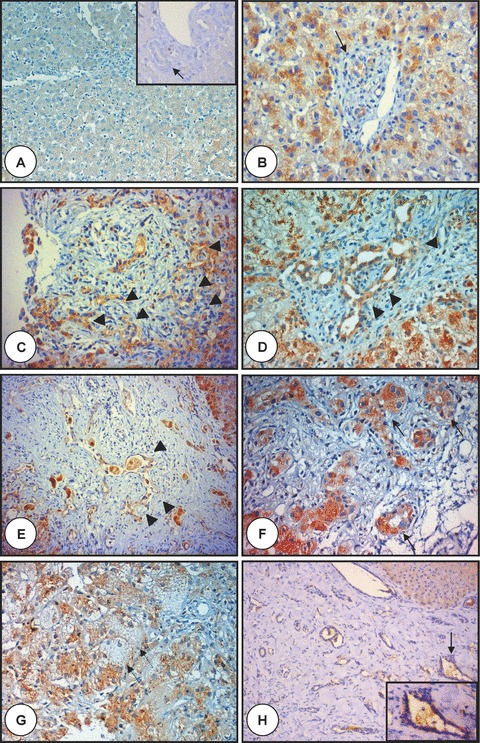
Polyductin expression in liver disease of newborns and infants. In (A), there is liver tissue with a baseline staining (anti-FP2, ×100) with no staining of the interlobular bile duct (arrow) (inset, anti-FP2, ×320); (B) and (C) show two examples of biliary atresia with ductular proliferation without ductal plate malformation at early (B) (the arrow points to a bile duct) and advanced (C) stage showing the ductular proliferation (arrowheads) of biliary atresia with a moderate FP2 staining (anti-FP2, ×400); (D) and (E) show two examples of biliary atresia with ductal plate malformation showing biliary structures (arrowheads) at early (D) and advanced (E) stage showing moderate FP2 staining (anti-FP2, d:×400, e:×200); (F) and (G) show liver tissue affected with neonatal hepatitis with mostly an intense staining of both pseudorosettes and neoductuli (arrows with continuous line), whereas giant cells show mostly very faint staining (arrow with dotted line) (anti-FP2, ×400). In (H) is shown liver tissue from an infant with congenital liver fibrosis (note the lack of ductular proliferation at the edges of the portal tracts) showing moderate diffuse FP2 staining of the biliary structures, whereas the hepatocytes show a mild diffuse staining. The arrows point to a selected region shown in inset. Note the enlarged biliary structure with moderate FP2 staining (anti-FP2, ×200 and ×320 in the inset).
Figure 3.
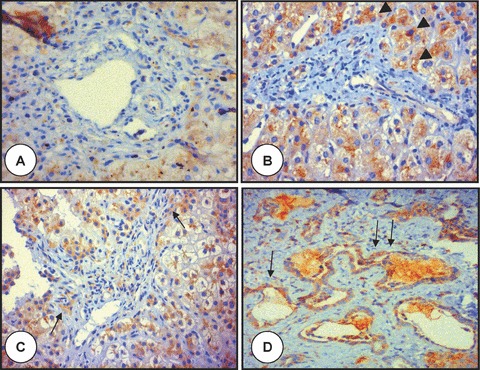
Polyductin expression in PIBD, AATD, PFIC I and Caroli’s disease. The microphotograph labelled with (A) shows a portal tract of a child with syndromatic paucity of the intrahepatic bile ducts (Alagille’s syndrome) showing practically baseline staining of the hepatocytes, but no staining in the portal area (anti-FP2, ×400); in (B) is shown the portal tract from an infant with alpha-1-antitrypsin deficiency with more intense FP-2 staining at level of the periportal area (arrowheads). The globules of accumulated alpha-1-antitrypsin are not seen because of the FP2 staining (anti-FP2, ×400); in (C) is shown the portal tract of an infant with progressive familiar intrahepatic cholestasis type I with moderate to intense staining of the biliary structures (arrows) and periportal hepatocytes (anti-FP2, ×400); (D) shows the liver tissue of a patient affected with Caroli’s disease. There is dilation of the bile ducts and intraluminal bulbar protrusions of the ductal wall with moderate to intense staining of the biliary structures (arrows) (anti-FP2, ×200).
Adult hepatobiliary carcinomas
Finally, we analysed two types of liver carcinomas: 16 cases of HCC and 23 cases of CCC. Hep Par 1 was negative in all CCC cases. Hep Par 1 was positive in 12 of 16 HCC. In addition, an extensive clinical radiological workup also helped to exclude metastatic adenocarcinoma. Twenty-two CCC cases showed positive staining for polyductin in malignant ductular proliferations (Fig. 4A and B), whereas the surrounding normal hepatocytes were negative or showed slight baseline staining only. FP2 staining was weak in nine cases (poorly differentiated CCC), moderate in seven (moderately differentiated CCC) and intense in six well-differentiated CCC. One CCC case did not show any staining, although different unmasking procedures were performed. This FP2 negative case showed very poor differentiation.
Figure 4.
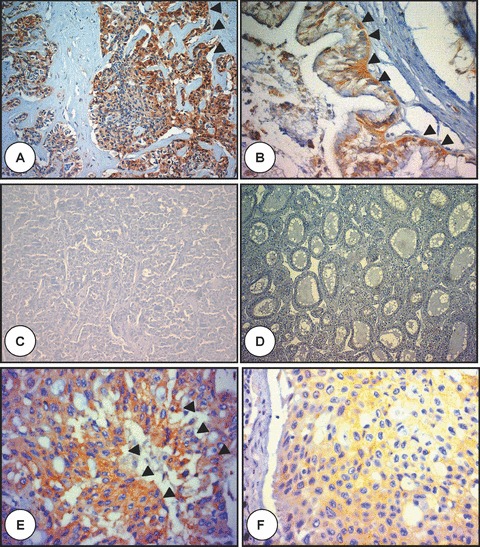
Polyductin expression in adult hepatobiliary carcinomas. (A) is the microphotograph of a cholangiocellular carcinoma (CCC) showing a moderate to intense FP2 staining of the neoplastic epithelium (arrowheads) embedded in a dense stroma (anti-FP2, ×200); in (B) there is a moderate FP2 staining in another CCC (arrowheads) (anti-FP2, ×400); in (C) no FP2 staining is demonstrated in a hepatocellular carcinoma (HCC) with trabecular growth pattern (anti-FP2, ×100), (D) no FP2 staining is also observed in another HCC with pseudoglandular growth pattern (anti-FP2, ×200), and (E and F) show tumour tissue metastasis to the lung from the liver combining cholangiocarcinomatous and hepatocellular features and showing FP2 staining mostly in cholangiocarcinomatous cells (E) (arrowheads) (anti-FP2, ×400).
In all 16 HCC cases (Fig. 4C) tumour cells were negative for FP2. Two of these cases showed a pseudoglandular growth pattern (Fig. 4D). Malignant hepatocytes were negative for poly-ductin expression, whereas reactive non-malignant proliferating bile ductules were positive. Regions close to neoplastic tissue showed reactive lymphocyte infiltration. In consideration of dichotomy of the values (positive/negative), the Fisher’s exact test was used as statistical test (binomial, comparison of two unpaired groups). The difference of FP2 labelling between CCC and HCC was therefore highly significant (P < 0.0001) (p = 4.50794 e10). An added case with combined features of hepatocellular and cholangiocellular differentiation showed positive FP2 staining in the cholangiocarcinomatous component (Fig. 4E and F). This patient died with multiple metastases (lung, brain) 1 month after diagnosis. An autopsy was performed. The diagnosis based on morphology and the absence of pre-existing liver disease was cholangiocarcinoma.
We have also performed double staining analyses in additional liver biopsies (Fig. 5A–D). A liver sample with Caroli’s disease, at a cirrhotic stage, showed moderate to intense Ep-CAM staining of the biliary epithelium and focal co-staining with FP2 at the limiting plate of the portal tract (Fig. 5A). A liver sample with chronic rejection with paucity of the interlobular bile ducts, in turn, revealed CK-19 staining of the bile duct epithelium and co-staining with FP2 at the limiting plate and in some biliary structures (Fig. 5B). The post-transplantation liver sample showing features of acute rejection revealed co-staining of CK19 and FP2 at the limiting plate of the portal tracts (Fig. 5C). In the same liver specimen, we could also detect FP2-staining with co-staining of the biliary structures with MUC-1 (Fig. 5D).
Figure 5.
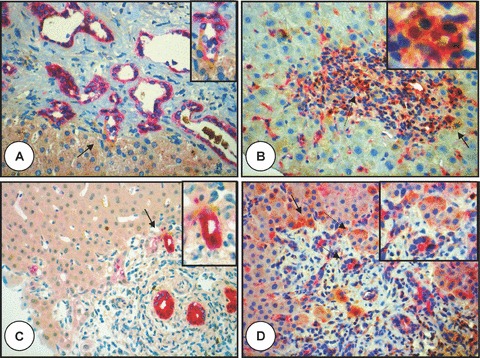
Polyductin expression and co-staining. (A) Microphotograph of the liver specimen with Caroli’s disease (stage: cirrhosis) showing a moderate to intense Ep-CAM staining of the biliary epithelium and focal co-staining with FP2 (arrow) at the limiting plate of the portal tract (anti-FP2 brown, anti-Ep-CAM red, ×400; the biliary structure indicated by the arrow is also shown in the inset); in (B) is shown a portal tract from a liver specimen with chronic rejection and paucity of the interlobular bile ducts (bile duct to portal tract ratio: 0.3) showing a portal tract with a residual bile duct and CK-19 staining of the bile duct epithelium and more co-staining at the limiting plate and in some biliary structures (orange staining, arrow) (anti-FP2 brown, anti-CK19 red, ×400). The segmented arrow shows a biliary duct with co-staining (also shown in the inset); in (C) co-CK19 and FP2 staining (arrow) is shown in the limiting plate of this portal tract from a liver specimen with acute rejection (anti-FP2 brown, anti-CK19 red, ×400; in the inset is the biliary structure shown by the arrow); in (D) is also shown the portal tract of a liver specimen with acute rejection showing some FP2 staining (brown) and co-staining of ductal plate cells (arrow with solid line) and biliary structures with MUC-1 (arrows with segmented lines, also demonstrated in the inset) (anti-FP2 brown, anti-MUC-1 red, ×400).
Discussion
Research on bile duct disorders has recently directed efforts to explore a potential common pathogenetic pathway for hepato-renal fibrocystic syndromes (HFRC). In the last few years, the PKHD1 gene was found to be mutated in patients affected with ARPKD, a disease characterized by biliary dysgenesis and fusiform dilatations of the renal collecting ducts. This gene’s product, polyductin, is expressed in kidney, liver, testis and pancreas, displaying both intracellular and membrane-bound distribution patterns. A proposed common model for the molecular mechanisms involved in HFRC includes mutated forms of proteins that contribute to default developmental pathways in ductal epithelium embryogenesis. In this study, we report extensively and in detail the expression profile of polyductin in foetal, perinatal, and infantile liver with normal and abnormal development of the intreahepatic biliary system, as well as in adult liver harbouring HCC and CCC. We demonstrated that polyductin is expressed in human foetal liver at the DP stage, the RDP stage and in DPM and disappears when bile ducts are remodelled. In addition, in a large series of neonatal and infantile liver diseases polyductin expression was mainly observed in infantile cholangiopathies with a neoductular reaction patterns (marked intraacinar pseudorosettes and/or marked periportal ductular proliferation). Finally, we provided evidence that anti-FP2 staining is a likely distinctive marker for CCC, as opposed to HCC.
Polyductin expression patterns have been investigated in several studies involving ARPKD. Previously, it has been localized in both collecting ducts and the thick ascending limb of Henle of human renal medulla [4–6]. Expression could also be found in murine branching of ureteric bud and intra and extrahepatic cholangiocytes. Here we show that polyductin expression can be traced from embryonic precursor cells to almost morphologically mature hepatocytes. We and others [7] detected an inconstant faint staining of the normal hepatocytes. These areas are not recognized sites of disease and will require further studies. Specific hepatocyte staining was initially detected only in foetal samples, whilst normal human adult hepatocytes showed no specific signal to FP2. It was therefore suggested that the stained foetal hepatocytes might be committed to a biliary phenotype and could potentially contribute to bile duct formation by later differentiation. This may support the idea of bipotential stem cells in liver development [31]. In some cases, a weak diffuse FP2 staining of hepato-cytes was observed. These data might correlate with the findings of soluble cytoplasmic and membrane-bound fractions of polyductin.
Fibrosis is a leading feature encountered in most of the pathologies included in this study and polyductin expression was associated with DPM and, particularly, in infantile cholangiopathies with marked intraacinar pseudorosettes and/or periportal ductular proliferation. Embryonic and early foetal liver is most likely composed of a population of bipotential progenitor or oval cells [32–35]. Thus, it would also be interesting to study the expression of oval cell markers in anti-FP2 stained ductular proliferating cells. De novo expression of polyductin in regenerating/proliferating hepatocytes might be explained by the fact that upon liver injury, stem cells receive a signal to replace damaged cells and this is usually accompanied by a loss of cell–cell adhesion and down-regulation of cadherin-mediated adhesion, which subsequently gives rise to enhanced expression of Ep-CAM with similar expression patterns [36, 37]. Interestingly, the same cells of proliferating ductular structures also seem to express some oval cell markers [38]. Thus, it can be postulated that polyductin has a functional role in development, differentiation and/or establishment of polarity of ductal epithelium. This is also compatible with our findings, showing different expression patterns in biliary cells during normal DP formation. Although the first two developmental stages of the intrahepatic biliary system, DP and RDP, showed positive expression for polyductin, no expression was detected in the third phase, the stage of the remodelled bile ducts. An interesting aspect to be further evaluated could be the hypothesis that polyductin is involved in regulating the development of lumen of the biliary structures. In fact, there is an intense expression of polyductin in the early stages of bile duct development when the lumen is formed. It is not unreasonable to propose that a reduction in full-length product levels would be linked and/or signal the transition from the second to the third stage of the intrahepatic biliary system. Thus, the absence of FP2 staining in uninvolved formed bile ducts and expression during DP stage may be due to polyductin’s possible role in the intrahepatic biliary system development.
The present study revealed that infantile cholangiopathies show an intense staining in association with ductular proliferation and biliary dysgenesis, whilst no or faint staining is found in cholestasis not otherwise specified or infantile hepatopathies. In this sense, it may support the close relationship of polyductin with biliary dysgenesis. In a previous study, Ward et al. reported that polyductin was detected in developing and mature intrahepatic bile ducts [7], but no staining was found in three liver samples from ARPKD patients. Our studies performed in two CHF liver samples, however, showed specific staining in ductular structures. Though the expression pattern observed by us is distinct from theirs, a few considerations might explain the differences: (1) It has been shown that PKHD1 encodes multiple protein isoforms [6]. In this context, different mutation combinations may lead to different isoform profiles in distinct patients, which may be differently recognized by a given antibody; (2) CHF is an invariable finding in ARPKD but may be also found in other diseases. In this scenario, the polyductin expression pattern may not be the same in ARPKD-related and ARPKD-unrelated CHF livers; (3) The severity of CHF may significantly vary among ARPKD patients and among ARPKD-unrelated CHF patients. This fact may support a potential heterogeneity in the polyductin expression profile in different patients. The fact that Ward et al. detected little staining in dilated collecting ducts in an ARPKD kidney specimen may support this possibility; and (4) Different antibodies may have distinct detection thresholds.
The absence of polyductin in HCC is of obvious interest. Primary liver carcinomas have been classified into either HCC or CCC, with a rare combined type of hepatocellular-cholangiocellular carcinoma [39]. HCC and CCC are thought to be derived from a single liver progenitor cell, thus expressing identical immunological markers, or to originate from different precursor cells, more differentiated towards hepatocytes or towards biliary epithelial cells, expressing various phenotypic markers. Because of the uncertainty as to the origin of HCC and CCC, no exclusive marker has yet been found to completely distinguish them [40, 41], particularly when only limited tissue is available for morphological classification. Our data indicate that anti-FP2 staining may be helpful in the differential diagnosis of HCC versus CCC. These data should be confirmed with larger number of cases and also including a number of metastatic adenocarcinoma from different origins.
In summary, immunohistochemical analysis of different hepatic and biliary foetal, neonatal, infantile and adult disorders with an anti-polyductin antibody revealed that this protein expression is a regular feature of hepatic cells and bile duct epithelium in early development stages. Later polyductin presence in such cells, on the other hand, may correlate with pathological manifestations. These results represent the first systematic study of polyductin expression in human pathologies associated with abnormal development of intrahepatic biliary tree, and support the following conclusions: (i) polyductin expression mirrors developmental properties of the primitive intrahepatic biliary system; (ii) polyductin is re-expressed in pathological conditions associated with DPM; and (iii) polyductin might be a potential marker to distinguish CCC from HCC.
Acknowledgments
We are thankful to Mr. L. Krause who found out the early conditions of the FP2 staining in liver tissue and both Ms. I. Jehart and Ms. I. Tschoerner for improving FP2 staining in all biopsies and surgical specimens. About the contribution, CS designed the experiments, and was responsible for the performance, control and interpretation of data. He was also responsible to get sponsor funding, coordinated the study and wrote the actual draft of the manuscript. LD performed part of the experiments, archived small samples, collected and interpreted data and wrote the first draft of the manuscript. LFM and LFO provided the anti-FP2 polyclonal antibody, supervised immunostaining electronically and critically reviewed the paper. HFO and GM were responsible for most of the diagnoses and GM critically reviewed the paper. This work has mostly been supported by the Tyrolean Perinatal Program, Innsbruck, Austria (Peripro) and Österreichische Krebshilfe Tirol (Austrian Cancer Research Tyrol) as extramural finding. The initial part of this work was supported by the Medical University of Innsbruck, Austria (intramural funding). The sponsors had no role in study design, data collection, data analysis, data interpretation or in writing of the report. There is no conflict of interest. Part of the work was presented at the 96th Annual Meeting of the United States & Canadian Academy of Pathology, March 24–30, 2007, San Diego, CA.
References
- 1.Zerres K, Mucher G, Becker J, et al. Prenatal diagnosis of autosomal recessive polycystic kidney disease (ARPKD): molecular genetics, clinical experience, and fetal morphology. Am J Med Genet. 1998;76:137–44. [PubMed] [Google Scholar]
- 2.Zerres K, Mucher G, Bachner L, et al. Mapping of the gene for autosomal recessive polycystic kidney disease (ARPKD) to chromosome 6p21-cen. Nat Genet. 1994;7:429–32. doi: 10.1038/ng0794-429. [DOI] [PubMed] [Google Scholar]
- 3.Guay-Woodford LM, Mucher G, Hopkins SD, et al. The severe perinatal form of autosomal recessive polycystic kidney disease maps to chromosome 6p21.1-p12: implications for genetic counseling. Am J Hum Genet. 1995;56:1101–7. [PMC free article] [PubMed] [Google Scholar]
- 4.Rossetti S, Torra R, Coto E, et al. A complete mutation screen of PKHD1 in autosomal-recessive polycystic kidney disease (ARPKD) pedigrees. Kidney Int. 2003;64:391–403. doi: 10.1046/j.1523-1755.2003.00111.x. [DOI] [PubMed] [Google Scholar]
- 5.Onuchic LF, Furu L, Nagasawa Y, et al. PKHD1, the polycystic kidney and hepatic disease 1 gene, encodes a novel large protein containing multiple immunoglobulin-like-plexin-transcription-factor domains and parallel beta-helix 1 repeats. Am J Hum Genet. 2002;70:1305–17. doi: 10.1086/340448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Menezes LF, Cai Y, Nagasawa Y, et al. Polyductin, the PKHD1 gene product, comprises isoforms expressed in plasma membrane, primary cilium, and cytoplasm. Kidney Int. 2004;66:1345–55. doi: 10.1111/j.1523-1755.2004.00844.x. [DOI] [PubMed] [Google Scholar]
- 7.Ward CJ, Yuan D, Masyuk TV, et al. Cellular and subcellular localization of the ARPKD protein; fibrocystin is expressed on primary cilia. Hum Mol Genet. 2003;12:2703–10. doi: 10.1093/hmg/ddg274. [DOI] [PubMed] [Google Scholar]
- 8.Masyuk TV, Huang BQ, Masyuk AI, et al. Biliary dysgenesis in the PCK rat, an orthologous model of autosomal recessive polycystic kidney disease. Am J Pathol. 2004;165:1719–30. doi: 10.1016/S0002-9440(10)63427-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sergi C, Kahl P, Otto HF. Contribution of apoptosis and apoptosis-related proteins to the malformation of the primitive intra-hepatic biliary system in Meckel syndrome. Am J Pathol. 2000;156:1589–98. doi: 10.1016/S0002-9440(10)65031-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oliveira NL, Kanawaty FR, Costa SC, Hessel G. Infection by cytomegalovirus in patients with neonatal cholestasis. Arch Gastroenterol. 2002;39:132–6. doi: 10.1590/s0004-28032002000200012. [DOI] [PubMed] [Google Scholar]
- 11.Goodman ZD. Neoplasms of the liver. Mod Pathol. 2007;20:S49–60. doi: 10.1038/modpathol.3800682. [DOI] [PubMed] [Google Scholar]
- 12.Soreide K, Korner H, Havnen J, Soreide JA. Bile duct cysts in adults. Br J Surg. 2004;91:1538–48. doi: 10.1002/bjs.4815. [DOI] [PubMed] [Google Scholar]
- 13.Clotman F, Lemaigre FP. Control of hepatic differentiation by activin/TGFbeta signaling. Cell Cycle. 2006;2:168–71. doi: 10.4161/cc.5.2.2341. [DOI] [PubMed] [Google Scholar]
- 14.Clotman F, Libbrecht L, Killingsworth MC, et al. Lack of cilia and differentiation defects in the liver of human foetuses with the Meckel syndrome. Liver Int. 2008;3:377–84. doi: 10.1111/j.1478-3231.2007.01617.x. [DOI] [PubMed] [Google Scholar]
- 15.Roskams T, Desmet V. Embryology of extra- and intrahepatic bile ducts, the ductal plate. Anat Rec. 2008;291:628–35. doi: 10.1002/ar.20710. [DOI] [PubMed] [Google Scholar]
- 16.Sergi C, Adam S, Kahl P, Otto HF. Study of the malformation of ductal plate of the liver in Meckel syndrome and review of other syndromes presenting with this anomaly. Pediatr Dev Pathol. 2000;3:568–83. doi: 10.1007/s100240010104. [DOI] [PubMed] [Google Scholar]
- 17.Desmet VJ. Intrahepatic bile ducts under the lens. J Hepatol. 1985;1:545–59. doi: 10.1016/s0168-8278(85)80752-2. [DOI] [PubMed] [Google Scholar]
- 18.Sergi C, Adam S, Kahl P, Otto HF. The remodeling of the primitive human biliary system. Early Hum Dev. 2000;58:167–78. doi: 10.1016/s0378-3782(00)00065-7. [DOI] [PubMed] [Google Scholar]
- 19.Salonen R, Paavola P. Meckel syndrome. J Med Genet. 1998;35:497–501. doi: 10.1136/jmg.35.6.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Desmet VJ. Ludwig symposium on biliary disorders – Part I. Pathogenesis of ductal plate abnormalities. Mayo Clin Proc. 1998;73:80–9. doi: 10.4065/73.1.80. [DOI] [PubMed] [Google Scholar]
- 21.Johnson CA, Gissen P, Sergi C. Molecular pathology and genetics of congenital hepatorenal fibrocystic syndromes. J Med Genet. 2003;40:311–9. doi: 10.1136/jmg.40.5.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Balistreri WF, Grand R, Hoofnagle JH, et al. Biliary atresia: current concepts and research directions. Summary of a symposium. Hepatology. 1996;23:1682–92. doi: 10.1002/hep.510230652. [DOI] [PubMed] [Google Scholar]
- 23.Kahn E. Biliary atresia revisited. Pediatr Dev Pathol. 2004;7:109–24. doi: 10.1007/s10024-003-0307-y. [DOI] [PubMed] [Google Scholar]
- 24.Azar G, Beneck D, Lane B, et al. Atypical morphological presentation of biliary atresia and value of serial liver biopsies. J Pediatr Gastroenterol Nutr. 2002;34:212–5. doi: 10.1097/00005176-200202000-00020. [DOI] [PubMed] [Google Scholar]
- 25.Ishak KG, Sharp HL. Developmental abnormalities and liver disease in childhood. In: MacSween RNM, Anthony PP, Scheurer PJ, Burt AD, Portmann BC, editors. Pathology of the liver. 3rd ed. Churchill Livingstone; 1994. pp. 88–103. Edinburgh: . [Google Scholar]
- 26.Danks DM, Campbell PE, Jack I, et al. Studies of the etiology of neonatal hepatitis and biliary atresia. Arch Dis Child. 1977;52:360–7. doi: 10.1136/adc.52.5.360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Snover DC. Biopsy diagnosis of liver disease. 1st ed. Williams & Wilkins; 1992. Biopsy in the evaluation of neonatal and infantile cholestasis. . In: . Baltimore, Hong Kong London: [Google Scholar]
- 28.Mueller RF. The Alagille syndrome (arteriohepatic dysplasia) J Med Genet. 1987;24:621–6. doi: 10.1136/jmg.24.10.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vogel M. Atlas der morphologischen Plazentadia-gnostik. 2nd. Springer-Verlag; 1996. German somatometric data; pp. 241–4. Berlin and Heidelberg: . [Google Scholar]
- 30.Terada T, Ueyama J, Ukita Y, Ohta T. Protein expression of double-stranded RNA-activated protein kinase (PKR) in intrahepatic bile ducts in normal adult livers, fetal livers, primary biliary cirrhosis, hepatolithiasis and intrahepatic cholangiocarcinoma. Liver. 2000;20:450–7. doi: 10.1034/j.1600-0676.2000.020006450.x. [DOI] [PubMed] [Google Scholar]
- 31.Haruna Y, Saito K, Spaulding S, et al. Identification of bipotential progenitor cells in human liver development. Hepatology. 1996;26:476–81. doi: 10.1002/hep.510230312. [DOI] [PubMed] [Google Scholar]
- 32.Marceau N, Blouin MJ, Germain L, Noel M. Role of different epithelial cell types in liver ontogenesis, regeneration and neoplasia. In Vitro Cell Dev Biol. 1989;25:336–41. doi: 10.1007/BF02624596. [DOI] [PubMed] [Google Scholar]
- 33.Ponder KP. Analysis of liver development, regeneration, and carcinogenesis by genetic marking studies. FASEB J. 1996;10:673–82. doi: 10.1096/fasebj.10.7.8635684. [DOI] [PubMed] [Google Scholar]
- 34.Desmet V, Roskams T, Van Eyken P. Ductular reaction in the liver. Pathol Res Pract. 1995;191:513–24. doi: 10.1016/s0344-0338(11)80870-8. [DOI] [PubMed] [Google Scholar]
- 35.Zimmermann A. Liver regeneration: the emergence of new pathways. Med Sci Monit. 2002;8:53–63. [PubMed] [Google Scholar]
- 36.Knittel T, Aurisch S, Neubauer K, et al. Cell-type-specific expression of neural cell adhesion molecule (N-CAM) in Ito cells of rat liver. Up-regulation during in vitro activation and in hepatic tissue repair. Am J Pathol. 1996;149:449–62. [PMC free article] [PubMed] [Google Scholar]
- 37.De Boer CJ, Van Krieken JH, Janssen-van Rhijn CM, Litvinov SV. Expression of Ep-CAM in normal, regenerating, metaplastic, and neoplastic liver. J Pathol. 1999;188:201–6. doi: 10.1002/(SICI)1096-9896(199906)188:2<201::AID-PATH339>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 38.Treem WR, Krzymowski GA, Cartun RW, et al. Cytokeratin immunohistochemical examination of liver biopsies in infants with Alagille syndrome and biliary atresia. J Pediatr Gastroenterol Nutr. 1992;15:73–80. doi: 10.1097/00005176-199207000-00011. [DOI] [PubMed] [Google Scholar]
- 39.Wu PC, Fang JW, Lau VK, et al. Classification of hepatocellular carcinoma according to hepatocellular and biliary differentiation markers. Clinical and biological implications. Am J Pathol. 1996;149:1167–75. [PMC free article] [PubMed] [Google Scholar]
- 40.Ihara A, Koizumi H, Hashizume R, Uchikoshi T. Expression of epithelial cadherin and alpha- and beta-catenins in non-tumoral livers and hepatocellular carcinomas. Hepatology. 1996;23:1441–7. doi: 10.1053/jhep.1996.v23.pm0008675162. [DOI] [PubMed] [Google Scholar]
- 41.Kozyraki R, Scoazec JY, Flejou JF, et al. Expression of cadherins and alpha-catenin in primary epithelial tumors of the liver. Gastroenterology. 1996;110:1137–49. doi: 10.1053/gast.1996.v110.pm8613003. [DOI] [PubMed] [Google Scholar]