

Abstract
Cellular myxoma and grade I myxofibrosarcoma are mesenchymal tumours that are characterized by their abundant myxoid extracellular matrix (ECM). Despite their histological overlap, they differ clinically. Diagnosis is therefore difficult though important. We investigated their (cyto) genetics and ECM. GNAS1-activating mutations have been described in intramuscular myxoma, and lead to downstream activation of cFos. KRAS and TP53 mutations are commonly involved in sarcomagenesis whereby KRAS subsequently activates c-Fos. A well-documented series of intramuscular myxoma (three typical cases and seven cases of the more challenging cellular variant) and grade I myxofibrosarcoma (n= 10) cases were karyotyped, analyzed for GNAS1, KRAS and TP53 mutations and downstream activation of c-Fos mRNA and protein expression. ECM was studied by liquid chromatography mass spectrometry and expression of proteins identified was validated by immunohistochemistry and qPCR. Grade I myxofibrosarcoma showed variable, non-specific cyto-genetic aberrations in 83,5% of cases (n= 6) whereas karyotypes of intramuscular myxoma were all normal (n= 7). GNAS1-activating mutations were exclusively found in 50% of intramuscular myxoma. Both tumour types showed over-expression of c-Fos mRNA and protein. No mutations in KRAS codon 12/13 or in TP53 were detected. Liquid chromatography mass spectrometry revealed structural proteins (collagen types I, VI, XII, XIV and decorin) in grade I myxofibrosarcoma lacking in intramuscular myxoma. This was confirmed by immunohistochemistry and qPCR. Intramuscular/cellular myxoma and grade I myxofibrosarcoma show different molecular genetic aberrations and different composition of their ECM that probably contribute to their diverse clinical behaviour. GNAS1 mutation analysis can be helpful to distinguish intramuscular myxoma from grade I myxofibrosarcoma in selected cases.
Keywords: GNAS, karyotype, myxoid, extracellular matrix, proteomics, LC-MS, MLPA, myxofibrosarcoma, intramuscular myxoma
Introduction
Myxoid tumours of soft tissue comprise a heterogeneous group of mesenchymal tumours characterized by abundant extracellular matrix (ECM). In this group, intramuscular myxoma and myxofibrosarcoma are most frequent, usually occurring in the extremities and at older age [1]. Histologically, intramuscular myxoma, and in particular its cellular variant (i.e. cellular myxoma), and grade I myxofibrosarcoma show considerable overlapping features [2]. Clinically, they differ as intramuscular myxoma shows no recurrence except for the cellular variant and never metastasizes. This is in contrast to myxofibrosarcoma that has a tendency to recur and, importantly, shows increase in tumour grade upon recurrence, thereby gaining metastatic potential [3]. Thus, the differential diagnosis between these two entities is important though can be challenging especially when presenting as an intramuscular tumour. Myxofibrosarcoma is usually characterized by complex, non-specific karyotypic aberrations. (Cyto)genetic data on intramuscular myxoma are sparse with only two cases described in the literature showing normal karyotypes [4, 5]. Recently, activating mutations in codon 201 of the GNAS1 gene have been described in intramuscular myxoma [6]. This gene encodes, among others, for the alpha-sub-unit of the heterotrimeric G-protein. This protein is involved in cell signalling and leads to the transcription of the protein c-Fos and subsequently activation of the cell cycle. Activating GNAS1 mutations and the subsequent activation of c-Fos are involved in the pathogenesis of fibrous dysplasia, which is a benign bone tumour associated with intramuscular myxoma in Mazabraud syndrome [7]. Activating mutations in codon 12/13 of KRAS also lead to downstream activation of c-Fos. KRAS-activating mutations have been described in both mouse and human sarcomas. Kirsch et al. showed that KRAS and TP53 mutations were sufficient to initiate high-grade sarcomas with myofibroblastic features in mice [8]. TP53 mutations are relatively common in sarcomas with non-specific genetic aberrations compared with sarcomas with reciprocal specific translocations [9]. Previous studies showed immunohistochemical p53 expression in 33% of myxofibrosarcoma, mainly occurring in grade II and grade III tumours [10]. We studied the genetic make-up of these tumours at levels of karyotype, GNAS1, TP53 and KRAS mutations and downstream expression of c-Fos in order to find a potential tool for differential diagnosis and to get more insight into the biology of these tumours. The constitution and function of their so-called myxoid ECM are poorly understood. Previously, we demonstrated that intramuscular myxoma and grade I myxofibrosarcoma showed no significant differences in the glycosaminoglycans present in their ECM [11]. To study the differences in ECM organization and association with clinical behaviour, we screened ECM lysates of intramuscular myxoma and grade I myxofibrosarcoma in a broad liquid chromatography mass spectrometry (LC-MS)–based survey.
Materials and methods
Patient data
The study included 10 intramuscular myxoma cases and 10 grade I myxofibrosarcoma cases that were collected retrospectively from the files of the Pathology Departments of the University Hospitals of Leuven and Leiden University Medical Center. In each case, 4-mm-thick sections of formalin-fixed, paraffin-embedded material were stained with haematoxylin and eosin (H&E). The histological diagnoses were revised (CDMF, PCWH, RS) and classified according to the 2002 WHO criteria. In case of myxofibrosarcoma, histological grading was performed according to the FNCLCC [12, 13]. Patient and tumour characteristics are shown in Table 1. Characteristic morphology of intramuscular myxoma, cellular myxoma and grade I myxofibrosarcoma is depicted in Fig. 1.
Table 1.
Patient and tumour characteristics
| Case | Diagnosis | P/R/M | Age | Gender | Site | Follow-up |
|---|---|---|---|---|---|---|
| 1 | IM | P | 55 | F | Left vastus lateralis muscle | NSR at 37 months |
| 2 | IM | P | 32 | M | Retroperitoneal right | NSR at 94 months |
| 3 | IM | P | 48 | F | Adductor muscles right leg | NSR at 34 months |
| 4 | IM* | P | 73 | F | Subcutaneous tight upper arm | NSR at 17 months |
| 5 | IM* | P | 59 | F | Right lateral upper leg | NSR at 31 months |
| 6 | IM* | P | 53 | F | Right psoas muscle | NSR at 62 months |
| 7 | IM* | P | 53 | F | Right hamstring muscle | NSR at 69 months |
| 8 | IM* | P | 59 | F | Left vastus lateralis muscle | NSR at 39 months |
| 9 | IM* | P | 61 | M | Right upper leg | NSR at 41 months |
| 10 | IM* | P | 62 | M | Left vastus medialis muscle | NSR at 68 months |
| 11 | MFS | P | 55 | M | Right vastus lateralis muscle | NSR at 66 months |
| 12 | MFS | P | 74 | M | Left lower arm | LR at 14 months |
| 13 | MFS | P | 71 | M | Left lower arm | LR at 36 months |
| 14 | MFS | P | 63 | F | Right hamstring muscles | NSR at 41 months |
| 15 | MFS | R | 84 | M | Subcutaneous left lower arm | LR at 12 months |
| 16 | MFS | P | 87 | M | Right upper arm | NSR at 18 months |
| 17 | MFS | P | 49 | F | Left gluteus muscle | LR at 2 months |
| 18 | MFS | P | 81 | M | Right upper arm | NSR at 60 months |
| 18 | MFS | P | 61 | M | Right gracilis muscle | LR at 42 months |
| 20 | MFS | P | 39 | F | Subcutaneous occiput | NSR at 25 months |
These were all cellular myxomas.
Abbreviations: IM, intramuscular myxoma; MFS, myxofibrosarcoma; P, primary lesion; R, local recurrence; M, metastasis, NSR, no sign of recurrence; LR, local recurrence. Intramuscular myxoma showed a female predominance and myxofibrosarcoma a slight male predominance. The median age of occurrence was 56 years for intramuscular myxoma and 66 years for myxofibrosarcoma. All cases were primaries except for case 15, which was a local recurrence. Except for case 2 and 20, all lesions occurred at the extremities. Out of the intramuscular myxomas, eight were truly intramuscular, whereas single cases were identified at other sites (case 2 and 4). None of the lesions were situated near articular surfaces. Clinical, radiological and histological follow-up (17–94 months) showed no local recurrence for intramuscular myxoma but did in half of the cases of myxofibrosarcoma. No patients had any clinical evidence for endocrinopathy or café-au-lait spots. One patient (case 1) suffered monostotic fibrous dysplasia of the distal femoral bone, diagnosed by conventional radiological examination and MRI, and was therefore suspected for Mazabraud or a partial form of McCune–Albright syndrome.
Figure 1.
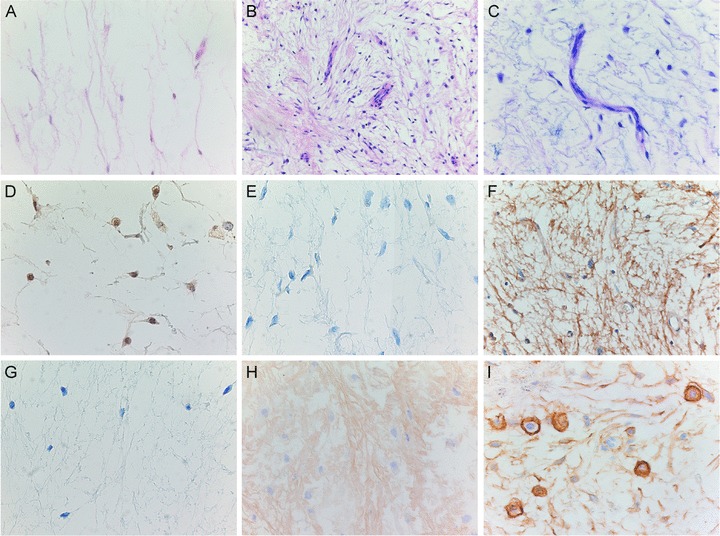
Overlapping histology of intramuscular myxoma, cellular myxoma and grade I myxofibrosarcoma and their immunohistochemical expression for c-Fos, decorin, collagen I, collagen VI and CD44. (A) Low-power view of intramuscular myxoma showing a hypocellular and hypovascular tumour. Its cellular variant is both more cellular and more vascular (B) but lacks the cytonuclear atypia and characteristic curvilinear vascular pattern of grade I myxofibrosarcoma (C). Low-power view of intramuscular myxoma (D) showing moderate cytoplasmic and nuclear expression for c-Fos in the majority (> 75%) of tumour cells. (E) Low-power view of intramuscular myxoma was completely negative for decorin, whereas grade I myxofibrosarcoma showed diffuse fibrillary staining for decorin in the ECM (F). Lack of collagen I expression in the ECM of intramuscular myxoma (G) and moderate staining for collagen VI in the ECM of grade I myxofibrosarcoma (H). A majority of tumour cells of grade I myxofibrosarcoma showed strong membranous staining for CD44 in the majority of tumour cells (I).
Karyotyping
Cells were harvested and 48-colour fluorescence in situ hybridization (FISH) staining was carried out, which stains every chromosome arm in a different colour combination. This was followed by digital imaging and analysis as previously described [5, 14]. Hybridizations with individual libraries labelled with single fluorochromes were used to confirm the detected re-arrangements. Break-points were assigned by using inverted DAPI counterstained images of the chromosomes.
GNAS1 direct sequencing
GNAS1 mutation analysis was performed for codon 201 (exon 8) and codon 227 (exon 9) using oligonucleotide primers extended with an M13-forward or reverse sequence (see supplementary Table 1). Tumour tissue was isolated from formalin-fixed, paraffin-embedded material (FFPE) by microdissection to enrich for tumour cells and avoid ‘contamination’ of non-neoplastic cells, that is, lymphocytes, endothelial cells/pericytes or muscular tissue at the periphery of the tumour. Genomic DNA was isolated from 10 consecutive 10-mm sections using a Chelex extraction method, as described earlier [15]. Polymerase chain reaction (PCR) was carried out to amplify the GNAS1 gene with the primers for exon 8 and exon 9, as previously reported [16]. PCR products were visualized and sequenced as previously described [17, 18].
GNAS1 multiplex ligation-dependent probe amplification (MLPA)
To detect mutations that are missed by direct sequencing due to contamination with normal cells, we set up GNAS1 MLPA for the mutations already detected by direct sequencing. MLPA was performed using the following oligonucleotide probes attached to M13-tail, depicted in supplementary Table 3. For the R201C mutant, probes with additional stuffer sequence were added: forward: 5′-CAGAC-3′ and reverse 5′-GATGTGCT-3′. MLPA was performed as previously described [19]. In short, adjacently annealing oligonucleotide probes were hybridized and ligated. After ligation, the common ends of the probes served as a template for PCR amplification using the primer pairs that are depicted in supplementary Table 3, with one of each pair fluorescently labelled. Products were separated on the ABI 3700 genetic analyser (Applied Biosystems, Foster City, CA) and analyzed.
KRAS mutation analysis
KRAS mutation analysis was performed on microdissected FFPE tumour tissue using two primer sets in a nested PCR which specifically amplifies a 114 bp fragment of KRAS exon 2, including codons 12 and 13 (supplementary Table 1). All primer sequences and amplification protocols have been previously described [20]. PCR products were purified with the QIAquick PCR Purification Kit (Qiagen, Germantown, MD) and sequenced with the fluorescent Big-Dyes Terminators Sequencing Kit (Applied Biosystems).
TP53 mutation analysis
TP53 hotspot mutation analysis was performed on microdissected FFPE tumour tissue using primer sets for exons 4a, 4b, 5c, 6, 7 and 8 in a nested PCR (supplementary Table 2). Samples were used in high-resolution melting curve analysis as previously described [46]. PCR products were purified with the QIAquick PCR Purification Kit (Qiagen) and sequenced with the fluorescent Big-Dyes Terminators Sequencing Kit (Applied Biosystems).
SDS-PAGE, in-gel tryptic digestion and mass spectrometry
Tumour lysates of intramuscular myxoma and grade I myxofibrosarcoma were prepared and depleted of albumin and IgG as previously described [11]. From each sample, 150 μg was separated by SDS-PAGE and stained with Colloidal Coomassie (Invitrogen, Leek, The Netherlands). The complete lanes from both intramuscular myxoma and grade I myxofibrosar-coma were cut into approximately 30 protein bands, and these samples were subsequently reduced, alkylated and in-gel digested using trypsin as described previously [21]. Peptides were subsequently collected using two rounds of extraction with 20 μl of 0.1% trifluoroacetic acid and were stored at −20°C prior to analysis by mass spectrometry. Samples were injected onto a nano-LC system (Ultimate, Dionex, Amsterdam, the Netherlands) equipped with a peptide trap column (Pepmap 100, 0.3 i.d. μ1 mm) and an analytical column (Pepmap 100, 0.075 i.d. 150 mm, Dionex). The mobile phases consisted of (A) 0.04% formic acid/0.4% acetonitrile and (B) 0.04% formic acid/90% acetonitrile. A 45-min. linear gradient from 0% to 60% B was applied at a flow rate of 0.2 ml/min. The outlet of the LC system was coupled to an HCT Ultra ion-trap mass spectrometer (Bruker Daltonics, Bremen, Germany) using a nano-electrospray ionization source. The spray voltage was set at 1.2 kV, and the temperature of the heated capillary was set to 165 °C. Eluting peptides were analyzed in the data-dependent MS/MS mode over a 400–1600 m/z range. The five most abundant fragments in each MS spectrum were selected for MS/MS analysis by collision-induced dissociation. Mass spectra were evaluated using the DataAnalysis 3.1 software package (Bruker Daltonics) and exported as a Mascot Generic File (MGF). MGF files were merged using the OMSSA Browser version 2.1.0. and searched against the human International Protein Index (IPI) database using the Mascot search algorithm (version 2.2, Matrixscience, London, UK), allowing mass tolerances of 0.5 Da for MS (#13C = 1) and 0.75 Da for MS/MS. One missed cleavage site was allowed for tryptic peptides.
Carbamidomethylcysteine was taken as a fixed modification and oxidation of methionine as a variable modification.
qPCR for mRNA expression
Following microdissection, RNA isolation and purification, qPCR was performed for c-Fos, decorin, collagen I alpha 1, collagen VI alpha 1, collagen XII alpha 1 and collagen XIV alpha 1, as described previously [11, 22, 23]. Primers (including those of the housekeeping genes) and controls are summarized in supplementary Table 1. A reference control panel, including a variety of neoplastic and non-neoplastic tissue was used as described previously [24]. After qPCR, the products were purified using QIAquick PCR Purification Kit (Qiagen). For sequence confirmation of the products, we used the Big Dyes Terminators Sequencing Kit (Applied Biosystems).
Immunohistochemistry
Immunohistochemistry was performed for c-Fos, decorin, collagen I-A1, collagen VI-A1 and CD44. Sections (4-μm-thick) were mounted on 3-aminopropylethoxysilane (Sigma, St. Louis, MO) and glutaraldehyde-coated slides and dried overnight at 37°C. Immunohistochemistry was performed as previously described, using the antibodies, conditions and controls described in supplementary Table 4 [11, 25]. For c-Fos and CD44, only nuclear and membranous staining were assessed, respectively, and scored as previously described [26]. For decorin, collagen I-A1 and collagen VI-A1, the ECM of whole sections were assessed. Scores of 0 (absent) and 1 (weak) were considered negative; scores of 2 (moderate) and 3 (strong) were considered positive.
Statistical analysis
Wilcoxon–Mann–Whitney test was used to calculate the differences for both the immunohistochemical and qPCR results.
Results
Karyotyping
Karyotypes were available for seven intramuscular myxomas (five cases were of the cellular variant) and six grade I myxofibrosarcomas. The karyotypes of the intramuscular myxomas (both typical and cellular cases) were all normal. Out of the grade I myxofibrosarcoma cases, only one case showed a normal karyotype, whereas five out of six cases showed variable, non-specific cytogenetic aberrations (Table 2).
Table 2.
Cytogenetic findings and GNAS1, KRAS and TP53 mutation analysis in intramuscular myxoma and grade I myxofibrosarcoma
| Case | Diagnosis | Karyotype | Mutation analysis | |||
|---|---|---|---|---|---|---|
| GNAS 1 | KRAS | TP53 a | ||||
| exon 8 | exon 9 | exon 2 | ||||
| 1 | IM | NA | R201S | wt | wt | wt |
| 2 | IM | 46, XY | wt | wt | wt | wt |
| 3 | IM | 46, XX | wt | wt | wt | wt |
| 4 | IMa | 46, XX | wt | wt | wt | wt |
| 5 | IMa | NA | R201H | wt | wt | wt |
| 6 | IMa | NA | wt | wt | wt | wt |
| 7 | IMa | 46, XY | wt | wt | wt | wt |
| 8 | IMa | 46, XY | R201C | wt | wt | wt |
| 9 | IMa | 46, XY | R201C | wt | wt | wt |
| 10 | IMa | 46, XY | R201H | wt | wt | wt |
| 11 | MFS | NA | wt | wt | wt | wt |
| 12 | MFS | NA 45, XX,-21/92,XXXX/92,XXXXb | wt | wt | wt | wt |
| 13 | MFS | 46, XY balanced translocation t(9,12) | wt | wt | wt | wt |
| 14 | MFS | NA | wt | wt | wt | wt |
| 15 | MFS | 46, XY | wt | wt | wt | wt |
| 16 | MFS | NA | wt | wt | wt | wt |
| 17 | MFS | 43,Y,-X,-19,-20/45,X,-Y/46,XY,+dmin/47,XY,/X/46,XY,del(4)(q22)/90,XXY,-Y,12/91,XXYY,add(l)(p36),del(2)(q31),-10,add(14)(p11),-16,-+16Mx2b | wt | wt | wt | wt |
| 18 | MFS | 82-l42,COMPLEX,1l-2Mb | wt | wt | wt | wt |
| 19 | MFS | NA | wt | wt | wt | wt |
| 20 | MFS | 46,XY,del(l)(p2l-p3l?),r(3)(q),del(5)(q),-l3,del(l5)(ql3–26.3?)b | wt | wt | wt | wt |
TP53 analysis was performed for exons 4a, 4b, 5c, 6, 7 and 8.
Karyotypes were previously published [5].
Abbreviation: NA, not available.
GNAS1, KRAS and TP53 mutation analysis
By direct sequencing, we found codon 201 activating GNAS1 mutations in 50% of intramuscular myxoma cases (including its cellular variant), but none in grade I myxofibrosarcoma cases. All GNAS1 mutations occurred in exon 8 with the base pair substitutions as shown in Table 2. GNAS1 MLPA confirmed the mutations detected by direct sequencing. In theory, MLPA should be able to detect mutant alleles in a background of wild-type alleles. Here, we show that this technique picks up mutations if only 10% of the alleles is mutated, which is superior to direct sequencing (requiring at least 25% mutated alleles). GNAS1 MLPA could not detect additional mutations. We did not observe any mutations in codon 12 or 13 of the KRAS gene in neither the intramuscular myxoma nor the grade I myxofibrosarcoma cases. Hotspot mutation analysis for TP53 showed wild-type sequences in all samples (Table 2).
LC-MS
The top 30 proteins identified in a proteomic screen of tumour lysates of grade I myxofibrosarcoma and intramuscular myxoma are shown in Table 3. Even after depletion, albumin was still the most prominent protein, and also other ‘classical’ serum proteins were among the most significant hits. We found five collagens (VI-A1, 2 and 3, XII-A1, XIV-A1) and five proteoglycans (lumican, PRG4, prolargin, decorin and biglycan) among the top 30 proteins in MFS. Only the collagen VI isoforms and lumican were identified in IM. Besides collagen VI isoforms and lumican, the other four collagens and four proteoglycans were not shown to be present in IM using mass spectrometry.
Table 3.
Proteins identified in MFS and IM
| IPI number | Protein name | Protein score | Matched queries | Seq. coverage (%) | Identified in IM |
|---|---|---|---|---|---|
| IPI00745872 | Serum albumin | 5315 | 231 | 40 | + |
| IPI00072917 | COL6A3 α 3 type VI collagen isoform 3 | 4263 | 201 | 24 | + |
| IPI00022463 | Serotransferrin | 4066 | 205 | 49 | + |
| IPI00553177 | α-1-anti-trypsin | 3593 | 164 | 40 | + |
| IPI 00845263 | Fibronectin 1 | 2443 | 123 | 21 | + |
| IPI00007960 | Periostin | 1230 | 62 | 37 | – |
| IPI00291136 | COL6A1 collagen α-1(VI) | 953 | 43 | 12 | + |
| IPI00465248 | ENO1 isoform α-enolase of α-enolase | 905 | 37 | 29 | + |
| IPI00020986 | LUM lumican precursor | 827 | 52 | 33 | + |
| IPI 00418471 | VIM vimentin | 803 | 42 | 31 | + |
| IPI00465028 | Triosephosphate isomerase | 645 | 26 | 47 | – |
| IPI 00021440 | Actin | 547 | 35 | 32 | + |
| IPI00329801 | Annexin A5 | 543 | 44 | 47 | + |
| IPI 00656111 | PRG4 | 436 | 30 | 12 | – |
| IPI 00020987 | Prolargin | 374 | 23 | 20 | – |
| IPI00022429 | ORM1 α-1-acid glycoprotein 1 precursor | 374 | 27 | 34 | + |
| IPI00418169 | Annexin A2 | 352 | 24 | 40 | + |
| IPI00302944 | COL12A1 isoform 4 of collagen α-1(XII) | 348 | 17 | 5 | – |
| IPI00304840 | COL6A2 isoform 2C2 of collagen α-2(VI) | 334 | 19 | 7 | + |
| IPI 00291262 | CLU clusterin precursor | 304 | 13 | 20 | – |
| IPI 00021841 | APOA1 apolipoprotein A-I precursor | 271 | 7 | 21 | – |
| IPI00176193 | COL14A1 isoform 1 of collagen α-1(XIV) | 264 | 12 | 9 | – |
| IPI 00793199 | Annexin IV | 257 | 10 | 30 | – |
| IPI 00013808 | α-actinin-4 | 212 | 6 | 5 | + |
| IPI 00012119 | Decorin | 194 | 23 | 25 | – |
| IPI 00010790 | Big lye an | 192 | 7 | 11 | – |
| IPI 00166729 | α-2-glycoprotein 1 | 192 | 6 | 12 | + |
| IPI 00026314 | Gelsolin | 186 | 7 | 9 | + |
Extracellular protein extracts from MFS and IM were depleted for albumin and IgG, separated by SDS-PAGE, in-gel digested with trypsin and analyzed by LC-MS. Shown are the top 30 proteins identified in MFS. Proteins that were also identified in IM are indicated with a + sign. Many forms of keratins and IgGs were also identified within the initial top 30 protein list, but for the sake of clarity, they have been removed.
qPCR for mRNA expression
Results of relative mRNA expression are shown in Table 4 and box-plots in Fig. 2. qPCR showed that c-Fos RNA was over-expressed in all tumours compared with control tissue, but not significantly differently expressed between intramuscular myxoma and grade I myxofibrosarcoma. No significant differences were seen between typical intramuscular myxoma and cellular myxoma. Grade I myxofibrosarcoma showed significant expression of decorin mRNA, whereas decorin mRNA was barely detectable in intramuscular myxoma. Grade I myxofibrosarcoma clearly showed significant over-expression of mRNA expression for collagens I-A1, VI-A1 and XIV-A1 compared with intramuscular myxoma (including cellular myxoma).
Table 4.
Log2-transformed relative expression data in qPCR
| Genes | IM (median ± S.D.) | Grade I MFS (median ± S.D.) | IM vs grade I MFS (P-value) |
|---|---|---|---|
| FOS | 4.06 ± 0.87 | 4.61 ± 0.89 | 0.105 |
| DCN | −6.92 ± 1 | 2.81 ± 0.79 | 0.001 |
| COL1A1 | 1.95 ± 1.84 | 6.71 ± 1.71 | 0.003 |
| COL6A1 | 0.14 ± 0.22 | 2.37 ± 0.47 | 0.023 |
| COL12A1 | −0.85 ± 1.11 | −0.22 ± 1.28 | 0.09 |
| COL14A1 | −0.29 ± 0.49 | 1.49 ± 0.59 | 0.001 |
Abbreviations: FOS, FBJ murine osteosarcoma viral oncogene homolog; DCN, decorin; COL1A1, collagen, type I, α1; COL6A1, collagen VI, α1; COL12A1, collagen, type XII, α1; COL14A1, collagen, type XIV, α1.
Figure 2.
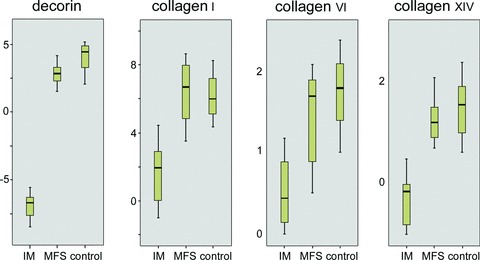
Box-plots showing qPCR results of structural ECM proteins. Abbreviations: IM, intramuscular myxoma; MFS, grade I myxofibrosar-coma. Intramuscular myx-oma showed significantly lower mRNA expression for decorin (P= 0.000), collagen I-A1 (P= 0.003), collagen VI-A1 (P= 0.023) and collagen XIV-A1 (P= 0.001).
Immunohistochemistry
The majority of intramuscular myxomas and grade I myxofibrosarcomas showed diffuse cytoplasmic and nuclear staining for c-Fos (Table 5). Both cytoplasmic and nuclear staining for c-Fos have been obtained in FFPE materials from other tumours [27]. However, the biological role of cytoplasmic c-Fos expression is not fully understood. And because c-Fos is a transcription factor that is active in the nucleus, we assessed only nuclear staining in scoring. Intramuscular myxoma and grade I myxofibrosarcoma did not show significantly different c-Fos expression (P = 0.648; Table 5). Decorin, collagen I-A1 and collagen VI-A1 were only present in the ECM. Strong positive staining for decorin was detected in all grade I myxofibrosarcomas but not in intramuscular myxoma, including its cellular variant (P = 0.0000). Collagen I-A1 expression was found to be equally present in the ECM of grade I myxofibrosarcoma and intramuscular myxoma (P = 1.000). Collagen VI-A1 expression was present in 40% of the ECM of intramuscular myxoma and all grade I myxofibrosarcoma (P = 0.004). Illustrations of collagen I-A1 and VI-A1 protein expression are shown in Fig. 1. Both grade I myxofibrosarcoma and intramuscular myxoma showed membranous and cytoplasmic staining for CD44 in the majority of cases, the latter most probably due to its production in the rough endoplasmic reticulum. As we were interested in the role of CD44 as a cell surface receptor for ECM molecules, only membranous staining was assessed in statistical analysis. Tumour cells of intramuscular myxoma and grade I myx-ofibrosarcoma showed no significantly different expression for CD44 (P= 0.0542; Table 5). Illustrations are shown in Fig. 1.
Table 5.
Results for immunohistochemical staining in intramuscular myxoma and grade I myxofibrosarcoma
| c-fos | dcn | col1a1 | col6a1 | cd44 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| pos* | pos# | pos* | pos# | pos* | pos# | pos* | pos# | pos* | pos# | |
| IM | 6/10 | 60 | 0/0 | 0 | 7/10 | 70 | 4/10 | 40 | 8/10 | 80 |
| Grade I MFS | 7/10 | 70 | 10/10 | 100 | 7/10 | 70 | 10/10 | 100 | 9/10 | 90 |
Abbreviations: IM, intramuscular myxoma; MFS, myxofibrosarcoma; dcn, decorin; pos*, number of positive tumours/total number of tumours that could be evaluated; pos#, percentage of cases that were positive.
Discussion
The cellular variant of intramuscular myxoma, also known as cellular myxoma, is the morphological intermediate between intramuscular myxoma and grade I myxofibrosarcoma. Compared with typical intramuscular myxoma, cellular myxoma is hypercellular, usually more vascular and shows increase in collagenous stroma. However, it lacks clear cytonuclear atypia and classical curvilinear blood vessels [2]. The risk of recurrence is rather low, which is sustained by the present data showing local recurrence in 50% of grade I myxofibrosarcoma but not in intramuscular myxoma. This includes the cases of the cellular variant, even after long-term follow-up, which is in accordance with the literature [2, 28]. It should be noted that not all cellular myxomas are truly intramuscular (see Table 1), which was already demonstrated by Van Roggen et al.[2]. In a previous study, we showed that myxofibrosarcoma is characterized by complex, non-specific cytogenetic aberrations probably reflecting genomic instability [5]. Chromosomal instability is often an early event in tumourigenesis, and there is a significant correlation between chromosomal instability phenotype and poor biological behaviour [29]. This is sustained by our data showing karyotypic aberrations only in myxofibrosarcoma but not in intramuscular myxoma. TP53 is an important mitotic checkpoint regulator, and p53 deficiency plays an important role in chromosomal instability and ploidy control [29]. Mutations in TP53 resulting in dysfunctional p53 occur mostly (93%) in exons 4–8, which are therefore called hotspot mutations [30]. We did not detect any TP53 hotspot mutations in intramuscular myxoma or in grade I myxofibrosarcoma. This means that TP53 mutations might not be involved in the tumourigenesis of either tumour. Otherwise, it might support the notion that structural loss of p53 is not always the cause of aneuploidy and that loss of p53 might represent a late event in tumourigenesis of myxofibrosarcoma, whereas aneuploidy occurs early [31]. This was sustained by previous data that p53 immunohistochemical staining was predominantly found in myxofibrosarcoma of grade II and III [10]. Cytogenetic data on intramuscular myxoma are sparse in the literature, with only two isolated cases described showing normal karyotypes [4, 32]. The present study confirms in a larger series that intramuscular myxoma has a normal karyotype. Cytogenetics might therefore be helpful in the differential diagnosis but are not always easily available in routine laboratories.
Okamoto et al. showed GNAS1-activating mutations in a small series of intramuscular myxoma [6]. We showed that GNAS1 codon 201 mutations were present in 50% of intramuscular myxoma and not in grade I myxofibrosarcoma. All mutations were heterozygous. Direct sequencing of codon 201 GNAS1 mutations is therefore a highly specific, not very sensitive but easy to perform method to distinguish intramuscular myxoma from grade I myxofibrosarcoma. Interestingly, here we report for the first time the R201S mutation in intramuscular myxoma. We could not detect any mutation in codon 227 (exon 9) of the GNAS1 gene, previously detected in fibrous dysplasia [33]. The GNAS1 gene encodes for the alpha sub-unit of the G-protein. Somatic-activating mutations in codon 201 and 227 of the GNAS1 gene have been described in fibrous dysplasia and the related Mazabraud and McCune Albright syndromes, leading to increased levels of cAMP and activation of protein kinase A and the MAPK pathway [7, 33–35]. This leads to increased transcription of the c-Fos protein, which is involved in growth and the inhibition of apoptosis [36]. We showed both RNA and protein over-expression for c-Fos in each of the tumour samples of intramuscular myxoma and grade I myxofibrosarcoma, suggesting that the MAPK signalling pathway is activated in these tumours. We did not detect GNAS1-activating mutations in myxofibrosarcoma nor did we detect codon 201 or 227 activating mutations in the other 50% of the intramuscular myxoma cases. As both are hypocellular tumours, we hypothesized that wild-type DNA of blood vessels and lymphocytes might interfere with the detection of this heterozygous mutation. Neither enrichment for mutated DNA by microdissection nor the use of more sensitive method to detect the mutations (MLPA) with a detection threshold of only 10% could increase the number of mutations found. Therefore, we believe that activation of the MAPK pathway in the tumours with no detectable mutations is caused by a different mechanism than GNAS1-activating mutations, possibly downstream of the G-protein. In vivo studies on mice showed that KRAS-activating mutations play an important role in sarcomagenesis [8]. In general, about 30% of solid tumours show activating mutations in codon 12 and 13 of the KRAS gene causing activation of the MAPK pathway and increased transcription of c-Fos [37, 38]. Moreover, KRAS mutations have been associated with chromosomal instability suggesting a potential role of KRAS in the tumourigenesis of grade I myxofibrosarcoma [39]. However, we could not detect any activating mutations in codon 12 and 13 of the KRAS gene in intramuscular myxoma nor grade I myxofibrosarcoma.
Intramuscular myxoma (including its cellular variant) and grade I myxofibrosarcoma are both characterized by their abundant so-called myxoid ECM. We have shown recently that glycosaminoglycans (e.g. hyaluronic acid) are major polysaccharides in the myxoid ECM of intramuscular myxoma and myxofibrosarcoma [11]. Based on the present, more detailed LC-MS–based survey, we showed that decorin, collagen VI-A1 and XIV-A1 were significantly over-expressed in the ECM of grade I myxofibrosarcoma compared with that of intramuscular myxoma. Decorin is a small leucine-rich proteoglycan (SLRP) in the ECM that ‘decorates’ collagens by interaction with their ‘d’ and ‘e’ bands. Decorin links these collagens (especially collagens I, VI and XIV) and plays an important role in fibrillogenesis and ECM formation. Decreased and impaired expression of decorin leads to abnormal ECM formation as in Ehlers–Danlos syndrome [40, 41]. Next to decorin, LC-MS revealed other SLRPs (i.e. lumican, PRG4, prolargin, biglycan) present in tumour lysates of grade I myxofibrosarcoma but not of intramuscular myxoma. Collagens XII and XIV are fibril-associated collagens with triple helices (FACIT) and modify the interactions between collagen I fibrils and the surrounding ECM [42]. Increased ECM rigidity activates integrins to promote focal adhesion families, leading to stimulation of the rho/Rock pathway and increased cell contractility, cell migration and invasion [43]. Intramuscular myxoma was found to express significantly less decorin and collagen VI and XIV (both protein and mRNA level) than grade I myxofibrosarcoma, suggesting that ECM formation in intramuscular myxoma is impaired compared with grade I myxofibrosarcoma. Validation of these immunohistochemical results in an independent series of both entities would be interesting though is hampered by the availability of well-characterized frozen material. Cell surface receptors facilitate the assembly and retention of the ECM and link changes in the ECM to activation of signal transduction pathways [44]. CD44 glycoprotein is a well-characterized cell adhesion molecule that is ubiquitously expressed on tumour cells of intramuscular myxoma and grade I myxofibrosarcoma. Its principal ligand is hyaluronic acid which was, however, not expressed significantly differently in intramuscular myxoma compared with grade I myxofibrosarcoma [11]. CD44 is also an important cell surface receptor for collagen I and XIV, suggesting that ECM-cell signalling via CD44 might play a role in these myxoid tumours of soft tissue [45]. Although grade I myxofibrosarcoma showed significantly higher mRNA expression for collagen I and XIV, this was not obvious at the protein level for collagen I. Whether differences in (the assembly of) the ECM of myxoid tumours of soft tissue effectively affect cell signalling and subsequent tumour growth and progression is still unknown. The evidence in our studies to support this theory is indirect and our results cannot exclude that the content and organization of the myxoid ECM might be just an epiphenomenon, driven by the initiating genetic event(s) or by the precise lineage or sub-type of the cell that is transformed. Based on our results, we suggest that molecular and cytogenetic aberrations as well as proper ECM organization might explain the different biology of these tumours.
Acknowledgments
We acknowledge Judith V.M.G. Bovée for critical comments and discussion, Marjo van Puijenbroek for performing the KRAS mutation analysis, Ronald van Eijk for help with TP53 mutation analysis and Marije IJszenga for performing the COBRA-FISH experiments. Rabbit polyclonal anti-decorin antibody was kindly provided by Annemiek van der Wal (Department of Pathology, Leiden University Medical Center, Leiden, the Netherlands). This project was financially supported by a grant from The Netherlands Organization for Health Research and Development (project number 920-03-403).
Supporting Information
Table S1 Primer sequences for qPCR and direct sequencing Abbreviations: FOS, FBJ murine osteosarcoma viral oncogene homolog; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; TBP, TATA box binding protein; GNAS1, GNAS complex locus; DCN, decorin; COL1A1, collagen type I 1; COL6A1, collagen VI 1; COL12A1, collagen type XII, 1; COL14A1, collagen type XIV, 1. * including additional M13 tail.
Table S2 Primers used for TP53 hotspot mutation analysis
Table S3 Primers and probes used for MLPA Abbreviation: GNAS1, GNAS complex locus.
Table S4 Details of the antibodies used and the immunohistochemical protocols applied
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
References
- 1.Graadt van Roggen JF, Hogendoorn PCW, Fletcher CDM. Myxoid tumours of soft tissue. Histopathology. 1999;35:291–12. doi: 10.1046/j.1365-2559.1999.00835.x. [DOI] [PubMed] [Google Scholar]
- 2.Graadt van Roggen JF, McMenamin ME, Fletcher CDM. Cellular myxoma: a clinicopathologic study of 38 cases confirming indolent clinical behavior. Mod Pathol. 2001;39:287–97. doi: 10.1046/j.1365-2559.2001.01209.x. [DOI] [PubMed] [Google Scholar]
- 3.Mentzel T, Calonje E, Wadden C, et al. Myxofibrosarcoma. Clinicopathologic analysis of 75 cases with emphasis on the low-grade variant. Am J Surg Pathol. 1996;20:391–05. doi: 10.1097/00000478-199604000-00001. [DOI] [PubMed] [Google Scholar]
- 4.Meis-Kindblom JM, Sjogren H, Kindblom LG, et al. Cytogenetic and molecular genetic analyses of liposarcoma and its soft tissue simulators: recognition of new variants and differential diagnosis. Virchows Arch. 2001;439:141–51. doi: 10.1007/s004280100423. [DOI] [PubMed] [Google Scholar]
- 5.Willems SM, Debiec-Rychter M, Szuhai K, et al. Local recurrence of myxofibrosarcoma is associated with increase in tumour grade and cytogenetic aberrations, suggesting a multistep tumour progression model. Mod Pathol. 2006;19:407–16. doi: 10.1038/modpathol.3800550. [DOI] [PubMed] [Google Scholar]
- 6.Okamoto S, Hisaoka M, Ushijima M, et al. Activating Gs(alpha) mutation in intramuscular myxomas with and without fibrous dysplasia of bone. Virchows Arch. 2000;437:133–37. doi: 10.1007/s004280000217. [DOI] [PubMed] [Google Scholar]
- 7.Candeliere GA, Glorieux FH, Prud’homme J, St.-Arnaud R. Increased expression of the c-fos proto-oncogene in bone from patients with fibrous dysplasia. N Engl J Med. 1995;332:1546–51. doi: 10.1056/NEJM199506083322304. [DOI] [PubMed] [Google Scholar]
- 8.Kirsch DG, Dinulescu DM, Miller JB, et al. A spatially and temporally restricted mouse model of soft tissue sarcoma. Nat Med. 2007;13:992–97. doi: 10.1038/nm1602. [DOI] [PubMed] [Google Scholar]
- 9.Borden EC, Baker LH, Bell RS, et al. Soft tissue sarcomas of adults: state of the translational science. Clin Cancer Res. 2003;9:1941–56. [PubMed] [Google Scholar]
- 10.Oda Y, Takahira T, Kawaguchi K, et al. Altered expression of cell cycle regulators in myxofibrosarcoma, with special emphasis on their prognostic implications. Hum Pathol. 2003;34:1035–42. doi: 10.1053/s0046-8177(03)00404-0. [DOI] [PubMed] [Google Scholar]
- 11.Willems SM, Schrage YM, Baelde JJ, et al. Myxoid soft tissue tumours have a heterogeneous composition of their extra-cellular matrix. Histopathology. 2008;52:465–74. doi: 10.1111/j.1365-2559.2008.02967.x. [DOI] [PubMed] [Google Scholar]
- 12.Mentzel T, Van Den Berg E, Molenaar WM. Myxofibrosarcoma. World Health Organization classification of tumours: pathology and genetics. In: Mertens FL, editor; Fletcher CDM, Unni KK, editors. Tumours of soft tissue and bone. IARC Press; 2004. pp. 102–03. Lyon. [Google Scholar]
- 13.Trojani M, Contesso G, Coindre JM, et al. Soft-tissue sarcomas of adults; study of pathological prognostic variables and definition of a histopathological grading system. Int J Cancer. 1984;33:37–42. doi: 10.1002/ijc.2910330108. [DOI] [PubMed] [Google Scholar]
- 14.Szuhai K, Tanke H. COBRA: combined binary ratio labeling of nucleic-acid probes for multi-color fluorescence in situ hybridization karyotyping. Nat. Protoc. 2006;1:264–75. doi: 10.1038/nprot.2006.41. [DOI] [PubMed] [Google Scholar]
- 15.De Leeuw WJ, Dierssen J, Vasen HF, et al. Prediction of a mismatch repair gene defect by microsatellite instability and immunohistochemical analysis in endometrial tumours from HNPCC patients. J Pathol. 2000;192:328–35. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH701>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 16.Candeliere GA, Roughley PJ, Glorieux FH. Polymerase chain reaction-based technique for the selective enrichment and analysis of mosaic arg201 mutations in G alpha s from patients with fibrous dysplasia of bone. Bone. 1997;21:201–06. doi: 10.1016/s8756-3282(97)00107-5. [DOI] [PubMed] [Google Scholar]
- 17.Bovée JVMG, Devilee P, Cornelisse CJ, et al. Identification of an EWS-pseudogene using translocation detection by RT-PCR in Ewing’s sarcoma. Biochem Biophys Res Commun. 1995;213:1051–60. doi: 10.1006/bbrc.1995.2234. [DOI] [PubMed] [Google Scholar]
- 18.Rozeman LB, Hameetman L, Cleton-Jansen AM, et al. Absence of IHH and retention of PTHrP signalling in enchon-dromas and central chondrosarcomas. J Pathol. 2005;205:476–82. doi: 10.1002/path.1723. [DOI] [PubMed] [Google Scholar]
- 19.White SJ, Vink GR, Kriek M, et al. Two-color multiplex ligation-dependent probe amplification: detecting genomic rearrangements in hereditary multiple exostoses. Hum Mutat. 2004;24:86–92. doi: 10.1002/humu.20054. [DOI] [PubMed] [Google Scholar]
- 20.Brink M, De Goeij AF, Weijenberg MP, et al. K-ras oncogene mutations in sporadic colorectal cancer in The Netherlands Cohort Study. Carcinogenesis. 2003;24:703–10. doi: 10.1093/carcin/bgg009. [DOI] [PubMed] [Google Scholar]
- 21.Steen H, Pandey A, Andersen JS, Mann M. Analysis of tyrosine phosphorylation sites in signaling molecules by a phosphotyrosine-specific immonium ion scanning method. Sci STKE. 2002;2002:L16. doi: 10.1126/stke.2002.154.pl16. [DOI] [PubMed] [Google Scholar]
- 22.Romeo S, Eyden B, Prins FA, et al. TGF-beta1 drives partial myofibroblastic differentiation in chondromyxoid fibroma of bone. J Pathol. 2006;208:26–34. doi: 10.1002/path.1887. [DOI] [PubMed] [Google Scholar]
- 23.Vandesompele J, De Preter K, Pattyn F, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-7-research0034. : research0034.1-.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rozeman LB, Hameetman L, Van Wezel T, et al. cDNA expression profiling of central chondrosarcomas: Ollier disease resembles solitary tumors and alteration in genes coding for energy metabolism with increasing grade. J Pathol. 2005;207:61–71. doi: 10.1002/path.1813. [DOI] [PubMed] [Google Scholar]
- 25.Koop K, Bakker RC, Eikmans M, et al. Differentiation between chronic rejection and chronic cyclosporine toxicity by analysis of renal cortical mRNA. Kidney Int. 2004;66:2038–46. doi: 10.1111/j.1523-1755.2004.00976.x. [DOI] [PubMed] [Google Scholar]
- 26.Bovée JVMG, Van Den Broek LJCM, et al. Up-regulation of PTHrP and Bcl-2 expression characterizes the progression of osteochondroma towards peripheral chondrosarcoma and is a late event in central chondrosarcoma. Lab Invest. 2000;80:1925–33. doi: 10.1038/labinvest.3780202. [DOI] [PubMed] [Google Scholar]
- 27.Hoyland J, Sharpe PT. Upregulation of c-fos protooncogene expression in pagetic osteoclasts. J Bone Miner Res. 1994;9:1191–4. doi: 10.1002/jbmr.5650090808. [DOI] [PubMed] [Google Scholar]
- 28.Nielsen GP, O’Connell JX, Rosenberg AE. Intramuscular myxoma: a clinicopathologic study of 51 cases with emphasis on hypercellular and hypervascular variants. Am J Surg Pathol. 1998;22:1222–7. doi: 10.1097/00000478-199810000-00007. [DOI] [PubMed] [Google Scholar]
- 29.Tomasini R, Mak TW, Melino G. The impact of p53 and p73 on aneuploidy and cancer. Trends Cell Biol. 2008;18:244–52. doi: 10.1016/j.tcb.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 30.Hernandez-Boussard T, Rodriguez-Tome P, Montesano R, Hainaut P. IARC p53 mutation database: a relational database to compile and analyze p53 mutations in human tumors and cell lines. International Agency for Research on Cancer. Hum Mutat. 1999;14:1–8. doi: 10.1002/(SICI)1098-1004(1999)14:1<1::AID-HUMU1>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 31.Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature. 1998;396:643–9. doi: 10.1038/25292. [DOI] [PubMed] [Google Scholar]
- 32.Lopez-Ben R, Pitt MJ, Jaffe KA, Siegal GP. Osteosarcoma in a patient with McCune-Albright syndrome and Mazabraud’s syndrome. Skeletal Radiol. 1999;28:522–6. doi: 10.1007/s002560050556. [DOI] [PubMed] [Google Scholar]
- 33.Weinstein LS, Liu J, Sakamoto A, et al. Minireview: GNAS: normal and abnormal functions. Endocrinology. 2004;145:5459–64. doi: 10.1210/en.2004-0865. [DOI] [PubMed] [Google Scholar]
- 34.Vallar L, Spada A, Giannattasio G. Altered Gs and adenylate cyclase activity in human GH-secreting pituitary adenomas. Nature. 1987;330:566–8. doi: 10.1038/330566a0. [DOI] [PubMed] [Google Scholar]
- 35.Weinstein LS, Shenker A, Gejman PV, et al. Activating mutations of the stimulatory G protein in the McCune-Albright syndrome. N Engl J Med. 1991;325:1688–95. doi: 10.1056/NEJM199112123252403. [DOI] [PubMed] [Google Scholar]
- 36.Wada T, Penninger JM. Mitogen-activated protein kinases in apoptosis regulation. Oncogene. 2004;23:2838–49. doi: 10.1038/sj.onc.1207556. [DOI] [PubMed] [Google Scholar]
- 37.Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007;26:3279–90. doi: 10.1038/sj.onc.1210421. [DOI] [PubMed] [Google Scholar]
- 38.Kranenburg O. The KRAS oncogene: past, present, and future. Biochim Biophys Acta. 2005;1756:81–2. doi: 10.1016/j.bbcan.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 39.Castagnola P, Giaretti W. Mutant KRAS, chromosomal instability and prognosis in colorectal cancer. Biochim Biophys Acta. 2005;1756:115–25. doi: 10.1016/j.bbcan.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 40.Ferdous Z, Wei VM, Iozzo R, et al. Decorin-transforming growth factor-interaction regulates matrix organization and mechanical characteristics of three-dimensional collagen matrices. J Biol Chem. 2007;282:35887–98. doi: 10.1074/jbc.M705180200. [DOI] [PubMed] [Google Scholar]
- 41.Ruhland C, Schonherr E, Robenek H, et al. The glycosaminoglycan chain of decorin plays an important role in collagen fibril formation at the early stages of fibrillo-genesis. FEBS J. 2007;274:4246–55. doi: 10.1111/j.1742-4658.2007.05951.x. [DOI] [PubMed] [Google Scholar]
- 42.Canty EG, Kadler KE. Procollagen trafficking, processing and fibrillogenesis. J Cell Sci. 2005;118:1341–53. doi: 10.1242/jcs.01731. [DOI] [PubMed] [Google Scholar]
- 43.Berrier AL, Yamada KM. Cell-matrix adhesion. J Cell Physiol. 2007;213:565–73. doi: 10.1002/jcp.21237. [DOI] [PubMed] [Google Scholar]
- 44.Toole BP. Hyaluronan: from extracellular glue to pericellular cue. Nat Rev Cancer. 2004;4:528–39. doi: 10.1038/nrc1391. [DOI] [PubMed] [Google Scholar]
- 45.Ehnis T, Dieterich W, Bauer M, et al. A chondroitin/dermatan sulfate form of CD44 is a receptor for collagen XIV (undulin) Exp Cell Res. 1996;229:388–97. doi: 10.1006/excr.1996.0384. [DOI] [PubMed] [Google Scholar]
- 46.Romeo S, Debiec-Rychter M, Van Glabbecke M, et al. Cell cycle/apoptosis molecule expression correlates with imatinib response in patients with advanced gastrointestinal stromal tumors. Clin Cancer Res. 2009;15:4191–8. doi: 10.1158/1078-0432.CCR-08-3297. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting info item