

Abstract
Pancreatitis-associated ascitic fluid (PAAF) is known to contribute to the progression of acute pancreatitis (AP). We have investigated the capability of PAAF to activate the expression of MCP-1 in pancreatic acinar cells and the involvement of MAPK, NF-κB and STAT3 as downstream signalling transduction pathways. The actions of dexamethasone (Dx) and N-acetylcysteine (NAC) on the PAAF’s acinar effects have also been evaluated. Acinar cells were incubated for 1 hr with PAAF collected from rats with severe AP induced by sodium taurocholate in the absence or presence of Dx (10−7 M) or NAC (30 mM). MCP-1 mRNA expression, phospho-p38-MAPK, IκBα, nuclear p65 levels and nuclear translocation of STAT3 were analysed. In response to PAAF, overexpression of MCP-1, phosphorylation of p38-MAPK, degradation of IκBα and increases in p65 nuclear levels and STAT3 activity were found in acinar cells. PAAF-mediated MCP-1 up-regulation was completely suppressed by Dx and NAC. MAPK activation was only inhibited by NAC, NF-κB activation was repressed by Dx and NAC, and STAT3 pathway was strongly blocked by Dx and significantly reduced by NAC. In conclusion, acinar cells were activated by PAAF to produce MCP-1, mainly via NF-κB and STAT3 pathways. Both downstream pathways were targeted by Dx and NAC to repress the PAAF-mediated acinar MCP-1 up-regulation.
Keywords: acinar cells, dexamethasone, MCP-1, N-acetylcysteine, pancreatitis-associated ascitic fluid, signal transduction
Introduction
Premature activation of pancreatic enzymes and secondary oxidative stress are associated with impairment in the pancreatic micro-circulation, which leads to the accumulation of ascitic fluid in the abdominal cavity [1, 2]. Pancreatitis-associated ascitic fluid (PAAF) is known to be important in the progression of acute pancreatitis (AP) [3, 4], because it contains mediators involved in the multiple organ failure, the main cause of death of the disease [5]. PAAF has been shown to up-regulate the expression of adhesion molecules in human endothelial cells [6], as well as cytokines in lung [7], leukocytes [8] and pancreatic acinar cells [9, 10]. Downstream signal transduction pathways have been demonstrated to mediate the expression of inflammatory mediators in acinar cells during AP, including nuclear factor-κB (NF-κB) [11–13] and mitogen-activated protein kinases (MAPK) [13–15]. Signal transducers and activators of transcription (STAT) family of transcription factors have similar profiles of target genes as NF-κB [16]. The STAT proteins are in a latent form in the cytoplasm and, on receptor activation by cytokines, become phosphorylated by members of the Janus kinase (JAK) family, which are physically associated with the receptor [17]. Upon phosphorylation, the STAT proteins dimerize and migrate to the nucleus where they bind to DNA and regulate specific gene expression [18]. Studies carried out in STAT knockout mice demonstrated that STATs are involved in cytokine signalling and immune responses [19]. Expression of JAK and STAT proteins has been demonstrated in pancreas of rats, acinar cells being the main source of these proteins rather than ductal, endocrine, vascular or blood cells [20]. In vitro experiments [20, 21] showed STAT1 and STAT3 activation in acinar cells induced by IFN-γ and TNF-α, respectively; however, their role in mediating the inflammatory response in AP is not well established.
Dexamethasone (Dx), a potent anti-inflammatory corticosteroid, and N-acetylcysteine (NAC), a powerful antioxidant, have shown to be beneficial in the treatment of AP by affecting the cytokine network [13, 22, 23]; however, the mechanisms of how this occurs remain unclear.
The aim of this study was to investigate the effect of PAAF on the expression of chemokine MCP-1 in pancreatic acinar cells as well as the involvement of MAPK, NF-κB and STAT3 as its signalling transduction pathways. The actions of Dx and NAC on PAAF-stimulated acinar cells were also investigated in order to find out whether the mechanisms mediating the acinar chemokine expression are sensitive to glucocorticoid and/or antioxidant treatments.
Methods
Reagents
N-Acetyl-l-cysteine (NAC), dexamethasone 21 phosphate disodium salt (Dx), sodium taurocholate (NaTc), buprenorphine, aminoacid mixture, bovine serum albumin (BSA), collagenase type XI, soybean trypsin inhibitor (STI), N-(2-hydroxyethyl) piperazine-N′-(2-ethanesulfonic acid) (HEPES), streptomycin and penicillin solution, Nonidet P-40, aprotinin, leupeptin, pepstatin, antipapain, chymostatin, phenylmethanesulfonyl fluoride (PMSF) and dithiothreitol (DTT) were supplied by Sigma Chemical Co (Madrid, Spain). Medium 199 (Gibco, Paisley, Scotland) and calf foetal serum (Biowhittaker, Walkersville, MD, USA) were also used. Other standard analytical grade laboratory reagents were obtained from Merck (Darmstadt, Germany).
Animals
Male Wistar rats (250–300 g) were housed individually in cages and maintained at 22 ± 1°C under a 12-hr light/dark cycle. The animals were fasted overnight before the experiment but they were allowed free access to water. All experiments were performed in accordance with European Community guidelines on ethical animal research, established by the European Community (86/609/EEC). The study was approved by the Institutional Animal Care and Use Committee of the University of Salamanca (Spain).
Production and preparation of PAAF
After 12 hrs of fasting, under strict sterile conditions and anaesthesia with 2–3% isofluorane, Forane® (Abott, Madrid, Spain) laparotomy was performed in order to induce AP in rats by the intraductal administration of a previously sterilized solution of 3.5% sodium taurocholate (NaTc). Following clamping of the proximal common bile duct 0.1 ml of NaTc solution was infused at 0.03 ml/min. After infusion, the bile duct clamp was removed and the abdominal wall was closed. Postoperative analgesia was maintained by intramuscular injections of buprenorphine (0.2 mg/Kg). PAAF was harvested by aspiration under sterile conditions from the peritoneal cavity 18 hrs after inducing AP. PAAFs from five different rats were pooled and centrifuged at 2500 g for 15 min. at 4°C, and the supernatants passed through a 0.2 μm filter (Millipore, Bedford, MA, USA). The resulting flowthrough was aliquoted and stored at −80°C until use.
Treatment of pancreatic acini
After 12 hrs of fasting and under anaesthesia with intraperitoneal injections of sodium pentobarbital (Roig Farma, Barcelona, Spain) at dose of 3 mg/100 g body weight, the pancreas of healthy rats was removed in order to isolate acinar cells by collagenase digestion at 20°C as previously described [13]. Afterwards, pancreatic acini were resuspended in Medium 199 supplemented with 10% of heat-inactivated calf foetal serum, streptomycin (0.1 mg/ml), and penicillin (100 U/ml) solution. In accordance with the results obtained in a separate set of experiments (Fig. 1), acinar cells were incubated for 1 hr in the absence (basal conditions) or presence of PAAF (20% v/v) containing 200 μg/ml STI. To investigate the effect of glucocorticoids and antioxidants, acinar cells were pre-incubated with Dx (10−7 M) or NAC (30 mM), respectively, for 30 min. before adding PAAF.
Figure 1.
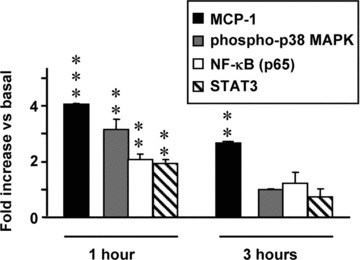
Time-course of acinar cells activation by pancreatitis-associated ascitic fluid (PAAF, 20% v/v). Results are mean ± S.E.M. Paired Student’s t-test showed significant differences versus non-PAAF-stimulated cells (basal conditions). (**P < 0.01, ***P < 0.001).
Analysis of MCP-1 mRNA expression by RT-PCR
Total RNA was extracted from acinar cells using RNAeasy kit treated with amplification grade DNase 1 (Quiagen, Valencia, Spain) according to the manufacturer’s instructions. Purity of RNA was verified by ethidium bromide staining on 1% agarose gels and its integrity by the presence of well-defined 28S and 18S rRNA bands.
Total RNA (1 μg) was reversed transcribed by using 1st Strand cDNA synthesis kit (Roche, Mannheim, Germany). The cDNA synthesized was used as template for PCR amplification by using Taq DNA polymerase, dNTPack (Roche) in the presence of 0.2 μM MCP-1 primers (Roche): MCP-1 (sense: 5′-CACTATGCAGGTCTCTGTCACG-3′, antisense; 5′-GACTCACTTGGTTCTG-GTCCA-3′, product size: 294 bp). Oligonucleotide primers for β-actin (sense: 5′-CACGGCATTGTAACCAACTG-3′, antisense: 5′-TCTCAGCTGTGGTGGT-GAAG-3′, product size: 400 bp) were used as the housekeeping gene. PCR was performed by the following procedures: MCP-1 (94°C for 30 sec., 56°C for 45 sec., 72°C for 45 sec., 30 cycles) and β-actin (94°C for 30 sec., 56°C for 30 sec., 68°C for 45 sec., 30 cycles). The amplified PCR products were separated on a 2% agarose gel stained with ethidium bromide. The MCP-1 and β-actin PCR products were run together on the same gel in order to normalize the band densities to the β-actin band, which were densitometrically quantified with a Gel Doc 1000/2000 image analysis system (BioRad, CA, USA) using the QuantityOne software programme.
Analysis of phospho p-38 MAPK and IκBα
Preparation of cytoplasmic cell lysates
Acinar cells were homogenized on ice in HEPES buffer, 10 mM, pH 7.9, containing 2 mM EDTA, 25 mM KCl and supplemented with 2 mM PMSF, DTT and a protease inhibitor cocktail containing aprotinin, leupeptin, pep-statin, antipapain and chymostatin (5 μg/ml each). The mixtures were maintained on ice for 20 min., after which Nonidet P-40 (0.4% v/v) was added for 2 min. and then centrifuged at 4°C for 5 min. at 14,000 g. The supernatants were collected and immediately stored at −80°C.
Western blot analysis
Cytoplasmic extracts from acinar cells (40 μg protein) were separated by 12% SDS-PAGE and electrophoretically transferred to a nitrocellulose membrane. Non-specific binding was blocked by incubating the blot in Tris-buffered saline (TBS) pH 7.6, containing 0.1% (v/v) Tween 20 and 5% (w/v) non-fat dry milk for 1 hr. Afterwards, blots were incubated with the primary antibody against either phospho-p38 MAPK or IκBα (Cell Signalling Technology, Beverly, MA, USA) at 1:1000 dilution in TBS buffer pH 7.6, containing 0.1% (v/v) Tween 20 and 5% (w/v) BSA overnight at 4°C. After washing for 1 hr with TBS containing 0.1% Tween 20, the blots were incubated for 1 hr with the respective horseradish peroxidase-conjugated secondary antibody at 1:2000 dilution in TBS buffer pH 7.6, containing 0.1% Tween 20 and 5% (w/v) non-fat dry milk and finally they were developed for visualization. The bands were detected with Phototope-HRP Detection kit (Cell Signalling Technology). Image J 1.32 software from http://rsbweb.nih.gov/ij/download.html was used to quantify the intensity of the bands.
Determination of NF-κB and STAT3 activation
Nuclear cell extract preparation
Nuclear protein extracts were obtained using a commercial nuclear extract kit following the recommendations of the manufacturer (Active Motif, Reixenxart, Belgium). Basically, isolated acinar cells were washed with ice-cold phosphate-buffered saline (PBS) containing phosphatase inhibitors and then lysed on ice in hypotonic buffer containing a cocktail of protease inhibitors. Nonidet P-40 (0.4% v/v) was added after 15 min. and incubated for 1 min. Nuclear fraction was collected in the pellet after centrifuging 30 sec. at 14,000 g and resuspended in lysis buffer containing DTT and protease inhibitor cocktail. After vortexing and incubating for 30 min. on ice, nuclear membranes were pelleted by centrifugation at 14,000 g for 10 min. and nuclear extract was collected from the supernatant and stored at −80°C.
NF-κB and STAT3 DNA binding was measured in nuclear extracts with the respective ELISA-based commercial kits (Trans AM NF-κB p65 and TransAM STAT3 activation assay, Active Motif, Reixenxart, Belgium). Nuclear proteins (5 μg) were added to each well coated with an oligonucleotide containing the consensus binding site for either NF-κB or STAT3 and incubated for 1 hr. Activation was detected by incubation for 1 hr with the respective primary antibody: anti-NF-kB, which specifically recognizes an epitope (p65) accessible only when the factor is activated and bound to its target DNA or anti-STAT3, which recognizes epitopes only accessible when STAT3 is activated. A secondary anti-IgG horseradish peroxidase conjugate allows detection of the activated NF-κB and STAT3 by a colorimetric reaction. Absorbance was read within 5 min. at 450 nm with a reference wavelength of 655 nm.
Statistical analysis
Data are expressed as mean ± S.E.M. Paired Student’s t-test was used in the comparison of data between non-PAAF-stimulated and PAAF-stimulated cells. Analysis of variance (ANOVA) followed by the Dunnett test was applied to evaluate the differences between more than two experimental groups. P-values lower than 0.05 were considered to be significant.
Results
Time-course of PAAF activation
As Fig. 1 shows, overexpression of MCP-1 was found in acinar cells incubated with PAAF for 1 hr and 3 hrs. However, phosphorylation of p38-MAPK and activation of NF-κB (measured by p65 nuclear levels) and STAT3 were only significantly (P < 0.01) increased in cells cultured with PAAF for 1 hr (time of PAAF activation in all subsequent experiments).
Viability of pancreatic acinar cells
Viability of the pancreatic acinar cells, determined 1 hr after cell culture by trypan blue dye exclusion assay, was 95%.
MCP-1 mRNA expression
Prominent MCP-1 RT-PCR products were found in acinar cells incubated with PAAF (Fig. 2). MCP-1/β-actin mRNA ratio indicated a four-fold increase in MCP-1 expression when compared with acinar cells cultured in the absence (basal conditions) and the presence of PAAF. Dx and NAC abrogated the PAAF-induced MCP-1 gene expression in pancreatic acinar cells, but they did not show any effect on non-stimulated acinar cells.
Figure 2.

MCP-1 mRNA expression in acinar cells under basal conditions and under stimulation with pancreatitis-associated ascitic fluid (PAAF, 20% v/v) in the absence or presence of dexamethasone (Dx, 10−7 M) and N-acetylcysteine (NAC, 30 mM). A representative RT-PCR and the mean values of five experiments are shown. Results are mean ± S.E.M. ANOVA followed by Dunnett’s test showed significant differences versus non-PAAF-stimulated cells (basal conditions) (***P < 0.001) and versus PAAF-stimulated cells (♦♦♦P < 0.001).
Downstream signalling pathways
Phosphorylation of p38-MAPK was analysed by Western blot (Fig. 3) to evaluate the activation of MAPK. Basal p38-MAPK phosphorylation did not vary by adding Dx or NAC. A significant (P < 0.01) increase in phospho-p38-MAPK was found in acinar cells cultured in the presence of PAAF. Pre-treatment with Dx did not prevent the PAAF-induced MAPK activation. However, a significant (P < 0.01) reduction in phospho-p38 was found in acinar cells cultured with PAAF in the presence of NAC.
Figure 3.
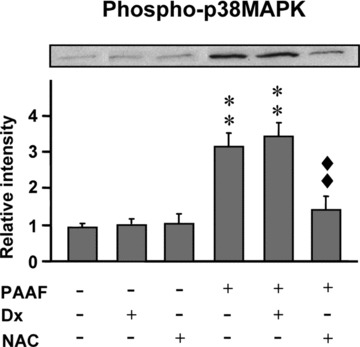
Phosphorylation of p38-MAPK expression in acinar cells under basal conditions and under stimulation with pancreatitis-associated ascitic fluid (PAAF, 20% v/v) in the absence or presence of dexamethasone (Dx, 10−7 M) and N-acetylcysteine (NAC, 30 mM). A representative Western blot and the mean values of five experiments are shown. Band intensity was determined by densitometry. Results are mean ± S.E.M. ANOVA followed by Dunnett’s test showed significant differences versus non-PAAF-stimulated cells (basal conditions) (**P < 0.01) and versus PAAF-stimulated cells (♦♦♦P < 0.01).
The activation of NF-κB was evaluated by analysis of IκBα (Fig. 4A) and p65 (Fig. 4B). Dx and NAC did not show any effect in acinar cells cultured under basal conditions. PAAF significantly activated NF-κB in acinar cells as indicated by the significant (P < 0.01) degradation of IκBα and the significant (P < 0.01) increase in p65 levels. Both Dx and NAC significantly inhibited NF-κB activation induced by PAAF, by preventing the degradation of IκBα and maintaining p65 values at similar levels to those that were found in acinar cells cultured in the absence of PAAF (basal conditions).
Figure 4.
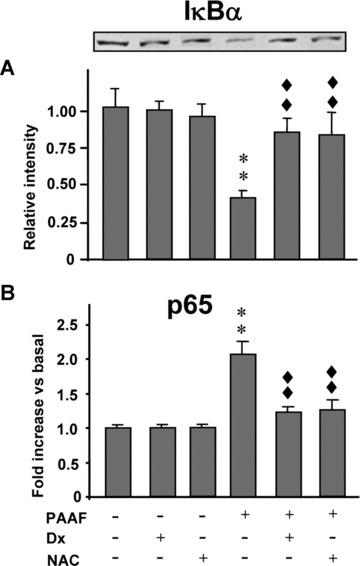
Activation of NF-κB in pancreatic acinar cells under basal conditions and under stimulation with pancreatitis-associated ascitic fluid (PAAF, 20% v/v) in the absence or presence of dexamethasone (Dx, 10−7 M) and N-acetylcysteine (NAC, 30 mM). (A) Representative Western blot of IκBα and the mean values of five experiments are shown. Band intensity was determined by densitometry. (B) p65 nuclear levels (NF-κB-DNA binding). Results are mean ± S.E.M. ANOVA followed by Dunnett’s test showed significant differences versus non-PAAF-stimulated cells (basal conditions) (**P < 0.01) and versus PAAF-stimulated cells (♦♦P < 0.01).
Figure 5 shows the STAT3 activity. A non-significant decrease was induced by Dx and NAC in the basal STAT3 activity. PAAF significantly (P < 0.01) up-regulated the activation of STAT3 in acinar cells. Dx significantly (P < 0.01) reduced the PAAF-induced STAT3 activation to values significantly (P < 0.05) lower than those found in acinar cells cultured in the absence of PAAF. NAC significantly (P < 0.05) diminished the STAT3 activation in PAAF-stimulated acinar cells, although the STAT3-DNA binding was found to be significantly (P < 0.05) higher than in PAAF non-stimulated cells.
Figure 5.
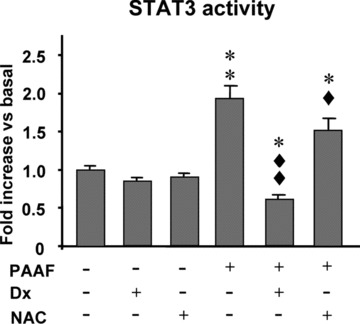
STAT3 activity in pancreatic acinar cells under basal conditions and in pancreatitis-associated ascitic fluid (PAAF)-stimulated acinar cells in the absence or presence of dexamethasone (Dx, 10−7 M) and N-acetylcysteine (NAC, 30 mM). Results are mean ± S.E.M. ANOVA followed by Dunnett’s test showed significant differences versus non-PAAF-stimulated cells (basal conditions) (*P < 0.05, **P < 0.01) and versus PAAF-stimulated cells (♦P < 0.05, ♦♦P < 0.01).
Discussion
In response to PAAF, up-regulation of cytokines and adhesion molecules in endothelial cells [6] and leukocytes [8] has been reported. In addition, PAAF has shown to be able to modulate the phenotype of pancreatic acinar cells to produce TNF-α[9, 24]. We hypothesized that PAAF might also activate acinar cells to produce chemokines, thereby maintaining continuous positive feedback during AP that would provide elevated levels of inflammatory factors with a key role in the attraction and subsequent recruitment of circulating leukocytes towards the tissues. It would support the removal of PAAF to improve the outcome of AP, because although there is some discrepancy between controlled animal studies and human trials, early peritoneal lavage has been reported to be beneficial in the progress of severe AP [25].
The present study has demonstrated that PAAF promotes the overexpression of MCP-1 in pancreatic acinar cells mediated by p38-MAPK, NF-κB and STAT3 activation. It has been further shown that Dx totally repressed the PAAF-induced MCP-1 expression via inhibition of NF-κB and STAT3 pathways. Down-regulation of MCP-1 expression in PAAF-activated acinar cells by NAC was mediated by inhibition of p38-MAPK, NF-κB and STAT-3 pathways.
In line with our results, phosphorylation of p38-MAPK was found in CCL-13 cells in response to PAAF [26]. On the other hand, activation of NF-κB has been reported by Ramudo et al.[9] in acinar cells cultured in the presence of ascitic fluid collected from rats with mild AP induced by bile-pancreatic duct obstruction. JAK/STAT activation has been reported in acinar cells cultured with cytokines [20, 21], but the involvement of the STAT signalling pathway in the PAAF-induced acinar cell inflammatory response has not been studied.
Several studies have tried to identify the PAAF factors responsible for the inflammatory response [6, 7, 9]. Although several candidates for specific components have been investigated, appropriate supporting evidence has not been found yet. Most likely, the activation of the inflammatory signalling pathways is caused not by an individual element but by the combined action of multiple factors. Accordingly, a multi-modal action could be defined for the PAAF in the activation of acinar cells. Because p38-MAPK has been demonstrated to be the main MAPK involved in AP [27] and its activation can occur in response to a variety of stressful stimuli [28], we have investigated the role of this MAPK as a potential upstream regulator of PAAF-mediated MCP-1 expression in acinar cells. On the other hand, up-regulation of inflammatory factors involves the activation of transcription factors. NF-κB appears as the primary activator of the expression of genes encoding interleukins, chemokines and adhesion molecules. It is activated by oxidative stress [29] and cytokines [30, 31]. The role of the STAT pathway in the transcriptional regulation of inflammatory factors is increasingly being appreciated. It provides a direct link between the cytokine receptors and cytokine-induced gene transcription [32]. Up-regulation of STAT3 in response to TNF-α has been reported in acinar cells [21], an interesting data due to the fact that acinar cells have been demonstrated to have TNF-α receptors, which are up-regulated by cerulein [33]. In addition, STAT has also been demonstrated to be a redox sensitive transcription factor [34]. Given that PAAF is a cytokine-rich biological fluid and under its presence acinar cells generate a high amount of reactive oxygen species (ROS), MAPKs as well as both transcription factors, NF-κB and STAT3, may be involved in the acinar MCP-1 up-regulation induced by PAAF. Data concerning this are of great interest in the consideration of the downstream signalling pathways as potential targets of therapeutical strategies designed to reduce both local and systemic inflammation during AP.
Glucocorticoids have a long history of use as anti-inflammatory agents by down-regulating inflammatory factors at transcriptional level. NF-κB and Activator protein-1 (AP-1) have been shown to be candidates as targets for the genomic action of glucocorticoids [35, 36]. On the other hand, interaction of glucocorticoids with the JAK/STAT pathway has been reported with positive [37] and negative [22, 23, 38] effects on cytokine signalling. Our results showed a negative cross-talk in acinar cells between Dx and either NF-κB or STAT3 pathway in parallel with a total repression of the PAAF-induced MCP-1 expression. This suggests that NF-κB and STAT3 may synergically act as downstream signalling transduction pathways of the PAAF-mediated inflammatory response in acinar cells. Dx prevented the phosphorylation and subsequent degradation of IκBα induced by PAAF in acinar cells.
Because IκBα acts as an inhibitory subunit [31], NF-κB remained in a latent form in the cytoplasm instead of being translocated to the nucleus. In fact, basal levels of p65 were found in acinar cells cultured with PAAF in the presence of Dx, thus indicating that NF-κB-DNA binding was inhibited. STAT3 pathway has shown to be especially sensitive to the Dx action. It was able to block the STAT3 activation to levels even lower than those detected in non-stimulated acinar cells (basal conditions), which may be the result of the oxidative stress triggered by the pancreas removal and during acini isolation, as Gallmeier et al. reported [20]. Inhibitory effects of Dx on the activation of STAT family members have also been reported in macrophages [22] and keratinocytes [23] stimulated by cytokines. Our results strongly suggest that STAT3 may be selected as a potential target for therapeutic intervention of AP and give new insight into the anti-inflammatory actions of glucocorticoids in AP. In contrast, Dx did not affect the acinar p38-MAPK phosphorylation induced by PAAF. As a possible non-genomic rapid action, glucocorticoids have been reported to cause inhibition [39] and activation [40, 41] of p38-MAPK. This indicates that the glucocorticoid action is cell type-dependent. Our results suggest that p38-MAPK activation does not seem to play a pivotal role in the acinar PAAF-mediated MCP-1 up-regulation, because Dx completely abrogated the acinar MCP-1 expression with no change in the PAAF-induced p38-MAPK phosphorylation.
NAC has demonstrated in in vitro[9] and in vivo[10, 13] studies to reduce the production of TNF-α in acinar cells. In line with this, we show that NAC down-regulated the PAAF-induced MCP-1 expression in acinar cells. In response to PAAF, acinar cells develop oxidative stress, which has been demonstrated to activate MAPK [42] and NF-κB [29, 43]. On the other hand, the inhibition of phosphatases induced by ROS allows the JAK-induced STAT phosphorylation required for STAT activation [18, 31]. Accordingly, the anti-inflammatory action of NAC may be explained by preventing the redox-activation of p38-MAPK and NF-κB and significantly reducing the STAT3 activity. The different effects of NAC versus Dx on the PAAF-activated signalling pathways may be explained by their specific actions as antioxidant and anti-inflammatory agents, respectively. Beacuse MAPK cascade is especially sensitive to the cellular redox state, NAC totally repressed p38-MAPK phosphorylation. However, NAC was not as effective as Dx to inhibit STAT3 activation, probably due to the fact that JAK may be activated not only by ROS but also by cytokines, whose presence, in turns, are especially sensitive to Dx action.
In summary, PAAF may be involved in the pathophysiology of AP by activating acinar cells to produce MCP-1, particularly via NF-κB and STAT3 signalling pathways. Dx and NAC suppressed the PAAF-mediated acinar MCP-1 cell up-regulation by inhibiting both downstream mechanisms. STAT3 activity was especially sensitive to Dx, thus becoming a key target in therapeutical interventions in AP.
Acknowledgments
This study was supported by a grant from FEDER-FIS (Fondo de Investigacion Sanitaria), Spain (PI05/0025). Thanks are due to Elizabeth Nestor for her linguistic assistance. Potential conflicts of interest do not exist.
References
- 1.Klar E, Messmer K, Warshaw AL, Herfarth C. Pancreatic ischaemia in experimental acute pancreatitis: mechanism, significance and therapy. Br J Surg. 1990;77:1205–10. doi: 10.1002/bjs.1800771104. [DOI] [PubMed] [Google Scholar]
- 2.Lehtola A. Peritoneal blood flow during acute experimental pancreatitis. The role of peritoneal exudate. Scand J Gastroenterol. 1986;21:756–60. doi: 10.3109/00365528609011113. [DOI] [PubMed] [Google Scholar]
- 3.Ranson JH, Spencer FC. The role of peritoneal lavage in severe acute pancreatitis. Ann Surg. 1978;187:565–75. doi: 10.1097/00000658-197805000-00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frey CF, Wong HN, Hickman D, Pullos T. Toxicity of hemorrhagic ascitic fluid associated with hemorrhagic pancreatitis. Arch Surg. 1982;117:401–4. doi: 10.1001/archsurg.1982.01380280007002. [DOI] [PubMed] [Google Scholar]
- 5.Beger HG, Rau B, Mayer J, Pralle U. Natural course of acute pancreatitis. World J Surg. 1997;21:130–5. doi: 10.1007/s002689900204. [DOI] [PubMed] [Google Scholar]
- 6.Masamune A, Shimosegawa T, Kimura K, et al. Specific induction of adhesion molecules in human vascular endothelial cells by rat experimental pancreatitis-associated ascitic fluids. Pancreas. 1999;18:141–50. doi: 10.1097/00006676-199903000-00005. [DOI] [PubMed] [Google Scholar]
- 7.Denham W, Yang J, Norman J. Evidence for an unknown component of pancreatic ascites that induces adult respiratory distress syndrome through an interleukin-1 and tumor necrosis factor-dependent mechanism. Surgery. 1997;122:295–301. doi: 10.1016/s0039-6060(97)90021-0. [DOI] [PubMed] [Google Scholar]
- 8.Masamune A, Shimosegawa T, Fujita M, et al. Ascites of severe acute pancreatitis in rats transcriptionally up-regulates expression of interleukin-6 and -8 in vascular endothelium and mononuclear leukocytes. Dig Dis Sci. 2000;45:429–37. doi: 10.1023/a:1005449601925. [DOI] [PubMed] [Google Scholar]
- 9.Ramudo L, Manso MA, De Dios I. Biliary pancreatitis-associated ascitic fluid activates the production of tumor necrosis factor-alpha in acinar cells. Crit Care Med. 2005;33:143–8. doi: 10.1097/01.ccm.0000150654.13653.5b. [DOI] [PubMed] [Google Scholar]
- 10.Ramudo L, Manso MA, Vicente S, De Dios I. Pro- and anti-inflammatory response of acinar cells during acute pancreatitis. Effect of N-acetyl cysteine. Cytokine. 2005;32:125–31. doi: 10.1016/j.cyto.2005.07.017. [DOI] [PubMed] [Google Scholar]
- 11.Gukovsky I, Gukovskaya AS, Blinman TA, et al. Early NF-kappaB activation is associated with hormone-induced pancreatitis. Am J Physiol. 1998;275:G1402–14. doi: 10.1152/ajpgi.1998.275.6.G1402. [DOI] [PubMed] [Google Scholar]
- 12.Steinle AU, Weidenbach H, Wagner M, et al. NF-kappaB/Rel activation in cerulein pancreatitis. Gastroenterology. 1999;116:420–30. doi: 10.1016/s0016-5085(99)70140-x. [DOI] [PubMed] [Google Scholar]
- 13.Ramudo L, Manso MA, Sevillano S, De Dios I. Kinetic study of TNF-alpha production and its regulatory mechanisms in acinar cells during acute pancreatitis induced by bile-pancreatic duct obstruction. J Pathol. 2005;206:9–16. doi: 10.1002/path.1747. [DOI] [PubMed] [Google Scholar]
- 14.Pereda J, Sabater L, Cassinello N, et al. Effect of simultaneous inhibition of TNF-alpha production and xanthine oxidase in experimental acute pancreatitis: the role of mitogen activated protein kinases. Ann Surg. 2004;240:108–16. doi: 10.1097/01.sla.0000129343.47774.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Samuel I, Zaheer S, Zaheer A. Bile-pancreatic juice exclusion increases p38MAPK activation and TNF-alpha production in ligation-induced acute pancreatitis in rats. Pancreatology. 2005;5:20–6. doi: 10.1159/000084486. [DOI] [PubMed] [Google Scholar]
- 16.Kisseleva T, Bhattacharya S, Braunstein J, Schindler CW. Signaling through the JAK/STAT pathway, recent advances and future challenges. Gene. 2002;285:1–24. doi: 10.1016/s0378-1119(02)00398-0. [DOI] [PubMed] [Google Scholar]
- 17.Wilks AF, Harpur AG, Kurban RR, et al. Two novel protein-tyrosine kinases, each with a second phosphotransferase-related catalytic domain, define a new class of protein kinase. Mol Cell Biol. 1991;11:2057–65. doi: 10.1128/mcb.11.4.2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Darnell JE., Jr STATs and gene regulation. Science. 1997;277:1630–5. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- 19.Imada K, Leonard WJ. The Jak-STAT pathway. Mol Immunol. 2000;37:1–11. doi: 10.1016/s0161-5890(00)00018-3. [DOI] [PubMed] [Google Scholar]
- 20.Gallmeier E, Schäfer C, Moubarak P, et al. JAK and STAT proteins are expressed and activated by IFN-gamma in rat pancreatic acinar cells. J Cell Physiol. 2005;203:209–16. doi: 10.1002/jcp.20216. [DOI] [PubMed] [Google Scholar]
- 21.Vona-Davis LC, Frankenberry KA, Waheed U, et al. Expression of STAT3 and SOCS3 in pancreatic acinar cells. J Surg Res. 2005;127:14–20. doi: 10.1016/j.jss.2005.03.019. [DOI] [PubMed] [Google Scholar]
- 22.Hu X, Li WP, Meng C, Ivashkiv LB. Inhibition of IFN-gamma signaling by glucocorticoids. J Immunol. 2003;170:4833–9. doi: 10.4049/jimmunol.170.9.4833. [DOI] [PubMed] [Google Scholar]
- 23.Stojadinovic O, Lee B, Vouthounis C, et al. Novel genomic effects of glucocorti-coids in epidermal keratinocytes: inhibition of apoptosis, interferon-gamma pathway, and wound healing along with promotion of terminal differentiation. J Biol Chem. 2007;282:4021–34. doi: 10.1074/jbc.M606262200. [DOI] [PubMed] [Google Scholar]
- 24.Denham W, Yang J, Fink G, et al. Pancreatic ascites as a powerful inducer of inflammatory cytokines. The role of known versus unknown factors. Arch Surg. 1997;132:1231–6. doi: 10.1001/archsurg.1997.01430350081013. [DOI] [PubMed] [Google Scholar]
- 25.Dugernier Th, Laterre PF, Reynaert MS. Ascites fluid in severe acute pancreatitis: from pathophysiology to therapy. Acta Gastroenterol Belg. 2000;63:264–8. [PubMed] [Google Scholar]
- 26.Yang J, Fier A, Carter Y, et al. Liver injury during acute pancreatitis: the role of pancreatitis-associated ascitic fluid (PAAF), p38-MAPK, and caspase-3 in inducing hepatocyte apoptosis. J Gastrointest Surg. 2003;7:200–7. doi: 10.1016/s1091-255x(02)00134-8. [DOI] [PubMed] [Google Scholar]
- 27.Yang J, Murphy C, Denham W, et al. Evidence of a central role for p38 map kinase induction of tumor necrosis factor alpha in pancreatitis-associated pulmonary injury. Surgery. 1999;126:216–22. [PubMed] [Google Scholar]
- 28.Dabrowski A, Boguslowicz C, Dabrowska M, et al. Reactive oxygen species activate mitogen-activated protein kinases in pancreatic acinar cells. Pancreas. 2000;21:376–84. doi: 10.1097/00006676-200011000-00008. [DOI] [PubMed] [Google Scholar]
- 29.Li N, Karin M. Is NF-kappaB the sensor of oxidative stress. FASEB J. 1999;13:1137–43. [PubMed] [Google Scholar]
- 30.Barnes PJ, Karin M. Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med. 1997;336:1066–71. doi: 10.1056/NEJM199704103361506. [DOI] [PubMed] [Google Scholar]
- 31.Wulczyn FG, Krappmann D, Scheidereit C. The NF-kappa B/Rel and I kappa B gene families: mediators of immune response and inflammation. J Mol Med. 1996;74:749–69. doi: 10.1007/s001090050078. [DOI] [PubMed] [Google Scholar]
- 32.Takeda K, Akira S. STAT family of transcription factors in cytokine-mediated biological responses. Cytokine Growth Factor Rev. 2000;11:199–207. doi: 10.1016/s1359-6101(00)00005-8. [DOI] [PubMed] [Google Scholar]
- 33.Gukovskaya AS, Gukovsky I, Zaninovic V, et al. Pancreatic acinar cells produce, release, and respond to tumor necrosis factor-alpha. Role in regulating cell death and pancreatitis. J Clin Invest. 1997;100:1853–62. doi: 10.1172/JCI119714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Simon AR, Rai U, Fanburg BL, Cochran BH. Activation of the JAK-STAT pathway by reactive oxygen species. Am J Physiol. 1998;275:C1640–52. doi: 10.1152/ajpcell.1998.275.6.C1640. [DOI] [PubMed] [Google Scholar]
- 35.Didonato JA, Saatcioglu F, Karin M. Molecular mechanisms of immunosup-pression and anti-inflammatory activities by glucocorticoids. Am J Respir Crit Care Med. 1996;154:S11–5. doi: 10.1164/ajrccm/154.2_Pt_2.S11. [DOI] [PubMed] [Google Scholar]
- 36.Jonat C, Rahmsdorf HJ, Park KK, et al. Antitumor promotion and antiinflammation: down-modulation of AP-1 (Fos/Jun) activity by glucocorticoid hormone. Cell. 1990;62:1189–204. doi: 10.1016/0092-8674(90)90395-u. [DOI] [PubMed] [Google Scholar]
- 37.Stöcklin E, Wissler M, Gouilleux F, Groner B. Functional interactions between Stat5 and the glucocorticoid receptor. Nature. 1996;383:726–8. doi: 10.1038/383726a0. [DOI] [PubMed] [Google Scholar]
- 38.Ishida-Takahashi R, Uotani S, Abe T, et al. Rapid inhibition of leptin signaling by glucocorticoids in vitro and in vivo. J Biol Chem. 2004;279:19658–64. doi: 10.1074/jbc.M310864200. [DOI] [PubMed] [Google Scholar]
- 39.Lasa M, Brook M, Saklatvala J, Clark AR. Dexamethasone destabilizes cyclooxygenase 2 mRNA by inhibiting mitogen-activated protein kinase p38. Mol Cell Biol. 2001;21:771–80. doi: 10.1128/MCB.21.3.771-780.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qi AQ, Qiu J, Xiao L, Chen YZ. Rapid activation of JNK and p38 by glucocorticoids in primary cultured hippocampal cells. J Neurosci Res. 2005;80:510–7. doi: 10.1002/jnr.20491. [DOI] [PubMed] [Google Scholar]
- 41.Li X, Qiu J, Wang J, et al. Corticosterone-induced rapid phosphorylation of p38 and JNK mitogen-activated protein kinases in PC12 cells. FEBS Lett. 2001;492:210–4. doi: 10.1016/s0014-5793(01)02254-2. [DOI] [PubMed] [Google Scholar]
- 42.Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001;81:807–69. doi: 10.1152/physrev.2001.81.2.807. [DOI] [PubMed] [Google Scholar]
- 43.Schreck R, Baeuerle PA. Assessing oxygen radicals as mediators in activation of inducible eukaryotic transcription factor NF-kappa B. Methods Enzymol. 1994;234:151–63. doi: 10.1016/0076-6879(94)34085-4. [DOI] [PubMed] [Google Scholar]