

Abstract
GFP-tagging is widely used as a molecular tool to localize and visualize the trafficking of proteins in cells but interpretation is frequently limited by the low resolution afforded by fluorescence light microscopy. Although complementary thin-section immunogold electron microscopic techniques go some way in aiding interpretation, major limitations, such as relatively poor structural preservation of membrane systems, low labelling efficiency and the two-dimensional nature of the images, remain. Here we demonstrate that the electron microscopic technique freeze-fracture replica immunogold labelling overcomes these disadvantages and can be used to define, at high resolution, the precise location of GFP-tagged proteins in specific membrane systems and organelles of the cell. Moreover, this technique provides information on the location of the protein within the phospholipid bilayer, potentially providing insight into mis-orientation of tagged proteins compared to their untagged counterparts. Complementary application of the freeze-fracture replica immunogold labelling technique alongside conventional fluorescence microscopy is seen as a novel and valuable approach to verification, clarification and extension of the data obtained using fluorescent-tagged proteins. The application of this approach is illustrated by new findings on PAT-family proteins tagged with GFP transfected into fibroblasts from patients with Niemann-Pick type C disease.
Keywords: GFP, immunocytochemistry, freeze-fracture electron microscopy, electron microscopy, PAT family proteins, Niemann-Pick disease
Introduction
Green fluorescent protein (GFP) has revolutionized the ability to track the location and movement of specific proteins in living cells [1–4]. When tagged to the protein of interest, the natural fluorescence of GFP provides a molecular label that is both innocuous to the cell and readily visualized by fluorescence light microscopy [5–7]. New variant fluorescent proteins and advances in the imaging technology associated with laser-scanning confocal microscopy continue to extend the scope of GFP as a tool to analyze protein function [8–10]. However, despite the major breakthroughs in understanding cell and molecular function that have been achieved through these approaches, certain inherent limitations need to be borne in mind, and where possible addressed, to maximize the value of the information obtained. Apart from awareness of the possible effects of the presence of a GFP tag on the properties of the protein, which may alter its function, trafficking route or orientation in the membrane, a key limitation centres on the resolution afforded by the imaging methods routinely employed. Even the most up-to-date developments in confocal imaging do not permit unambiguous identification of all cellular organelles and the precise location of a fluorescent signal within or adjacent to their membranes [6].
One approach aimed at addressing this major drawback involves parallel examination of fluorescently labelled samples with those processed for the higher resolution technique of thin-section electron microscopy combined with immunogold labelling of GFP. Though this has met with some success, key limitations nevertheless remain [11–15]. First, the reliance on aldehyde fixation (in both the low-temperature resin and cryosection procedures) has adverse effects on epitope preservation, reducing the efficiency of labelling. Second, structural preservation of cells, their organelles and membranes is of a much lower quality than that associated with conventional thin-section electron microscopy of glutaraldehyde and osmium tetroxide-fixed samples. Third, and especially crucially, the two-dimensional views provided by sections do not permit determination of whether a protein is located in or adjacent to a given membrane.
We here demonstrate that the electron-microscopic technique of freeze-fracture replica immunolabelling [16] effectively overcomes these drawbacks. This technique has all the advantages of freeze-fracture electron microscopy in imaging en face the membranes of identified organelles at macromolecular resolution, combined with the ability to localize immunogold labelled GFP with precision in the membrane plane [17–21]. Because freeze-fracture splits membranes along an interior hydrophobic plane, label can be assigned to one or other of the half-membrane leaflets that correspond to each of the phospholipid monolayers. Moreover, the membrane splitting process creates three-dimensional perspectives of cellular organization in which the spatial relationships of the various membrane-bound organelles are readily appreciated. Finally, the specimens are stabilized by a purely physical means (i.e. rapid freezing), thus avoiding the need for chemical fixation. Freeze-fracture replica immunolabelling is thus uniquely equipped to discriminate, at high resolution, the spatial relationship between closely associated membranes and organelles, and to define the precise location of GFP-tagged proteins in relation to them.
To illustrate the potential of this approach, we present here image data on the localization of PAT-family proteins tagged with GFP transfected into fibroblasts from patients with Niemann-Pick type C (NPC) disease. In this disease, mutations of either the NPC1 or NPC2 gene lead to accumulation of unesterified cholesterol and glycolipids in the lysosomes and severely reduced ability of the cells to make lipid droplets. The PAT family proteins, perilipin and adipophilin, are targeted to lipid droplets and play important roles in their formation and function.
Materials and Methods
Cell culture and transfection
Human Niemann-Pick C fibroblasts were cultured in DMEM with 10% foetal calf serum. Transfection of detached cells was performed using the nucleofection method of Amaxa (Cologne, Germany). After resuspension, 1 × 106 cells were mixed with vector DNA in nucleofector solution and electroporated in the nucleofector device. The following vectors were used: CD4-GFP vector, GFP-perilipin vector and adipophilin-GFP vector. We tried both possible adipophilin-GFP constructs. The C-terminal adipophilin-GFP vector was used in this study. We found no expression of the N-terminal adipophilin-GFP vector in THP-1 macrophages as well as in NPC fibrob-lasts. We attribute this to a disturbed translation and protein folding process. The cell suspension was removed immediately from the cuvette by adding prewarmed medium and added to a six-well plate. The transfection efficiency of GFP-perilipin expression vector was approximately 60%.
Human monocytic THP-1 cells from the American Type Culture Collection (ATCC, Manassas, VA) were cultured in suspension in RPMI 1640 medium containing the supplements recommended by Iwashima et al.[22], and differentiated to macrophages by adding 100 μM phorbol 12-myristate 13-acetate to the medium. THP-1 cells were transfected following a similar procedure to that described above. The transfection efficiency of GFP-CD4 expression vector was approximately 50%.
The cells were processed 24 hrs after transfection as described below.
Freeze-fracture replica immunolabelling
Cells were mounted in 30% glycerol on gold-nickel alloy carriers and immediately rapidly frozen in Freon 22 cooled with liquid nitrogen. The samples were fractured using a Balzers BA 310 freeze-fracture unit at −100°C. Replicas of the freshly fractured cells were immediately made by electron beam evaporation of platinum-carbon and carbon at angles of 38° and 90° and to thicknesses of 2 nm and 20 nm, respectively. The replicas were incubated overnight in 5% sodium dodecyl sulphate in 10 mm Tris/30 mM sucrose (pH 8.3), conditions designed to remove the cells from the replicas apart from a layer of molecules immediately adherent to the platinum-carbon.
The replicas were washed in distilled water and incubated as follows: (i) 5% BSA blocking solution (15 min.), (ii) primary antibody in blocking solution (1 hr), (iii) PBS, three changes (total 30 min.),(iv) secondary antibody/gold complex in blocking solution (1 hr), (v) PBS, three changes (total 30 min.), (vi) 0.5% glutaraldehyde in PBS (10 min.), (vii) distilled water, two changes. The primary antibodies used were: (i) rabbit poly-clonal antibody raised against the entire sequence of GFP (ab290, abcam, Cambridge, UK), (ii) guinea pig polyclonal antibody raised against portions of the amino termini of human perilipin A and B (GP30; Progen Biotechnik, Heidelberg, Germany), (iii) mouse monoclonal antibody raised against a peptide matching amino acids 5–27 from the amino terminus of human adipophilin (AP 125; Progen Biotechnik, Heidelberg, Germany). All primary antibodies were checked for specificity by Western blotting. The Western blot characterization of the GFP antibody is shown in Figure 1. The secondary antibodies used were (i) goat anti-rabbit, (ii) donkey anti-guinea pig and (iii) goat antimouse, coupled to 12 nm or 18 nm colloidal gold (Jackson Immunoresearch, West Grove, PA, USA). Double labelling to detect the target GFP-tagged protein by labelling both GFP and the protein itself was conducted by simultaneous incubation in the GFP plus perilipin or GFP plus adipophilin primary antibodies followed by mixtures of appropriate, matching secondary antibodies, one coupled to 18 nm gold and the other to 12 nm gold. In all experiments, controls were conducted in parallel (secondary antibody alone to check for non-specific binding to the replica, irrelevant primary antibody etc.). The labelled replicas were mounted on grids, and examined in a Philips 410 transmission electron microscope.
Figure 1.
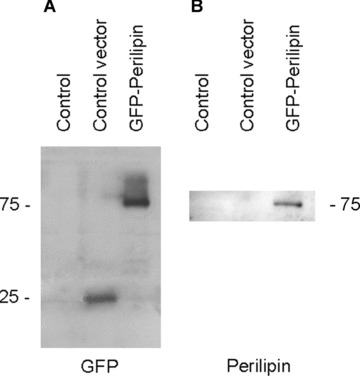
Western blot characterization of the GFP and perilipin antibodies. (A) Using the GFP antibody, no signal is detected in non-transfected THP-1 macrophages (control). In cells overexpressing the control vector, a single band of ∼25 kD is seen (representing GFP). Cells expressing GFP-perilipin give a band at ∼75 kD representing the GFP-perilipin construct. (B) The antibody to perilipin detects band at ∼75 kD in GFP-perilipin overexpressing macrophages. Secondary antibody detection: A, peroxidase-conjugated anti-rabbit; B, peroxidase-conjugated anti-guinea pig.
Immunofluorescence light microscopy
Parallel immunofluorescence studies were conducted using the same primary antibodies to perilipin, adipophilin and CD4 as detailed above, detected with Cy3-conjugated antimouse secondary antibody (Chemicon, Temecula, CA, USA) for adipophilin and CD4, and with Cy3-conjugated anti-guinea pig antibody (Chemicon, Temecula, CA, USA) for perilipin. Oil red has been used to show lipid droplets.
Results and discussion
NPC fibroblasts do not express perilipin and, in the few lipid droplets present, only low amounts of adipophilin are detected (Fig. 2). Cultured NPC fibroblasts were transfected with GFP-adipophilin and GFP-perilipin vectors as part of a series of experiments to determine whether enhanced expression of PAT family proteins alters the ability of NPC fibroblasts to form lipid droplets. The principle of the freeze-fracture replica immunolabelling technique used to localize these proteins in the transfected cells is illustrated in Figure 3. In brief, after rapid freezing, fracturing under high-vacuum and preparation of platinum-carbon replicas, the replicated samples are treated with sodium dodecyl sulphate to remove the bulk of the cellular material, leaving just a fine layer of directly adherent molecules. In this way, epitopes are retained for immunolabelling without compromising the visibility of structural detail in the replicas. Immunolabelling was carried out with anti-GFP, anti-adipophilin or anti-perilipin antibodies followed by species-matched secondary antibodies coupled to 18 nm gold. Double labelling was carried out by using mixtures of the primary antibodies selected followed by incubation with mixtures of appropriate, matching 18 nm and 12 nm gold conjugated secondary antibodies. For comparison, standard immunofluorescence light microscopy was conducted in parallel.
Figure 2.
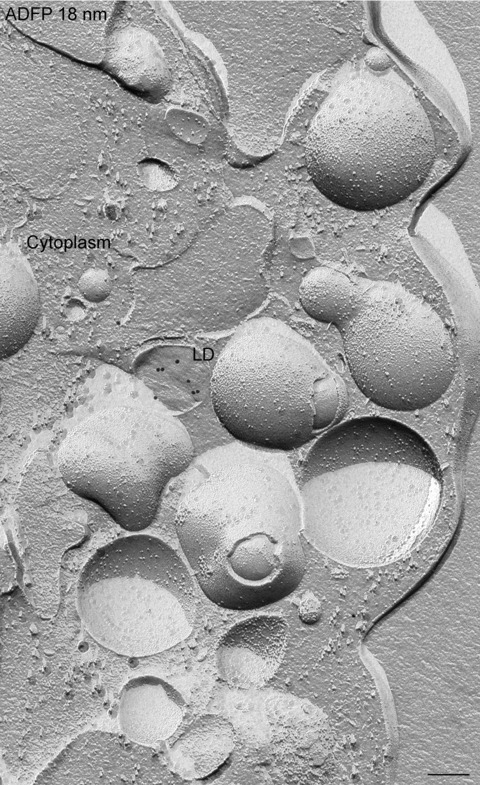
Freeze-fracture image of a non-transfected NPC fibroblast. Only a few single lipid droplets (LD) are present and a low amount of adipophilin (ADFP) is detected after freeze-fracture replica immunolabelling using adipophilin antibodies and 18 nm gold secondary antibodies. Bar: 0.2 μm
Figure 3.
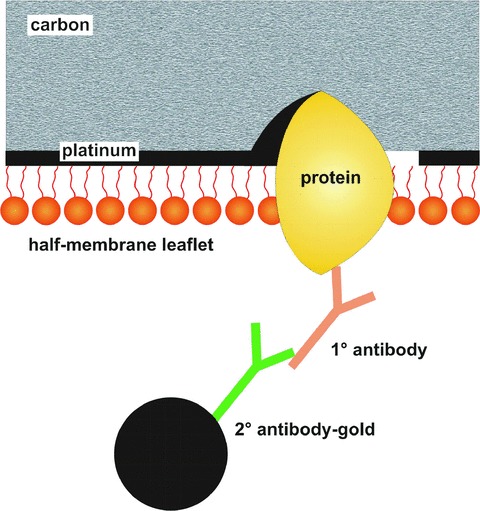
Diagram illustrating the principle of freeze-fracture replica immunolabelling to visualize the location of GFP-tagged and non-tagged proteins at high resolution in membranes. The technique permits retention of a layer of molecules attached to the replica. In this way, membrane proteins can be labelled using a primary antibody followed by a secondary antibody coupled to colloidal gold. The platinum-carbon replica permits visualization of membrane structural detail, as in a standard freeze-fracture replica. Superimposed on this planar view of the membrane interior, the gold marker, labelling the targeted protein at the membrane surface, is clearly visible.
Figure 4A–D illustrates immunofluorescence localization of perilipin in NPC fibroblasts after transfection with GFP-perilipin. Identical labelling patterns are observed whether the perilipin is detected using the anti-perilipin antibody (Fig. 4A) or directly by GFP fluorescence (Fig. 4B), as confirmed by merging the images (Fig. 4C). The appearance of the punctate cytoplasmic labelling is strongly suggestive of lipid droplets. The fluorescence image in Figure 4D shows lipid droplets labelled with oil red (red) and GFP (green). GFP is seen at the periphery of lipid droplets. This identification is confirmed when the GFP-perilipin is localized using freeze-fracture replica immunolabelling with anti-GFP (Fig. 4E). In freeze-fracture, lipid droplets have a unique smooth appearance enabling their unambiguous discrimination from other organelles. Where the fracture follows the outermost layer of the droplet to give a concave fracture, the enveloping outer phospholipid monolayer is seen en face; convex fractures give mirror image (complementary) views. Some fractures skip along successive layers of the lipid, revealing a multilayered appearance. Others cross fracture the droplet to give what is essentially a cross-section of the interior. The gold label, seen as sharply defined black particles, can be seen in abundance on the outer phospholipid monolayer of the droplet, and some label is also present deeper in the interior. Similar results are obtained irrespective of whether immunogold labelling is for GFP or perilipin (Fig. 4E and F), a conclusion confirmed by conducting double labelling using gold markers of two sizes to discriminate GFP and perilipin labels (Fig. 4G). Thus, antibodies to GFP are as effective as those to the protein in providing high-resolution localization data. No label with either antibody is apparent on the cytoplasmic matrix or the membranes of the endoplasmic reticulum or other organelles.
Figure 4.
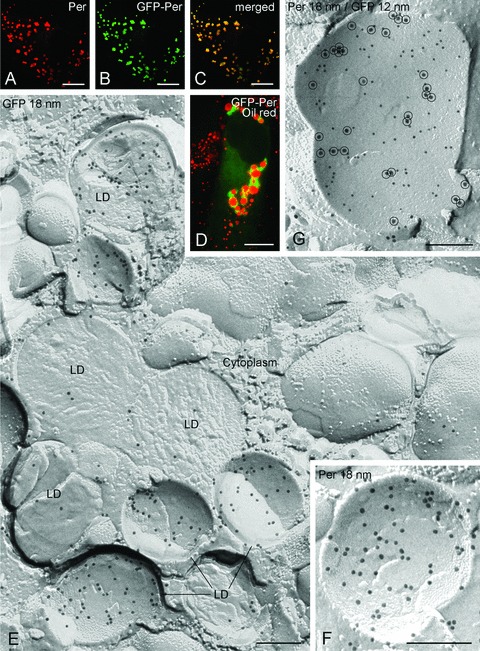
Comparative detection of GFP-perilipin (GFP-Per) by immunofluorescence microscopy (A–D) and freeze-fracture replica immunolabelling (E–G) in NPC fibroblasts. The punctate cytoplasmic labelling seen using perilipin antibodies (A) or from GFP fluorescence (B) at light microscopy is demonstrated to reside in lipid droplets (LD) by fracture replica immunolabelling using anti-GFP antibodies followed by 18 nm gold secondary antibodies (E). Identical labelling of the lipid droplets is apparent using perilipin antibody with 18 nm gold secondary (F), and when simultaneous labelling using GFP antibody with 12 nm gold secondary and perilipin antibody with 18 nm gold secondary(encircled) (G). Bars: A–D 5 μm; E-G 0.2 μm
In NPC cells transfected with adipophilin-GFP, the pattern of labelling on lipid droplets is similar to that observed with the GFP-perilipin transfected cells, irrespective of whether anti-GFP or anti-adipophilin antibodies are applied (Fig. 5). However, in addition, prominent labelling for adipophilin-GFP is apparent in the plasma membrane and nuclear membranes (Figs. 6 and 7). In the plasma membrane, a high density of labelling is present on the P-half of the membrane (the half membrane leaflet adjacent to the protoplasm) though significant labelling is also apparent on the E-half (the half membrane leaflet adjacent to the extracellular space) (Fig. 6). This finding suggests that the presence of the GFP-tag results in an abnormal orientation of adipophilin in the plasma membrane, for normally this protein only occupies the P-half [18, 20]. A similar observation was made in the nuclear membranes, suggesting that GFP induces mis-orientation of the adipophilin irrespective of the membrane destination. Thus, the freeze-fracture immunolabelling approach, in contrast to other high-resolution localization techniques, has the ability to identify alterations in the orientation of tagged membrane proteins.
Figure 5.
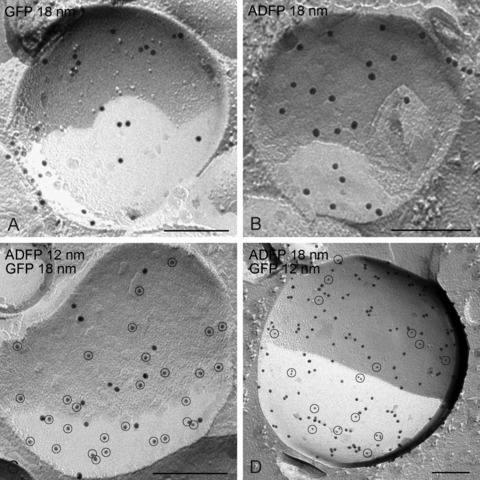
Demonstration of the validity of the GFP-adipophilin (ADFP) freeze-fracture replica immunolabelling technique. In GFP-adipophilin transfected cells, the labelling of lipid droplets obtained using anti-GFP immunogold (A) is identical to that obtained when applying anti-adipophilin antibodies/immunogold (B). When double labelling is carried out using both approaches simultaneously, the results are the same, irrespective of whether 18-nm diameter gold or 12-nm diameter gold is used. Bars: 0.2 μm
Figure 6.
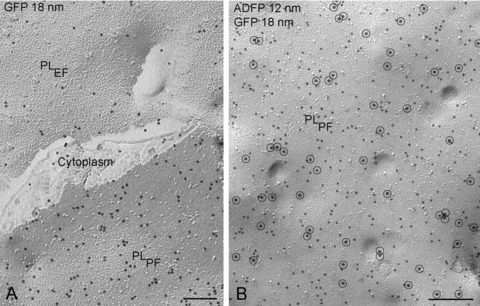
GFP-adipophilin (ADFP) is demonstrated in the plasma membrane of NPC fibroblasts by freeze-fracture replica immunolabelling using GFP antibody and 18-nm gold secondary antibody (A) and with double labelling using GFP antibody/18 nm gold secondary antibody (encircled) and adipophilin antibody/12 nm gold (B). Both fracture faces of the plasma membrane (PL) are labelled (fracture face of half-membrane leaflet adjacent to the protoplasm = PLPF; fracture face of half-membrane leaflet adjacent to the extracellular space = PLEF). Bars: 0.2 μm
Figure 7.
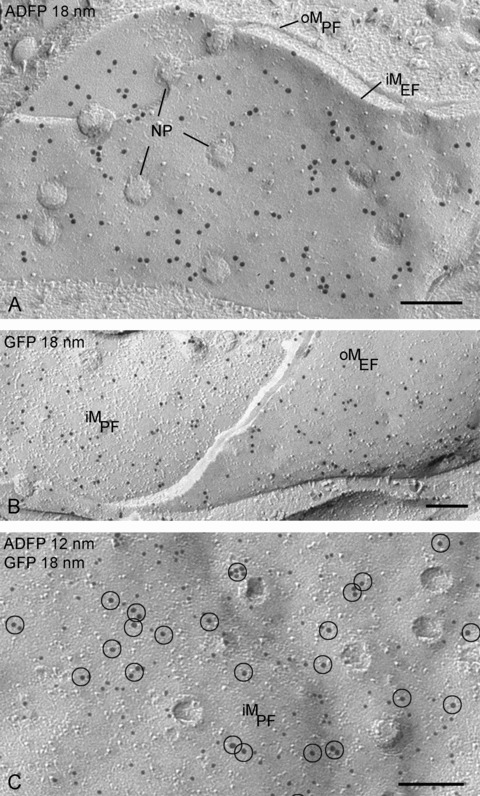
The GFP-adipophilin (ADFP) is also detected in the nuclear membranes by labelling with adipophilin antibodies (A), GFP antibodies (B) and both antibodies (18 nm gold particles encircled) (C). NP, nuclear pores; oM, outer nuclear membrane; iM, inner nuclear membrane; PF, fracture face of half-membrane leaflet attached to protoplasm; EF, fracture face of half-membrane leaflet attached to endo-plasmic space (lumen between inner and outer nuclear membranes or endoplasmic reticulum). Bars: 0.2 μm
To establish the utility of freeze-fracture replica immunolabelling for localizing other GFP-tagged proteins expressed using different vectors and hence the general applicability of the approach, we further examined GFP-tagged CD4, an endoplasmic reticulum-specific protein. Immunofluorescence microscopy of CD4-GFP transfected macrophages using anti-CD4 antibodies gives a corresponding cytoplasmic labelling pattern to that obtained with direct GFP fluorescence (Fig. 8A–C). The freeze-fracture replica immunolabelling technique with immunogold labelling of GFP demonstrates specific labelling of the endoplas-mic reticulum membranes (Fig. 8D). This label is confined to the protoplasmic membrane half. Owing to the continuity between endoplasmic reticulum and nuclear envelope, CD4-GFP is also detected on the nuclear membranes (both on the protoplasmic and on the endoplasmic membrane halves) (Fig. 8E and F). These findings highlight the power of the freeze-fracture replica immunolabelling technique in identifying the precise membrane locations of membrane proteins where other techniques furnish no more than clues.
Figure 8.
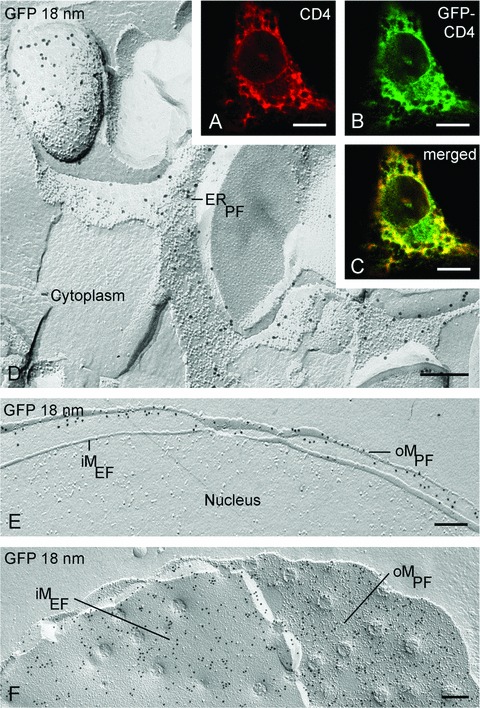
Localization of GFP-CD4 by immunofluorescence in macrophages shows extensive labelling throughout the cytoplasm (A–C). Freeze-fracture replica immunolabelling with GFP antibodies shows that this label resides in the endoplasmic reticulum (ER) (D) and nuclear membranes (E, F). oM, outer nuclear membrane; iM, inner nuclear membrane; PF, fracture face of half-membrane leaflet attached to protoplasm; EF, fracture face of half-membrane leaflet attached to endoplasmic space. Bars: A–C 5 μm; D–F 0.2 μm
In summary, the images presented here demonstrate the ability of the freeze-fracture replica immunolabelling technique to localize GFP-tagged proteins at high resolution in a range of readily identified membrane systems and organelles in different cell types. In these examples, freeze-fracture replica immunolabelling demonstrates that each GFP-tagged protein has a distinctive intracellular distribution, perilipin being confined to lipid droplets, adipophilin occurring in the plasma membrane and nuclear membranes as well as lipid droplets, and CD4 within endoplasmic reticulum and nuclear membranes. While use of anti-GFP antibodies combined with freeze-fracture replica immunogold labelling expands the range of proteins that can be localized at high resolution within the cell to those for which antibodies may not be available, the contribution of the freeze-fracture replica labelling technique to interpretation of conventional fluorescence imaging of GFP is potentially of greater impact. This is because freeze-fracture replica immunolabelling has the unique ability to determine the specific half-membrane leaflet in which a protein or interest resides, and whether this localization and hence orientation in the membrane is altered by the presence of the tag. It should be emphasized that freeze-fracture replica immunolabelling should not be considered as a substitute for GFP-fluorescence light microscopy, but to complement it by resolving interpretative uncertainties and extending and thereby maximizing the information obtained.
Acknowledgments
We thank K. Schlattmann, C. Köppler and M. Opalka for competent and indispensable technical assistance, Dr. B. Schwappach for providing the specific ER-vector, Dr. H. Heid for the perilipin vector and Dr G. Schmitz for the human Niemann-Pick C fibroblasts. This work was supported by the Deutsche Forschungsgemeinschaft, Sonderforschungsbereich 492.
References
- 1.Chalfie M, Tu Y, Euskirchen G, et al. Green fluorescent protein as a marker for gene expression. Science. 1984;263:802–5. doi: 10.1126/science.8303295. [DOI] [PubMed] [Google Scholar]
- 2.Prior IA, Harding A, Yan J, et al. GTP-dependent segregation of H-ras from lipid rafts is required for biological activity. Nat Cell Biol. 2001;3:368–75. doi: 10.1038/35070050. [DOI] [PubMed] [Google Scholar]
- 3.Misteli T, Spector DL. Applications of the green fluorescent protein in cell biology and biotechnology. Nat Biotech. 1997;15:961–64. doi: 10.1038/nbt1097-961. [DOI] [PubMed] [Google Scholar]
- 4.Hanson MR, Köhler RH. GFP imaging: methodology and application to investigate cellular compartmentation in plants. J Exp Bot. 2001;52:529–39. [PubMed] [Google Scholar]
- 5.Van Roessel P, Brand AH. Imaging into the future: visualizing gene expression and protein interactions with fluorescent proteins. Nat Cell Biol. 2002;4:E15–20. doi: 10.1038/ncb0102-e15. [DOI] [PubMed] [Google Scholar]
- 6.Chudakov DM, Lukyanov S, Lukyanov KA. Fluorescent proteins as a toolkit for in vivo imaging. Trends Biotech. 2005;23:605–13. doi: 10.1016/j.tibtech.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 7.Verkusha VV, Lukyanov KA. The molecular properties and applications of Anthozoa fluorescent proteins and chromoproteins. Nat Biotech. 2004;22:289–96. doi: 10.1038/nbt943. [DOI] [PubMed] [Google Scholar]
- 8.Terskikh A, Fradkov A, Ermakova G, et al. “Fluorescent timer”: Protein that changes color with time. Science. 2000;290:1585–8. doi: 10.1126/science.290.5496.1585. [DOI] [PubMed] [Google Scholar]
- 9.Forster A, Pannell R, Drynan LF, et al. The invertor knock-in conditional chromosomal translocation mimic. Nature Meth. 2005;2:27–30. doi: 10.1038/nmeth727. [DOI] [PubMed] [Google Scholar]
- 10.Ward BM, Moss B. Visualization of intracellular movement of vaccinia virus virions containing a green fluorescent protein-B5R membrane protein chimera. J Virol. 2001;75:4802–13. doi: 10.1128/JVI.75.10.4802-4813.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thomsen P, Roepstorff K, Stahlhut M, Van Deurs B. Caveolae are highly immobile plasma membrane microdomains, which are not involved in constitutive endocytic trafficking. Mol Biol Cell. 2002;13:238–50. doi: 10.1091/mbc.01-06-0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martin S, Driessen K, Nixon SJ, et al. Regulated localization of Rab18 to lipid droplets. J Biol Chem. 2005;280:42325–35. doi: 10.1074/jbc.M506651200. [DOI] [PubMed] [Google Scholar]
- 13.Follet-Gueye ML, Pagny S, Faye L, et al. An improved chemical fixation method suitable for immunogold localization of green fluorescent protein in the golgi apparatus of tobacco bright yellow (BY-2) cells. J Histochem Cytochem. 2003;51:931–40. doi: 10.1177/002215540305100708. [DOI] [PubMed] [Google Scholar]
- 14.Luby-Phelps K, Ning G, Fogerty J, Besharse JC. Visualization of identified GFP-expressing cells by light and electron microscopy. J Histochem Cytochem. 2003;51:271–4. doi: 10.1177/002215540305100301. [DOI] [PubMed] [Google Scholar]
- 15.Paupard MC, Miller A, Grant B, et al. Immuno-EM localization of GFP-tagged yolk proteins in C. elegans using microwave fixation. J Histochem Cytochem. 2001;49:949–56. doi: 10.1177/002215540104900803. [DOI] [PubMed] [Google Scholar]
- 16.Fujimoto K. Freeze-fracture replica electron microscopy combined with SDS digestion for cytochemical labeling of integral membrane proteins. Application to the immunogold labeling of intercellular junctional complexes. J Cell Sci. 1995;108:3443–9. doi: 10.1242/jcs.108.11.3443. [DOI] [PubMed] [Google Scholar]
- 17.Robenek H, Hofnagel O, Buers I, et al. Butyrophilin controls milk fat globule secretion. Proc Natl Acad Sci USA. 2006;103:10385–90. doi: 10.1073/pnas.0600795103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Robenek H, Hofnagel O, Buers I, et al. Adipophilin-enriched domains in the ER membrane are sites of lipid droplet biogenesis. J Cell Sci. 2006;119:4215–24. doi: 10.1242/jcs.03191. [DOI] [PubMed] [Google Scholar]
- 19.Robenek H, Lorkowski S, Schnoor M, Troyer D. Spatial integration of TIP47 and adipophilin in macrophage lipid bodies. J Biol Chem. 2005;280:5789–94. doi: 10.1074/jbc.M407194200. [DOI] [PubMed] [Google Scholar]
- 20.Robenek H, Robenek MJ, Buers I, et al. Lipid droplets gain PAT family proteins by interaction with specialized plasma membrane domains. J Biol Chem. 2005;280:26330–8. doi: 10.1074/jbc.M413312200. [DOI] [PubMed] [Google Scholar]
- 21.Severs NJ. Freeze-fracture electron microscopy. Nat Protocols. 2007;2:547–76. doi: 10.1038/nprot.2007.55. [DOI] [PubMed] [Google Scholar]
- 22.Iwashima Y, Eto M, Hata A, et al. Advanced glycation end products-induced gene expression of scavenger receptors in cultured human monocyte-derived macrophages. Biochem Biophys Res Commun. 277:529–39. doi: 10.1006/bbrc.2000.3685. 200; [DOI] [PubMed] [Google Scholar]