

Abstract
Urothelial carcinoma, or transitional cell carcinoma, is the most common urologic malignancy that carries significant morbidity, mortality, recurrence risk and associated health care costs. Despite use of current chemotherapies and immunotherapies, long-term remission in patients with muscle-invasive or metastatic disease remains low, and disease recurrence is common. The molecular chaperone Heat Shock Protein-90 (Hsp90) may offer an ideal treatment target, as it is a critical signaling hub in urothelial carcinoma pathogenesis and potentiates chemoradiation. Preclinical testing with Hsp90 inhibitors has demonstrated reduced proliferation, enhanced apoptosis and synergism with chemotherapies and radiation. Despite promising preclinical data, clinical trials utilizing Hsp90 inhibitors for other malignancies had modest efficacy. Therefore, we propose that Hsp90 inhibition would best serve as an adjuvant treatment in advanced muscle-invasive or metastatic bladder cancers to potentiate other therapies. An overview of bladder cancer biology, current treatments, molecular targeted therapies, and the role for Hsp90 inhibitors in the treatment of urothelial carcinoma is the focus of this review.
Keywords: urothelial carcinoma, pathogenesis, bladder cancer treatments, heat shock protein-90, Hsp90 inhibitors
INTRODUCTION
Epidemiology
Bladder cancer is the fifth most common type of cancer and the second most frequent urologic malignancy in males after prostate cancer. It is the ninth leading cause of cancer death in the United States. The median age at diagnosis and death are 73 and 79 years respectively [1]. In 2014, there were an estimated 74, 690 new cases of bladder cancer and 15, 580 deaths [1]. In the United States, the estimated lifetime risk of urothelial carcinoma is 1 of 25 in men and 1 of 80 in women [2]. In fact, there is a 3:1 male-to-female predominance in urothelial carcinoma, likely related to protective effect of estrogens and increased androgen-receptor signaling in males [3–5]. Health care costs associated with treatment and surveillance of bladder cancer exceed that of all other malignancies, making the design of effective therapies essential not only for patients, but also for public health [6].
More than 90 percent of bladder cancers are urothelial carcinomas (transitional cell carcinomas). Squamous cell carcinomas comprise approximately 5 percent of bladder cancers, and less common neoplasms include micropapillary urothelial carcinoma, small cell carcinoma, sarcomas, and other rare tumors [7]. In addition, other solid tumors may metastasize to the bladder through local spread (prostate, testicular, ovarian, cervical, and endometrial cancers) [7, 8].
Of all newly diagnosed urothelial carcinomas, nearly 70 percent are non-muscle invasive and are treated surgically with transurethral resection with or without intravesical therapies [9, 10]. Non-muscle invasive urothelial carcinoma frequently recurs, in 50 to 70 percent of patients, while muscle-invasive disease has a propensity to metastasize [11]. There is a risk of progression to muscle invasive disease, occurring in up to 15 percent of non-muscle invasive tumors [11].
Risk factors
Advanced age and smoking are major risk factors for developing bladder cancer. A meta-analysis of epidemiologic studies showed that cigarette smokers have a four-fold increased risk than that of nonsmokers and the number of pack-years further increased that risk. More than 50 percent of urothelial carcinomas occur in smokers, which make up 14.5 to 21 percent of the population [12]. This equates to one-half of male urinary tract cancers and one-third of female urinary tract cancers that are attributed to cigarette smoking [13, 14]. Smoking promotes progression of non-invasive to muscle-invasive tumors, and cessation improves long-term prognosis when compared to chronic smokers [15]. Additional risk factors in urothelial carcinoma include chronic exposure to aromatic amine and aniline dyes, Schistosomiasis infection, prior pelvic irradiation, arsenic exposure, phenacetin-containing analgesics and chemotherapy drugs (particularly alkylating agents) [16].
Pathology
Bladder cancers are staged and prognosticated according to the tumor-node-metastasis (TNM) staging system [7]. Non-muscle invasive bladder cancers and muscle-invasive bladder cancers have distinct phenotypic, etiologic, and prognostic characteristics. Non-muscle invasive bladder cancers are, by definition, confined to the mucosa or submucosa, while muscle invasive bladder cancers invade into the muscularis propria or serosal surface of the bladder. Non-muscle invasive urothelial carcinoma develops with hyperplasia of the epithelium with development of branching vessels to form a papillary pattern [17]. Urothelial hyperplasia can progress to form low-grade urothelial carcinoma, which has a high recurrence risk, or can progress to a high-grade tumor [18]. Muscle invasive urothelial carcinoma involves dysplasia of the urothelium and occasionally progresses from carcinoma in situ (CIS) [17]. CIS is high grade, and has the propensity to progress to an invasive carcinoma, and muscle invasive tumors with a higher risk of metastasis [7].
Urothelial carcinoma pathogenesis
The molecular pathogenesis of urothelial carcinomas requires deregulation of multiple signal transduction pathways, therefore, it is a malignancy in which molecular targeted therapies will be useful to block key signaling events involved in bladder cancer biology [19]. Urothelial carcinomas are genetically complex with various oncogenic drivers, numerous mutations within a single tumor, copy number alterations, gene fusion transcripts, and cytogenetic aberrations (Figure 1). Muscle invasive urothelial carcinomas have more mutations, chromosomal aberrations, and aneuploidy than the non-invasive tumors, however, there are common genes implicated in the pathogenesis of both types.
Figure 1. Signaling networks and treatment targets in muscle-invasive and metastatic urothelial carcinomas.
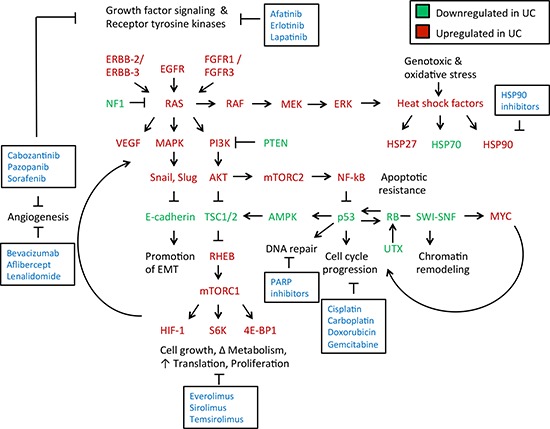
Growth factor signaling is increased in urothelial carcinoma [60]. This results in triggering of growth factor receptors (ERBB-2, ERBB-3, EGFR, FGFR1, FGFR3) leading to Ras activation. Hyperactivation of Ras is a key transition from a non-invasive to an invasive phenotype in urothelial carcinomas [18]. Ras hyperactivation results in phosphotidylinositol-3-kinase (PI3K) signaling, that leads to Akt and mTOR activation downstream. Ras hyperactivation also increases activity of MAP kinases, which activate key regulators of the epithelial-mesenchymal transition [81]. This ultimately leads to an inhibition of E-cadherin expression, promoting local invasion of the tumor through a loss of appropriate cell-cell adhesion [189]. Ras also induces RAF-MEK-ERK signaling, which impacts cytoskeletal dynamics as well as induces a heat shock factor response with increased activity of Hsp27 and Hsp90, as well as other components [155]. Ras is negatively regulated by NF1, which is deficient in some urothelial carcinomas, allowing for uninhibited Ras activation. PI3K activity is inhibited by PTEN, which is also deficient in some urothelial carcinomas due to mutation, leading to increased activation of Akt by PI3K [60, 190]. Akt inhibits the tuberous sclerosis complex (TSC) that acts as a negative regulator of mTORC1 activity. PI3K-Akt activation, as well as mutation within a TSC component (TSC1 or TSC2), leads to inappropriate mTORC1 activation by Rheb GTPase [191]. mTORC1 promotes numerous anabolic processes, including cell growth, metabolism, protein translation, and hypoxic signaling through increased production of hypoxia-inducible factor-1 (HIF-1) [192]. HIF-1 and vascular endothelial growth factor (VEGF) promote angiogenesis and support an intratumor vasculature. Akt also stimulates the mechanistic target of rapamycin (mTOR) complex 2 to activate NF-kB and promote cytoskeletal growth [193]. NF-kB in turn inhibits p53, which promotes apoptotic resistance [194]. Loss of p53 expression leads to uninhibited cell cycle progression, as does loss of the retinoblastoma (RB1) tumor suppressor gene [195]. Reduced RB1 expression results from mutation of its locus as well as through reduced accessibility of chromatin to transcribe its locus from inactivation of the SWI-SNF chromatin remodeling complex [84]. Increased cell cycle progression, paired with an increase in anabolic processes, promotes survival and growth of the tumor. *Molecules in red are upregulated in urothelial carcinomas, while those in green are downregulated. Molecular targeted therapies to disrupt these key processes implicated in urothelial carcinomas growth and progression are highlighted in boxes.
Heat shock proteins (Hsp) are over-expressed in both non-muscle invasive and muscle invasive bladder cancers [20]. They allow bladder cancer cells to survive and progress despite various sources of cellular stress. The heat shock response prevents cancer cells from undergoing apoptosis, despite an accumulation of genomic mutations, and hostile hypoxic and/or acidotic tumor environments [20]. Several proteins involved in bladder cancer biology are regulated by the Hsp90 chaperone complex, which aids in their stabilization, maintains their protein expression and promotes oncogenesis.
Hsp90: a signaling hub in urothelial carcinoma biology
Structure and function
Hsp90 plays an important role in urothelial carcinoma biology, as well as in carcinogenesis of other tumors, by its function as a molecular chaperone that cancer cells utilize to protect over-expressed or mutated oncoproteins from misfolding and degradation [21–25]. Proteins chaperoned by Hsp90, also known as clients, control numerous cellular processes that support tumor growth and metastasis, including signal transduction, angiogenesis, anti-apoptotic pathways and tumor invasion [26]. Hsp90 is a homo-dimeric protein that comprises of three domains: i) the N-terminal domain, containing nucleotide, drug and co-chaperone (proteins that regulate Hsp90 function) binding sites; ii) the middle (M) domain, which provides binding motif for client proteins and other co-chaperones, and iii) the C-terminal domain containing a dimerization motif and binding sites for yet other co-chaperones. An unstructured charged-linker region connects N and M domains and, therefore, provides conformational flexibility to the Hsp90 protein [27–37] (Figure 2). Hsp90 function is coupled to its ATPase activity [38] and this, in turn, provides conformational cycle that is “fine-tuned” by co-chaperones and post-translational modifications such phosphorylation, acetylation, ubiquitination, oxidation, methylation, S-nitrosylation and SUMOylation [39–44] (Figure 2). Clinically evaluated Hsp90 inhibitors disrupt the chaperone cycle by occupying the nucleotide-binding pocket in the N-domain, therefore, inhibiting the ATPase activity [32, 45]. As a result, Hsp90-dependent client proteins are ubiquitinated and degraded in the proteasome [43, 46–48].
Figure 2. Hsp90 chaperone cycle.
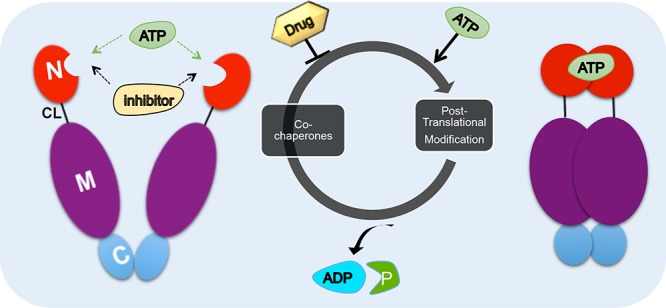
ATP binding to the N-terminal domain of Hsp90 (red) in an “open” conformation promotes transient dimerization of the N-domains “closed” conformation leading to ATP hydrolysis [38]. The co-chaperones such as Aha1, Cdc37, HOP and p23 and post-translational modification influence the rate of ATP hydrolysis. Domain labeling is as follows: N, N-domain (red); CL, charged linker (black); M, M-domain (purple); C, C-domain (blue).
Protein clients
Several Hsp90 client proteins act as drivers of urothelial carcinoma, and thus inhibitors or modulators of Hsp90 function may impede urothelial carcinoma pathogenesis (Figures 1 and 3). A recent comprehensive study, by the Cancer Genome Atlas Project, identified novel protein determinants involved in urothelial carcinoma including: tumor suppressors (TP53, Rb) [49, 50], oncogenes (ErbB2/HER2, ErbB3, Myc) [49, 51, 52], cell cycle regulatory proteins (by Hsp90 and Cdc37 co-chaperone, cyclins and cyclin-dependent kinases, including Cdk2) [53, 54], Ras-MAPK pathway proteins (HRas and multiple MAP kinases) [55], mTOR pathway components (Akt) [56], growth factor receptors (EGFR, FGFR1/3/4) [57–59] and transcriptional regulators which include proteins involved in histone modification (SWI/SNF complex members), STAT3, MLL/3, SP1 and FOXA2 [60–62] (Figure 3).
Figure 3. Hsp90 is a central hub to bladder cancer signaling.
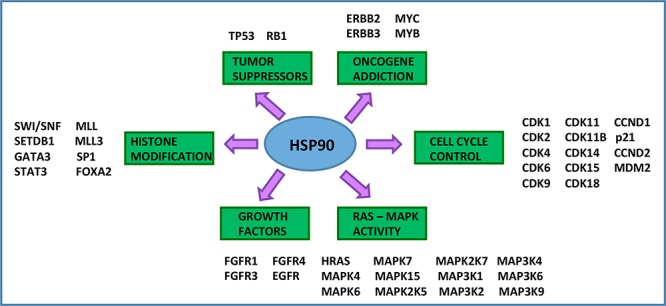
Hsp90 is a critical signaling hub in the etiopathogenesis of urothelial carcinoma. Hsp90 clients include tumor suppressors, oncogenes, growth factors, cell cycle regulators, histone modifying enzymes, and signal transducers [21, 49–55, 57, 58, 60–62]. All of the listed genes are subject to mutation, gene amplification, or deletion in urothelial carcinoma and are Hsp90 client proteins.
Hsp90 and oncogenic signaling pathways in urothelial carcinoma pathogenesis
Urothelial carcinomas have somatic mutations of tumor suppressor genes, which include, but are not limited to TP53, RB1, PTEN, TSC1, and p16, and activation of oncogenic drivers [60, 63] (Figure 1). While mutations in tumor suppressor genes allow for uncontrolled cell proliferation and cell cycle progression, the driver mutation present in many urothelial carcinomas is mutation or gene fusion of the fibroblast growth factor receptor 3 (FGFR3) [58].
Fibroblast growth factor signaling
Fibroblast growth factor receptor 3 (FGFR3) is a tyrosine kinase receptor and key regulator of cellular growth and differentiation. FGFR3 depends on Hsp90 for its stability and function and treatment with Hsp90 inhibitors target FGFR3 for degradation [58]. FGFR3 is shown to be over-expressed in 75 percent of non-muscle invasive bladder cancers. It is upregulated by multiple mechanisms in bladder cancer cells: 1) somatic mutation, 2) over-expression of the wild-type protein, or 3) gene fusion with transforming acid coiled coil (TACC3) or BAI-1 associated protein 2-like 1 (BAI1AP2L1) partners [64]. FGFR3-TACC3 or FGFR3-BAI1AP2L1 gene fusions can serve as the drivers of oncogenesis in bladder cancer cells, especially in non-muscle invasive bladder cancers, as they activate numerous downstream signaling pathways, including PI3K-Akt-mTOR, Ras, MAP kinases, STATs, and phospholipase-Cγ [65].
Cell cycle deregulation
Loss of cell cycle control is characteristic of carcinogenesis in urothelial carcinoma, as in many other cancers, due to the inactivation of cell cycle-regulating tumor suppressor genes (including TP53, RB, ATM), mutation of cell cycle progression regulators (CDKN1A, CDKN2A, CCND1, and CCNE1), or activating mutations of genes promoting cell cycle progression (including MDM2 and E2F3). Inactivation of the cell cycle regulator p53 results in G1 to S cell cycle progression and uncontrolled cell growth in urothelial carcinomas. p53 is inactivated in muscle invasive bladder cancers to a greater extent than non-muscle invasive bladder cancers (24 versus 76 percent) [17, 60]. Wild type p53 is also a known client of Hsp90, and interacts with Hsp90 through its DNA binding domain [66]. Hsp90 stabilizes mutant p53 through inhibiting the proto-oncogene MDM2 to function as an E3 ubiquitin protein ligase [67, 68]. Although many urothelial carcinomas exhibit p53 inactivation, Hsp90 inhibitors can still promote growth arrest. In lymphocytic leukaemia cells, inhibition of Hsp90 was found to have opposing effects on wild-type and mutant p53 proteins, with stabilization and increased expression of the wild-type protein [69]. Upregulation of p53 results in increased p21 expression, which induces cell cycle arrest at the G1 to S transition point [69]. Hsp90 inhibition induces apoptosis through p53-dependent induction, mediated by PUMA and Bax [68]. Inactivation of the retinoblastoma (RB) tumor suppressor gene is present in approximately 50 percent of high grade and muscle-invasive urothelial carcinomas [70]. Additionally, urothelial carcinomas are the most common carcinomas in survivors with RB cancer [71]. RB loss alone cannot accelerate urothelial proliferation, but inhibits p53. In an animal model for urothelial carcinoma, mice deficient in both RB and P53, but not either tumor suppressor gene alone, were the most sensitive to carcinogen exposure. Growth inhibition induced by Hsp90 inhibitors, occurs through G1 to S phase arrest through reducing RB phosphorylation. RB inhibition reduces expression of cyclin-associated kinases D and E, to prevent cell cycle progression. This was supported by the absence of growth arrest in cells lacking RB, compared to wild-type RB expression [72].
P16 (CDKN2) is a tumor suppressor gene encoded on chromosome 9p21, a region frequently deleted in urothelial carcinomas. P16 arrests the cell cycle through inhibition of cyclin dependent kinases (CDK) 4 and 6. CDK4 and CDK6 (both are Hsp90 clients) phosphorylate RB to stimulate G1 to S phase transition in the cell cycle, therefore, loss of p16 results in uninhibited cell growth [73, 74]. Loss of p16 secondary to deletion occurs in 54 percent of urothelial carcinomas. A study of minimally invasive (T1a) urothelial carcinomas identified that loss of p16 was associated with a reduction in progression-free survival, but did not affect recurrence rates. The loss of p16 was identified as an independent predictor of tumor progression at a given tumor stage and grade [74]. Unlike invasive urothelial carcinomas, p16INK4a is over-expressed in urothelial carcinoma in situ, which is secondary to enhanced Ras/MAPK signaling [75].
DNA repair
Cells have evolved DNA repair mechanisms essential for the removal of damaged DNA caused by several endogenous (hydrolysis, reactive oxygen species, alkylation, DNA mismatches, insertions or deletions, strand breaks) and exogenous (ultraviolet light, ionizing radiation, chemotherapeutic drugs) sources. As tobacco smoking is considered a risk factor for bladder cancer it is also a source of carcinogens that damage DNA in urothelial cells [76]. Whole-exome sequencing studies identified somatic mutations in genes related to DNA repair pathways including P53, KDM6A, ATM, ERCC2, FANCD2, PALB2, BAP1, BRCA1 and -2) [77, 78]. Interestingly, these mutations seem to be correlated with recurrence-free survival in patients with muscle invasive bladder cancer [77]. As more studies identify alterations in the DNA repair mechanism, poly (ADP-ribose) polymerase (PARP) inhibitors combined with DNA damaging agents may be a good therapeutic strategy in some patients [78].
PI3K-Akt-mTOR activation
Activation of the phosphoinositide-3-kinase (PI3K)-Akt-mTOR (mammalian target of rapamycin) pathway occurs in urothelial carcinomas, particularly in muscle-invasive tumors. Its activation is associated with reduced survival and tumor progression [79]. This usually occurs secondary to PTEN (phosphatase and tensin homolog) deletion, which results in increased activation of Akt by uninhibited PI3K activity. Akt activation results in mTORC2 (mTOR complex 2) activation and subsequent changes in cytoskeletal dynamics. It also promotes mTORC1 (mTOR complex 1) activation through inhibition of a negative regulator, the tuberous sclerosis complex (TSC1 and TSC2). TSC1/TSC2 complex inhibits Rheb (Ras homolog enriched in brain) GTPase, which activates mTORC1. mTORC1 stimulates cellular growth through regulation of protein translation by its targets S6 kinase and 4E-BP1. Akt, which activates both mTORC1 and 2, is an Hsp90 client [56].
PTEN, a tumor suppressor gene that negatively regulates activation of the PI3K/Akt/mTOR pathway, is deficient in over 50 percent of urothelial carcinomas due to loss of heterozygosity (LOH). Microdeletions at the 10q23 locus is often the source of PTEN LOH. Reduced PTEN levels leads to hyperactive Akt through PDK1 (3-phosphoinositide-dependent kinase 1)-mediated phosphorylation. Hsp90 stabilizes Akt and under Hsp90 inhibition, Akt is rapidly ubiquitinated, greatly shortening its half-life (to one-third) within treated cells [56].
Epithelial-mesenchymal transition
There are several signaling pathways that cooperate to induce epithelial-to-mesenchymal transition (EMT) in cancer [80]. One of them is the activation of the Ras-MAPK pathway [18]. A low level of Ras-MAPK activation is present in non-invasive urothelial carcinomas, while hyperactivation occurs in high-grade tumors [18]. The Ras-MAPK pathway is activated by receptor tyrosine kinases, as well as by fibroblast growth factor signaling. In addition, Src signaling (c-Src depends on Hsp90 for its maturation) stimulates EMT through cytoskeletal remodeling and inhibiting formation of adherens junctions [81].
Epigenetic control and transcriptional regulation
Reduced histone modification with inactivation of acetyltransferases, methyltransferases, deubiquitinases, and demethylating enzymes occurs in 89 percent of urothelial carcinomas [60]. There are numerous chromatin modifying or remodeling enzymes subject to frequent mutations in urothelial carcinoma, which include BAP1, UTX, MLL, NCOR1, ARID1A, CHD1 and 6, CREBBp, EP300, and the SWI/SNF complex [78, 82]. Mutation causes inactivation of chromatin modifying enzymes in over 60 percent of urothelial carcinomas, with the most common mutations within members of the SWI/SNF complex [60]. The SWI/SNF complex maintains an environment of tumor suppression, and is frequently lost in urothelial carcinomas [83, 84].
Current and investigational treatments for urothelial carcinoma
Treatment of metastatic urothelial carcinoma remains a major challenge, with no improvement in overall survival achieved in the past 20 years [85]. Patients with advanced urothelial carcinoma have a poor overall survival of less than 30 percent at 5 years post-diagnosis [86]. Yet, chemotherapy still provides benefit over cystectomy, alone [87, 88]. Generally, 70 percent of patients with metastatic urothelial carcinoma respond to systemic chemotherapy, however more than 90 percent of them relapse and eventually die from the disease [89].
Although several molecular targeted therapies for urothelial carcinoma are under investigation, no new agents have been approved by the Food and Drug Administration (FDA) in the past 3 decades. The only FDA approved therapies to date for muscle invasive bladder cancer are cisplatin, doxorubicin hydrochloride, and gemcitabine hydrochloride, all of which are cytotoxic chemotherapies. Non-muscle invasive bladder cancers are primarily treated with transurethral resection and intravesical therapies.
Intravesical therapies
Currently, there are several intravesical therapies for the treatment of non-invasive urothelial carcinomas, including Mitomycin C, epirubicin, doxorubicin, and instillation of live attenuated Mycobacterium bovis bacillus Calmette-Guérin (BCG) [90]. BCG reduces the risk of progression and recurrence and is used to treat patients with carcinoma in situ as well as non-invasive urothelial carcinomas (T1a tumors) [91–96]. Despite the success of intravesical treatments, disease progression and recurrence are common, with a lifetime recurrence risk of 88 percent in some studies [97]. Additionally, with the need for frequent surveillance, repeat biopsies, treatment of recurrence, and management of complications, the costs associated with treatment of urothelial carcinoma is very high, often exceeding $150, 000 over the patient's lifetime [6].
Chemotherapies
The alkylating agent cisplatin, the topoisomerase II inhibitor, doxorubicin hydrochloride and the nucleoside analog gemcitabine hydrochloride, all are non-specific blockers of DNA synthesis in proliferating cells [98–100]. There are many toxicities associated with these drugs, which include but are not limited to bone marrow suppression with pancytopenia, peripheral edema and capillary leak syndrome, pulmonary toxicity including respiratory distress syndrome and pulmonary fibrosis, gastrointestinal toxicities with weight loss, hepatotoxicity that can lead to liver failure, renal toxicity and hemolytic-uremic syndrome and infections [101, 102].
One of the most commonly used combination regimens for advanced bladder cancer, includes methotrexate, vinblastine, doxorubicin, and cisplatin, only leads to a complete response in 1 of 4 patients [103]. Cisplatin is a chemotherapy agent that binds to purine DNA bases causing cross-linking and triggering DNA damage response [104]. Overcoming the DNA damage response can lead to drug resistance [104, 105]. This high rate of treatment failure has been attributed, in part, to the development of cisplatin resistance, which occurs through multiple mechanisms, such as reduced cisplatin binding to DNA (pre-target resistance), reduced formation of DNA-cisplatin adducts (on-target resistance), altered DNA damage response (post-target resistance), and influenced signaling pathways that reduce the DNA damage response (off-target resistance) [104, 106]. More recently, Choi et al. identified a p53-like subtype of invasive bladder cancer associated with cisplatin-based chemotherapy resistance to apoptosis [107]. In addition to resistance, many patients with urothelial carcinoma have impaired renal function, and cisplatin is nephrotoxic [108]. Therefore, individuals with impaired renal function are not candidates for cisplatin treatment [109]. Given the resistance and toxicity of cisplatin, alternative strategies to sensitize bladder cancer cells to chemotherapy and allow decreased dose of cisplatin, while increasing the response and durability, are being explored.
Several clinical trials are ongoing for the treatment of advanced, unresectable, or metastatic urothelial carcinoma (Table 1). These include new chemotherapeutics targeting the DNA damage response or histone deacetylases, inhibitors of the mammalian target of rapamycin (mTOR), tyrosine kinase inhibitors (TKIs), HER2 blockade, anti-angiogenic therapies, and immunotherapies. Immunotherapy approaches include BCG [110], cytokine-based treatments (recombinant IL-2 or IL-7) to elicit cellular immune responses [111], or dendritic cell vaccines [112].
Table 1. FDA-approved and investigational therapies for urothelial carcinoma.
| Drug category | Drug urothelial carcinoma | FDA approved or clinical trial phase | Type / stage of cancer |
|---|---|---|---|
| Chemotherapy | Cisplatin (alkylating agent) | Approved | T4b and metastatic urothelial carcinoma |
| Doxorubicin hydrochloride (topoisomerase II inhibitor) | Approved | Stage IV and recurrent bladder cancer | |
| Gemcitabine hydrochloride | Approved | Advanced bladder cancer | |
| 5-fluoro-2′-deoxcytidine + tetrahydrouridine | Phase II | Advanced bladder cancer | |
| Eribulin mesylate (E7389) | Phase I / II | Locally advanced or metastatic bladder cancer | |
| Veliparib / ABT-888 | Phase I | Non-resectable or metastatic | |
| Romidepsin (histone deacetylase inhibition) | Phase I | Advanced urothelial carcinoma | |
| Anti-tumor immunity | rhIL-7 vaccine | Phase II | Metastatic urothelial carcinoma |
| DC205-NY-ESO-1 fusion protein vaccine +/− sirolimus | Phase I | Metastatic urothelial carcinoma | |
| Ad/HER2/Neu dendritic cell vaccine | Phase I | T3a and above HER2+ bladder cancer | |
| Bacillus Calmette-Guerin + PANVAC | Phase II | Non-muscle invasive bladder cancer | |
| ALT-801 (IL-2 recombinant fusion protein) post-chemotherapy with cisplatin and gemcitabine | Phase I / II | Advanced stage muscle-invasive bladder cancer | |
| mTOR inhibitors | ABI-009 (nab-rapamycin) | Phase I / II | Advanced non-muscle invasive bladder cancer |
| Sirolimus, post-chemotherapy with cisplatin and gemcitabine hydrochloride | Phase I / II | T2 to T4 tumors | |
| Anti-angiogenic therapy | Cabozantinib | Phase II | Advanced stage bladder cancer |
| Bevacizumab, post-chemotherapy with gemcitabine hydrochloride + cisplatin | Phase III | Metastatic, unresectable, or locally advanced bladder cancer | |
| Lenalidomide, post-chemotherapy with gemcitabine hydrochloride and carboplatin | Phase I | Unresectable or metastatic bladder cancer | |
| Tyrosine kinase inhibitors | Afatinib | Phase II | Ureteral cancer, stage III, stage IV urothelial carcinoma |
| Dovitinib | Phase II | BCG-refactory urothelial carcinoma, FGFR3-mutated urothelial carcinoma | |
| Erlotinib | Phase II | Stage I, II, III and recurrent urothelial carcinoma | |
| Gefitinib | Phase II (completed) | Locally advanced and metastatic bladder cancer | |
| Pazopanib | Phase II (completed) | Locally advanced and metastatic bladder cancer | |
| Sorafenib | Phase II (completed) | Locally advanced and metastatic bladder cancer | |
| Sunitinib | Phase II | BCG-refactory bladder cancer | |
| HER2 blockade | Afatinib (targets EGF and HER2) | Phase II | Refactory bladder cancer |
| MGAH22, a human chimeric antibody against HER2 | Phase I | HER2 positive bladder cancer |
New chemotherapies
Additional chemotherapies under early stage investigation for urothelial carcinoma include the combination of 5-fluoro-2′-deoxycytidine and tetrahydrouridine (antimetabolites), eribulin mesylate (an inhibitor of microtubule dynamics), veliparib (an inhibitor of PARP 1 and 2, which prevents DNA repair within cancer cells to enhance chemoradiosensitivity), and ropidepsin (a histone deacetylase inhibitor). As many bladder cancer tumors possess cells with somatic mutations in genes associated with DNA repair, they should be especially sensitive to chemotherapeutic drugs that promote DNA damage [78]. Chemotherapy remains the first-line therapy for metastatic urothelial carcinoma, but several factors make some patients poor treatment candidates, including poor performance status, renal insufficiency, neuropathy, ototoxicity, and heart failure (New York Heart Association grade 3 and above) [113].
Tyrosine kinase inhibitors
Tyrosine kinase inhibitors (TKIs) evaluated in clinical studies for urothelial carcinoma include afatinib, erlotinib, dovitinib, sunitinib, gefitinib, pazopanib, and sorafenib. Erlotinib primarily targets the epidermal growth factor receptor (EGFR), and was shown to have optimal use in the neoadjuvant setting, resulting in downstaging of the tumor prior to surgery [114]. Sunitinib is an oral, small-molecule, multi-targeted tyrosine kinase inhibitor, with significant activity against VEGFR, PDGFR, stem cell factor receptor, fms-like tyrosine kinase 3 (Flt3) and the tyrosine kinase receptor encoded by the RET proto-oncogene. A double-blind, randomized, phase 2 trial of maintenance sunitinib versus placebo was tested in MIBC patients who have previously undergone chemotherapy, and was found to have anti-tumor activity [115], however maintenance sunitinib did not appear to improve the 6-month progression rate [116], therefore, it is not an ideal second-line option for patients unable to tolerate cisplatin-based chemotherapy regimens [113]. Sorafenib is an oral, multi-kinase inhibitor which blocks tumor cell proliferation by targeting the Ras/Raf/ERK pathway at the level of Raf kinase, and blocks angiogenesis by targeting the VEGFR and PDGFR families. It was found to have insufficient activity as a first-line treatment for advanced urothelial carcinoma [117]. Pazopanib is an oral multi-kinase angiogenesis inhibitor that targets VEGFR, PDGFR and stem cell receptor factor. Synergistic efficacy of pazopanib with docetaxel was demonstrated in docetaxel-resistant bladder cancer cells [118]. However, in a phase 2 trial, Pazopanib did not show significant activity against metastatic urothelial carcinoma [119]. Gefitinib is an oral, small-molecule inhibitor of the intracellular domain of EGFR [120]. Gefitinib was found to have a growth-inhibitory and anti-invasive effect in urothelial carcinoma cell lines [120]. However, gefitinib demonstrated minimal anti-tumor activity in patients with metastatic urothelial carcinoma in patients with prior chemotherapy and is ineffective as a second-line agent for urothelial carcinoma or in combination therapy with cisplatin and gemcitabine [121]. Dovitinib (TKI258) is another TKI with promising pre-clinical data, significantly retarding the growth of bladder tumor xenografts in vitro and in vivo [122], however, it had limited single agent activity in previously treated muscle invasive bladder cancer patients in a phase II clinical study [123]. Lapatinib is a TKI with dual targeting of EGFR and ERBB2, most suitable for metastatic urothelial carcinoma patients with EGFR or ERBB2-overexpressing tumors [124], and can be used post-chemotherapy or in chemoresistant patients [121].
Angiogenesis inhibitors
Anti-angiogenic therapies include bevacizumab, aflibercept, lenalidomide, and the anti-angiogenic TKIs (cabozantinib, pazopanib, and sorafenib). Bevacizumab is used in conjunction with cisplatin and gemcitabine as a first-line treatment for metastatic urothelial carcinoma. A recent phase II clinical trial showed an increase in overall survival and had an acceptable toxicity profile [125]. A phase III study is currently ongoing (Gemcitabine Hydrochloride and Cisplatin With or Without Bevacizumab in Treating Patients With Advanced Urinary Tract Cancer (NCT00942331)). Another anti-angiogenic therapy, Aflibercept, is a human recombinant fusion protein that acts as a soluble decoy receptor, also known as a “VEGF Trap” [126]. It contains vascular endothelial growth factor receptor-1 and 2 (VEGFR-1 and 2) fused to the Fc fragment of IgG1, which inhibits VEGF signaling. Aflibercept fusion protein is used after platinum-based chemotherapy, and has limited single-agent activity, based on a phase II clinical study [126].
HER-2 blockade
Expression of the HER-2/neu oncogene is associated with tumor invasion and metastatic potential and is expressed on 28 percent of urothelial carcinomas overall, and greater than 50 percent of muscle-invasive tumors [127]. Lapatinib, a tyrosine kinase inhibitor, elicits dual blockade of EGFR and HER-2/neu [128]. Although both of these targets are over-expressed on urothelial carcinomas and play pathogenic roles, treatment with lapatinib as a second-line therapy for metastatic urothelial carcinoma in combination with paclitaxel was poorly-tolerated [129].
Mechanistic target of rapamycin (mTOR) inhibitors
Inhibitors of the mTOR have demonstrated anti-tumor activity alone and in combination with chemotherapy, as mTOR inhibition enhances chemosensitivity of urothelial carcinoma cells [130]. mTOR inhibitors induce cell cycle arrest at the G0 to G1 growth phase and inhibit VEGF production [131]. Everolimus (also known as RAD001) has synergistic activity with cisplatin, and provides anti-tumor activity in a subset of patients with muscle invasive bladder cancer [132, 133]. Everolimus evoked an anti-angiogenic response, but requires functional PTEN, as PTEN loss was associated with resistance to everolimus and other mTOR inhibitors [134, 135]. Long-term rapamycin (also known as Sirolimus) treatment reduced the incidence of urothelial carcinoma in renal transplant recipients [136], who are at increased risk of malignancy due to a reduced ability to elicit anti-tumor immune responses, however the protection against incidence and recurrence was not 100 percent [137]. Temsirolimus was tested in a phase II clinical trial after failure of platinum based therapy for muscle invasive bladder cancer, but yielded poor treatment responses [138].
Immunotherapies
Immunotherapies for urothelial carcinoma include instillation of live but attenuated BCG, dendritic cell-based vaccines, recombinant IL-7, and recombinant IL-2 fusion protein. These are all aimed at improving anti-tumor immune responses. BCG elicits production of hydrogen peroxide, superoxide, and free radical oxygen, promoting oxidative stress within urothelial carcinoma cells [139]. Oxidative stress promotes DNA damage and lipid peroxidation, which leads to tumor cell death and release of the “danger signal” HMGB1 (high-mobility group protein 1) that stimulates the innate immune response [140]. AdCD40L adenoviral vaccine is used to upregulate CD40 ligand to improve anti-tumor immune responses as a neoadjuvant treatment for muscle invasive bladder cancer undergoing cystectomy, and has successful gene transfer causing enhanced immune activation [141]. A completed phase I/II trial demonstrated safety and induction of an immune response (identified by increased numbers of IFNγ-producing T lymphocytes) [141], and is currently under evaluation in a larger phase II trial for evaluation of efficacy. NY-ESO-1 vaccine, in conjunction with BCG and sargramostim is under investigation in muscle invasive bladder cancer patients post-cystectomy that have tumors that express NY-ESO-1 or LAGE-1 antigens (expressed in nearly 50 percent of muscle invasive bladder cancers) [142]. Immune responses against these antigens were present in patients having received the vaccination for advanced urothelial carcinoma [143]. CDX-1307, a monoclonal antibody that targets the mannose receptor and beta-hCG (human chorionic gonadotropin), is also under evaluation in clinical trials [144]. Increased numbers of CD8+ cytotoxic lymphocytes infiltrating the tumor is associated with beneficial anti-tumor immune responses and serves as a good prognostic indicator [145]. These current and investigational treatments for urothelial carcinoma are summarized in Table 2.
Table 2. Preclinical studies of Hsp90 inhibitors in bladder cancer.
| Hsp90 inhibitor | Combination therapy | Model system | Outcome | Mechanism | Reference |
|---|---|---|---|---|---|
| 17-AAG | Cisplatin | JTC-30 (low grade papillary), RT4 (grade 1), KK47 (grade 1), 5637 (grade 2), 1376 (grade 3), and T24 (grade 3) bladder cancer cell lines | Synergistic reduction in cell survival | ↓ Activation of Erk1/2, Akt, PI3K ↑ Apoptosis ↓ Cell cycle progression |
[160, 174] |
| 17-AAG | Docetaxel | RT4 (grade 1), KK47 (grade 1), 5637 (grade 2), 1376 (grade 3), and T24 (grade 3) bladder cancer cell lines | Synergistic reduction in cell survival | ↑ Apoptosis ↓ Cell cycle progression |
[160] |
| 17-AAG | Gemcitabine | RT4 (grade 1), KK47 (grade 1), 5637 (grade 2), 1376 (grade 3), and T24 (grade 3) bladder cancer cell lines | Synergistic reduction in cell survival | ↓ Chk1 ↑ Apoptosis ↓ Cell cycle progression |
[160] |
| 17-AAG | Pifitrhrin-μ | RT4 (grade 1), KK47 (grade 1), 5637 (grade 2), 1376 (grade 3), and T24 (grade 3) bladder cancer cell lines | Synergistic reduction in cell survival | ↓ p-Akt, p-Bad ↑ Apoptosis |
[160] |
| 17-AAG | Cisplatin + radiotherapy | 5637 (grade 2), T24 (grade 3), and UM-UC-3 bladder cancer cell lines | Chemoradio-sensitization | Inactivation of anti-apoptotic proteins erbB2, Akt, NF-kB | [161] |
| 17-AAG | Cisplatin | SCID xenografts with 5637 bladder cancer cells Bladder cancer initiating cell xenograft model |
Increased reduction in tumor size compared to cisplatin alone No treatment related death or weight loss |
Inactivation of anti-apoptotic proteins erbB2, Akt, NF-kB | [174] [161] |
| 17-DMAG | Cisplatin + radiotherapy | 5637 (grade 2), T24 (grade 3), and UM-UC-3 bladder cancer cell lines | Chemoradio-sensitization | Inactivation of Akt and Erk ↓ Survival of bladder cancer cells |
[161] |
Hsp90 inhibitors in treating urothelial carcinoma
One promising target to enhance clinical responses is treatment with Hsp90 inhibitors. There are currently sixteen Hsp90 inhibitors evaluated in clinical trials for numerous hematopoietic and solid malignancies [22]. These include melanoma, small cell lung cancer, non-small cell lung cancer, breast cancer, gastrointestinal stromal tumor, gastric cancer, colon adenocarcinoma, ovarian cancer, primary peritoneal cancer in women, fallopian tube cancer, prostate adenocarcinoma, leukemia, lymphoma, myeloproliferative disorders, and myelodysplastic syndromes [146–148]. Several Hsp90 inhibitors have demonstrated safety, while the second generation Hsp90 inhibitor, ganetespib, is currently in phase III clinical trials (for non-small cell lung cancer) and already has shown manageable side-effects in phase II clinical trials [149]. We do not expect that molecular targeted therapy will replace current treatments for urothelial carcinoma, rather it may be used adjunctively with other therapies to heighten the clinical response and reduce residual disease or recurrence.
As numerous client proteins participate directly in the pathogenesis of urothelial carcinoma, Hsp90 could be an ideal target [21] (Figures 1 and 3). Targeting Hsp90 also inhibits pathways involved in tumor invasion and metastasis, by affecting cellular migration and angiogenesis [150]. For instance, Hsp90 blockade by geldanamycin in bladder cancer cells was shown to inhibit signaling by the hepatocyte growth factor and its target oncogene c-Met, which participates in tumor cell migration by disruption of extracellular matrix components [150]. Over-expression of c-Met is associated with poor prognosis in urothelial carcinoma and can be blocked by Hsp90 inhibitors [151, 152]. Hsp90 inhibition also inhibits hypoxia-inducible factor signaling, another factor associated with poor prognosis in urothelial carcinoma due to induction of angiogenesis promoted by vascular endothelial growth factor signaling [153, 154].
In urothelial carcinoma, no distinct correlation exists between Hsp90 protein levels and grade or pathologic stage of the cancer [155, 156], yet its inhibition results in destabilization of ErbB2, NF-κB, and phosphorylated Akt [157]. These proteins are Hsp90 clients [158, 159], and their collective inhibition prevents the usual deregulated cell growth and desensitization to pro-apoptotic signals [160, 161].
More recently, bladder cancer cell lines with FGFR3 somatic mutation, FGFR3-TACC3 and FGFR3-BAI1AP2L1 gene fusions were all found to be sensitive to Hsp90 inhibition. Urothelial carcinoma cells with FGFR3 mutations that are insensitive to pan-FGFR inhibitors (those containing the FGFR3S249C or FGFR3Y375C mutations) were found to be sensitive to Hsp90 inhibitors [162]. While both ganetespib and an FGFR inhibitor promoted regression of bladder cancer xenografts harboring the FGFR3-TACC3 fusion, combination of the two agents led to a further, significant reduction in tumor volume [162]. Thus, the use of HSP90 inhibitors alone or in combination with targeted therapies may serve as a therapeutic strategy for genetically defined bladder cancers.
The effects of Hsp90 inhibitors in combination with chemotherapy in treating urothelial carcinoma
In the etiopathogenesis of bladder cancer and other solid tumors, there is increased expression of Hsp90 [163]. Hsp90 inhibition alone has cytostatic effects in bladder cancer cell lines in vitro and it also provides synergistic effects with various chemotherapy drugs [160], including those used in the clinic for urothelial carcinoma treatment. Treatment of head and neck cancer cells with Hsp90 inhibitor 17-AAG was shown to activate apoptosis by restoring wild type p53 function through the disruption of p53 interaction with its negative regulator murine double minute proteins X (MDMX) [164]. When cells were treated with a combination of Hsp90 inhibitor 17-AAG and cisplatin there was a more prominent apoptotic effect in vitro, tumor growth inhibition in vivo and restoration of wild type p53 levels. Hsp90 inhibitors provide synergism with cisplatin, and likewise, cisplatin improves clinical responses to Hsp90 inhibitors by preventing the compensatory heat shock response due to Hsp90 inhibition [165]. The first generation Hsp90 inhibitor 17-AAG was found to have synergistic anti-tumor activity when combined with cisplatin and/or gemcitabine in treating refractory and metastatic solid tumors, including urothelial carcinoma in patients with unresectable or metastatic disease [166]. However, the ability to escalate the dose of 17-AAG when used with chemotherapy drugs was limited, due to hematologic toxicities [166]. Second generation Hsp90 inhibitor ganetespib potentiated the cytotoxicity of doxorubicin and improved shrinkage of metastatic lesions when used in combination therapy in a study of triple negative breast cancer [167]. This could be due to induction of DNA damage, resulting in mitotic arrest and enhanced apoptosis in cancer cells. Thus, Hsp90 inhibitors have been investigated in combinatorial use for all current FDA-approved treatments for urothelial carcinoma and show promise, as they provide synergy with chemotherapy.
Hsp90 inhibitors are also radio-sensitizing compounds [101, 168]. They prevent DNA damage response after exposure to radiation to induce apoptosis in treated cells, providing synergism with radiation therapy [169, 170]. Radiation therapy is used adjunctively with multi-agent chemotherapy regimens in bladder-sparing protocols and in those with muscle-invasive disease [171] and, thus, Hsp90 inhibitors may provide added benefit. In acting synergistically with chemotherapy, Hsp90 inhibitors may provide an alternative to adjunctive radiotherapy and may reduce morbidities, as a standard radiation dose (54 – 64 Gray) promotes radiation cystitis [161].
Therefore, Hsp90 inhibitors can sensitize tumor cells to chemotherapy or radiotherapy [172, 173]. They can also provide anti-tumorigenic defense at low non-cytocidal doses when combined with chemoradiation [174]. CD44-expressing tumor-initiating cells in bladder cancer have been found to confer resistance to cisplatin compared to CD44- negative cells [174]. Combination of Hsp90 inhibition and chemoradiotherapy potentiated significant apoptosis of Grade III T24 bladder cancer cells compared to cells received single treatments. This elevated sensitization by 17-AAG may overcome cisplatin resistance [161, 174]. One mechanism of chemoradiosensitization by Hsp90 inhibitors is through reducing expression of the oncoproteins ErbB2 and NF-kB as they participate in resistance to chemoradiotherapy [175] and are Hsp90 clients [21].
Resistance to Hsp90 inhibitors
Hsp90 inhibition activates the heat shock response, which has been shown to limit efficacy of Hsp90 inhibitors in cancer therapy [176–179]. Of note, Hsp27, 40, and 70 are upregulated when Hsp90 is inhibited [180]. Hsp27 prevents protein aggregation of Hsp90 client proteins; Hsp40 acts as an ATPase modulator for both Hsp90 and Hsp70 [180, 181]. Blocking the compensatory heat shock response by Hsp27, 40, and 70 can reduce resistance to Hsp90 inhibitors [180]. Increased expression of heat shock proteins in urothelial carcinoma is associated with poor prognosis and treatment resistance [20, 182]. Multi-targeted Hsp inhibition increases apoptosis, induces G2/M cell cycle arrest, and inhibits autophagy in cancer cells [160, 179, 183]. Inhibition of mTOR inhibits upregulation of heat shock factor (HSF) in response to Hsp90 inhibition, and would also serve as a means to reduce resistance [184].
Further, it was noted that bladder cancer cell lines which express UGT1A (UDP-glucuronosyltransferase) enzymes were insensitive to resorcinol-based HSP90 inhibitors such as ganetespib and NVP-AUY922 (both UGT1A substrates), but sensitive to the ansamycin-based HSP90 inhibitors suggesting that intratumoral metabolism plays a role in drug resistance. Thus, UGT1A expression in bladder cancer may represent a predictive biomarker for what appears to be the most clinically advanced HSP90 inhibitors [162].
Hsp90 has a second drug-binding site in the C-domain [185] and coumarin derivative antibiotics target this site without activating HSF [186, 187]. This is unlike the effect linked to the Hsp90 N-domain inhibitors. Existing data strongly supports further medicinal chemistry optimization and preclinical evaluation of C-terminal Hsp90 inhibitors in urothelial carcinoma [188].
In summary, HSP90 inhibition provides novel opportunities for targeting common pathways evolved in urothelial carcinoma of the bladder, increasing sensitivities to known chemotherapeutic agents, and potentially optimizing sensitivity to radiation therapy.
CONCLUSION
Urothelial carcinoma is driven largely by loss of the tumor suppressor genes p53, PTEN, RB, and p16, requiring restoration of loss of function versus gain of function, which can more easily be targeted. There are also limited options available for patients with advanced disease who progress on or are ineligible for chemotherapy, resulting in poor outcomes. Our increased understanding of tumor biology and molecular pathways involved in urothelial carcinoma has allowed us to explore unique treatment targets. The inhibitors of the molecular chaperone Hsp90 have shown promise in clinical trials for other epithelial malignancies. Combination of Hsp90 inhibitors with chemotherapeutic agents provides better response rates in urothelial carcinoma in vitro. Further studies with new-generation Hsp90 inhibitors in the treatment of urothelial carcinomas will unravel the optimal combinational therapy, with a possible decrease in drug resistance. This may ultimately provide a long-term survival benefit and disease remission.
Acknowledgments
We would like to thank our collaborators Len Neckers, Jane Trepel, Weiwen Ying, Chris Prodromou, Laurence H. Pearl, Gabriella Choisis, Brian Blagg and Timothy Haystead for their scientific contributions. This work was supported by funds from SUNY Upstate Medical University.
REFERENCES
- 1.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics. CA Cancer Journal for Clinicians. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 2.Ploeg M, Aben KK, Kiemeney LA. The present and future burden of urinary bladder cancer in the world. World journal of urology. 2009;27:289–293. doi: 10.1007/s00345-009-0383-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fleshner NE, Herr HW, Stewart AK, Murphy GP, Mettlin C, Menck HR. The National Cancer Data Base report on bladder carcinoma. Cancer. 1996;78:1505–1513. doi: 10.1002/(sici)1097-0142(19961001)78:7<1505::aid-cncr19>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 4.Lucca I, Fajkovic H, Klatte T. Sex steroids and gender differences in nonmuscle invasive bladder cancer. Current opinion in urology. 2014 doi: 10.1097/MOU.0000000000000092. [DOI] [PubMed] [Google Scholar]
- 5.Chang C, Lee SO, Yeh S, Chang TM. Androgen receptor (AR) differential roles in hormone-related tumors including prostate, bladder, kidney, lung, breast and liver. Oncogene. 2014;33:3225–3234. doi: 10.1038/onc.2013.274. [DOI] [PubMed] [Google Scholar]
- 6.Svatek RS, Hollenbeck BK, Holmang S, Lee R, Kim SP, Stenzl A, Lotan Y. The economics of bladder cancer: costs and considerations of caring for this disease. European urology. 2014;66:253–262. doi: 10.1016/j.eururo.2014.01.006. [DOI] [PubMed] [Google Scholar]
- 7.Eble JS, G, Epstein JI, Sesterhemm IA. Cancer IAfRo; Chapter 2: tumors of the urinary system. Lyon: IARC Press; 2004. World Health Organization classification of tumors: tumors of the urinary system and male genital organs; pp. 86–154. [Google Scholar]
- 8.Van Hemelrijck M, Feller A, Garmo H, Valeri F, Korol D, Dehler S, Rohrmann S. Incidence of second malignancies for prostate cancer. PloS one. 2014;9 doi: 10.1371/journal.pone.0102596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaufman DS, Shipley WU, Feldman AS. Bladder cancer. Lancet. 2009;374:239–249. doi: 10.1016/S0140-6736(09)60491-8. [DOI] [PubMed] [Google Scholar]
- 10.Richter S, Sridhar SS. New directions for biologic targets in urothelial carcinoma. Molecular Cancer Therapeutics. 2012;11:1226–1235. doi: 10.1158/1535-7163.MCT-11-0756. [DOI] [PubMed] [Google Scholar]
- 11.van der Heijden AG, Witjes JA. Recurrence, Progression, and Follow-Up in Non-Muscle-Invasive Bladder Cancer. European Urology. 2009;8:556–562. [Google Scholar]
- 12.Crivelli JJ, Xylinas E, Kluth LA, Rieken M, Rink M, Shariat SF. Effect of smoking on outcomes of urothelial carcinoma: A systematic review of the literature. European urology. 2014;65:742–754. doi: 10.1016/j.eururo.2013.06.010. [DOI] [PubMed] [Google Scholar]
- 13.Zeegers MP, Tan FE, Dorant E, van Den Brandt PA. The impact of characteristics of cigarette smoking on urinary tract cancer risk: a meta-analysis of epidemiologic studies. Cancer. 2000;89:630–639. doi: 10.1002/1097-0142(20000801)89:3<630::aid-cncr19>3.3.co;2-h. [DOI] [PubMed] [Google Scholar]
- 14.Brait M, Munari E, LeBron C, Noordhuis MG, Begum S, Michailidi C, Gonzalez-Roibon N, Maldonado L, Sen T, Guerrero-Preston R, Cope L, Parrella P, Fazio VM, Ha PK, Netto GJ, Sidransky D, et al. Genome-wide methylation profiling and the PI3K-AKT pathway analysis associated with smoking in urothelial cell carcinoma. Cell Cycle. 2013;12:1058–1070. doi: 10.4161/cc.24050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Simonis K, Shariat SF, Rink M. Smoking and smoking cessation effects on oncological outcomes in nonmuscle invasive bladder cancer. Current opinion in urology. 2014;24:492–499. doi: 10.1097/MOU.0000000000000079. [DOI] [PubMed] [Google Scholar]
- 16.Johansson SL, Cohen SM. Epidemiology and etiology of bladder cancer. Seminars in surgical oncology. 1997;13:291–298. doi: 10.1002/(sici)1098-2388(199709/10)13:5<291::aid-ssu2>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 17.Knowles MA, Hurst CD. Molecular biology of bladder cancer: new insights into pathogenesis and clinical diversity. Nature reviews Cancer. 2015;15:25–41. doi: 10.1038/nrc3817. [DOI] [PubMed] [Google Scholar]
- 18.Netto GJ. Molecular biomarkers in urothelial carcinoma of the bladder: are we there yet? Nature reviews Urology. 2012;9:41–51. doi: 10.1038/nrurol.2011.193. [DOI] [PubMed] [Google Scholar]
- 19.Zhou H, Huang HY, Shapiro E, Lepor H, Huang WC, Mohammadi M, Mohr I, Tang MS, Huang C, Wu XR. Urothelial tumor initiation requires deregulation of multiple signaling pathways: implications in target-based therapies. Carcinogenesis. 2012;33:770–780. doi: 10.1093/carcin/bgs025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ischia J, So AI. The role of heat shock proteins in bladder cancer. Nature Reviews Urology. 2013;10:386–395. doi: 10.1038/nrurol.2013.108. [DOI] [PubMed] [Google Scholar]
- 21.Trepel J, Mollapour M, Giaccone G, Neckers L. Targeting the dynamic HSP90 complex in cancer. Nature Reviews Cancer. 2010;10:537–549. doi: 10.1038/nrc2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jhaveri K, Taldone T, Modi S, Chiosis G. Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. Biochimica et Biophysica Acta - Molecular Cell Research. 2012;1823:742–755. doi: 10.1016/j.bbamcr.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Neckers L, Workman P. Hsp90 molecular chaperone inhibitors: Are we there yet? Clinical Cancer Research. 2012;18:64–76. doi: 10.1158/1078-0432.CCR-11-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jhaveri K, Ochiana SO, Dunphy MPS, Gerecitano JF, Corben AD, Peter RI, Janjigian YY, Gomes-Dagama EM, Koren Iii J, Modi S, Chiosis G. Heat shock protein 90 inhibitors in the treatment of cancer: Current status and future directions. Expert Opinion on Investigational Drugs. 2014;23:611–628. doi: 10.1517/13543784.2014.902442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miyata Y, Nakamoto H, Neckers L. The therapeutic target Hsp90 and cancer hallmarks. Current Pharmaceutical Design. 2013;19:347–365. doi: 10.2174/138161213804143725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hong DS, Banerji U, Tavana B, George GC, Aaron J, Kurzrock R. Targeting the molecular chaperone heat shock protein 90 (HSP90): Lessons learned and future directions. Cancer Treatment Reviews. 2013;39:375–387. doi: 10.1016/j.ctrv.2012.10.001. [DOI] [PubMed] [Google Scholar]
- 27.Ali MMU, Mark Roe S, Vaughan CK, Meyer P, Panaretou B, Piper PW, Prodromou C, Pearl LH. Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature. 2006;440:1013–1017. doi: 10.1038/nature04716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Prodromou C, Pearl LH. Structure and functional relationships of Hsp90. Curr Cancer Drug Targets. 2003;3:301–323. doi: 10.2174/1568009033481877. [DOI] [PubMed] [Google Scholar]
- 29.Ratzke C, Mickler M, Hellenkamp B, Buchner J, Hugel T. Dynamics of heat shock protein 90 C-terminal dimerization is an important part of its conformational cycle. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:16101–16106. doi: 10.1073/pnas.1000916107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lavery LA, Partridge JR, Ramelot TA, Elnatan D, Kennedy MA, Agard DA. Structural Asymmetry in the Closed State of Mitochondrial Hsp90 (TRAP1) Supports a Two-Step ATP Hydrolysis Mechanism. Molecular Cell. 2014;53:330–343. doi: 10.1016/j.molcel.2013.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shiau AK, Harris SF, Southworth DR, Agard DA. Structural Analysis of Ecoli hsp90 Reveals Dramatic Nucleotide-Dependent Conformational Rearrangements. Cell. 2006;127:329–340. doi: 10.1016/j.cell.2006.09.027. [DOI] [PubMed] [Google Scholar]
- 32.Stebbins CE, Russo AA, Schneider C, Rosen N, Hartl FU, Pavletich NP. Crystal structure of an Hsp90-geldanamycin complex: Targeting of a protein chaperone by an antitumor agent. Cell. 1997;89:239–250. doi: 10.1016/s0092-8674(00)80203-2. [DOI] [PubMed] [Google Scholar]
- 33.Hainzl O, Lapina MC, Buchner J, Richter K. The charged linker region is an important regulator of Hsp90 function. Journal of Biological Chemistry. 2009;284:22559–22567. doi: 10.1074/jbc.M109.031658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hessling M, Richter K, Buchner J. Dissection of the ATP-induced conformational cycle of the molecular chaperone Hsp90. Nature Structural and Molecular Biology. 2009;16:287–293. doi: 10.1038/nsmb.1565. [DOI] [PubMed] [Google Scholar]
- 35.Mickler M, Hessling M, Ratzke C, Buchner J, Hugel T. The large conformational changes of Hsp90 are only weakly coupled to ATP hydrolysis. Nature Structural and Molecular Biology. 2009;16:281–286. doi: 10.1038/nsmb.1557. [DOI] [PubMed] [Google Scholar]
- 36.Retzlaff M, Stahl M, Eberl HC, Lagleder S, Beck J, Kessler H, Buchner J. Hsp90 is regulated by a switch point in the C-terminal domain. EMBO Reports. 2009;10:1147–1153. doi: 10.1038/embor.2009.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsutsumi S, Mollapour M, Graf C, Lee CT, Scroggins BT, Xu W, Haslerova L, Hessling M, Konstantinova AA, Trepel JB, Panaretou B, Buchner J, Mayer MP, Prodromou C, Neckers L. Hsp90 charged-linker truncation reverses the functional consequences of weakened hydrophobic contacts in the N domain. Nature Structural and Molecular Biology. 2009;16:1141–1147. doi: 10.1038/nsmb.1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Panaretou B, Prodromou C, Roe SM, O'Brien R, Ladbury JE, Piper PW, Pearl LH. ATP binding and hydrolysis are essential to the function of the Hsp90 molecular chaperone in vivo. EMBO Journal. 1998;17:4829–4836. doi: 10.1093/emboj/17.16.4829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Walton-Diaz A, Khan S, Bourboulia D, Trepel JB, Neckers L, Mollapour M. Contributions of co-chaperones and post-translational modifications towards Hsp90 drug sensitivity. Future Medicinal Chemistry. 2013;5:1059–1071. doi: 10.4155/fmc.13.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li J, Richter K, Buchner J. Mixed Hsp90-cochaperone complexes are important for the progression of the reaction cycle. Nature Structural and Molecular Biology. 2011;18:61–67. doi: 10.1038/nsmb.1965. [DOI] [PubMed] [Google Scholar]
- 41.Soroka J, Wandinger SK, Mäusbacher N, Schreiber T, Richter K, Daub H, Buchner J. Conformational Switching of the Molecular Chaperone Hsp90 via Regulated Phosphorylation. Molecular Cell. 2012;45:517–528. doi: 10.1016/j.molcel.2011.12.031. [DOI] [PubMed] [Google Scholar]
- 42.Mollapour M, Bourboulia D, Beebe K, Woodford MR, Polier S, Hoang A, Chelluri R, Li Y, Guo A, Lee MJ, Fotooh-Abadi E, Khan S, Prince T, Miyajima N, Yoshida S, Tsutsumi S, et al. Asymmetric Hsp90 N Domain SUMOylation Recruits Aha1 and ATP-Competitive Inhibitors. Molecular Cell. 2014;53:317–329. doi: 10.1016/j.molcel.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mollapour M, Tsutsumi S, Donnelly AC, Beebe K, Tokita MJ, Lee MJ, Lee S, Morra G, Bourboulia D, Scroggins BT, Colombo G, Blagg BS, Panaretou B, Stetler-Stevenson WG, Trepel JB, Piper PW, et al. Swe1Wee1-Dependent Tyrosine Phosphorylation of Hsp90 Regulates Distinct Facets of Chaperone Function. Molecular Cell. 2010;37:333–343. doi: 10.1016/j.molcel.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mollapour M, Tsutsumi S, Neckers L. Hsp0 phosphorylation, Wee1 and the cell cycle. Cell Cycle. 2010;9:2310–2316. doi: 10.4161/cc.9.12.12054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roe SM, Prodromou C, O'Brien R, Ladbury JE, Piper PW, Pearl LH. Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. Journal of Medicinal Chemistry. 1999;42:260–266. doi: 10.1021/jm980403y. [DOI] [PubMed] [Google Scholar]
- 46.Xu W, Mimnaugh EG, Kim JS, Trepel JB, Neckers LM. Hsp90, not grp94, regulates the intracellular trafficking and stability of nascent ErbB2. Cell Stress and Chaperones. 2002;7:91–96. doi: 10.1379/1466-1268(2002)007<0091:hngrti>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Caplan AJ, Mandal AK, Theodoraki MA. Molecular chaperones and protein kinase quality control. Trends in Cell Biology. 2007;17:87–92. doi: 10.1016/j.tcb.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 48.Garcia-Carbonero R, Carnero A, Paz-Ares L. Inhibition of HSP90 molecular chaperones: Moving into the clinic. The Lancet Oncology. 2013;14:e358–e369. doi: 10.1016/S1470-2045(13)70169-4. [DOI] [PubMed] [Google Scholar]
- 49.Yamaki H, Nakajima M, Shimotohno KW, Tanaka N. Molecular basis for the actions of Hsp90 inhibitors and cancer therapy. The Journal of antibiotics. 2011;64:635–644. doi: 10.1038/ja.2011.60. [DOI] [PubMed] [Google Scholar]
- 50.Park SJ, Kostic M, Dyson HJ. Dynamic Interaction of Hsp90 with Its Client Protein p53. Journal of molecular biology. 2011;411:158–173. doi: 10.1016/j.jmb.2011.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sidera K, Gaitanou M, Stellas D, Matsas R, Patsavoudi E. A critical role for HSP90 in cancer cell invasion involves interaction with the extracellular domain of HER-2. The Journal of biological chemistry. 2008;283:2031–2041. doi: 10.1074/jbc.M701803200. [DOI] [PubMed] [Google Scholar]
- 52.Gerbin CS, Landgraf R. Geldanamycin selectively targets the nascent form of ERBB3 for degradation. Cell stress & chaperones. 2010;15:529–544. doi: 10.1007/s12192-009-0166-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Prince T, Sun L, Matts RL. Cdk2: a genuine protein kinase client of Hsp90 and Cdc37. Biochemistry. 2005;44:15287–15295. doi: 10.1021/bi051423m. [DOI] [PubMed] [Google Scholar]
- 54.Stepanova L, Leng X, Parker SB, Harper JW. Mammalian p50Cdc37 is a protein kinase-targeting subunit of Hsp90 that binds and stabilizes Cdk4. Genes & development. 1996;10:1491–1502. doi: 10.1101/gad.10.12.1491. [DOI] [PubMed] [Google Scholar]
- 55.Mosser DD, Morimoto RI. Molecular chaperones and the stress of oncogenesis. Oncogene. 2004;23:2907–2918. doi: 10.1038/sj.onc.1207529. [DOI] [PubMed] [Google Scholar]
- 56.Basso AD, Solit DB, Chiosis G, Giri B, Tsichlis P, Rosen N. Akt forms an intracellular complex with heat shock protein 90 (Hsp90) and Cdc37 and is destabilized by inhibitors of Hsp90 function. The Journal of biological chemistry. 2002;277:39858–39866. doi: 10.1074/jbc.M206322200. [DOI] [PubMed] [Google Scholar]
- 57.Ahsan A, Ramanand SG, Whitehead C, Hiniker SM, Rehemtulla A, Pratt WB, Jolly S, Gouveia C, Truong K, Van Waes C, Ray D, Lawrence TS, Nyati MK. Wild-type EGFR is stabilized by direct interaction with HSP90 in cancer cells and tumors. Neoplasia. 2012;14:670–677. doi: 10.1593/neo.12986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Laederich MB, Degnin CR, Lunstrum GP, Holden P, Horton WA. Fibroblast growth factor receptor 3 (FGFR3) is a strong heat shock protein 90 (Hsp90) client: implications for therapeutic manipulation. The Journal of biological chemistry. 2011;286:19597–19604. doi: 10.1074/jbc.M110.206151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sawai A, Chandarlapaty S, Greulich H, Gonen M, Ye Q, Arteaga CL, Sellers W, Rosen N, Solit DB. Inhibition of Hsp90 down-regulates mutant epidermal growth factor receptor (EGFR) expression and sensitizes EGFR mutant tumors to paclitaxel. Cancer research. 2008;68:589–596. doi: 10.1158/0008-5472.CAN-07-1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Weinstein JN, Akbani R, Broom BM, Wang W, Verhaak RGW, McConkey D, Lerner S, Morgan M, Creighton CJ, Smith C, Cherniack AD, Kim J, Pedamallu CS, Noble MS, Al-Ahmadie HA, Reuter VE, et al. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507:315–322. doi: 10.1038/nature12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Prinsloo E, Kramer AH, Edkins AL, Blatch GL. STAT3 interacts directly with Hsp90. IUBMB life. 2012;64:266–273. doi: 10.1002/iub.607. [DOI] [PubMed] [Google Scholar]
- 62.Wang SA, Chuang JY, Yeh SH, Wang YT, Liu YW, Chang WC, Hung JJ. Heat shock protein 90 is important for Sp1 stability during mitosis. Journal of molecular biology. 2009;387:1106–1119. doi: 10.1016/j.jmb.2009.02.040. [DOI] [PubMed] [Google Scholar]
- 63.Abat D, Demirhan O, Inandiklioglu N, Tunc E, Erdogan S, Tastemir D, Uslu IN, Tansug Z. Genetic alterations of chromosomes, p53 and p16 genes in low- and high-grade bladder cancer. Oncology Letters. 2014;8:25–32. doi: 10.3892/ol.2014.2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.di Martino E, Tomlinson DC, Knowles MA. A Decade of FGF Receptor Research in Bladder Cancer: Past, Present, and Future Challenges. Advances in urology. 2012;2012:429213. doi: 10.1155/2012/429213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Williams SV, Hurst CD, Knowles MA. Oncogenic FGFR3 gene fusions in bladder cancer. Human molecular genetics. 2013;22:795–803. doi: 10.1093/hmg/dds486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hagn F, Lagleder S, Retzlaff M, Rohrberg J, Demmer O, Richter K, Buchner J, Kessler H. Structural analysis of the interaction between Hsp90 and the tumor suppressor protein p53. Nature structural & molecular biology. 2011;18:1086–1093. doi: 10.1038/nsmb.2114. [DOI] [PubMed] [Google Scholar]
- 67.Peng Y, Chen L, Li C, Lu W, Chen J. Inhibition of MDM2 by hsp90 contributes to mutant p53 stabilization. The Journal of biological chemistry. 2001;276:40583–40590. doi: 10.1074/jbc.M102817200. [DOI] [PubMed] [Google Scholar]
- 68.He K, Zheng X, Zhang L, Yu J. Hsp90 inhibitors promote p53-dependent apoptosis through PUMA and bax. Molecular Cancer Therapeutics. 2013;12:2559–2568. doi: 10.1158/1535-7163.MCT-13-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lin K, Rockliffe N, Johnson GG, Sherrington PD, Pettitt AR. Hsp90 inhibition has opposing effects on wild-type and mutant p53 and induces p21 expression and cytotoxicity irrespective of p53/ATM status in chronic lymphocytic leukaemia cells. Oncogene. 2008;27:2445–2455. doi: 10.1038/sj.onc.1210893. [DOI] [PubMed] [Google Scholar]
- 70.Xu HJ, Cairns P, Hu SX, Knowles MA, Benedict WF. Loss of RB protein expression in primary bladder cancer correlates with loss of heterozygosity at the RB locus and tumor progression. International journal of cancer Journal international du cancer. 1993;53:781–784. doi: 10.1002/ijc.2910530513. [DOI] [PubMed] [Google Scholar]
- 71.Fletcher O, Easton D, Anderson K, Gilham C, Jay M, Peto J. Lifetime risks of common cancers among retinoblastoma survivors. Journal of the National Cancer Institute. 2004;96:357–363. doi: 10.1093/jnci/djh058. [DOI] [PubMed] [Google Scholar]
- 72.Srethapakdi M, Liu F, Tavorath R, Rosen N. Inhibition of Hsp90 function by ansamycins causes retinoblastoma gene product-dependent G1 arrest. Cancer research. 2000;60:3940–3946. [PubMed] [Google Scholar]
- 73.Yeager T, Stadler W, Belair C, Puthenveettil J, Olopade O, Reznikoff C. Increased p16 levels correlate with pRb alterations in human urothelial cells. Cancer research. 1995;55:493–497. [PubMed] [Google Scholar]
- 74.Shariat SF, Tokunaga H, Zhou J, Kim J, Ayala GE, Benedict WF, Lerner SP. p53, p21, pRB, and p16 expression predict clinical outcome in cystectomy with bladder cancer. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2004;22:1014–1024. doi: 10.1200/JCO.2004.03.118. [DOI] [PubMed] [Google Scholar]
- 75.Steinestel J, Cronauer MV, Muller J, Al Ghazal A, Skowronek P, Arndt A, Kraft K, Schrader M, Schrader AJ, Steinestel K. Overexpression of p16(INK4a) in urothelial carcinoma in situ is a marker for MAPK-mediated epithelial-mesenchymal transition but is not related to human papillomavirus infection. PloS one. 2013;8:e65189. doi: 10.1371/journal.pone.0065189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Corral R, Lewinger JP, Van Den Berg D, Joshi AD, Yuan JM, Gago-Dominguez M, Cortessis VK, Pike MC, Conti DV, Thomas DC, Edlund CK, Gao YT, Xiang YB, Zhang W, Su YC, Stern MC. Comprehensive analyses of DNA repair pathways, smoking and bladder cancer risk in Los Angeles and Shanghai. International journal of cancer Journal international du cancer. 2014;135:335–347. doi: 10.1002/ijc.28693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yap KL, Kiyotani K, Tamura K, Antic T, Jang M, Montoya M, Campanile A, Yew PY, Ganshert C, Fujioka T, Steinberg GD, O'Donnell PH, Nakamura Y. Whole-exome sequencing of muscle-invasive bladder cancer identifies recurrent mutations of UNC5C and prognostic importance of DNA repair gene mutations on survival. Clinical cancer research: an official journal of the American Association for Cancer Research. 2014;20:6605–6617. doi: 10.1158/1078-0432.CCR-14-0257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nickerson ML, Dancik GM, Im KM, Edwards MG, Turan S, Brown J, Ruiz-Rodriguez C, Owens C, Costello JC, Guo G, Tsang SX, Li Y, Zhou Q, Cai Z, Moore LE, Lucia MS, et al. Concurrent alterations in TERT, KDM6A, and the BRCA pathway in bladder cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2014;20:4935–4948. doi: 10.1158/1078-0432.CCR-14-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sun CH, Chang YH, Pan CC. Activation of the PI3K/Akt/mTOR pathway correlates with tumour progression and reduced survival in patients with urothelial carcinoma of the urinary bladder. Histopathology. 2011;58:1054–1063. doi: 10.1111/j.1365-2559.2011.03856.x. [DOI] [PubMed] [Google Scholar]
- 80.Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nature reviews Molecular cell biology. 2014;15:178–196. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nature reviews Cancer. 2002;2:442–454. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 82.Gui Y, Guo G, Huang Y, Hu X, Tang A, Gao S, Wu R, Chen C, Li X, Zhou L, He M, Li Z, Sun X, Jia W, Chen J, Yang S, et al. Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nature genetics. 2011;43:875–878. doi: 10.1038/ng.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wilson BG, Roberts CW. SWI/SNF nucleosome remodellers and cancer. Nature reviews Cancer. 2011;11:481–492. doi: 10.1038/nrc3068. [DOI] [PubMed] [Google Scholar]
- 84.Roberts CW, Orkin SH. The SWI/SNF complex—chromatin and cancer. Nature reviews Cancer. 2004;4:133–142. doi: 10.1038/nrc1273. [DOI] [PubMed] [Google Scholar]
- 85.Lei AQ, Cheng L, Pan CX. Current treatment of metastatic bladder cancer and future directions. Expert Review of Anticancer Therapy. 2011;11:1851–1862. doi: 10.1586/era.11.181. [DOI] [PubMed] [Google Scholar]
- 86.Sternberg CN. Muscle invasive and metastatic bladder cancer. Annals of Oncology. 2006;17:x23–x30. doi: 10.1093/annonc/mdl231. [DOI] [PubMed] [Google Scholar]
- 87.Winquist E, Kirchner TS, Segal R, Chin J, Lukka H. Neoadjuvant chemotherapy for transitional cell carcinoma of the bladder: A systematic review and meta-analysis. Journal of Urology. 2004;171:561–569. doi: 10.1097/01.ju.0000090967.08622.33. [DOI] [PubMed] [Google Scholar]
- 88.Tanji N, Ozawa A, Miura N, Yanagihara Y, Sasaki T, Nishida T, Kikugawa T, Ikeda T, Ochi T, Shimamoto K, Aoki K, Yokoyama M. Long-term results of combined chemotherapy with gemcitabine and cisplatin for metastatic urothelial carcinomas. International Journal of Clinical Oncology. 2010;15:369–375. doi: 10.1007/s10147-010-0069-2. [DOI] [PubMed] [Google Scholar]
- 89.Saxman SB, Propert KJ, Einhorn LH, Crawford ED, Tannock I, Raghavan D, Loehrer PJ, Sr, Trump D. Long-term follow-up of a phase III intergroup study of cisplatin alone or in combination with methotrexate, vinblastine, and doxorubicin in patients with metastatic urothelial carcinoma: a cooperative group study. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 1997;15:2564–2569. doi: 10.1200/JCO.1997.15.7.2564. [DOI] [PubMed] [Google Scholar]
- 90.Tomaszewski JJ, Smaldone MC. Emerging intravesical therapies for management of nonmuscle invasive bladder cancer. Open Access Journal of Urology. 2010:67–84. [PMC free article] [PubMed] [Google Scholar]
- 91.Babjuk M, Burger M, Zigeuner R, Shariat SF, Van Rhijn BWG, Compérat E, Sylvester RJ, Kaasinen E, Böhle A, Palou Redorta J, Rouprêt M. EAU guidelines on non-muscle-invasive Urothelial carcinoma of the bladder: Update (2013) European urology. 2013;64:639–653. doi: 10.1016/j.eururo.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 92.Meyer JP, Persad R, Gillatt DA. Use of bacille Calmette-Guérin in superficial bladder cancer. Postgraduate Medical Journal. 2002;78:449–454. doi: 10.1136/pmj.78.922.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mungan NA, Witjes JA. Bacille Calmette-Guérin in superficial transitional cell carcinoma. British Journal of Urology. 1998;82:213–223. doi: 10.1046/j.1464-410x.1998.00720.x. [DOI] [PubMed] [Google Scholar]
- 94.Gontero P, Oderda M, Mehnert A, Gurioli A, Marson F, Lucca I, Rink M, Schmid M, Kluth LA, Pappagallo G, Sogni F, Sanguedolce F, Schiavina R, Martorana G, Shariat SF, Chun F. The impact of intravesical gemcitabine and 1/3 dose bacillus Calmette-Guérin instillation therapy on the quality of life in patients with nonmuscle invasive bladder cancer: Results of a prospective, randomized, phase II trial. Journal of Urology. 2013;190:857–862. doi: 10.1016/j.juro.2013.03.097. [DOI] [PubMed] [Google Scholar]
- 95.Liu XD, AC, Patel P, Viney RP, Foster MC, Porfiri E, James ND, Bryan RT. Cytokines as effectors and predictors of responses in the treatment of bladder cancer by bacillus Calmette-Guerin. Future Oncology. 2014;10:1443–1456. doi: 10.2217/fon.14.79. [DOI] [PubMed] [Google Scholar]
- 96.Prescott S, Jackson AM, Hawkyard SJ, Alexandroff AB, James K. Mechanisms of action of intravesical bacille Calmette-Guérin: Local immune mechanisms. Clinical Infectious Diseases. 2000;31:S91–S93. doi: 10.1086/314066. [DOI] [PubMed] [Google Scholar]
- 97.Lamm DL. Efficacy and safety of bacille Calmette-Guérin immunotherapy in superficial bladder cancer. Clinical Infectious Diseases. 2000;31:S86–S90. doi: 10.1086/314064. [DOI] [PubMed] [Google Scholar]
- 98.Siddik ZH. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene. 2003;22:7265–7279. doi: 10.1038/sj.onc.1206933. [DOI] [PubMed] [Google Scholar]
- 99.Jakse G, Hofstadter F. Mechanisms of action of intravesical treatment - Effect on the ABH surface antigens of urothelial cells. Cancer Chemotherapy and Pharmacology. 1983;11:S74–S78. [PubMed] [Google Scholar]
- 100.Plunkett W, Huang P, Xu YZ, Heinemann V, Grunewald R, Gandhi V. Gemcitabine: Metabolism, mechanisms of action, and self-potentiation. Seminars in Oncology. 1995;22:3–10. [PubMed] [Google Scholar]
- 101.James ND, Hussain SA, Hall E, Jenkins P, Tremlett J, Rawlings C, Crundwell M, Sizer B, Sreenivasan T, Hendron C, Lewis R, Waters R, Huddart RA. Radiotherapy with or without chemotherapy in muscle-invasive bladder cancer. New England Journal of Medicine. 2012;366:1477–1488. doi: 10.1056/NEJMoa1106106. [DOI] [PubMed] [Google Scholar]
- 102.Tsang RY, Al-Fayea T, Au HJ. Cisplatin overdose: Toxicities and management. Drug Safety. 2009;32:1109–1122. doi: 10.2165/11316640-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 103.Sternberg CN, Yagoda A, Scher HI, Watson RC, Ahmed T, Weiselberg LR, Geller N, Hollander PS, Herr HW, Sogani PC. Preliminary results of M-VAC (methotrexate, vinblastine, doxorubicin and cisplatin) for transitional cell carcinoma of the urothelium. Journal of Urology. 1985;133:403–407. doi: 10.1016/s0022-5347(17)48996-8. [DOI] [PubMed] [Google Scholar]
- 104.Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, Castedo M, Kroemer G. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31:1869–1883. doi: 10.1038/onc.2011.384. [DOI] [PubMed] [Google Scholar]
- 105.Hurst CD, Knowles MA. Molecular subtyping of invasive bladder cancer: time to divide and rule? Cancer cell. 2014;25:135–136. doi: 10.1016/j.ccr.2014.01.026. [DOI] [PubMed] [Google Scholar]
- 106.Galluzzi L, Vitale I, Michels J, Brenner C, Szabadkai G, Harel-Bellan A, Castedo M, Kroemer G. Systems biology of cisplatin resistance: past, present and future. Cell Death Dis. 2014;5:e1257. doi: 10.1038/cddis.2013.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Choi W, Porten S, Kim S, Willis D, Plimack ER, Hoffman-Censits J, Roth B, Cheng T, Tran M, Lee IL, Melquist J, Bondaruk J, Majewski T, Zhang S, Pretzsch S, Baggerly K, et al. Identification of distinct basal and luminal subtypes of muscle-invasive bladder cancer with different sensitivities to frontline chemotherapy. Cancer cell. 2014;25:152–165. doi: 10.1016/j.ccr.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mitazaki S, Kato N, Suto M, Hiraiwa K, Abe S. Interleukin-6 deficiency accelerates cisplatin-induced acute renal failure but not systemic injury. Toxicology. 2009;265:115–121. doi: 10.1016/j.tox.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 109.Nicholson S. Chemotherapy for bladder cancer in patients with impaired renal function. Nature Reviews Urology. 2012;9:52–57. doi: 10.1038/nrurol.2011.176. [DOI] [PubMed] [Google Scholar]
- 110.Redelman-Sidi G, Glickman MS, Bochner BH. The mechanism of action of BCG therapy for bladder cancer-A current perspective. Nature Reviews Urology. 2014;11:153–162. doi: 10.1038/nrurol.2014.15. [DOI] [PubMed] [Google Scholar]
- 111.Chevalier MF, Nardelli-Haefliger D, Domingos-Pereira S, Jichlinski P, Derré L. Immunotherapeutic strategies for bladder cancer. Human Vaccines and Immunotherapeutics. 2014;10:977–981. doi: 10.4161/hv.27621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Dhodapkar MV, Sznol M, Zhao B, Wang D, Carvajal RD, Keohan ML, Chuang E, Sanborn RE, Lutzky J, Powderly J, Kluger H, Tejwani S, Green J, Ramakrishna V, Crocker A, Vitale L, et al. Induction of antigen-specific immunity with a vaccine targeting NY-ESO-1 to the dendritic cell receptor DEC-205. Science Translational Medicine. 2014;6 doi: 10.1126/scitranslmed.3008068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Galsky MD, Hahn NM, Rosenberg J, Sonpavde G, Hutson T, Oh WK, Dreicer R, Vogelzang N, Sternberg CN, Bajorin DF, Bellmunt J. Treatment of patients with metastatic urothelial cancer “unfit” for Cisplatin-based chemotherapy. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2011;29:2432–2438. doi: 10.1200/JCO.2011.34.8433. [DOI] [PubMed] [Google Scholar]
- 114.Pruthi RS, Nielsen M, Heathcote S, Wallen EM, Rathmell WK, Godley P, Whang Y, Fielding J, Schultz H, Grigson G, Smith A, Kim W. A phase II trial of neoadjuvant erlotinib in patients with muscle-invasive bladder cancer undergoing radical cystectomy: clinical and pathological results. BJU international. 2010;106:349–354. doi: 10.1111/j.1464-410X.2009.09101.x. [DOI] [PubMed] [Google Scholar]
- 115.Gallagher DJ, Milowsky MI, Gerst SR, Ishill N, Riches J, Regazzi A, Boyle MG, Trout A, Flaherty AM, Bajorin DF. Phase II study of sunitinib in patients with metastatic urothelial cancer. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2010;28:1373–1379. doi: 10.1200/JCO.2009.25.3922. [DOI] [PubMed] [Google Scholar]
- 116.Grivas PD, Daignault S, Tagawa ST, Nanus DM, Stadler WM, Dreicer R, Kohli M, Petrylak DP, Vaughn DJ, Bylow KA, Wong SG, Sottnik JL, Keller ET, Al-Hawary M, Smith DC, Hussain M. Double-blind, randomized, phase 2 trial of maintenance sunitinib versus placebo after response to chemotherapy in patients with advanced urothelial carcinoma. Cancer. 2014;120:692–701. doi: 10.1002/cncr.28477. [DOI] [PubMed] [Google Scholar]
- 117.Sridhar SS, Winquist E, Eisen A, Hotte SJ, McWhirter E, Tannock IF, Mukherjee SD, Wang L, Blattler C, Wright JJ, Moore MJ. A phase II trial of sorafenib in first-line metastatic urothelial cancer: a study of the PMH Phase II Consortium. Investigational new drugs. 2011;29:1045–1049. doi: 10.1007/s10637-010-9408-4. [DOI] [PubMed] [Google Scholar]
- 118.Li Y, Yang X, Su LJ, Flaig TW. Pazopanib synergizes with docetaxel in the treatment of bladder cancer cells. Urology. 2011;78:233. doi: 10.1016/j.urology.2011.02.041. e237–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Pili R, Qin R, Flynn PJ, Picus J, Millward M, Ho WM, Pitot H, Tan W, Miles KM, Erlichman C, Vaishampayan U. A phase II safety and efficacy study of the vascular endothelial growth factor receptor tyrosine kinase inhibitor pazopanib in patients with metastatic urothelial cancer. Clinical genitourinary cancer. 2013;11:477–483. doi: 10.1016/j.clgc.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Nicolle G, Daher A, Maille P, Vermey M, Loric S, Bakkar A, Wallerand H, Vordos D, Vacherot F, de Medina SG, Abbou CC, Van der Kwast T, Thiery JP, Radvanyi F, Chopin DK. Gefitinib inhibits the growth and invasion of urothelial carcinoma cell lines in which Akt and MAPK activation is dependent on constitutive epidermal growth factor receptor activation. Clinical cancer research: an official journal of the American Association for Cancer Research. 2006;12:2937–2943. doi: 10.1158/1078-0432.CCR-05-2148. [DOI] [PubMed] [Google Scholar]
- 121.Philips GK, Halabi S, Sanford BL, Bajorin D, Small EJ, Cancer B. A phase II trial of cisplatin (C), gemcitabine (G) and gefitinib for advanced urothelial tract carcinoma: results of Cancer and Leukemia Group B (CALGB) 90102. Annals of oncology: official journal of the European Society for Medical Oncology / ESMO. 2009;20:1074–1079. doi: 10.1093/annonc/mdn749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lamont FR, Tomlinson DC, Cooper PA, Shnyder SD, Chester JD, Knowles MA. Small molecule FGF receptor inhibitors block FGFR-dependent urothelial carcinoma growth in vitro and in vivo. British journal of cancer. 2011;104:75–82. doi: 10.1038/sj.bjc.6606016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Iyer G, Milowsky MI. Fibroblast growth factor receptor-3 in urothelial tumorigenesis. Urologic oncology. 2013;31:303–311. doi: 10.1016/j.urolonc.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 124.Petrylak DP, Tangen CM, Van Veldhuizen PJ, Jr, Goodwin JW, Twardowski PW, Atkins JN, Kakhil SR, Lange MK, Mansukhani M, Crawford ED. Results of the Southwest Oncology Group phase II evaluation (study S0031) of ZD1839 for advanced transitional cell carcinoma of the urothelium. BJU international. 2010;105:317–321. doi: 10.1111/j.1464-410X.2009.08799.x. [DOI] [PubMed] [Google Scholar]
- 125.Hahn NM, Stadler WM, Zon RT, Waterhouse D, Picus J, Nattam S, Johnson CS, Perkins SM, Waddell MJ, Sweeney CJ, Hoosier Oncology G. Phase II trial of cisplatin, gemcitabine, and bevacizumab as first-line therapy for metastatic urothelial carcinoma: Hoosier Oncology Group GU 04–75. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2011;29:1525–1530. doi: 10.1200/JCO.2010.31.6067. [DOI] [PubMed] [Google Scholar]
- 126.Twardowski P, Stadler WM, Frankel P, Lara PN, Ruel C, Chatta G, Heath E, Quinn DI, Gandara DR. Phase II study of Aflibercept (VEGF-Trap) in patients with recurrent or metastatic urothelial cancer, a California Cancer Consortium Trial. Urology. 2010;76:923–926. doi: 10.1016/j.urology.2010.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Jimenez RE, Hussain M, Bianco FJ,, Jr, Vaishampayan U, Tabazcka P, Sakr WA, Pontes JE, Wood DP, Jr, Grignon DJ. Her-2/neu overexpression in muscle-invasive urothelial carcinoma of the bladder: prognostic significance and comparative analysis in primary and metastatic tumors. Clinical cancer research: an official journal of the American Association for Cancer Research. 2001;7:2440–2447. [PubMed] [Google Scholar]
- 128.Marin AP, Arranz EE, Sanchez AR, Aunon PZ, Baron MG. Role of anti-Her-2 therapy in bladder carcinoma. Journal of cancer research and clinical oncology. 2010;136:1915–1920. doi: 10.1007/s00432-010-0850-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Culine S, Sellam Z, Bouaita L, Assaf E, Delbaldo C, Verlinde-Carvalho M, Pouessel D. Combining paclitaxel and lapatinib as second-line treatment for patients with metastatic transitional cell carcinoma: a case series. Anticancer research. 2012;32:3949–3952. [PubMed] [Google Scholar]
- 130.Makhlin I, Zhang J, Long CJ, Devarajan K, Zhou Y, Klein-Szanto AJ, Huang M, Chernoff J, Boorjian SA. The mTOR pathway affects proliferation and chemosensitivity of urothelial carcinoma cells and is upregulated in a subset of human bladder cancers. BJU international. 2011;108:E84–90. doi: 10.1111/j.1464-410X.2010.09844.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Mansure JJ, Nassim R, Chevalier S, Rocha J, Scarlata E, Kassouf W. Inhibition of mammalian target of rapamycin as a therapeutic strategy in the management of bladder cancer. Cancer biology & therapy. 2009;8:2339–2347. doi: 10.4161/cbt.8.24.9987. [DOI] [PubMed] [Google Scholar]
- 132.Pinto-Leite R, Arantes-Rodrigues R, Palmeira C, Colaco B, Lopes C, Colaco A, Costa C, da Silva VM, Oliveira P, Santos L. Everolimus combined with cisplatin has a potential role in treatment of urothelial bladder cancer. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2013;67:116–121. doi: 10.1016/j.biopha.2012.11.007. [DOI] [PubMed] [Google Scholar]
- 133.Milowsky MI, Iyer G, Regazzi AM, Al-Ahmadie H, Gerst SR, Ostrovnaya I, Gellert LL, Kaplan R, Garcia-Grossman IR, Pendse D, Balar AV, Flaherty AM, Trout A, Solit DB, Bajorin DF. Phase II study of everolimus in metastatic urothelial cancer. BJU international. 2013;112:462–470. doi: 10.1111/j.1464-410X.2012.11720.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Seront E, Rottey S, Sautois B, Kerger J, D'Hondt LA, Verschaeve V, Canon JL, Dopchie C, Vandenbulcke JM, Whenham N, Goeminne JC, Clausse M, Verhoeven D, Glorieux P, Branders S, Dupont P, et al. Phase II study of everolimus in patients with locally advanced or metastatic transitional cell carcinoma of the urothelial tract: clinical activity, molecular response, and biomarkers. Annals of oncology: official journal of the European Society for Medical Oncology / ESMO. 2012;23:2663–2670. doi: 10.1093/annonc/mds057. [DOI] [PubMed] [Google Scholar]
- 135.Seront E, Pinto A, Bouzin C, Bertrand L, Machiels JP, Feron O. PTEN deficiency is associated with reduced sensitivity to mTOR inhibitor in human bladder cancer through the unhampered feedback loop driving PI3K/Akt activation. British journal of cancer. 2013;109:1586–1592. doi: 10.1038/bjc.2013.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hu XP, Ma LL, Wang Y, Yin H, Wang W, Yang XY, Zhang XD. Rapamycin instead of mycophenolate mofetil or azathioprine in treatment of post-renal transplantation urothelial carcinoma. Chinese medical journal. 2009;122:35–38. [PubMed] [Google Scholar]
- 137.Wang SM, Tai HC, Chueh SC, Chung SD, Lai MK. Sirolimus does not absolutely abolish the occurrence/recurrence of urothelial carcinoma in renal transplant recipients. Transplantation proceedings. 2008;40:2395–2396. doi: 10.1016/j.transproceed.2008.06.031. [DOI] [PubMed] [Google Scholar]
- 138.Gerullis H, Eimer C, Ecke TH, Georgas E, Freitas C, Kastenholz S, Arndt C, Heusch C, Otto T. A phase II trial of temsirolimus in second-line metastatic urothelial cancer. Medical oncology. 2012;29:2870–2876. doi: 10.1007/s12032-012-0216-x. [DOI] [PubMed] [Google Scholar]
- 139.Shah G, Zielonka J, Chen F, Zhang G, Cao Y, Kalyanaraman B, See W. H2O2 generation by bacillus Calmette-Guerin induces the cellular oxidative stress response required for bacillus Calmette-Guerin direct effects on urothelial carcinoma biology. The Journal of urology. 2014;192:1238–1248. doi: 10.1016/j.juro.2014.05.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Zhang G, Chen F, Cao Y, Amos JV, Shah G, See WA. HMGB1 release by urothelial carcinoma cells in response to Bacillus Calmette-Guerin functions as a paracrine factor to potentiate the direct cellular effects of Bacillus Calmette-Guerin. The Journal of urology. 2013;190:1076–1082. doi: 10.1016/j.juro.2013.01.050. [DOI] [PubMed] [Google Scholar]
- 141.Malmstrom PU, Loskog AS, Lindqvist CA, Mangsbo SM, Fransson M, Wanders A, Gardmark T, Totterman TH. AdCD40L immunogene therapy for bladder carcinoma—the first phase I/IIa trial. Clinical cancer research: an official journal of the American Association for Cancer Research. 2010;16:3279–3287. doi: 10.1158/1078-0432.CCR-10-0385. [DOI] [PubMed] [Google Scholar]
- 142.Dyrskjot L, Zieger K, Kissow Lildal T, Reinert T, Gruselle O, Coche T, Borre M, Orntoft TF. Expression of MAGE-A3, NY-ESO-1, LAGE-1 and PRAME in urothelial carcinoma. British journal of cancer. 2012;107:116–122. doi: 10.1038/bjc.2012.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Sharma P, Bajorin DF, Jungbluth AA, Herr H, Old LJ, Gnjatic S. Immune responses detected in urothelial carcinoma patients after vaccination with NY-ESO-1 protein plus BCG and GM-CSF. Journal of immunotherapy. 2008;31:849–857. doi: 10.1097/CJI.0b013e3181891574. [DOI] [PubMed] [Google Scholar]
- 144.Jana BR, Galsky MD, Hahn NM, Milowsky MI, Sonpavde G. Novel molecular targets for the therapy of urothelial carcinoma. Expert opinion on therapeutic targets. 2012;16:499–513. doi: 10.1517/14728222.2012.677441. [DOI] [PubMed] [Google Scholar]
- 145.Sharma P, Shen Y, Wen S, Yamada S, Jungbluth AA, Gnjatic S, Bajorin DF, Reuter VE, Herr H, Old LJ, Sato E. CD8 tumor-infiltrating lymphocytes are predictive of survival in muscle-invasive urothelial carcinoma. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:3967–3972. doi: 10.1073/pnas.0611618104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Baselga J, Swain SM. Novel anticancer targets: revisiting ERBB2 and discovering ERBB3. Nature reviews Cancer. 2009;9:463–475. doi: 10.1038/nrc2656. [DOI] [PubMed] [Google Scholar]
- 147.Solit DB, Chiosis G. Development and application of Hsp90 inhibitors. Drug discovery today. 2008;13:38–43. doi: 10.1016/j.drudis.2007.10.007. [DOI] [PubMed] [Google Scholar]
- 148.Solit DB, Osman I, Polsky D, Panageas KS, Daud A, Goydos JS, Teitcher J, Wolchok JD, Germino FJ, Krown SE, Coit D, Rosen N, Chapman PB. Phase II trial of 17-allylamino-17-demethoxygeldanamycin in patients with metastatic melanoma. Clinical cancer research: an official journal of the American Association for Cancer Research. 2008;14:8302–8307. doi: 10.1158/1078-0432.CCR-08-1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Socinski MA, Goldman J, El-Hariry I, Koczywas M, Vukovic V, Horn L, Paschold E, Salgia R, West H, Sequist LV, Bonomi P, Brahmer J, Chen LC, Sandler A, Belani CP, Webb T, et al. A multicenter phase II study of ganetespib monotherapy in patients with genotypically defined advanced non-small cell lung cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2013;19:3068–3077. doi: 10.1158/1078-0432.CCR-12-3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Koga F, Tsutsumi S, Neckers LM. Low dose geldanamycin inhibits hepatocyte growth factor and hypoxia-stimulated invasion of cancer cells. Cell Cycle. 2007;6:1393–1402. doi: 10.4161/cc.6.11.4296. [DOI] [PubMed] [Google Scholar]
- 151.Cheng HL, Trink B, Tzai TS, Liu HS, Chan SH, Ho CL, Sidransky D, Chow NH. Overexpression of c-met as a prognostic indicator for transitional cell carcinoma of the urinary bladder: A comparison with p53 nuclear accumulation. Journal of Clinical Oncology. 2002;20:1544–1550. doi: 10.1200/JCO.2002.20.6.1544. [DOI] [PubMed] [Google Scholar]
- 152.Cheng HL, Liu HS, Lin YJ, Chen HHW, Hsu PY, Chang TY, Ho CL, Tzai TS, Chow NH. Co-expression of RON and MET is a prognostic indicator for patients with transitional-cell carcinoma of the bladder. British journal of cancer. 2005;92:1906–1914. doi: 10.1038/sj.bjc.6602593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Theodoropoulos VE, Lazaris AC, Sofras F, Gerzelis I, Tsoukala V, Ghikonti I, Manikas K, Kastriotis I. Hypoxia-inducible factor 1α expression correlates with angiogenesis and unfavorable prognosis in bladder cancer. European urology. 2004;46:200–208. doi: 10.1016/j.eururo.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 154.Chai CY, Chen WT, Hung WC, Kang WY, Huang YC, Su YC, Yang CH. Hypoxia-inducible factor-1α expression correlates with focal macrophage infiltration, angiogenesis and unfavourable prognosis in urothelial carcinoma. Journal of Clinical Pathology. 2008;61:658–664. doi: 10.1136/jcp.2007.050666. [DOI] [PubMed] [Google Scholar]
- 155.Lebret T, Watson RWG, Molinié V, O'Neill A, Gabriel C, Fitzpatrick JM, Botto H. Heat shock proteins HSP27, HSP60, HSP70, and HSP90: Expression in bladder carcinoma. Cancer. 2003;98:970–977. doi: 10.1002/cncr.11594. [DOI] [PubMed] [Google Scholar]
- 156.Yu HJ, Chang YH, Pan CC. Prognostic significance of heat shock proteins in urothelial carcinoma of the urinary bladder. Histopathology. 2013;62:788–798. doi: 10.1111/his.12087. [DOI] [PubMed] [Google Scholar]
- 157.Karkoulis PK, Stravopodis DJ, Margaritis LH, Voutsinas GE. 17-Allylamino-17-demethoxygeldanamycin induces downregulation of critical Hsp90 protein clients and results in cell cycle arrest and apoptosis of human urinary bladder cancer cells. BMC Cancer. 2010;10:481. doi: 10.1186/1471-2407-10-481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Sato S, Fujita N, Tsuruo T. Modulation of Akt kinase activity by binding to Hsp90. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:10832–10837. doi: 10.1073/pnas.170276797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Barksdale KA, Bijur GN. The basal flux of Akt in the mitochondria is mediated by heat shock protein 90. Journal of Neurochemistry. 2009;108:1289–1299. doi: 10.1111/j.1471-4159.2009.05878.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Ma L, Sato F, Sato R, Matsubara T, Hirai K, Yamasaki M, Shin T, Shimada T, Nomura T, Mori K, Sumino Y, Mimata H. Dual targeting of heat shock proteins 90 and 70 promotes cell death and enhances the anticancer effect of chemotherapeutic agents in bladder cancer. Oncol Rep. 2014;31:2482–2492. doi: 10.3892/or.2014.3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Yoshida S, Koga F, Tatokoro M, Kawakami S, Fujii Y, Kumagai J, Neckers L, Kihara K. Low-dose Hsp90 inhibitors tumor-selectively sensitize bladder cancer cells to chemoradiotherapy. Cell Cycle. 2011;10:4291–4299. doi: 10.4161/cc.10.24.18616. [DOI] [PubMed] [Google Scholar]
- 162.Acquaviva J, He S, Zhang C, Jimenez JP, Nagai M, Sang J, Sequeira M, Smith DL, Ogawa LS, Inoue T, Tatsuta N, Knowles MA, Bates RC, Proia DA. FGFR3 translocations in bladder cancer: differential sensitivity to HSP90 inhibition based on drug metabolism. Molecular cancer research: MCR. 2014;12:1042–1054. doi: 10.1158/1541-7786.MCR-14-0004. [DOI] [PubMed] [Google Scholar]
- 163.McDowell CL, Bryan Sutton R, Obermann WMJ. Expression of Hsp90 chaperome proteins in human tumor tissue. International Journal of Biological Macromolecules. 2009;45:310–314. doi: 10.1016/j.ijbiomac.2009.06.012. [DOI] [PubMed] [Google Scholar]
- 164.Roh JL, Kim EH, Park HB, Park JY. The Hsp90 inhibitor 17-(allylamino)-17-demethoxygeldanamycin increases cisplatin antitumor activity by inducing p53-mediated apoptosis in head and neck cancer. Cell Death Dis. 2013;4:e956. doi: 10.1038/cddis.2013.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.McCollum AK, Lukasiewicz KB, TenEyck CJ, Lingle WL, Toft DO, Erlichman C. Cisplatin abrogates the geldanamycin-induced heat shock response. Molecular Cancer Therapeutics. 2008;7:3256–3264. doi: 10.1158/1535-7163.MCT-08-0157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Hubbard J, Erlichman C, Toft DO, Qin R, Stensgard BA, Felten S, Ten Eyck C, Batzel G, Ivy SP, Haluska P. Phase I study of 17-allylamino-17 demethoxygeldanamycin, gemcitabine and/or cisplatin in patients with refractory solid tumors. Investigational new drugs. 2011;29:473–480. doi: 10.1007/s10637-009-9381-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Proia DA, Bates RC. Ganetespib and HSP90: Translating Preclinical Hypotheses into Clinical Promise. Cancer research. 2014;74:1294–1300. doi: 10.1158/0008-5472.CAN-13-3263. [DOI] [PubMed] [Google Scholar]
- 168.Machida H, Matsumoto Y, Shirai M, Kubota N. Geldanamycin, an inhibitor of Hsp90, sensitizes human tumour cells to radiation. International Journal of Radiation Biology. 2003;79:973–980. doi: 10.1080/09553000310001626135. [DOI] [PubMed] [Google Scholar]
- 169.Dote H, Burgan WE, Camphausen K, Tofilon PJ. Inhibition of Hsp90 compromises the DNA damage response to radiation. Cancer research. 2006;66:9211–9220. doi: 10.1158/0008-5472.CAN-06-2181. [DOI] [PubMed] [Google Scholar]
- 170.Koll TT, Feis SS, Wright MH, Teniola MM, Richardson MM, Robles AI, Bradsher J, Capala J, Varticovski L. HSP90 inhibitor, DMAG, synergizes with radiation of lung cancer cells by interfering with base excision and ATM-mediated DNA repair. Molecular Cancer Therapeutics. 2008;7:1985–1992. doi: 10.1158/1535-7163.MCT-07-2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Pos F, Remeijer P. Adaptive Management of Bladder Cancer Radiotherapy. Seminars in Radiation Oncology. 2010;20:116–120. doi: 10.1016/j.semradonc.2009.11.005. [DOI] [PubMed] [Google Scholar]
- 172.Kabakov AE, Kudryavtsev VA, Gabai VL. Hsp90 inhibitors as promising agents for radiotherapy. Journal of Molecular Medicine. 2010;88:241–247. doi: 10.1007/s00109-009-0562-0. [DOI] [PubMed] [Google Scholar]
- 173.Kabakov AE, Kudryavtsev VA, Makarova YM. Inhibitors of heat shock protein 90 activity: A novel class of tumor radiosensitizers. Biophysics. 2011;56:339–345. [PubMed] [Google Scholar]
- 174.Tatokoro M, Koga F, Yoshida S, Kawakami S, Fujii Y, Neckers L, Kihara K. Potential role of Hsp90 inhibitors in overcoming cisplatin resistance of bladder cancer-initiating cells. International journal of cancer Journal international du cancer. 2012;131:987–996. doi: 10.1002/ijc.26475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Koga F, Yoshida S, Tatokoro M, Kawakami S, Fujii Y, Kumagai J, Neckers L, Kihara K. ErbB2 and NFκB overexpression as predictors of chemoradiation resistance and putative targets to overcome resistance in Muscle-Invasive bladder cancer. PloS one. 2011;6 doi: 10.1371/journal.pone.0027616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Guo F, Rocha K, Bali P, Pranpat M, Fiskus W, Boyapalle S, Kumaraswamy S, Balasis M, Greedy B, Armitage ESM, Lawrence N, Bhalla K. Abrogation of heat shock protein 70 induction as a strategy to increase antileukemia activity of heat shock protein 90 inhibitor 17-allylamino-demethoxy geldanamycin. Cancer research. 2005;65:10536–10544. doi: 10.1158/0008-5472.CAN-05-1799. [DOI] [PubMed] [Google Scholar]
- 177.Zaarur N, Gabai VL, Porco JA, Jr, Calderwood S, Sherman MY. Targeting heat shock response to sensitize cancer cells to proteasome and Hsp90 inhibitors. Cancer research. 2006;66:1783–1791. doi: 10.1158/0008-5472.CAN-05-3692. [DOI] [PubMed] [Google Scholar]
- 178.Sato A, Asano T, Ito K, Asano T. 17-Allylamino-17-demethoxygeldanamycin and ritonavir inhibit renal cancer growth by inhibiting the expression of heat shock factor-1. International Journal of Oncology. 2012;41:46–52. doi: 10.3892/ijo.2012.1419. [DOI] [PubMed] [Google Scholar]
- 179.Budina-Kolomets A, Balaburski GM, Bondar A, Beeharry N, Yen T, Murphy ME. Comparison of the activity of three different HSP70 inhibitors on apoptosis, cell cycle arrest, autophagy inhibition, and HSP90 inhibition. Cancer Biology and Therapy. 2014;15:194–199. doi: 10.4161/cbt.26720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.McConnell JR, McAlpine SR. Heat shock proteins 27, 40, and 70 as combinational and dual therapeutic cancer targets. Bioorganic and Medicinal Chemistry Letters. 2013;23:1923–1928. doi: 10.1016/j.bmcl.2013.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Horne BE, Li T, Genevaux P, Georgopoulos C, Landry SJ. The Hsp40 J-domain stimulates Hsp70 when tethered by the client to the ATPase domain. Journal of Biological Chemistry. 2010;285:21679–21688. doi: 10.1074/jbc.M110.113118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Lebret T, Watson RWG, Fitzpatrick JM. Heat shock proteins: Their role in urological tumors. Journal of Urology. 2003;169:338–346. doi: 10.1016/S0022-5347(05)64123-7. [DOI] [PubMed] [Google Scholar]
- 183.Powers MV, Clarke PA, Workman P. Death by chaperone: HSP90, HSP70 or both? Cell Cycle. 2009;8:518–526. doi: 10.4161/cc.8.4.7583. [DOI] [PubMed] [Google Scholar]
- 184.Acquaviva J, He S, Sang J, Smith DL, Sequeira M, Zhang C, Bates RC, Proia DA. mTOR inhibition potentiates HSP90 inhibitor activity via cessation of HSP synthesis. Molecular Cancer Research. 2014;12:703–713. doi: 10.1158/1541-7786.MCR-13-0605. [DOI] [PubMed] [Google Scholar]
- 185.Marcu MG, Chadli A, Bouhouche I, Catelli M, Neckers LM. The heat shock protein 90 antagonist novobiocin interacts with a previously unrecognized ATP-binding domain in the carboxyl terminus of the chaperone. The Journal of biological chemistry. 2000;275:37181–37186. doi: 10.1074/jbc.M003701200. [DOI] [PubMed] [Google Scholar]
- 186.Donnelly A, Blagg BSJ. Novobiocin and additional inhibitors of the Hsp90 C-terminal nucleotide-binding pocket. Current Medicinal Chemistry. 2008;15:2702–2717. doi: 10.2174/092986708786242895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Matts RL, Dixit A, Peterson LB, Sun L, Voruganti S, Kalyanaraman P, Hartson SD, Verkhivker GM, Blagg BSJ. Elucidation of the Hsp90 C-terminal inhibitor binding site. ACS Chemical Biology. 2011;6:800–807. doi: 10.1021/cb200052x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Gavenonis J, Jonas NE, Kritzer JA. Potential C-terminal-domain inhibitors of heat shock protein 90 derived from a C-terminal peptide helix. Bioorganic and Medicinal Chemistry. 2014 doi: 10.1016/j.bmc.2014.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Yun SJ, Kim WJ. Role of the epithelial-mesenchymal transition in bladder cancer: from prognosis to therapeutic target. Korean journal of urology. 2013;54:645–650. doi: 10.4111/kju.2013.54.10.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Wang DS, Rieger-Christ K, Latini JM, Moinzadeh A, Stoffel J, Pezza JA, Saini K, Libertino JA, Summerhayes IC. Molecular analysis of PTEN and MXI1 in primary bladder carcinoma. International journal of cancer Journal international du cancer. 2000;88:620–625. doi: 10.1002/1097-0215(20001115)88:4<620::aid-ijc16>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 191.Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 192.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 193.Dreesen O, Brivanlou AH. Signaling pathways in cancer and embryonic stem cells. Stem cell reviews. 2007;3:7–17. doi: 10.1007/s12015-007-0004-8. [DOI] [PubMed] [Google Scholar]
- 194.Xia Y, Padre RC, De Mendoza TH, Bottero V, Tergaonkar VB, Verma IM. Phosphorylation of p53 by IkappaB kinase 2 promotes its degradation by beta-TrCP. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:2629–2634. doi: 10.1073/pnas.0812256106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Schulz WA. Understanding urothelial carcinoma through cancer pathways. International journal of cancer Journal international du cancer. 2006;119:1513–1518. doi: 10.1002/ijc.21852. [DOI] [PubMed] [Google Scholar]