

Abstract
Firefly luciferases, which emit visible light in a highly specific ATP-dependent process, have been adapted for a variety of applications including gene reporter assays, whole-cell biosensor measurements and in vivo imaging. We have previously reported the ~2-fold enhanced activity and 1.4-greater bioluminescence quantum yield properties of a chimeric enzyme that contains the N-domain of Photinus pyralis luciferase joined to the C-domain of Luciola italica luciferase. Subsequently, we identified 5 amino acid changes based on L. italica that are the main determinants of the improved bioluminescence properties. Further engineering to enhance thermal and pH stability produced a novel luciferase called PLG2. We present here a systematic comparison of the spectral and physical properties of the new protein with P. pyralis luciferase and demonstrate the potential of PLG2 for use in assays based on the detection of femtomol levels of ATP. Additionally, we compared the performance of a mammalian codon-optimized version of the cDNA for PLG2 with the luc2 gene in HEK293T cells. Using an optimized low-cost assay system, PLG2 activity can be monitored in mammalian cell lysates and in living cells offering an improved alternative to Promega’s luc2 for reporter and imaging applications.
Keywords: Bioluminescence, firefly, luciferase, ATP, reporter, chimera, luc2, imaging
Introduction
Bioluminescence-based methods are firmly established research tools in cell biology, molecular biology and analytical chemistry [1, 2]. As a result of the pioneering work of McElroy, White and Seliger [3–5], beetle luciferases, particularly those found in fireflies, are used in numerous applications that include probes for detecting bacteria and fungi [6–9], whole-cell based biosensors [10–15] and gene expression assays [16–19]. Firefly luciferases have also been expressed in live mammals to visualize disease states [20–23], tumor growth and metastasis [24–27], cell trafficking [28, 29] and genetic regulation [30]. Many of these applications can be applied in high-throughput screening format in support of drug development [31–34]. One of the distinctive properties of the beetle luciferases is the highly specific dependence of the Luc1-catalyzed light emission process on ATP (Eqs (1–3)). The Photinus pyralis Luc
| (1) |
| (2) |
| (3) |
bioluminescence system is highly efficient with a quantum yield of 41.0 ± 7.4% [35]. Bioluminescence is especially advantageous when high sensitivity is required because, unlike fluorescence systems that need an external light source, there is virtually no non-specific light production induced in the cellular environment. It has been estimated that attomole levels of luciferase can be quantified using photomultiplier tubes or charge-coupled devices [36], making Luc an attractive candidate for bioanalytical uses that demand high sensitivity.
Recently, we reported [37] the ~2-fold enhanced specific activity and 1.4-fold greater bioluminescence quantum yield properties of PpyLit,2 a chimeric enzyme that contains the N-domain of P. pyralis luciferase joined to the C-domain of L. italica luciferase. In effect, the L. italica luciferase C-domain sequence introduced 27 changes, 23 amino acid substitutions and 4 deletions, into the full 550 amino acid P. pyralis sequence. Subsequently, we extended our investigation of PpyLit by identifying 5 amino acids that are the main determinants of the improved bioluminescence properties. Starting with PLG12, which contains the 5 amino acid changes, and building on our experience producing thermostable P. pyralis luciferase variants with altered emission kinetics [38, 39], we sought to develop a novel enzyme that would provide improved performance in all applications in which ATP levels are measured and the commercial luc2 gene is used in a lysed cell format.
We describe here a new luciferase called PLG2 and present a systematic comparison of the spectral and physical properties of the new protein with PpyWT and demonstrate the potential of PLG2 for use in assays based on the detection of low levels of ATP. Additionally, we compared the performance of a codon-optimized version of the cDNA for PLG2 with the luc2 gene in HEK293T cells. Using optimized low-cost assay systems, PLG2 activity can be monitored in mammalian cell lysates and living cells offering an improved alternative to Promega’s luc2 for reporter and imaging applications.
Materials and methods
Materials
The following materials were obtained from the sources indicated: ATP (disodium salt hydrate), MgSO4, and Triton X-100 from Sigma-Aldrich (St. Louis, MO); dithiothreitol (DTT) from Acros; beetle luciferin (LH2), Bright-Glo™ Luciferase Assay system, Passive Lysis Buffer (PLB), and pF4Ag containing the luc2 gene (non-commercial expression vector driven by a CMV promoter) were from Promega (Madison, WI). A human codon optimized version of the PLG2 gene2 was constructed by modifying the portion corresponding to the N-terminus as previously described [39] and synthesis of the C-domain by GenScript according to the OptimumGene™ algorithm. The codon optimized PLG2 gene was prepared by ligation of the 2 optimized domains and cloned into the Sgf1/Pme1sites of the Promega pF4Ag vector.
General methods
Concentrations of purified proteins were determined with the Bio-Rad Protein Assay system using bovine serum albumin as the standard. Sequences of luciferase genes in the pGEX-6P-2 and pF4Ag vectors were verified by DNA sequencing at the W. M. Keck Biotechnology Laboratory (Yale University, New Haven, CT). Methods for mutagenesis, protein expression and purification, bioluminescence spectra and specific activity were previously reported [37].
ATP detection using PLG2
Assays were performed in triplicate in white 96-well plates (Greiner Bio-One North America, Inc., Monroe, NC) using a Luminoskan Ascent microplate luminometer (Thermo, Waltham, MA) equipped with an automatic injector cleaned with 70% aqueous ethanol prior to use. The Tricine buffer, plasticware and pipet tips were autoclaved prior to use. A stock solution of ATP (10 mM) in 50 mM Tricine, pH 8 was prepared and serially diluted into the same buffer producing solutions of ATP with final concentrations of 0.1 μM to 0.1 pM. Aliquots (0.05 ml) of ATP dilutions corresponding to 1 × 10−11 to 1 × 10−17 mol of ATP were assayed by automatic injection of 0.05 ml of a single reagent solution (PLG2 assay mix) in 50 mM Tricine, pH 8 containing 2.5–3.3 μM PLG2 enzyme, 0.8 mM LH2 and 10 mM MgSO4. Light emission was recorded for 1 s after a 1 s delay following injection of the reagent. Using this methodology a representative ATP standard curve (Fig. 2) was constructed using the mean values from 3 independent trials. To detect ATP levels below ~1 × 10−14 mol, it was necessary to reduce the bioluminescence background by storing the PLG2 assay mix overnight at 4°C prior to use.
Fig. 2.
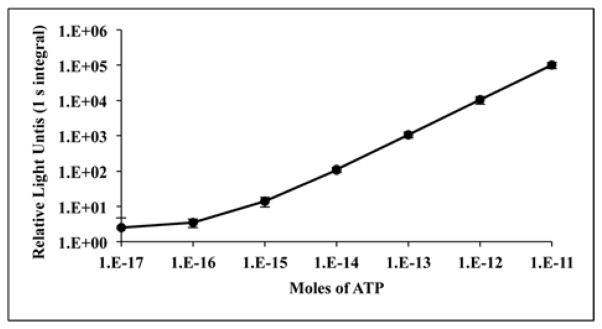
ATP standard curve generated from the bioluminescent signal of the PLG2 enzyme. The mean relative light units (RLUs) for 1 s integrals (from 3 independent experiments) performed as described in detail (Materials and methods) are plotted as a function of ATP levels and the standard deviations are indicated by error bars. The correlation coefficient (r) for the portion of the curve from 1E−16–1E−11 moles of ATP is 0.999 and the associated significance is p<0.001.
For analysis of ATP content in HEK293T cells, aliquots of cells were washed twice with phosphate buffered saline (PBS) and then a cell suspension in PBS was counted using a TC10 automated cell counter (Bio-Rad). Triplicate cell suspensions (200,000 cells in 0.1 ml of PBS) were serially diluted in PBS and lysed by 5 cycles of freezing at −80 °C and thawing at 30 °C. The lysed cell suspensions were diluted 10-fold in 50 mM Tricine buffer, pH 8 and aliquots (0.05 ml) were assayed in triplicate as described above. An ATP standard curve was generated as described by preparing the ATP solutions in 50 mM Tricine buffer, pH 8 containing 10% PBS.
For analysis of ATP content in Escherichia coli, a glycerol stock of BL21(DE3)pLysS cells was used to inoculate duplicate 5 ml cultures in Luria Bertani media (LB) that were incubated at 37 °C overnight with shaking. Aliquots of each culture were diluted 100-fold in fresh LB and grown for 2 h at 37 °C with shaking and 1 ml portions were removed and centrifuged at 4000 × g to pellet the cells. The supernatants were discarded to remove any extracellular ATP and the cells were suspended in 0.5 ml of fresh LB. The cell suspensions were serially diluted in LB and a portion of each dilution was plated on LB agar to determine the number of colony forming units (cfu). The remaining volume (0.45 ml) was centrifuged at 4000 × g to pellet cells, which were suspended in 10 mM Tris-Cl pH 8 containing 1 mM EDTA and lysed by heating for 5 min at 98 °C in a thermal cycler. After incubation on ice for 5 min, aliquots (0.05 ml) of the cell suspensions were assayed by automatic injection of 0.05 ml PLG2 assay mix as described above.
Transfection of HEK293T mammalian cells with luc2 and PLG2
HEK293T cells were plated in 24 well plates at a density of 250,000 cells/well in Dulbecco’s Modified Eagle’s Medium (Corning) + 10% fetal bovine serum. The same day cells were transfected with 200 ng of each DNA construct (transfection mix included 500 ng total DNA including empty vector and 1.5 μl Lipofectamine 2000 in 100 μl OptiMEM) and grown overnight at 37 °C with CO2. The next day, growth media was removed, the cells (~600,000) were washed with PBS and lysed in 100 μl PLB (Promega). Each construct was transfected in triplicate and three independent trials were performed. Transfection efficiencies were estimated by co-transfecting with eCFP and measuring the percentage of fluorescent cells.
Model reporter assays in mammalian cell lysates
Luminescence measurements of soluble cell lysates from HEK293T mammalian cells (prepared in triplicate) were performed in triplicate assays in white 96-well plates (Greiner Bio-One North America, Inc., Monroe, NC) using a Luminoskan Ascent microplate luminometer (Thermo Scientific, Waltham, MA). Each well contained 10 μl of soluble cell lysate diluted (1:10) in PBS containing 1% Triton. Assays were initiated by automatic injection of 0.1 ml of a solution containing LH2 (0.3 mM), ATP (0.7 mM), MgSO4 (2.5 mM), and DTT (15 mM) in 50 mM Tricine, pH 7.6 or alternatively with Bright-Glo™ assay reagent. After a 1 min delay, bioluminescence signals were integrated for 10 s and recorded. To estimate the concentration of expressed active enzymes, total protein was estimated from bioluminescence activity using standard curves generated from purified luciferases and corrected for minor differences in transfection efficiencies.
Model reporter assays in living mammalian cells
HEK293T cells were transfected in triplicate with pF4Ag plasmids containing the luc2 or PLG2 gene. The next day, media was removed and cells were washed with PBS, removed with trypsin, counted and diluted to 10,000 cells/ml in media. Cells (100 μl) were pipetted in triplicate into opaque 96-well tissue culture treated plates (Corning Life Sciences, Tewksbury, MA) and grown overnight at 37 °C with CO2. The next day, 10 mM D-LH2 was added to CO2 independent media (Life Technologies, Carlsbad, CA) containing 10% FBS at 37 °C and 100 μl of the mixture was added to each well. The plate was immediately placed in the Luminoskan luminometer heated to 37 °C and bioluminescence was monitored.
Statistical analysis
Curve fitting and data analysis were performed using the regression function of the Analysis ToolPack of Microsoft Excel version 14.0. To detect statistical correlations, Pearson’s correlation coefficient (r) was calculated between the luciferase activity and moles of ATP or cell number. The associated probability (p) was determined using the Student t-distribution function of Excel. Each result is represented as the mean ± standard deviation.
Results and Discussion
PLG2 luciferase
A new firefly luciferase called PLG22 has been developed by incorporating the 5 amino acid changes of PLG12 that provided ~90% of the activity enhancement of chimeric PpyLit [37] (Table 1) and similar partial protection against red-shifting of bioluminescence at low pH. Additional improvements in stability to heat and pH were realized by the inclusion of 5 mutations previously employed to fortify Luc variants [38]. Also, extended glow emission kinetics were engineered into PLG2 (Table 1 and Fig. 1) mainly by the inclusion of the Ile351Val change, which had been shown to restore activity in a red light-emitting variant of PpyWT [39]. The specific activities of the purified luciferases shown in Table 1 were determined from bioluminescence activity measurements using saturating levels of LH2 and Mg-ATP. The Km value of PLG2 for LH2 was ~3-fold higher than the value of PpyWT, while the Mg-ATP values were very similar. Replacing P. pyralis luciferase with PLG2 in in vitro assays should provide greater sensitivity based on the ~1.4-fold brighter flash intensity and longer lived signal that provides an ~4-fold greater integrated signal. The specific activity properties of PLG2 are remarkable because thermostable Lucs like UltraGlo typically suffer from considerably decreased bioluminescence activity. Additionally, the lack of emission color change at low pH (~6.5) and the much greater thermostability of PLG2 compared to PpyWT (24 h versus 20 min at 37 °C) are significant because stability to acidic pH and temperature are important for reliable in vitro assays, especially for reporter experiments with lysed mammalian cells using photomultiplier tube-based detection devices.
Table 1.
Properties of luciferasesa
| Enzyme | Relative Specific Activityb | Km (μM) | Bioluminescence λmaxc (nm) | Thermal Inactivationd (h) at 37°C | |||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Flash Height | Integrated | LH2 | Mg-ATP | pH 7.8 | pH 6.5 | ||
| PpyWT | 100 ± 4 | 100 ± 2 | 15 ± 2 | 86 ± 7 | 560 (70) | 609, 560 sh (92) | 0.3 ± 0.03 |
| PpyLit | 180 ± 10 | 200 ± 12 | 25 ± 2 | 53 ± 6 | 560 (69) | 561, 610 sh (90) | 0.07 ± 0.02 |
| PLG2 | 135 ± 5 | 425 ± 2 | 52 ± 6 | 79 ± 5 | 559 (67) | 561 (71) | > 24 ± 5 |
The data for PpyWT and PpyLit and the methods used to produce the values for PLG2 are described in reference [37].
Specific activities were obtained at pH 7.8 with LH2 (400 μM) and Mg-ATP (2 mM) and are expressed relative to PpyWT values, which are defined as 100. Integrated activities are based on total light emission in 15 min.
Bioluminescence emission maxima were obtained from two trials and the standard deviation is ± 1 nm. The bandwidth at half-maximal intensity is shown in parentheses and sh indicates a shoulder.
Time for the maximum initial activity to decay to 50% at 37 °C. All activity values were obtained from at least three trials and are reported as means ± standard deviation.
Fig. 1.

Bioluminescence time course of reactions of PLG2 (solid line) and PpyWT (dashed line). Reactions (0.4 m1) in 25 mM glycylglycine buffer, pH 7.8 containing 1 μg of protein and 400 μM LH2 were initiated by the injection of 0.12 ml of a solution of 9 mM Mg-ATP in the same buffer.
ATP analysis
We explored the possibility that PLG2 could offer improved sensitivity at lower cost in applications in which wild-type P. pyralis luciferase is used in: (1) assays based on ATP detection; (2) specialized applications for detecting very low levels of ATP, e.g. with the Promega Enlighten System [40]; and (3) genetic reporter applications in mammalian cells in which luc2 is employed in a lysis format [41, 42]. First, we developed PLG2 assay mix, a minimal single reagent cocktail comprised of 50 mM Tricine buffer, pH 8.0 containing 2.5–3.3 μM PLG2, 0.8 mM LH2, and 10 mM MgSO4. The reagent was typically kept at ambient temperature without loss of activity over a 6–8 h period. PLG2 assay mix could be stored at 4 °C for 48 h without measureable loss of activity and after 2 weeks 85% activity remained. Additionally, the reagent can be flash frozen in liquid N2, lyophilized, and reconstituted with minimal (< 10%) activity loss that can be compensated for by adding additional enzyme. An ATP assay method was established with a plate reading luminometer using a single injector. Reactions were initiated by the addition of either 50 or 100 μL of PLG2 assay mix into equal volumes of ATP standards and light emission was monitored for 1 s after a 1 s delay. With this simple rapid method, a standard curve based on 3 independent trials was generated (Fig. 2) that demonstrated the linearity of the method over 6 orders of magnitude with a detection limit of ~1 × 10−16 mol of ATP. This result is consistent with the femtomol detection capabilities of the Promega ENLITEN® kit and represents detection of ATP at a level limited by instrument noise. The sensitivity improvement that is possible with PLG2 equates to using less enzyme to reach femtomol detection. However, the enhanced stability of PLG2 should provide performance advantages compared to currently available standard luciferases. While results in individual labs may vary, especially depending on the type of light measuring device that is used, the standard curve presented here illustrates that femtomol detection of ATP is straightforward and feasible with a stable, inexpensive single assay reagent and a basic detection device.
To demonstrate the potential of PLG2 for practical application, we applied our methodology to assessing ATP levels in mammalian and bacterial cells. The cell number and relative light units (RLUs) of bioluminescence were proportional over 5 orders of magnitude, from 1 to 10,000 mammalian cells (Fig. 3A, r=0.999, p<0.001). Using a standard curve similar to the one in Fig. 2 with the buffer described in Materials and methods generated by assaying ATP dilutions with the PLG2 assay mix, we determined there was also a strong correlation between cell number and moles of ATP (Fig. 3B) and that there were 5.5 ± 0.4 fmol of ATP in a single HEK293T cell. This value represents the mean and standard deviation from triplicate experiments. This is in good agreement with the reported value of 2 – 6 fmol ATP per viable mammalian cell [44]. With E. coli, the relationships between cell number and both bioluminescence (Fig. 3A) and ATP content (Fig. 3B) also correlated strongly over 5 orders of magnitude (r=0.999, p<0.001). We were able to detect as little as 50 cells (Fig. 3A) and determined the mean level of ATP/cell was 1.6 ± 0.3 amol, which is consistent with the literature values of 0.2 – 2 amol per bacterial cell [45, 46].
Fig. 3.
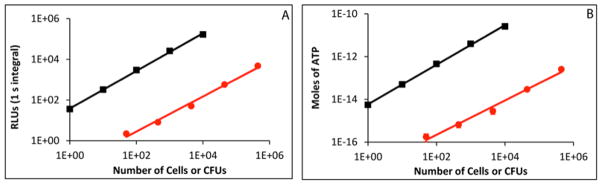
The correlations between the number of HEK293T cells (black lines) or E. coli colony forming units (CFUs, red lines) and (A) relative activity units (RLUs) and (B) the corresponding moles of ATP. The RLUs were determined from bioluminescence assays using the PLG2 assay mix, as described in the Materials and methods and standard curves (similar to Fig. 2) were used to calculate the corresponding moles of ATP. Assays were performed in triplicate. Symbols represent mean values, and lines represent the linear data fit. Error bars indicate the standard deviations among replicates. Pearson’s correlation coefficient (r) and the associated significance (p) values were: r=0.999, p<0.001 for the number of HEK293T cells vs. RLUs or moles of ATP; r=0.999, p<0.001 for the number of E. coli CFUs vs. RLUs or moles of ATP.
Model reporter assays in mammalian cells
We also investigated whether the PLG2 enzyme might provide greater sensitivity than the widely used luc2, a P. pyralis luciferase containing 2 neutral substitutions, in cell lysis-based reporter applications using codon optimized genes for PLG2 and luc2 expressed in mammalian cells. The proteins were expressed at 37 °C in HEK293T cells, and lysates of equivalent cell count were prepared and assayed in 96-well format using a Luminoskan instrument. For convenience, the cells were lysed with PLB. Bioluminescence activity was monitored with a single solution minimal assay reagent (0.3 mM LH2, 0.7 mM ATP, 2.5 mM MgSO4 and 15 mM DTT in 50 mM Tricine, pH 7.6) and the relative intensities of the signals produced by PLG2 and luc2 were 9.2 ± 0.6 × 104 and 2.1 ± 0.2 × 104 RLU, respectively (Fig. 4) based on 10 s integrals obtained after a min delay. The 4.4-fold higher activity realized with PLG2 will likely enable greater sensitivity in reporter applications. We estimated that the basis of the greater signal produced by PLG2 is ~2.2-fold higher protein expression yield and ~2-fold greater specific activity. The daily reproducibility of the method was ~10% and we found that the lysates could be frozen at −80 °C and assayed 1 day to 3 months later without significant activity loss.
Fig. 4.
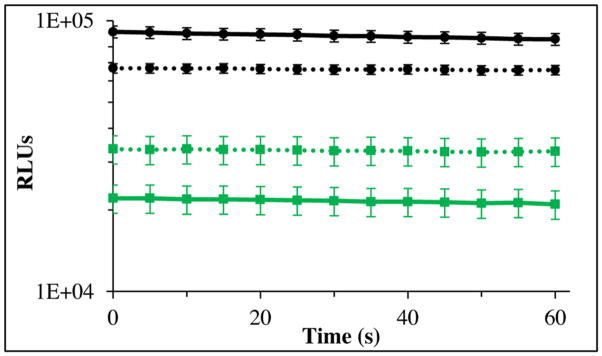
Relative bioluminescence activities of soluble cell lysates from equivalent numbers of HEK293T cells expressing mammalian codon-optimized PLG2 (black) and luc2 (green) at 37 °C. Lysates were diluted 1:10 in PBS containing 2% Triton and 10 μl was assayed using a Luminoskan Ascent microplate luminometer. Bioluminescence was initiated by the injection of: (solid lines) 0.1 ml of a solution containing 0.7 mM ATP, 0.3 mM LH2, 15 mM DTT, and 2.5 mM MgSO4 in 50 mM Tricine buffer (pH 7.6); or (dashed lines) 0.1 ml Bright-Glo™ assay reagent. Data collection began 1 min after the injection of substrates.
Alternatively, reporter assays with the novel codon-optimized P. Pyralis variant were conducted conveniently by substituting the Promega Bright-Glo™ assay reagent for our minimal assay reagent, as evidenced in a second HEK293T cell lysate trial (Fig. 4). Similar to their performance in the first model assay, PLG2 and luc2 emitted strong signals with negligible decay rates over the short duration of the analyses (Fig. 4). However, while the luc2 activity increased ~1.6 times, the PLG2 activity decreased 1.4-fold in Bright-Glo™. For assays with equipment lacking an automatic injector creating a situation where long term stability of the signals is important, Bright-Glo™ is the preferred reagent because the signals decay much more slowly over a 30 min period.
We also preliminarily evaluated the potential of PLG2 for use in live cell imaging applications in which bioluminescence is dependent on the cellular availability of ATP and exogenously added LH2. As demonstrated by the results in Fig. 5 that were produced by introducing LH2 into live HEK293T cells, the bioluminescence emission signal produced by PLG2 was ~2.5 to 3.5 times greater than that obtained from luc2. The difference in the relative bioluminescence in the cell lysate and live cell experiments was not due to transfection, efficiencies, protein yields or affinities for ATP. Possibly because of limited permeability of the cells to LH2, the difference is related to the greater affinity of luc2 for LH2 based on its 3.5-fold lower Km value for this substrate.
Fig. 5.
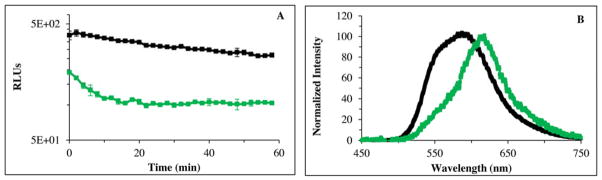
Relative bioluminescence activity and emission spectra of living HEK293T cells expressing mammalian codon-optimized PLG2 (black) and luc2 (green). (A) Bioluminescence was initiated by the addition of 0.1 ml of 10 mM LH2 to wells containing equivalent numbers of cells in 0.1 ml of media and then monitored (triplicate assays) at 37 °C. (B) Normalized bioluminescence spectra of living cells at 37 °C. The spectra were collected 1 min after the addition of 10 mM LH2 (0.25 ml) to a cuvette containing 0.25 ml of cells at 37 °C.
The results presented here are positive indicators that PLG2 is an excellent candidate for in vitro ATP analysis and bioanalytical applications in mammalian cells. As luc2 has proven suitable for dual-reporter and cell-sensor assays, we expect that PLG2 should perform as well or better because this enzyme produces brighter signals, has similar bioluminescence color that is resistant to low pH shifting, is much more stable at 37 °C, and appears to express in higher yield in mammalian cells. Substituting PLG2 for luc2 should provide great benefit to established applications and new ones such as a promising method for high-throughput drug screening based on the measurement of intracellular ATP levels with a luciferase containing an appended protein transduction domain [47].
Acknowledgments
This project was supported by the Air Force Office of Scientific Research under Grant FA9550-14-1-0100 (BRB), the National Science Foundation under Grants MCB 0842831 and MCB 1410390 (BRB), the National Institutes of Health under Grant R15CA125731-02 (MJG), FARB Project RFBO128121 (AR), and the Hans & Ella McCollum’21 Vahlteich Endowment (BRB). We thank Lance Encell and Mary Hall for stimulating and helpful discussions.
Footnotes
Abbreviations used: LB, Luria Bertani media; LH2, D-firefly luciferin; Luc, firefly luciferase; PBS, phosphate-buffered saline containing 40 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 1.8 mM KH2PO4 (pH 7.3); PLB, Passive Lysis Buffer; PLG1, PpyWT containing Ile457Val/Ala482Gly/His489Lys/Ala503Asn/Lys543Gln; PLG2, PLG1 luciferase containing Thr214Ala/Ala215Leu/Ile232Ala/Phe295Leu/Ile351Val/Glu354Lys; PLG2 assay mix, a single reagent solution in 50 mM Tricine, pH 8 containing 2.5–3.3 μM PLG2 enzyme, 0.8 mM LH2, and 10 mM MgSO4; PpyLit, chimeric protein comprised of PpyWT residues 1–439 and L. italica luciferase residues 442–548; PpyWT, recombinant P. pyralis luciferase containing the additional N-terminal peptide GlyProLeuGlySer; and RLU, relative light units.
The construction, expression, purification and characterization of PpyLit, PLG1 and PLG2 are described in US provisional patent No. PCT/US2013/042178 where they are referred to as CCC1, CCC2 and CCC4, respectively.
References
- 1.Roda A. Chemiluminescence and bioluminescence : past, present and future. Royal Society of Chemistry; Cambridge, UK: 2011. [Google Scholar]
- 2.Widder EA, Falls B. Review of Bioluminescence for Engineers and Scientists in Biophotonics. Ieee Journal of Selected Topics in Quantum Electronics. 2014;20 [Google Scholar]
- 3.McElroy WD. The Energy Source for Bioluminescence in an Isolated System. Proc Natl Acad Sci U S A. 1947;33:342–345. doi: 10.1073/pnas.33.11.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.White EH, Rapaport E, Seliger HH, Hopkins TA. Chemi- and bioluminescence of firefly luciferin. Efficient chemical production of electronically excited states. Bioorg Chem. 1971;1:92–122. [Google Scholar]
- 5.DeLuca M. Firefly luciferase. Adv Enzymol. 1976;44:37–68. doi: 10.1002/9780470122891.ch2. [DOI] [PubMed] [Google Scholar]
- 6.Squirrel DJ, Price RL, Murphy MJ. Rapid and specific detection of bacteria using bioluminescence. Anal Chim Acta. 2002;457:109–114. [Google Scholar]
- 7.Foucault ML, Thomas L, Goussard S, Branchini BR, Grillot-Courvalin C. In Vivo Bioluminescence Imaging for the Study of Intestinal Colonization by Escherichia coli in Mice. Appl Environ Microbiol. 2010;76:264–274. doi: 10.1128/AEM.01686-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vecchiarelli A, d’Enfert C. Shedding natural light on fungal infections. Virulence. 2012;3:15–17. doi: 10.4161/viru.3.1.19247. [DOI] [PubMed] [Google Scholar]
- 9.Shinozaki Y, Sato J, Igarashi T, Suzuki S, Nishimoto K, Harada Y. Evaluation of an Improved Bioluminescence Assay for the Detection of Bacteria in Soy Milk. Biocontrol Science. 2013;18:1–7. doi: 10.4265/bio.18.1. [DOI] [PubMed] [Google Scholar]
- 10.Hakkila K, Maksimow M, Karp M, Virta M. Reporter genes lucFF, luxCDABE, gfp, and dsred have different characteristics in whole-cell bacterial sensors. Anal Biochem. 2002;301:235–242. doi: 10.1006/abio.2001.5517. [DOI] [PubMed] [Google Scholar]
- 11.Gu M, Mitchell R, Kim B. Whole-cell-based biosensors for environmental biomonitoring and application. Adv Biochem Eng Biotechnol. 2004;87:269–305. doi: 10.1007/b13533. [DOI] [PubMed] [Google Scholar]
- 12.Urban A, Eckermann S, Fast B, Metzger S, Gehling M, Ziegelbauer K, Rubsamen-Waigmann H, Freiberg C. Novel whole-cell antibiotic biosensors for compound discovery. Appl Environ Microbiol. 2007;73:6436–6443. doi: 10.1128/AEM.00586-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Binkowski BF, Butler BL, Stecha PF, Eggers CT, Otto P, Zimmerman K, Vidugiris G, Wood MG, Encell LP, Fan F, Wood KV. A Luminescent Biosensor with Increased Dynamic Range for Intracellular cAMP. Acs Chemical Biology. 2011;6:1193–1197. doi: 10.1021/cb200248h. [DOI] [PubMed] [Google Scholar]
- 14.Roda A, Cevenini L, Michelini E, Branchini BR. A portable bioluminescence engineered cell-based biosensor for on-site applications. Biosens Bioelectron. 2011;26:3647–3653. doi: 10.1016/j.bios.2011.02.022. [DOI] [PubMed] [Google Scholar]
- 15.Shi JW, Zhang H, Fang LR, Xi YQ, Zhou YR, Luo R, Wang D, Xiao SB, Chen HC. A novel firefly luciferase biosensor enhances the detection of apoptosis induced by ESAT-6 family proteins of Mycobacterium tuberculosis. Biochem Biophys Res Commun. 2014;452:1046–1053. doi: 10.1016/j.bbrc.2014.09.047. [DOI] [PubMed] [Google Scholar]
- 16.Gould SJ, Subramani S. Firefly luciferase as a tool in molecular and cell biology. Anal Biochem. 1988;175:5–13. doi: 10.1016/0003-2697(88)90353-3. [DOI] [PubMed] [Google Scholar]
- 17.Leclerc GM, Boockfor FR, Faught WJ, Frawley LS. Development of a destabilized firefly luciferase enzyme for measurement of gene expression. Biotechniques. 2000;29:590–591. 594–596. doi: 10.2144/00293rr02. 598 passim. [DOI] [PubMed] [Google Scholar]
- 18.Paddison PJ, Caudy AA, Hannon GJ. Stable suppression of gene expression by RNAi in mammalian cells. Proc Natl Acad Sci USA. 2002;99:1443–1448. doi: 10.1073/pnas.032652399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vannini A, Agriesti F, Mosca F, Roncarati D, Scarlato V, Danielli A. A Convenient and Robust In Vivo Reporter System To Monitor Gene Expression in the Human Pathogen Helicobacter pylori. Appl Environ Microbiol. 2012;78:6524–6533. doi: 10.1128/AEM.01252-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lewis MD, Francisco AF, Taylor MC, Burrell-Saward H, McLatchie AP, Miles MA, Kelly JM. Bioluminescence imaging of chronic Trypanosoma cruzi infections reveals tissue-specific parasite dynamics and heart disease in the absence of locally persistent infection. Cellular Microbiology. 2014;16:1285–1300. doi: 10.1111/cmi.12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Taylor MC, Kelly JM. Optimizing bioluminescence imaging to study protozoan parasite infections. Trends in Parasitology. 2014;30:161–162. doi: 10.1016/j.pt.2014.01.006. [DOI] [PubMed] [Google Scholar]
- 22.Kuo MS, Auriau J, Pierre-Eugene C, Issad T. Development of a Human Breast-Cancer Derived Cell Line Stably Expressing a Bioluminescence Resonance Energy Transfer (BRET)-Based Phosphatidyl Inositol-3 Phosphate (PIP3) Biosensor. Plos One. 2014;9 doi: 10.1371/journal.pone.0092737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cevenini L, Camarda G, Michelini E, Siciliano G, Calabretta MM, Bona R, Kumar TRS, Cara A, Branchini BR, Fidock DA, Roda A, Alano P. Multicolor Bioluminescence Boosts Malaria Research: Quantitative Dual-Color Assay and Single-Cell Imaging in Plasmodium falciparum Parasites. Anal Chem. 2014;86:8814–8821. doi: 10.1021/ac502098w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rehemtulla A, Stegman LD, Cardozo SJ, Gupta S, Hall DE, Contag CH, Ross BD. Rapid and quantitative assessment of cancer treatment response using in vivo bioluminescence imaging. Neoplasia. 2000;2:491–495. doi: 10.1038/sj.neo.7900121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brutkiewicz S, Mendonca M, Stantz K, Comerford K, Bigsby R, Hutchins G, Goebl M, Harrington M. The expression level of luciferase within tumour cells can alter tumour growth upon in vivo bioluminescence imaging. Luminescence. 2007;22:221–228. doi: 10.1002/bio.953. [DOI] [PubMed] [Google Scholar]
- 26.Koo V, Hamilton PW, Williamson K. Non-invasive in vivo imaging in small animal research. Cellular Oncology. 2006;28:127–139. doi: 10.1155/2006/245619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Soling A, Rainov NG. Bioluminescence imaging in vivo - application to cancer research. Expert Opinion on Biological Therapy. 2003;3:1163–1172. doi: 10.1517/14712598.3.7.1163. [DOI] [PubMed] [Google Scholar]
- 28.Hardy J, Edinger M, Bachmann MH, Negrin RS, Fathman CG, Contag CH. Bioluminescence imaging of lymphocyte trafficking in vivo. Exp Hematol. 2001;29:1353–1360. doi: 10.1016/s0301-472x(01)00756-1. [DOI] [PubMed] [Google Scholar]
- 29.Luker KE, Luker GD. Bioluminescence imaging of reporter mice for studies of infection and inflammation. Antiviral Research. 2010;86:93–100. doi: 10.1016/j.antiviral.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Contag CH, Bachmann MH. Advances in vivo bioluminescence imaging of gene expression. Annual Review of Biomedical Engineering. 2002;4:235–260. doi: 10.1146/annurev.bioeng.4.111901.093336. [DOI] [PubMed] [Google Scholar]
- 31.Roda A, Guardigli M, Michelini E, Mirasoli M. Bioluminescence in analytical chemistry and in vivo imaging. Trac-Trends in Analytical Chemistry. 2009;28:307–322. [Google Scholar]
- 32.Che PL, Cui L, Kutsch O, Cui LW, Li QJ. Validating a Firefly Luciferase-Based High-Throughput Screening Assay for Antimalarial Drug Discovery. Assay and Drug Development Technologies. 2012;10:61–68. doi: 10.1089/adt.2011.0378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thorne N, Shen M, Lea WA, Simeonov A, Lovell S, Auld DS, Inglese J. Firefly Luciferase in Chemical Biology: A Compendium of Inhibitors, Mechanistic Evaluation of Chemotypes, and Suggested Use As a Reporter. Chemistry & Biology. 2012;19:1060–1072. doi: 10.1016/j.chembiol.2012.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yoon SM, Namkung W, Lee J. A Comparison of Ypet and Firefly Luciferase as Reporter Proteins for High-Throughput Screening. Biosci, Biotechnol, Biochem. 2013;77:2328–2330. doi: 10.1271/bbb.130537. [DOI] [PubMed] [Google Scholar]
- 35.Ando Y, Niwa K, Yamada N, Enomot T, Irie T, Kubota H, Ohmiya Y, Akiyama H. Firefly bioluminescence quantum yield and colour change by pH-sensitive green emission. Nat Photonics. 2008;2:44–47. [Google Scholar]
- 36.Lundin A. Optimized assay of firefly luciferase with stable light emission. In: Szalay A, Kricka LJ, Stanley PE, editors. Bioluminescence and Chemiluminescence: Status Report. John Wiley and Sons; Chichester: 1993. pp. 291–295. [Google Scholar]
- 37.Branchini BR, Southworth TL, Fontaine DM, Davis AL, Behney CE, Murtiashaw MH. A Photinus pyralis and Luciola italica Chimeric Firefly Luciferase Produces Enhanced Bioluminescence. Biochemistry. 2014;53:6287–6289. doi: 10.1021/bi501202u. [DOI] [PubMed] [Google Scholar]
- 38.Branchini BR, Ablamsky DM, Murtiashaw MH, Uzasci L, Fraga H, Southworth TL. Thermostable red and green light-producing firefly luciferase mutants for bioluminescent reporter applications. Anal Biochem. 2007;361:253–262. doi: 10.1016/j.ab.2006.10.043. [DOI] [PubMed] [Google Scholar]
- 39.Branchini BR, Ablamsky DM, Davis AL, Southworth TL, Butler B, Fan F, Jathoul AP, Pule MA. Red-emitting luciferases for bioluminescence reporter and imaging applications. Anal Biochem. 2010;396:290–297. doi: 10.1016/j.ab.2009.09.009. [DOI] [PubMed] [Google Scholar]
- 40.P. Corporation, editor. ENLIGHTEN ATP Assay System Bioluminescence Detection Kit for ATP Measurement. Madison, Wi USA: 2009. [Google Scholar]
- 41.Yang HC, Shen L, Siliciano RF, Pomerantz JL. Isolation of a cellular factor that can reactivate latent HIV-1 without T cell activation. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:6321–6326. doi: 10.1073/pnas.0809536106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xie Y, Rosser JM, Thompson TL, Boeke JD, An WF. Characterization of L1 retrotransposition with high-throughput dual-luciferase assays. Nucleic Acids Res. 2011;39 doi: 10.1093/nar/gkq1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Siebring-van Olst E, Vermeulen C, de Menezes RX, Howell M, Smit EF, van Beusechem VW. Affordable Luciferase Reporter Assay for Cell-Based High-Throughput Screening. Journal of Biomolecular Screening. 2013;18:453–461. doi: 10.1177/1087057112465184. [DOI] [PubMed] [Google Scholar]
- 44.Sonderhoff SA, Kilburn DG, Piret JM. Analysis of Mammalian Viable Cell Biomass Based on Cellular Atp. Biotechnol Bioeng. 1992;39:859–864. doi: 10.1002/bit.260390807. [DOI] [PubMed] [Google Scholar]
- 45.Karl DM, Nealson KH. Regulation of Cellular-Metabolism during Synthesis and Expression of the Luminous System in Beneckea and Photobacterium. J Gen Microbiol. 1980;117:357–368. [Google Scholar]
- 46.Aleskerova LE, Alenina KA, Efremenko EN, Mazhul’ MM, Piskunkova NA, Ismailov AD. ATP pool and bioluminescence in psychrophilic bacteria Photobacterium phosphoreum. Microbiology+ 2014;83:315–321. [PubMed] [Google Scholar]
- 47.Lee MS, Park WS, Kim YH, Ahn WG, Kwon SH, Her S. Intracellular ATP Assay of Live Cells Using PTD-Conjugated Luciferase. Sensors. 2012;12:15628–15637. doi: 10.3390/s121115628. [DOI] [PMC free article] [PubMed] [Google Scholar]