

Abstract
Th2 cytokine IL-4 has been previously shown to suppress the production of proinflammatory cytokines in monocytes. However, the underlying molecular mechanism by which IL-4 signaling antagonizes proinflammatory responses is poorly characterized. In particular, whether IL-4 can modulate inflammasome signaling is unknown. Here, we provide evidence that IL-4 suppresses NLRP3-dependent caspase-1 activation and the subsequent IL-1β secretion but does not inhibit AIM2- or NLRC4-dependent caspase-1 activation in THP-1 and mouse bone marrow-derived macrophages. Upon LPS or LPS/ATP stimulation, IL-4 markedly inhibited the assembly of NLRP3 inflammasome, including NLRP3-dependent ASC oligomerization, NLRP3-ASC interaction, and NLRP3 speck-like oligomeric structure formation. The negative regulation of NLRP3 inflammasome by IL-4 was not due to the impaired mRNA or protein production of NLRP3 and proinflammatory cytokines. Supporting this observation, IL-4 attenuated NLRP3 inflammasome activation even in reconstituted NLRP3-expressing macrophages in which NLRP3 expression is not transcriptionally regulated by TLR-NF-κB signaling. Furthermore, the IL-4-mediated suppression of NLRP3 inflammasome was independent of STAT6-dependent transcription and mitochondrial ROS. Instead, IL-4 inhibited subcellular redistribution of NLRP3 into mitochondria and microtubule polymerization upon NLRP3-activating stimulation. Our results collectively suggest that IL-4 could suppress NLRP3 inflammasome activation in a transcription-independent manner, thus providing an endogenous regulatory machinery to prevent excessive inflammasome activation.
INTRODUCTION
The inflammasome complex is assembled and activated upon sensing of various cytoplasmic pathogen- or danger-associated molecular patterns.1,2 Assembly of the inflammasome complex leads to the activation of caspase-1, which induces the maturation and secretion of proinflammatory cytokines, such as IL-1β and IL-18.1 At the sites of microbial infection or tissue injury, inflammasome signaling initiates the local inflammatory response by secreting these cytokines to maintain host homeostasis. However, recent accumulating evidence also demonstrated that the inflammasome plays a crucial role in potentiating many metabolic disorders, including atherosclerosis, obesity, and type II diabetes.3,4 In this regard, the activation of inflammasome signaling, under stress conditions, should be properly regulated to prevent excessive and chronic inflammation.
Among the identified inflammasome components, NOD-like receptor family, pyrin domain-containing 3 (NLRP3) is the main focus of interest, as it is potentially implicated in chronic inflammatory disorders.4 Indeed, Nlrp3−/− mice showed a remarkable reduction in the development of atherosclerosis and type II diabetes.5,6 Moreover, many stimuli triggering chronic metabolic disorders, such as cholesterol crystals, saturated fatty acids, uric acid crystals, and amyloid β, were shown to activate the NLRP3 inflammasome in bone marrow-derived macrophages (BMMs).5,7–9 In spite of the emerging physiological importance of the NLRP3 inflammasome, the molecular mechanism by which NLRP3 is activated and regulated in response to a wide range of stimuli is still poorly understood. In particular, the host endogenous suppression mechanism of the NLRP3 inflammasome, which might be essential to maintaining host immune homeostasis, is largely unknown.
Macrophages play a critical role in triggering an inflammatory response via the secretion of proinflammatory cytokines, such as IL-1β and TNF-α. Interestingly, macrophages are polarized into two different subpopulations: classically activated (M1) macrophages and alternatively activated (M2) macrophages.10 This fine-tuning of macrophage activation depends on the context of surrounding cytokines. For example, the Th1 cytokine, IFN-γ, promotes M1 macrophage polarization, which is specialized for antimicrobial and proinflammatory responses. On the other hand, the Th2 cytokine, IL-4, induces M2 macrophage polarization, which is important for helminth immunity, allergic responses, and tissue repair.10,11 In addition, IL-4 treatment diminishes the production of proinflammatory cytokines upon LPS stimulation12,13, suggesting that type 2 immune responses, including M2 polarization, are able to suppress excessive inflammation to maintain metabolic homeostasis. However, aside from its role in downregulating proinflammatory genes, the anti-inflammatory mechanism of IL-4 has not been clarified well.
In addition to its potential anti-inflammatory role, IL-4 has been proposed to attenuate the pathogenesis of metabolic disorders, such as type II diabetes and Alzheimer’s disease.14,15 Considering these observations, we attempted to examine whether IL-4 could function as an endogenous modulator of inflammasome activation. In the present study, we investigated the potential anti-inflammatory role of IL-4 with regard to inflammasome signaling pathways.
RESULTS
IL-4 suppresses NLRP3-dependent caspase-1 activation
To explore whether IL-4 could regulate the inflammasome activity of macrophages, we first examined the effect of IL-4 on NLRP3 inflammasome signaling in PMA-differentiated THP-1 macrophages. LPS treatment, which activates the NLRP3 inflammasome in PMA-primed THP-1 cells16, caused a robust activation of caspase-1, as determined by extracellular secretion of caspase-1 p20, and a subsequent secretion of IL-1β, but IL-4 pretreatment remarkably reduced LPS-stimulated caspase-1 and IL-1β activation in THP-1 cells (Figure 1a). IL-4 treatment also attenuated the extracellular release of caspase-1 p20 and IL-1β in LPS-primed mouse BMMs that were stimulated with ATP (Figure 1b).
Figure 1. Inhibition of NLRP3-dependent caspase-1 signaling by IL-4.
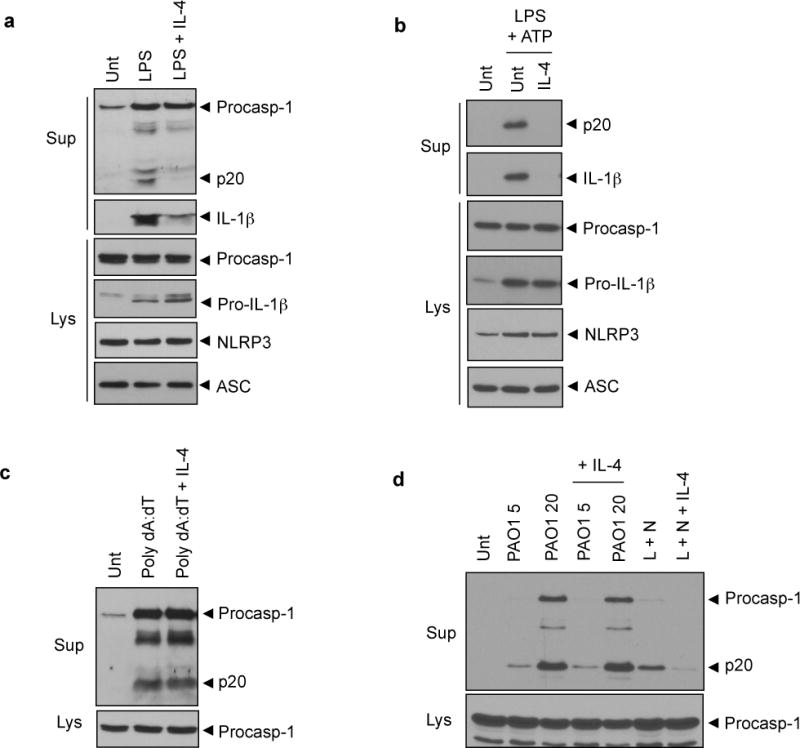
(a) PMA-differentiated THP-1 cells were untreated (Unt) or treated with LPS (0.5 μg ml−1, 6h) in the presence or absence of IL-4 pretreatment (20 ng ml−1, 3h) as indicated. (b) Murine BMMs were untreated (Unt) or primed with LPS (0.25 μg ml−1, 3h) in the presence or absence of IL-4 pretreatment (20 ng ml−1, 3h), followed with ATP treatment (3 mM, 45 min). (c) PMA-primed THP-1 cells were untreated (Unt) or transfected with poly dA:dT (1.5 μg, 7h) in the presence or absence of IL-4 pretreatment (20 ng ml−1, 3h). (d) Murine BMMs were untreated (Unt) or infected with Pseudomonas aeruginosa (PAO1; indicated multiplicity of infection (MOI), 2h) in the presence or absence of IL-4 pretreatment (20 ng ml−1, 3h) or primed with LPS (L, 0.25 μg ml−1, 3h) with or without IL-4 pretreatment, followed by nigericin treatment (N, 2.5 μM, 40 min). Culture supernatants (Sup) and lysates (Lys) were immunoblotted with the indicated antibodies. Procasp-1, procaspase-1.
Similarly, IL-1β secretion from THP-1 cells that were stimulated with alum or monosodium urate (MSU), which are crystalline stimuli for NLRP3 inflammasome, was clearly diminished by IL-4 treatment (Supplementary Figure 1a), suggesting that IL-4 suppresses NLRP3 inflammasome activation in human and mouse macrophages. Unlike IL-4 treatment, Th1 cytokine IFN-γ treatment slightly increased NLRP3 inflammasome activation from THP-1 cells upon stimulated with LPS or alum (Supplementary Figure 1b).
Next, we examined whether IL-4 could regulate absent in melanoma 2 (AIM2) and NOD-like receptor family, CARD domain-containing 4 (NLRC4) inflammasome activation. IL-4 treatment showed no inhibition on the caspase-1 activation in PMA-differentiated THP-1 cells (Figure 1c) and mouse BMMs (Supplementary Figure 1c) upon transfection with poly dA:dT, which activates AIM2 inflammasome.17 Furthermore, IL-4 failed to attenuate NLRC4-mediated caspase-1 activation in mouse BMMs that were infected with Pseudomonas aeruginosa (Figure 1d) or Salmonella typhimurium (Supplementary Figure 1d). These results collectively demonstrate that IL-4 negatively modulates NLRP3-dependent, but not AIM2- or NLRC4-dependent, caspase-1 processing and IL-1β secretion.
IL-4 does not inhibit LPS-stimulated transcriptional induction of proinflammatory cytokines
IL-4 has been shown to reduce the production of proinflammatory cytokines, including IL-1β.12,13 To gain a molecular insight to the anti-inflammatory function of IL-4, we determined the extracellular secretion levels of IL-1β and IL-6 from LPS-stimulated macrophages. Consistent with the aforementioned results, IL-1β secretion was significantly reduced by IL-4 treatment in PMA-differentiated THP-1 cells that were stimulated with LPS (Figure 2a, left) and in LPS-primed BMMs that were stimulated with ATP (Figure 2b, left). However, the secretion of another proinflammatory cytokine, IL-6, was unaffected by IL-4 treatment in THP-1 cells (Figure 2a, right) and BMMs (Figure 2b, right). These observations demonstrate that IL-4 modulates NLRP3 inflammasome-dependent IL-1β secretion but has no effect on LPS-stimulated IL-6 production.
Figure 2. NF-κB signaling-independent inhibition of NLRP3 inflammasome by IL-4.
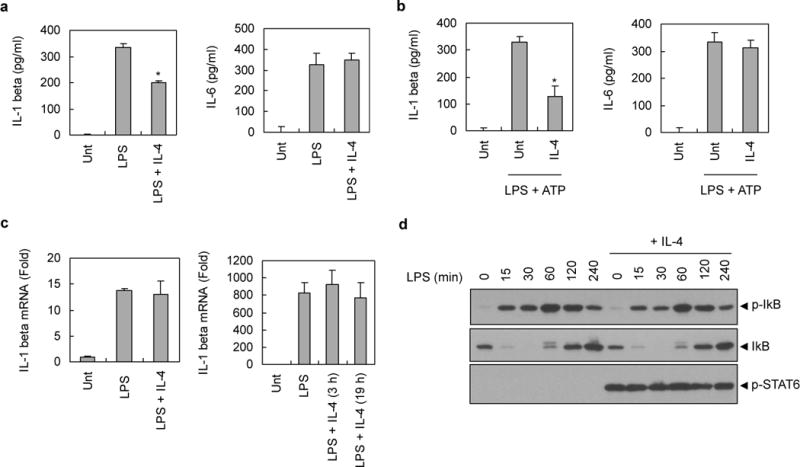
(a) PMA-differentiated THP-1 cells were untreated (Unt) or treated with LPS in the presence or absence of IL-4 pretreatment, as described in Fig. 1a. Culture supernatants were assayed for extracellular IL-1β or IL-6 by ELISA. The asterisk indicates a significant difference compared with LPS-treated samples (n = 4, *P<0.01) (b) Murine BMMs were untreated (Unt) or treated with LPS (0.25 μg ml−1, 3h) in the presence or absence of IL-4 pretreatment (20 ng ml−1, 3h), followed with ATP (3 mM, 45 min). Culture supernatants were assayed for extracellular IL-1β or IL-6 by ELISA. The asterisk indicates a significant difference compared with LPS/ATP-treated samples (n = 4, *P<0.05) (c) PMA-primed THP-1 cells (left panel) or murine BMMs (right panel) were untreated (Unt) or treated with LPS (0.5 μg ml−1 or 0.25 μg ml−1, respectively) for 3h in the presence or absence of IL-4 pretreatment (20 ng ml−1, 3h or 19h) as indicated. Then, mRNA production of IL-1β was determined by quantitative real-time PCR. Four (left panel) or three (right panel) independently treated wells were analyzed for quantification of mRNA. (d) PMA-differentiated THP-1 cells were untreated or treated with LPS (0.5 μg ml−1) for the indicated times in the presence or absence of IL-4 pretreatment (20 ng ml−1, 3h). Soluble lysates were immunoblotted for the indicated antibodies.
To examine whether the attenuated secretion of IL-1β by IL-4 is due to a decrease in the transcriptional induction of pro-IL-1β, we determined the mRNA levels of pro-IL-1β upon LPS stimulation by quantitative real-time PCR. Contrary to the expectation, IL-4 showed no significant inhibition on the mRNA production of pro-IL-1β in LPS-stimulated THP-1 cells (Figure 2c, left) and BMMs (Figure 2c, right). This finding was also verified by reverse transcription PCR (Supplementary Figure 2a). Similarly, IL-4 did not suppress the mRNA induction of IL-6 in both THP-1 cells and BMMs stimulated with LPS (Supplementary Figure 2b and 2c, respectively). These data suggest that IL-4 does not inhibit the LPS-stimulated transcription of proinflammatory cytokines in our experimental conditions. Supporting these observations, IL-4 did not affect the LPS-induced NF-κB signaling pathway as determined by phosphorylation and degradation of IκB in PMA-differentiated THP-1 cells (Figure 2d). Likewise, LPS-stimulated IκB phosphorylation and pro-IL-1β induction were not impaired by IL-4 pretreatment in BMMs (Supplementary Figure 2d), indicating that the IL-4-mediated suppression of NLRP3 inflammasome is independent of NF-κB-mediated transcription.
IL-4 inhibits the assembly of NLRP3 inflammasome
To provide a molecular insight into the IL-4-mediated suppression of the NLRP3 inflammasome, we examined the oligomerization of the inflammasome adaptor protein, apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC). ASC oligomerization is the most critical event for the activation of caspase-1.16,18,19 In PMA-differentiated THP-1 cells, LPS treatment induced a robust formation of the ASC dimer and oligomers, which was consistent with a previous report.16 However, much weaker ASC oligomerization was observed in the presence of IL-4 (Figure 3a, left). Contrary to IL-4, treatment with the Th1 cytokine, IFN-γ, caused a slightly stronger ASC oligomerization that was triggered by LPS. Poly dA:dT transfection, which activates the AIM2 inflammasome, also induced ASC oligomerization in agreement with a previous report17, but this poly dA:dT-induced ASC oligomerization was unaffected by IL-4 treatment in THP-1 cells (Figure 3a, right). These data further suggest that the inflammasome-modulating role of IL-4 is limited to NLRP3-mediated inflammasome signaling and also indicate that IL-4 may regulate the upstream event of ASC oligomerization mediated by NLRP3.
Figure 3. Inhibition of NLRP3 inflammasome assembly by IL-4.
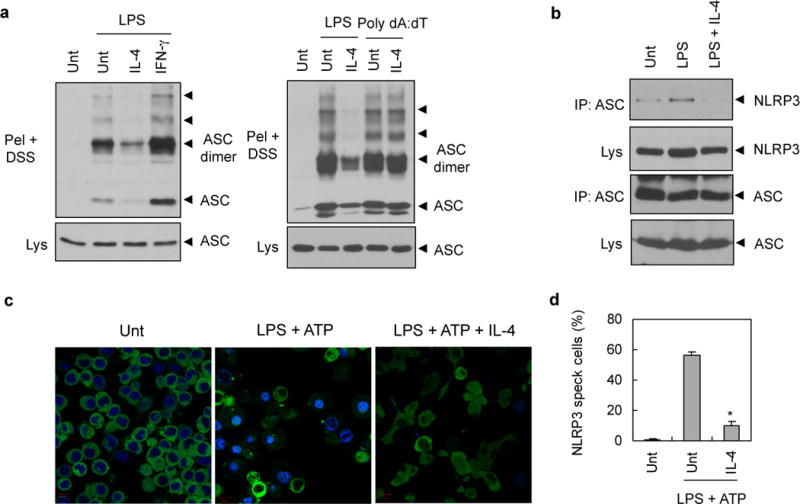
(a) PMA-differentiated THP-1 cells were untreated (Unt) or treated with LPS (0.5 μg ml−1, 6h) or transfected with poly dA:dT (2 μg, 6h) in the presence or absence of IL-4 or IFN-γ pretreatment (20 ng ml−1, 3h). Chemical crosslinking was then performed as described in Materials and methods. DSS-crosslinked NP-40 insoluble pellets (Pel + DSS) or soluble lysates (Lys) were immunoblotted with anti-ASC antibody. (b) PMA-differentiated THP-1 cells were untreated (Unt) or treated with LPS (0.5 μg ml−1, 3h) in the presence or absence of IL-4 pretreatment (20 ng ml−1, 3h). Soluble lysates were immunoprecipitated with anti-ASC antibody, and the immunoprecipitates were then immunoblotted with anti-NLRP3 antibody. (c) NLRP3-GFP-expressing BMMs were untreated (Unt) or treated with LPS (0.25 μg ml−1, 3h) in the presence or absence of IL-4 pretreatment for 3h (20 ng ml−1), followed with ATP (2.5 mM, 30 min). Cells were then observed by confocal microscope. Scale bar, 10 μM. (d) Relative percentages of NLRP3-speck containing cells as treated in Fig. 3c. Asterisk indicates significant difference from LPS plus ATP-treated sample. (n = 3, *P<0.005)
Then, we examined whether IL-4 could affect the association of NLRP3 with ASC, which may be essential for ASC oligomerization, although the interaction was rarely shown in the previous study. In PMA-differentiated THP-1 cells, LPS stimulation slightly increased the molecular interaction between NLRP3 and ASC, whereas IL-4 treatment largely abolished the NLRP3-ASC association (Figure 3b). This finding raises the possibility that IL-4 signaling may directly interfere with NLRP3-ASC association or inhibit the conversion of inactive NLRP3 to its active form.
Next, we examined the effect of IL-4 on the self-oligomerization of NLRP3, which is also required for ASC oligomerization.20 Upon stimulation with LPS plus ATP, NLRP3 protein was oligomerized to form speck-like aggregates in some BMMs (Figure 3c, middle). Notably, IL-4 treatment largely abolished the formation of NLRP3 speck-like structures (Figure 3c, right and Figure 3d). Considering that self-oligomerization of NLRP3 could be triggered by the conversion of inactive NLRP3 into its active form, IL-4 signaling may regulate the activation state of NLRP3 upon stimulation with LPS plus ATP. Overall, our data clearly demonstrate that IL-4 negatively modulate the assembly of NLRP3 inflammasome in response to LPS or LPS/ATP stimulation.
IL-4-mediated suppression of NLRP3 inflammasome is independent of M2 macrophage polarization and transcriptional induction of NLRP3
Alternative macrophage activation by IL-4 is the central phenomenon that mediates many physiological functions of IL-4.10,11 To test whether M2 macrophage polarization by IL-4 is required for the suppression of NLRP3 inflammasome, PMA-differentiated THP-1 cells were differentially treated with IL-4 for 16h before LPS stimulation to promote M2 polarization or together with LPS. The co-treatment of IL-4 with LPS, as well as 16h pretreatment with IL-4, showed a significant reduction in IL-1β secretion (Figure 4a and 4b, left), but not in IL-6 secretion (Figure 4b, right). This result suggests that M2 macrophage activation by IL-4 is not critical for its suppression of NLRP3 inflammasome signaling.
Figure 4. IL-4 inhibits NLRP3 inflammasome in a M2 polarization- and NLRP3 transcription-independent manner.
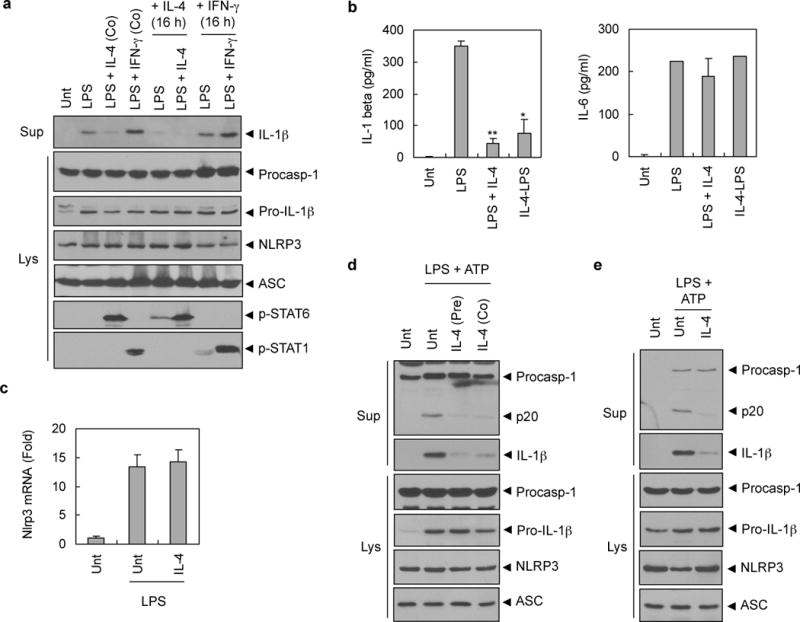
(a) PMA-differentiated THP-1 cells were untreated (Unt) or pretreated with IL-4 (lane 5, 6) or IFN-γ (lane 7, 8) (20 ng ml−1) for 16h, and then stimulated with LPS (0.5 μg ml−1) together with IL-4 (lane 3, 6) or IFN-γ (lane 4, 8) for 6h. Culture supernatants or lysates were immunoblotted with the indicated antibodies. (b) PMA-differentiated THP-1 cells were untreated (Unt) or treated with LPS (0.5 μg ml−1) alone (LPS) or together with IL-4 (20 ng ml−1, LPS+IL-4) for 6h, or pretreated with IL-4 (20 ng ml−1) for 16h before LPS stimulation (IL-4-LPS). Culture supernatants were assayed for extracellular IL-1β or IL-6 by ELISA. The asterisks indicate significant differences from LPS-treated cells. (n = 2, *P<0.05; **P<0.01) (c) Murine BMMs were untreated (Unt) or treated with LPS (0.25 μg ml−1, 3h) in the presence or absence of IL-4 pretreatment (20 ng ml−1, 3h). The mRNA level of Nlrp3 was determined by quantitative real-time PCR. Three independently treated wells were analyzed. (d) Murine BMMs were untreated (Unt), or treated with LPS (0.25 μg ml−1, 3h) in the presence or absence of IL-4 pretreatment (pre) for 3h followed with ATP (3 mM, 45 min), or treated with LPS (0.25 μg ml−1, 3h) followed with co-treatment of ATP and IL-4 (Co; 20 ng ml−1) for 45 min. (e) NLRP3-reconstituted Nlrp3-knockout macrophages (N1–8) were untreated (Unt) or treated with LPS (0.25 μg ml−1, 3h) in the presence or absence of IL-4 pretreatment (20 ng ml−1, 3h) followed with ATP (3 mM, 45 min). (d–e) Culture supernatants (Sup) or lysates (Lys) were immunoblotted with the indicated antibodies.
To confirm that M2 macrophage polarization is not responsible for IL-4-mediated modulation of NLRP3 inflammasome, we determined the mRNA levels of M2 marker genes in THP-1 cells upon IL-4 treatment. After 3 days of PMA-differentiation in THP-1 cells, 24h treatment of IL-4 induced a remarkable increase in the mRNA levels of M2 markers, such as MRC2 and CCL13 (Supplementary Figure 3a). However, after 20h of PMA treatment as in most our experimental condition, THP-1 cells showed a different basal expression of M2 genes (Supplementary Figure 3b). Indeed, PMA treatment induced an mRNA induction of not only TNF-α (M1) but MRC2 (M2) after 20h of PMA treatment in THP-1 cells (Supplementary Figure 3c). In this our experimental condition, the presence of IL-4 had no effect on the M2 gene expression compared with LPS-treated control (Supplementary Figure 3b). These data indicate that IL-4 treatment in our experimental setting did not induce M2 macrophage activation. Therefore, we conclude that the IL-4-mediated suppression of NLRP3 inflammasome is independent of macrophage polarization status.
Then, to examine the possibility that the IL-4-mediated suppression of NLRP3 inflammasome is due to a reduction in NLRP3 expression by IL-4. We first determined the mRNA level of Nlrp3 in mouse BMMs. LPS stimulation increased mRNA level of Nlrp3, but IL-4 pretreatment did not affect the LPS-induced transcription of Nlrp3 (Figure 4c). To provide more evidence to support the non-transcriptional effect of IL-4, we treated IL-4 with ATP for 45 min to LPS-primed BMMs. As well as 3h pretreatment, IL-4 treatment for the last 45 min with ATP also reduced caspase-1 activation and IL-1β secretion in BMMs (Figure 4d). To support this observation, we performed similar experiment in the stably NLRP3-reconstituted Nlrp3-knockout macrophages (N 1–8) which express no endogenous NLRP3 but reconstituted NLRP3.21 The expression level of NLRP3 in N1–8 macrophages is not transcriptionally regulated by TLR signaling. In NLRP3-reconstituted N1–8 macrophages, IL-4 markedly suppressed LPS/ATP-stimulated caspase-1 activation and IL-1β secretion (Figure 4e). These evidences collectively indicate that the attenuated NLRP3 inflammasome activation by IL-4 is non-transcriptional phenomenon.
IL-4-mediated suppression of NLRP3 inflammasome is independent of STAT6 and ROS
The engagement of IL-4 to the α chain of the IL-4 receptor recruits the gamma chain of the receptor or IL-13 receptor-α chain 1.22 The dimerization of IL-4R subunits activates the downstream JAK, which leads to the activation of STAT6.22 Subsequently, STAT6 induces a variety of target genes. In addition to the STAT6-inducible genes, STAT6 exhibits anti-inflammatory functions by suppressing IFN-γ/STAT1-dependent transcription.23
To assess whether STAT6 is necessary for the IL-4-mediated suppression of NLRP3 signaling, we examined inflammasome activation in Stat6-deficient BMMs from Stat6−/− mice. As shown in Figure 5a and 5b, IL-4 significantly attenuated NLRP3-dependent caspase-1 activation and IL-1β secretion in both wild-type and Stat6−/− BMMs. On the other hand, IL-4 treatment had no effect on LPS-stimulated IL-6 production in not only wild-type but also Stat6-deficient BMMs (Figure 5b, right). Contrary to PMA-differentiated THP-1 cells, IL-4 induced a M2 marker gene, such as arginase 1, in wild-type mouse BMMs, but not in Stat6-deficient BMMs (Figure 5c). Nevertheless, the IL-4-mediated suppression of NLRP3 inflammasome activation was also observed in Stat6-deficient BMMs. This result indicates that STAT6 or STAT6-mediated transcription, including M2 marker genes, is not required for the suppression of the NLRP3 inflammasome by IL-4.
Figure 5. STAT6- or mitochondrial ROS-independent inhibition of NLRP3 inflammasome by IL-4.
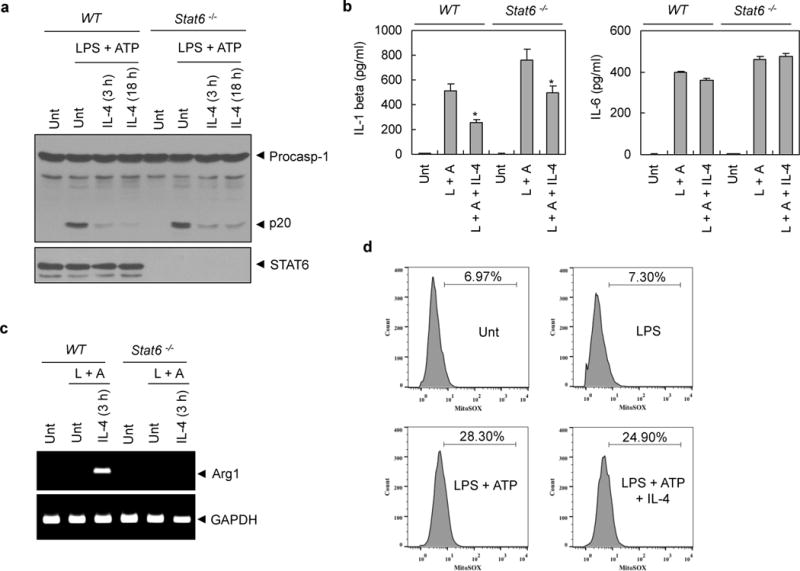
(a) Murine BMMs from wild-type (WT) or Stat6-deficient mice were untreated (Unt) or primed with LPS (0.25 μg ml−1, 3h) in the presence or absence of IL-4 pretreatment (20 ng ml−1, 3h or 18h) followed with ATP (2.5 mM, 30 min). Soluble lysates were immunoblotted as indicated. (b) Culture supernatants from (a) were assayed for extracellular IL-1β or IL-6 by ELISA. (c) RNA extracts from (a) were assayed for mRNA production of arginase1 (Arg1) by RT-PCR. (d) Murine BMMs were untreated (Unt) or treated with LPS (0.25 μg ml−1, 3h) in the presence or absence of IL-4 pretreatment (20 ng ml−1, 3h) followed with ATP stimulation (2.5 mM, 30 min). Cells were stained with MitoSox and analyzed by flow cytometry.
The underlying mechanism, by which inactive NLRP3 is converted into active form, is still unclear, and the assembly of NLRP3 inflammasome in response to a variety of stimuli requires further elucidation. However, mitochondrial ROS have been shown to be a major candidate for activating NLRP3.24 Therefore, we determined whether IL-4 could affect mitochondrial ROS production. ATP stimulation in LPS-primed BMMs increased a mitochondrial ROS production in consistent with previous report24 (Figure 5d, lower left). However, IL-4 treatment showed little effect on the mitochondrial ROS production triggered by LPS plus ATP, which indicates that the IL-4-mediated suppression of NLRP3 inflammasome seems not due to the decrease in mitochondrial ROS production.
IL-4 regulates the subcellular localization of NLRP3
Recent studies proposed that the mitochondrial localization of NLRP3 is important for the assembly and activation of NLRP3 inflammasome.20,25 To examine whether IL-4 could affect the subcellular redistribution of NLRP3, we carefully observed NLRP3 expression by confocal microscope. NLRP3 was evenly distributed in the cytosol and rarely colocalized with mitochondria in resting macrophages (Figure 6a, upper panel). Upon LPS plus ATP stimulation, many NLRP3 speck-like aggregates were observed similarly as in Fig. 3C. Of particular interest, some swelling cells were appeared only in response to LPS plus ATP stimulation, and in these cells, NLRP3 was relocated to the perinuclear region (Figure 6a, middle panel), which was in agreement with a previous report.24 Also, NLRP3 noticeably colocalized with mitochondria in these swelling macrophages. This suggests that the mitochondrial redistribution of NLRP3 is implicated in its activation. However, IL-4 markedly blocked the redistribution of NLRP3 into perinuclear region and its colocalization with mitochondria (Figure 6a, lower panel). To provide more evidence to support this phenomenon, we performed subcellular fractionation experiment. LPS plus ATP stimulation substantially increased translocation of NLRP3 into mitochondrial fraction and IL-4 diminished this mitochondrial redistribution of NLRP3 (Figure 6b). These findings further suggest that IL-4 may inhibit NLRP3 redistribution, thereby suppressing the assembly and the activation of NLRP3 inflammasome.
Figure 6. IL-4 modulates the subcellular localization of NLRP3 and microtubule polymerization.
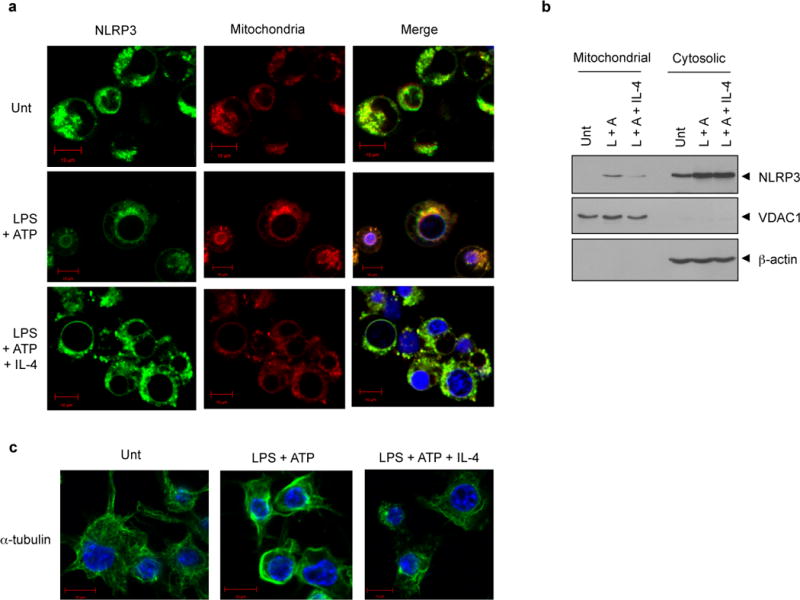
(a) NLRP3-GFP-expressing BMMs were untreated (Unt) or primed with LPS (0.25 μg ml−1, 3h) in the presence or absence of IL-4 pretreatment (20 ng ml−1, 3h), followed with ATP (2.5 mM, 30 min). Cells were stained with MitoTracker and observed by confocal microscope. Scale bar, 10 μM. (b) Murine BMMs were treated as in (a) and the cell lysates were fractionated into cytosolic and mitochondrial fraction, and then immunoblotted with the indicated antibodies. (L, LPS; A, ATP) (c) Murine BMMs were treated as in (a) and stained with the anti-α-tubulin antibody. Then, the immunofluorescence assay was performed as described in Materials and methods. Blue signals represent nuclear fluorescence. Scale bar, 10 μM.
Previous studies also demonstrated that microtubule polymerization is implicated in inflammasome activation, based on the finding that colchicine or nocodazole, an inhibitor of microtubule polymerization, clearly attenuated NLRP3 or pyrin inflammasome activation.18,25 Considering that microtubule network is also important for NLRP3 or mitochondria translocation,25 we examined whether IL-4 treatment could affect microtubule dynamics under NLRP3-stimulating conditions. Interestingly, ATP treatment into LPS-primed BMMs robustly induced a polymerization of microtubule network (Figure 6c, middle panel), but IL-4 greatly diminished microtubule polymerization (Figure 6c, right panel). This observation suggests that IL-4 inhibits microtubule polymerization triggered by NLRP3-activating stimulation. Although it is unclear whether microtubule polymerization is critical for the relocation of NLRP3 into the perinuclear region, our results demonstrate that IL-4 negatively modulates NLRP3 inflammasome signaling by way of regulating microtubule network and NLRP3 subcellular redistribution.
DISCUSSION
Inflammasome signaling provides a primary defense against microbial infection or tissue injury, but deregulation of inflammasome activation might lead to chronic inflammatory disorders.3 In this regard, inflammasome signaling needs to be properly controlled to prevent excessive inflammation and maintain immune homeostasis. However, the host endogenous feedback inhibition of the inflammasome is still poorly elucidated. Activation of the NLRP3 inflammasome requires two independent processes: the initial transcriptional induction of NLRP3 and the subsequent posttranslational activation of NLRP3 for the inflammasome assembly.26 Because NLRP3 transcription relies on NF-κB activity upon pattern-recognition receptor signaling, the second posttranslational activation step is of particular importance to modulate NLRP3 activity.
In the present study, we show that the Th2 cytokine, IL-4, could function as the endogenous regulator for the assembly of NLRP3 inflammasome. IL-4 is produced by mature Th2 cells and induces Th2 responses, such as the production of IgE-secreting B cells and the alternative activation of macrophages.10,11 Previous reports have demonstrated that effector CD4+ T cells can suppress innate immune responses.27,28 Our data also propose that IL-4 produced from activated adaptive immune system can regulate early innate inflammasome activation.
IL-4 exerts anti-inflammatory effects via transcriptional regulation in monocytes and macrophages.12,13,29 For example, IL-4 stimulates the expression of anti-inflammatory cytokines, such as IL-1R antagonist, and suppresses the production of pro-inflammatory cytokines, including IL-1β, IL-6, and TNF-α. These IL-4-induced changes in transcriptional profiling lead to the M2 polarization of macrophages.22 Indeed, M2 macrophages are implicated in allergic inflammation, helminth immunity, and tissue repair, whereas M1 macrophages are specialized in proinflammatory responses.11
Interestingly, our results suggest that IL-4 can suppress NLRP3-mediated caspase-1 activation and IL-1β secretion in macrophages without transcriptional modulation. In our experimental conditions, IL-4 treatment reduced NLRP3 inflammasome signaling, but had no effect on the LPS-induced transcription of pro-IL-1β and NLRP3. To support our findings, IL-4 treatment did not impair LPS-induced NF-κB activation. In this regard, it is possible that IL-4 did not suppress the LPS-induced secretion of IL-6 in our study because of intact NF-κB signaling in the presence of IL-4. More importantly, the suppressive effect of IL-4 on the NLRP3 inflammasome was independent of STAT6 and the STAT6-dependent expression of M2 genes. These findings collectively suggest that IL-4 can function as an endogenous anti-inflammatory molecule not only by regulating protein expression as reported previously and but also by modulating NLRP3 inflammasome signaling.
The underlying mechanism by which IL-4 inhibits the assembly and activation of NLRP3 inflammasome is still unclear. However, the inflammasome-modulating effect of IL-4 was not involved in the STAT6-dependent transcription of regulatory proteins, and IL-4 did not affect mitochondrial ROS production. Instead, our results show that IL-4 blocked the mitochondrial redistribution of NLRP3 and microtubule polymerization upon stimulation with LPS and ATP. Considering that microtubule network is possibly implicated in the transport of NLRP3,25 we could infer that the IL-4-driven less polymerized microtubule network might affect NLRP3 redistribution leading to the attenuated assembly of NLRP3 inflammasome. It will be, therefore, of interest to elucidate whether microtubule polymerization is responsible for the transport of NLRP3 into mitochondria. In conclusion, our data suggest that IL-4 could control NLRP3 inflammasome activation via a transcription-independent manner to provide an innate immune suppressive mechanism.
MATERIALS AND METHODS
Reagents and antibodies
Human or murine recombinant IL-4 and IFN-γ were purchased from Invitrogen (Carlsbad, CA, USA). LPS, ATP, nigericin, and poly dA:dT were purchased from Sigma (St. Louis, MI, USA). Human or mouse IL-1β and IL-6 ELISA kits were obtained from R&D (Minneapolis, MN, USA). Mito-SOX and MitoTracker Red were purchased from Invitrogen. The anti-human caspase-1 antibody was generated in the laboratory (p20), as described previously,17 or purchased from Santa Cruz (p10) (Santa Cruz, CA, USA). The anti-mouse IL-1β antibody was obtained from R&D. Anti-mouse caspase-1 and NLRP3 antibodies were from Adipogen (San Diego, CA, USA). Anti-VDAC1 antibody was from Abcam (Cambridge, UK). All other antibodies detecting human IL-1β, phosphorylated (phospho)-IκB, IκB, phospho-STAT6, STAT6, and phospho-STAT1 were obtained from Cell Signaling (Beverly, MA, USA).
Cell culture
THP-1 cells were grown in RPMI 1640 medium supplemented with 10% FBS, 2 mM glutamine, 10 mM HEPES, 1 mM sodium pyruvate, 0.05 mM 2-mercaptoethanol, and 100 U ml−1 penicillin/streptomycin. To induce differentiation of THP-1 cells into macrophages, THP-1 cells were treated with 0.4 μM PMA for 2h and incubated in growth medium for an additional 20h before the appropriate treatments. Mouse BMMs were prepared from C57BL/6 or Stat6-deficient mice as described previously.30 All mice were maintained in specific pathogen-free conditions. Protocols for mice experiments were approved by institutional ethical committee, Yonsei University College of Medicine. Immortalized NLRP3-GFP-expressing BMMs and NLRP3-reconstituted BMMs (N1–8) were generated by the retroviral infection of Nlrp3-deficient BMMs as described previously.21,31 BMMs were maintained in L929-conditioned DMEM, which was supplemented with 10% FBS and 100 U ml−1 penicillin/streptomycin.
Immunoblot analysis
Cells were lysed in 20 mM HEPES (pH 7.5) buffer containing 0.5% Nonidet P-40 (NP-40), 50 mM KCl, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, and protease inhibitors. Soluble lysates were subjected to SDS-PAGE, and then immunoblotted with the appropriate antibodies. Secreted proteins from cell culture supernatants were concentrated for immunoblotting as described previously.17 All the blots were the representative image of at least three-independent experiments. For the coimmunoprecipitation experiment, cells were lysed in 10 mM HEPES buffer (pH 7.4) containing 0.2% NP-40, 100 mM KCl, 5 mM MgCl2, 0.5 mM EGTA, 1 mM DTT, and protease inhibitors. Lysates were pre-cleared with Protein G sepharose 4 beads (GE) and immunoprecipitated with the indicated antibodies, and the bead-bound proteins were immunoblotted with the appropriate antibodies.
Determination of ASC oligomerization
To detect ASC oligomerization, chemical cross-linking assay was performed as described previously.18,32 Briefly, cells were lysed and centrifuged at 6000 rpm for 10 min. The pellets were washed with PBS and then resuspended in PBS. The resuspended pellets were cross-linked with 1 mM discuccinimidyl suberate (DSS, Pierce) for 30 min. The cross-linked pellets and soluble lysates were fractionated on SDS-PAGE and immunoblotted with anti-ASC antibody.
Bacterial infections
Mouse BMMs were infected with Salmonella typhimurium and Pseudomonas aeruginosa PAO1 (kindly gifted from Dr. Yoon SS, Yonsei University College of Medicine) as described previously.33 Bacteria were grown overnight with aeration, diluted with fresh medium (1:20 or 1:50) and then grown for additional 2h. BMMs were infected with Salmonella typhimurium at the indicated multiplicity of infection (MOI) for 30 min, washed to remove extracellular bacteria and incubated with gentamicin (100 μg/ml)-containing medium for 2.5h before harvesting. BMMs were also infected with the P. aeruginosa PAO1 at the indicated MOI for 2h before harvest.
Quantification of mRNA production and cytokine secretion
To measure mRNA production, quantitative real-time PCR or reverse transcription (RT)-PCR assay was performed. Briefly, total cellular RNA was isolated using the TRIzol reagent (Invitrogen) and reverse transcribed using PrimeScript™ RT Master Mix (Takara) according to the manufacturer’s instructions. Template DNA was amplified by quantitative real-time PCR using SYBR Premix Ex Taq™II (Takara). RT-PCR was performed with the AccuPower HotStart PCR PreMix (Bioneer). Primers were as follows: 5′-GGG CCT CAA GGA AAA GAA TC-3′ and 5′-TTC TGC TTG AGA GGT GCT GA-3′ (human IL-β); 5′-TAC CCC CAG GAG AAG ATT CC-3′ and 5′-TTT TCT GCC AGT GCC TCT TT-3′ (human IL-6); 5′-GAG TCA ACG GAT TTG GTC GT-3′ and 5′-TTG ATT TTG GAG GGA TCT CG-3′ (human GAPDH); 5′-GCC CAT CCT CTG TGA CTC AT-3′ and 5′-AGG CCA CAG GTA TTT TGT CG-3′ (mouse IL-1β); 5′-AGT TGC CTT CTT GGG ACT GA-3′ and 5′-TCC ACG ATT TCC CAG AGA AC-3′ (mouse IL-6); 5′-AAC TTT GGC ATT GTG GAA GG-3′ and 5′-ACA CAT TGG GGG TAG GAA CA-3 (mouse GAPDH); 5′-GTG AAG AAC CCA CGG TCT GT-3′ and 5′-CTG GTT GTC AGG GGA GTG TT-3′ (mouse arginase 1); 5′-ATG CTG CTT CGA CAT CTC CT-3′ and 5′-AAC CAA TGC GAG ATC CTG AC-3′ (mouse Nlrp3). To quantify the amounts of secreted cytokines, culture supernatants were assayed using Quantikine ELISA kits for IL-1β or IL-6 (R&D), according to the manufacturer’s protocol.
Confocal microscopy
For the immunofluorescence assay, cells were grown on cover slips in 12-well plates and treated as described in the Figure legends. Cells were fixed with 4% formaldehyde and permeabilized with 0.2% Triton X-100. After blocking with 4% BSA (Sigma), cells were incubated with anti-α-tubulin antibody (Santa Cruz), followed by incubation with fluorescein-conjugated anti-mouse IgG (Jackson). Cells on cover slips were then examined using a confocal microscope (Zeiss LSM 700). Confocal microscopy of NLRP3-GFP-expressing BMMs was performed as described previously.20 To quantify NLRP3 speck formation, relative percentage of NLRP speck-containing cells was determined by dividing the number of NLRP3 speck-containing cells by the number of total cells. Three independently treated wells were counted and analyzed.
Measurement of mitochondrial ROS
Measurements of mitochondrial ROS production were performed using MitoSox (Invitrogen) as described previously.34 Briefly, cells were resuspended in HBSS, and MitoSox (2.5 μM) was added and incubated at 37°C for 10 min. The fluorescence of the cells was monitored by flow cytometry.
Subcellular fractionation
Subcellular fractionation experiments were performed as described previously.20 Briefly, cells were lysed in buffer A containing 250 mM sucrose, 10 mM HEPES (pH 7.8), 10 mM KCl, 2 mM MgCl2, 0.1 mM EGTA, 1 mM DTT, 0.1% NP-40 and protease inhibitors. The lysates were centrifuged for 10 min at 700 g, and the supernatants were centrifuged for 10 min at 12500 g. The resultant supernatants were used for cytosolic fraction and the pellets were resuspended in 10 mM HEPES buffer (pH 7.4) containing 0.5% NP-40, 50 mM KCl, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA and protease inhibitors. Then, the resuspended pellets were recentrifuged and the supernatants were used for mitochondria-enriched fraction.
Statistical analysis
All values were expressed as the mean and SE of individual samples. Data were analyzed with Student t test. The P values ≤ 0.05 were considered significant.
Supplementary Material
Acknowledgments
This work was supported by the National Research Foundation of Korea Grant funded by the Korean Government (2012R1A1A2041532 and 2013R1A2A2A01067985), a faculty research grant of Yonsei University College of Medicine for 2014 (6-2014-0037), and by the National Institute of Health grant AR055398 to E.S.A.
References
- 1.Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 2.Franchi L, Munoz-Planillo R, Nunez G. Sensing and reacting to microbes through the inflammasomes. Nat Immunol. 2012;13:325–332. doi: 10.1038/ni.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Henao-Mejia J, Elinav E, Thaiss CA, Flavell RA. Inflammasomes and metabolic disease. Annu Rev Physiol. 2014;76:57–78. doi: 10.1146/annurev-physiol-021113-170324. [DOI] [PubMed] [Google Scholar]
- 4.Wen H, Ting JP, O’Neill LA. A role for the NLRP3 inflammasome in metabolic diseases–did Warburg miss inflammation? Nat Immunol. 2012;13:352–357. doi: 10.1038/ni.2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–1361. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011;17:179–188. doi: 10.1038/nm.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT, et al. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol. 2011;12:408–415. doi: 10.1038/ni.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 9.Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol. 2008;9:857–865. doi: 10.1038/ni.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol. 2009;27:451–483. doi: 10.1146/annurev.immunol.021908.132532. [DOI] [PubMed] [Google Scholar]
- 11.Van Dyken SJ, Locksley RM. Interleukin-4- and interleukin-13-mediated alternatively activated macrophages: roles in homeostasis and disease. Annu Rev Immunol. 2013;31:317–343. doi: 10.1146/annurev-immunol-032712-095906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.te Velde AA, Huijbens RJ, Heije K, de Vries JE, Figdor CG. Interleukin-4 (IL-4) inhibits secretion of IL-1 beta, tumor necrosis factor alpha, and IL-6 by human monocytes. Blood. 1990;76:1392–1397. [PubMed] [Google Scholar]
- 13.Vannier E, Miller LC, Dinarello CA. Coordinated antiinflammatory effects of interleukin 4: interleukin 4 suppresses interleukin 1 production but up-regulates gene expression and synthesis of interleukin 1 receptor antagonist. Proc Natl Acad Sci U S A. 1992;89:4076–4080. doi: 10.1073/pnas.89.9.4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O’Connor JC, Sherry CL, Guest CB, Freund GG. Type 2 diabetes impairs insulin receptor substrate-2-mediated phosphatidylinositol 3-kinase activity in primary macrophages to induce a state of cytokine resistance to IL-4 in association with overexpression of suppressor of cytokine signaling-3. J Immunol. 2007;178:6886–6893. doi: 10.4049/jimmunol.178.11.6886. [DOI] [PubMed] [Google Scholar]
- 15.Kiyota T, Okuyama S, Swan RJ, Jacobsen MT, Gendelman HE, Ikezu T. CNS expression of anti-inflammatory cytokine interleukin-4 attenuates Alzheimer’s disease-like pathogenesis in APP+PS1 bigenic mice. FASEB J. 2010;24:3093–3102. doi: 10.1096/fj.10-155317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fernandes-Alnemri T, Wu J, Yu JW, Datta P, Miller B, Jankowski W, et al. The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 2007;14:1590–1604. doi: 10.1038/sj.cdd.4402194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature. 2009;458:509–513. doi: 10.1038/nature07710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu JW, Fernandes-Alnemri T, Datta P, Wu J, Juliana C, Solorzano L, et al. Pyrin activates the ASC pyroptosome in response to engagement by autoinflammatory PSTPIP1 mutants. Mol Cell. 2007;28:214–227. doi: 10.1016/j.molcel.2007.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR, et al. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell. 2014;156:1193–1206. doi: 10.1016/j.cell.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Park S, Juliana C, Hong S, Datta P, Hwang I, Fernandes-Alnemri T, et al. The mitochondrial antiviral protein MAVS associates with NLRP3 and regulates its inflammasome activity. J Immunol. 2013;191:4358–4366. doi: 10.4049/jimmunol.1301170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Juliana C, Fernandes-Alnemri T, Kang S, Farias A, Qin F, Alnemri ES. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J Biol Chem. 2012;287:36617–36622. doi: 10.1074/jbc.M112.407130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang HW, Joyce JA. Alternative activation of tumor-associated macrophages by IL-4: priming for protumoral functions. Cell Cycle. 2010;9:4824–4835. doi: 10.4161/cc.9.24.14322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ohmori Y, Hamilton TA. STAT6 is required for the anti-inflammatory activity of interleukin-4 in mouse peritoneal macrophages. J Biol Chem. 1998;273:29202–29209. doi: 10.1074/jbc.273.44.29202. [DOI] [PubMed] [Google Scholar]
- 24.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 25.Misawa T, Takahama M, Kozaki T, Lee H, Zou J, Saitoh T, et al. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat Immunol. 2013;14:454–460. doi: 10.1038/ni.2550. [DOI] [PubMed] [Google Scholar]
- 26.Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013;13:397–411. doi: 10.1038/nri3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim KD, Zhao J, Auh S, Yang X, Du P, Tang H, et al. Adaptive immune cells temper initial innate responses. Nat Med. 2007;13:1248–1252. doi: 10.1038/nm1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guarda G, Dostert C, Staehli F, Cabalzar K, Castillo R, Tardivel A, et al. T cells dampen innate immune responses through inhibition of NLRP1 and NLRP3 inflammasomes. Nature. 2009;460:269–273. doi: 10.1038/nature08100. [DOI] [PubMed] [Google Scholar]
- 29.Fenton MJ, Buras JA, Donnelly RP. IL-4 reciprocally regulates IL-1 and IL-1 receptor antagonist expression in human monocytes. J Immunol. 1992;149:1283–1288. [PubMed] [Google Scholar]
- 30.Fernandes-Alnemri T, Yu JW, Juliana C, Solorzano L, Kang S, Wu J, et al. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat Immunol. 2010;11:385–393. doi: 10.1038/ni.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Juliana C, Fernandes-Alnemri T, Wu J, Datta P, Solorzano L, Yu JW, et al. Anti-inflammatory compounds parthenolide and Bay 11–7082 are direct inhibitors of the inflammasome. J Biol Chem. 2010;285:9792–9802. doi: 10.1074/jbc.M109.082305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu JW, Farias A, Hwang I, Fernandes-Alnemri T, Alnemri ES. Ribotoxic stress through p38 mitogen-activated protein kinase activates in vitro the human pyrin inflammasome. J Biol Chem. 2013;288:11378–11383. doi: 10.1074/jbc.M112.448795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hwang I, Park S, Hong S, Kim EH, Yu JW. Salmonella Promotes ASC Oligomerization-dependent Caspase-1 Activation. Immune Netw. 2012;12:284–290. doi: 10.4110/in.2012.12.6.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hong S, Hwang I, Lee YS, Park S, Lee WK, Fernandes-Alnemri T, et al. Restoration of ASC expression sensitizes colorectal cancer cells to genotoxic stress-induced caspase-independent cell death. Cancer Lett. 2013;331:183–191. doi: 10.1016/j.canlet.2012.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.