

Summary
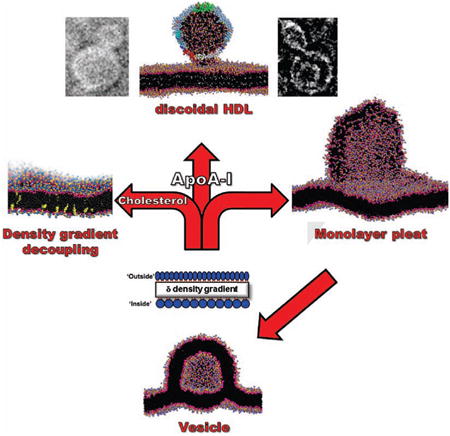
Biogenesis of high density lipoproteins (HDL) is coupled to the transmembrane protein, ATP-binding cassette transporter A1 (ABCA1), which transports phospholipid (PL) from the inner to the outer membrane monolayer. Using a combination of computational and experimental approaches, we show that increased outer lipid monolayer surface density driven by excess PL or by membrane insertion of amphipathic helices, results in pleating of the outer monolayer to form membrane-attached discoidal bilayers. Apolipoprotein (apo)A-I accelerates and stabilizes the pleats. In the absence of apoA-I, pleats collapse to form vesicles. These results mimic cells overexpressing ABCA1 that, in the absence of apoA-I, form and release vesicles. We conclude that the basic driving force for nascent discoidal HDL assembly is a PL pump-induced surface density increase that produces lipid monolayer pleating. We then argue that ABCA1 forms an extracellular reservoir containing an isolated pressurized lipid monolayer decoupled from the transbilayer density buffering of cholesterol.
Graphical abstract
Introduction
Clinical and epidemiological studies show a robust, inverse association of high density lipoprotein cholesterol (HDL-C) levels with risk for coronary vascular disease (CVD) (Wilson et al., 1988). The relationship of HDL-C to CVD is complex, however, because lower HDL-C levels do not uniformly associate with increased cardiovascular risk (Voight et al., 2012). Moreover, several recent clinical studies have failed to show any benefit from pharmacological elevation of HDL-C levels in statin-treated humans with established CVD (Rader and Tall, 2012). These observations suggest that HDL-C levels provide limited information about HDL's proposed cardioprotective effects. Therefore, the rational design of therapies and assessments of their benefits demand detailed knowledge of the assembly and molecular structure of the cardioprotective form of HDL.
ApoA-I, the major HDL protein, is secreted into the bloodstream as a lipid-free or lipid-poor monomer (pre-β HDL), which is widely believed to be the most common acceptor of sterol transported out of cells (Kane and Malloy, 2012). The circulating form of mature human apoA-I contains 243 residues encoded by exon-3 (1-43) and exon-4 (44-243). The region encoded by exon-4 contains 10 tandem 11/22 residue repeats, termed helical repeats 1 to 10, which form a series of lipid-binding amphipathic α helices.
The standard model for nascent HDL particles is a discoidal particle (dHDL) appoximately 100 Å in diameter and the thickness of a PL bilayer (Atkinson et al., 1980; Forte et al., 1971; Sloop et al., 1983; Wlodawer et al., 1979). Several years ago our lab, inspired by a lipid-free crystal structure for N-terminally truncated apoA-I (Borhani et al., 1997), derived a detailed amphipathic α helical double belt model for dHDL (Li et al., 2004; Segrest et al., 1999). The general features of this model have been confirmed by several laboratories using a variety of physical chemical methods (Bhat et al., 2007; Davidson and Thompson, 2007; Martin et al., 2006; Tricerri et al., 2001).
The antiatherogenic nature of HDL has been most firmly related to the process of reverse cholesterol transport (RCT)—the removal of excess cholesterol from cells. RCT is linked to the generation of HDL particles through apoA-I-mediated lipid removal (Figure S1), a process tightly coupled to activity of ATP-binding cassette transporter A1 (ABCA1), a transmembrane ATPase (Figure 1). Tangier disease is a familial HDL deficiency syndrome caused by mutations in the gene encoding ABCA1 (Bodzioch et al., 1999; Brooks-Wilson et al., 1999; Lawn et al., 1999; Rust et al., 1999) that results in the failure of apoA-I to acquire cellular lipids (Figure 1) and in an increased prevalence of atherosclerosis (Francis et al., 1995). ApoA-I accepts PL from ABCA1 to become dHDL. In this configuration, two anti-parallel molecules of apoA-I form a belt around the edge of a disc of PL and unesterified cholesterol (UC) (Borhani et al., 1997; Koppaka et al., 1999; Segrest et al., 1999); dimerization is driven in part by the formation of salt bridges between the 10 helical repeats (Segrest et al., 1999).
Figure 1. Role of ABCA1 and apoA-I in assembly of HDL from bilayers.

Diagram of the role of the transmembrane protein ABCA1 in assembly of nascent HDL. Red ovals, transmembrane domains of ABCA1. See also Figure S1.
ABCA1 has been shown to be a PL pump, transporting palmitoyloleoyl phosphatidylcholine (POPC), palmitoyloleoyl phosphatidylserine (POPS) and sphingomyelin (SM) (Quazi and Molday, 2013) from the inner to the outer membrane monolayer (Figure 1). Cells overexpressing ABCA1, in the absence of apoA-I, have been shown to extrude vesicles (microparticles) from their membrane surfaces into the cell media (Duong et al., 2006). The product of ABCA1, HDL, is a supramolecular assembly of lipid and apoA-I plus a heterogeneous collection of other proteins. In the current study, we use a combination of computational approaches, such as molecular dynamics (MD), and experimental approaches, to explore the hypothesis that increased outer lipid monolayer surface pressure—for clarity, since density is what we quantify, surface pressure will be referred to as surface density—created by ABCA1-mediated pumping of PL drives apoA-I lipidation.
In an important breakthrough in understanding the mechanism of nascent HDL formation by ABCA1, Nagata, et al (Nagata et al., 2013) recently proposed a dimeric ABCA1 model for HDL assembly. They show, rather convincingly, that: i) ABCA1 monomers translocate lipids into a reservoir formed by its large extracellular domains; ii) lipidated ABCA1 forms a cytoskeleton-fixed dimer that binds two lipid-free apoA-I; iii) lipids are translocated to apoA-I; iv) ABCA1 dissociates to monomers; and finally, v) the cycle repeats.
Here we use our differential surface density results to expand upon the model of Nagata, et al (Nagata et al., 2013) and propose a physics-based model for HDL assembly. In the model, we propose that the lipid reservoir formed by dimeric ABCA1: i) binds apoA-I, ii) encapsulates a monolayer created during PL translocation, iii) forms monolayer pleats under PL pump-induced increased lipid monolayer surface density, and iv) interacts with and is stabilized by apoA-I to form dHDL particles.
Results
Effects of lipid-induced increased surface density on periodic POPC bilayers
Vedhachalam et al (Vedhachalam et al., 2007) proposed that increased surface pressure on the outer monolayer (OM), created when ABCA1 pumps PL, drives apoA-I lipidation. To examine this hypothesis, we used coarse grained (CG) MD to explore the physical forces that might drive HDL biogenesis.
We first developed a method for modeling lipid-driven surface density increases created by PL-pumping of ABCA1 using CGMD of periodic planar bilayers and increasing the molecular density of the OM (Figure S2). PL molecules were uniformly removed from the inner monolayer (IM) and sufficient water molecules from the periodic box to reduce its size but maintain the IM at a constant lateral pressure of one atm (determined by measuring average area per POPC). Under these conditions the OM has an excess of PL (decreased average area per POPC) and therefore increased lateral packing density. We use δ to quantify this outer to inner surface density gradient as a ratio of the molecular densities.
We first simulated a periodic bilayer of pure POPC. As the OM surface density increased in the absence of apoA-I, a bilayer hemi-disc with reoriented polar headgroups covering its edge was gradually extruded from the OM (Figures 2A-C); the greater the increase, the larger the disc. During the transition from bilayer to bilayer disc at δ = 1.33, undulations formed that ultimately buckled (Figure 2B) and folded through a metastable intermediate (Figure 2C) to form a bilayer extending from the OM (Figure 2D), a transition completed in 1-2 ns. When d was increased to 1.54, the bilayer disc edge began rolling over to minimize the energy cost of the exposed lipids on the edge (Figure 2E) leading to the formation of an intermediate igloo-like structure (Figure 2F). Increasing δ to 1.82, the igloo-like intermediate gradually converted in 100 ns to a closed but still attached vesicle (Figures 2F-G). Similar results have been reported for CGMD monolayers at air-water interfaces subjected to increased surface density (Baoukina et al., 2008).
Figure 2. CGMD of a planar periodic bilayer showing OM pleating leading to formation of attached vesicle, driven by an increased outer surface density.
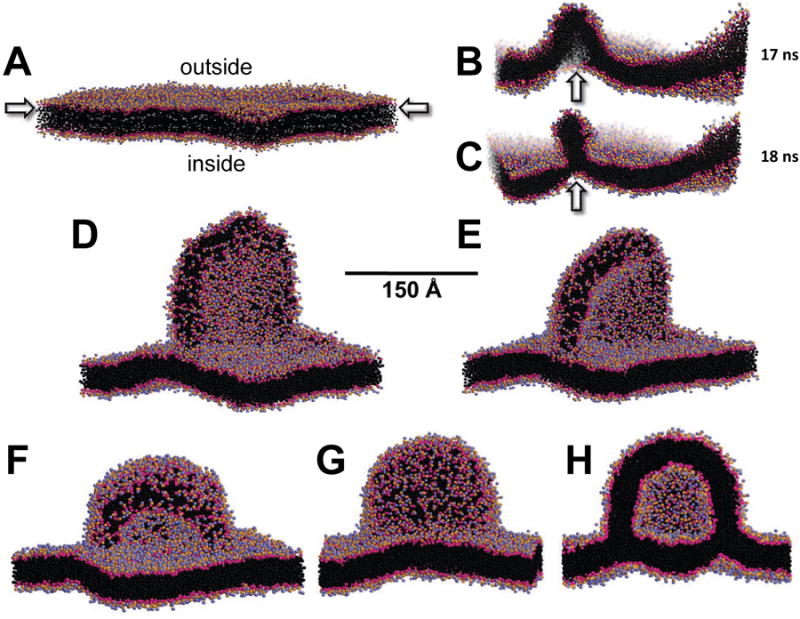
(A) Starting periodic bilayer containing 3840 POPC. (B-C) After creation of a TBDG of δ = 1.33, the OM buckles at 68 ns (B) to create a metastable intermediate that collapses at 72 ns (C) to form a budding disc. (D-F) The OM when subjected to increased δ = 1.54 and forms a hemi-disc pleat followed by rolling up the disc edges to create an igloo-like structure after (D) 80 ns, (E) 200 ns and (F) 300 ns. (G-H) The igloo-like structure, after increasing to δ = 1.82 and additional simulation for 100 ns, closes to create a bilayer-attached vesicle formed entirely from the OM: (G) surface view; (H) cross-sectional view. Polar moieties of POPC are shown as red and blue spheres and hydrocarbon tails as black spheres; water and ions are not shown. See also Figure S3.
We performed lipid bilayer pressure profile calculations on a POPC bilayer simulated at δ = 1.10 and, although the bilayer undulates to some extent, the results (Figure S3) clearly show that the OM is at a substantially higher pressure than the IM, supporting our assumption that an increased δ follows from an increased pressure gradient. We were not able to perform lipid bilayer pressure profile calculations at δ producing pleating (e.g. δ = 1.30) because under these conditions bilayer surfaces undulate significantly before buckling and pleating.
Effects of UC on bilayer pleating
Plasma membranes generally have a high UC content. For example, membranes from liver, epithelium, intestine and platelets have an UC to total lipid mole fraction, χc,ranging from 0.1-0.5 and an average χc of 0.28 (Sampaio et al., 2011). Since UC is thought both to increase the bending rigidity of bilayers and to act as a surface area buffer by flipping from one side of the bilayer to the other (Choubey et al., 2013), and since nascent dHDL contains UC (Duong et al., 2006), we examined the effects of UC on POPC bilayer pleating using MD. We created three periodic POPC-UC bilayers containing χc of 0.13, 0.26 and 0.36. To sample the effects of this range of UC content upon the ability of increased δ to create a bilayer pleat, we created two replicas of each. One replica was subjected to MD for 320 ns in the absence of a transbilayer density gradient (TBDG)—a symmetric bilayer (δ = 0)—and the second replica to MD for 200 ns with d = 1.33 and the results are summarized in Table 1 and Figure 3.
Table 1. Properties of Surface Density-Induced Monolayer Pleats.
| Mole fraction UC (χc) | Density differential (δ) | Simulation time (ns) | Number POPC | Number UC | Mole fraction UC (χc) | ΔUC inside | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
||||||||||
| pleat | outside | inside | pleat | outside | inside | pleat | outside | inside | ||||
| 0.0 | 1.00 | 320 | 0 | 1920 | 1920 | 0 | 0 | 0 | - | - | - | 0 |
| 0.0 | 1.33 | 200 | 515 | 1418 | 1427 | 0 | 0 | 0 | 0.00 | 0.00 | 0.00 | 0 |
| 0.13 | 1.00 | 320 | 0 | 1664 | 1664 | 0 | 256 | 256 | - | - | - | 0 |
| 0.13 | 1.33 | 200 | 266 | 1398 | 1248 | 22 | 201 | 223 | 0.08 | 0.13 | 0.15 | 33 |
| 0.26 | 1.00 | 320 | 0 | 1408 | 1408 | 0 | 506 | 518 | - | - | - | 0 |
| 0.26 | 1.33 | 200 | 124 | 1284 | 1056 | 40 | 431 | 426 | 0.24 | 0.25 | 0.29 | 35 |
| 0.36 | 1.00 | 320 | 0 | 1235 | 1235 | 0 | 674 | 696 | - | - | - | 0 |
| 0.36 | 1.33 | 200 | 72 | 1163 | 926 | 26 | 595 | 583 | 0.27 | 0.34 | 0.39 | 53 |
|
| ||||||||||||
| 0.36 | 1.00 | 1000 | 0 | 1235 | 1235 | 0 | 679 | 691 | - | - | - | 0 |
| 0.36 | 1.33 | 1000 | 109 | 1126 | 926 | 28 | 527 | 649 | 0.20 | 0.32 | 0.41 | 119 |
| 0.36 | 1.33 | 1400 | 119 | 1116 | 926 | 32 | 523 | 649 | 0.21 | 0.32 | 0.41 | 119 |
|
| ||||||||||||
| 0.13 | 1.25 | 0 | 0 | 1664 | 1331 | 0 | 256 | 202 | 0.00 | 0.13 | 0.13 | 0 |
| 0.13 | 1.25 | 400 | 0 | 1664 | 1331 | 0 | 190 | 268 | 0.00 | 0.10 | 0.17 | 66 |
| 0.13 | 1.25 | 800 | 0 | 1664 | 1331 | 0 | 179 | 279 | 0.00 | 0.10 | 0.17 | 77 |
| 0.13 | 1.25 | 1200 | 0 | 1664 | 1331 | 0 | 164 | 294 | 0.00 | 0.09 | 0.18 | 92 |
| 0.13 | 1.25 | 1600 | 0 | 1664 | 1331 | 0 | 148 | 310 | 0.,00 | 0.08 | 0.19 | 108 |
Figure 3. Effects on OM pleating of addition of UC to a POPC planar periodic bilayer.
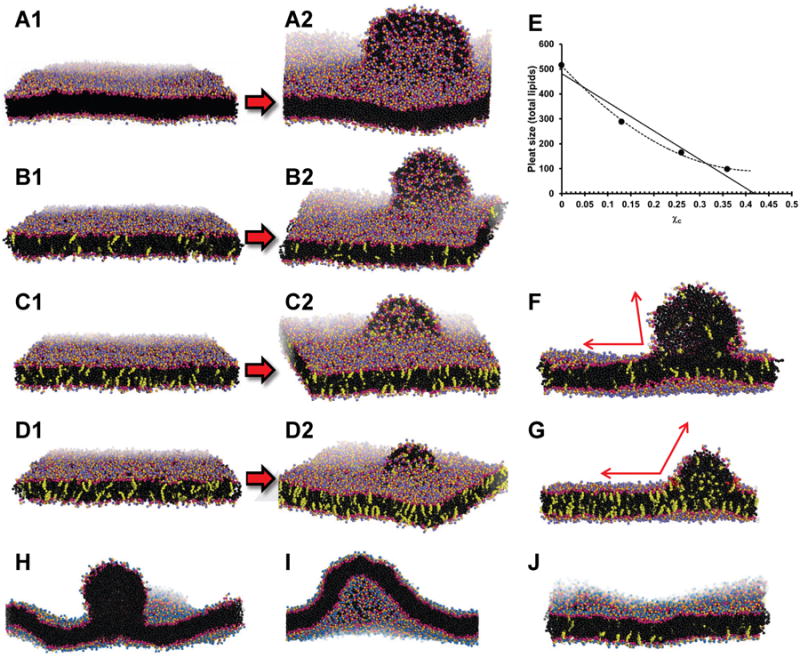
1 (A1 for example) shows the initial bilayer and 2 (A2 for example) shows the bilayer pleat created after simulation. (A) Pure POPC bilayer, δ = 1.33, at 200 ns. (B) POPC-UC bilayer, δ = 1.33, χc = 0.13, at 200 ns. (C) POPC-UC bilayer, δ = 1.33, containing χc = 0.26, at 200 ns. (D) POPC-UC bilayer, δ = 1.33, χc = 0.36, at 200 ns. (E) Plot of pleat size (total molecules of lipid) versus χc. The solid line is a linear trendline fit to data and the dotted line is a polynomial trendline fit to data. (F) Cross-section of a POPC-UC bilayer, δ = 1.33, χc = 0.13 at 200 ns. (G) Cross-section of a POPC bilayer, δ = 1.33, χc = 0.13 at 1000 ns. The angle of the pleat at its junction with the OM is indicated by red arrows. (H) Pleating of a POPC-alone bilayer at 200 ns with δ = 1.30. (I) Protuberance created in a POPC-alone bilayer at 600 ns with δ = 1.25. (J) Flattening of the protuberance in a POPC bilayer by UC flipping from OM to IM at 1600 ns with δ = 1.25 and χc = 0.13. UC is shown as yellow spheres; POPC and solvent as in Figure 2.
For a pure POPC bilayer with δ = 1.33, at the time of the metastable transition at 76 ns the pleat contained approximately 160 POPC, and after full monolayer pleat formation at 200 ns the full pleat contained 515 POPC (Table 1). After initial 320 ns of MD of each of the three replicas containing differing concentrations of UC, as the replicas subjected to increased δ also increased χc from 0 to 0.36, the size of the bilayer pleat formed after 200 ns of MD progressively decreased with increasing χc (Figure 3) from 515 POPC at χc = 0, to 72 POPC at χc = 0.36 (Table 1). Of particular note, in response to an increasing δ, an increasing number of UC, 33 to 53, flipped from the OM to the IM (Table 1).
Because condensation of the area per POPC by UC in our MD was likely not equilibrated (Marrink et al., 2007), we increased the simulation time for the bilayers containing χc = 0.36 —the symmetric bilayer and the bilayer with the TBDG—to 1 μs. With longer simulations, both bilayers condensed further and the size of the bilayer pleat in the replica with a TBDG increased from 72 to 109 POPC. Of special note, there was an additional flipping of 66 UC to the IM. After an additional 400 ns there was no additional UC flipping and the pleat increased only slightly in size, from 109 to 119 POPC (Table 1). This suggests that the UC concentration in the IM, χc = 0.41, is saturated to the point that additional flipping of UC has a high energy cost, i. e., is kinetically trapped. In Figure 3E we plotted pleat size as total lipid count against χc. When the data points are fitted to a linear trendline, the χc axis is intercepted at approximately 0.41; when the data points are fitted to a polynomial trendline, the slope of the curve approaches 0 at approximately χc = 0.41. These results are compatible with kinetic trapping of UC in the inner bilayer at χc ≈ 0.41, an observation that, to the best of our knowledge, has not been made before.
The membrane UC also affects bilayer bending rigidity. Figure 3F shows a cross-section of the pleat induced by δ = 1.33, on a bilayer with χc = 0.13; the pleat can bend enough to form an acute angle (red arrows) with the bilayer surface. However, Figure 3G shows that increasing χc to 0.36 increases the bending rigidity of the bilayer so that the pleat can only bend enough to form an obtuse angle (red arrows) with the bilayer surface.
To test whether UC flipping can prevent pleating in bilayers at lower χc, we simulated bilayers at increasing δ and compared pleating in the presence and absence of UC. An UC concentration of χc = 0.13 was used since nascent dHDL produced by ABCA1 overexpressed in cultured cells have an UC concentration of approximately χc = 0.10 (Duong et al., 2006).
We found a break in the behavior of the UC-free bilayer between δ = 1.25 and 1.30. After 50 ns simulation, the UC-free bilayer pleated at δ = 1.30 (Figure 3H) but, although after 600 ns it formed a pronounced protuberance (Figure 3I), the UC-free bilayer did not pleat at δ = 1.25. As proof of concept that the flipping of UC from the OM to the IM can dampen or decouple a TBDG, the UC-containing bilayer, χc = 0.13, at δ = 1.25 was essentially flat after 1600 ns (Figure 3J). This inhibition of a bilayer protuberance was the result of 108 UC flipping to the IM (Table 1), producing a net difference of 216 UC between OM and IM to partially decouple the TBDG.
Effects of apoA-I on bilayer pleating
We then examined the effects of apoA-I on pleating of bilayers. First, we inserted a linear antiparallel α helical double belt of apoA-I (Segrest et al., 1999) on the outer surface of the periodic POPC bilayer (Figure 4A and Figure S4A). Using δ = 1.18, substantially lower than the δ = 1.30 required for pleating of POPC-alone bilayers, the OM pleated after 400 ns to form a disc whose edge was lined by the apoA-I double belt, creating a nascent HDL-like hemi-disc (Figures 4B-C) that converged by approximately 300 ns (Figure S4B). This lowering of the δ required for pleating from 1.30 to 1.18 represents the surface density increase produced by the amphipathic helix effect (see below).
Figure 4. CGMD showing stabilization by apoA-I of OM pleats created from a planar periodic bilayer by an increased TBDG δ.
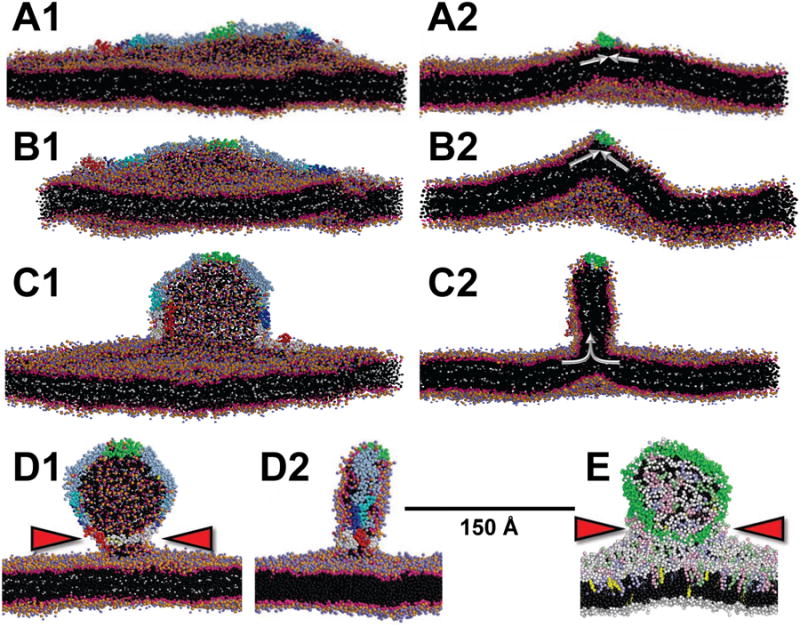
(A) 100 ns simulation of the initial bilayer (without deletions) showing bending created by the wedge-effect of OM insertion of the amphipathic helical domains of apoA-I. (B) At 100 ns with d = 1.18 showing increased bilayer bending. (C) At400 ns with δ = 1.18 showing sudden transformation of increased bilayer bending into bilayer pleating and formation of a hemi-disc whose edges are lined by the apoA-I belt. (D) θ-apoA-I model (see Figure S4C) attached to the OM of a bilayer at 8 μs with δ = 1.18. Red arrowheads indicate the lipid stalk. Left hand panels are viewed orthogonal to the long axis and right hand panels are cross-sectional views down the long axis of apoA-I; white arrows indicate the OM surface density vector. POPC and solvent as in Figure 2. Tandem helical repeats of apoA-I are: repeat 1 (residues 44-65), blue; repeat 5 (121-142), green; repeat 8 (187-208), cyan; C-terminal repeat 10 (220-241), red. The N-terminal domain (1-43) is white and the remainder of apoA-I is gray. (E) θ apoA-I model attached to the OM of a mixed lipid bilayer at 12.8 μs with δ = 1.33. Polar moieties of POPC are shown as white spheres and hydrocarbon tails as black spheres; POPE, pale blue spheres; POPS, pink spheres; SM, pale green spheres; UC, yellow spheres; protein, green spheres. Red arrowheads indicate the lipid stalk. See also Figure S4 and Movie S1.
Since neither longer simulations nor higher δ was able to achieve a pleat more extensive than a hemi-disc (Figure S4E), we explored the effects of a series of different starting apoA-I conformations. These various models, excepting one, did not progress beyond formation of hemi-discs. In one exception, a circular double belt form (Borhani et al., 1997) whose flat surface was parallel to the surface of the outside monolayer (Figure S4F) produced bizarre structures attached to the OM that were divided into two topologically distinct lipid monolayers: inside (i) and exterior to (e) the double belt ring (Figure S4F). Although interesting, because of topological complexities we excluded the circular structures from current consideration.
The most successful model was what we refer to as the theta (θ) conformation in which the N- and C termini of the apoA-I double belt were held together by harmonic constraints and the dimer was laid out on the bilayer surface like the Greek letter θ (Figure S4C). The logic of this starting structure was that the N- and C termini of each end of the apoA-I double belt did not, with the exception of the circular double belt model, remain in contact without a constraining force. As an example, simulation of the θ structure without harmonic constraints at δ = 1.25 resulted in formation of a distorted hemi-disc (Figure S4G). Significantly, the θ conformation with the N- and C termini of the apoA-I double belt held together by harmonic constraints, at δ = 1.18, resulted in a complete apoA-I double belt dHDL attached by a lipid stalk (Figure 4D and Movie S1); this simulation converged by 3 μs (Figure S4D).
The constrained θ model would be topologically equivalent to strong bonding of the N- and C-termini of the apoA-I double belt to extracellular domains of dimeric ABCA1. The two-fold axis of symmetry created might be related to the observation that apoA-I binding requires ABCA1 dimerization (Nagata et al., 2013).
Since nascent dHDL particles created by cells over-expressing ABCA1 possess a mixed lipid composition that include, in addition to PC and UC, phosphatidylserine (PS), phosphatidylethanolamine (PE) and SM, we created a bilayer with a pure POPC inside monolayer and a mixed lipid outside monolayer molar composition of POPC:POPS:POPE:UC:SM = 60:10:10:10:10—similar to the nascent dHDL particles (Duong et al., 2006)—for CGMD with apoA-I and a δ > 0. The mixed lipid bilayer required δ = 1.33 for pleating, substantially greater than the δ = 1.18 required for pleating of apoA-I/POPC bilayers (Figure 4E). Further, it is worth noting that, in spite of the presence of SM, UC flipping occurred (90 flipped inside and 92 remained outside). However, the final hemi-discs were essentially equal in size. Although, in our studies, dHDL size is more a function of δ than lipid composition, the size of nascent HDL particles assembled by ABCA1 could be regulated both by lipid composition and by ATP-driven pumping pressure.
In vitro studies of the effects of amphipathic helix insertion on PL bilayers
Insertion of amphipathic helices into bilayer surfaces is known to increase the bilayer surface pressure or density (Datta et al., 2001). Incubation of certain amphipathic helical proteins and peptides with dimyristoyl PC (DMPC) vesicles spontaneously disrupt the vesicles to form bilayer discs (Li et al., 2004). A number of years ago we postulated that an increase in surface density is directly responsible for disruption of intact DMPC vesicles by amphipathic helices (Segrest, 1976).
Although apoA-I is generally assumed not to disrupt POPC vesicles, this assertion has not been studied systematically. To explore the relationship of amphipathic helices to the increased surface density and bilayer pleating observed in our in silico CGMD, we performed in vitro studies of the interactions of apoA-I and a synthetic amphipathic peptide mimetic of apoA-I, 4F (Datta et al., 2001), with lipid vesicles. In these studies, we incubated apoA-I—with a monolayer surface exclusion pressure of 34 dyn/cm (Palgunachari et al., 1996)—and 4F— with a higher monolayer surface exclusion pressure of 40 dyn/cm (Datta et al., 2001)—with large unilamellar vesicles (LUV) of two compositions: pure POPC and mixed lipids (Duong et al., 2006).
Size-exclusion column chromatography showed that apoA-I associated with both types of LUV (Figure 5A). As expected from the difference in monolayer exclusion pressures, 4F causes significantly more disruption—measured by carboxyfluorescein leakage—upon interaction with both types of LUV than apoA-I (Figures 5B-C). The leakage induced in POPC LUV by 4F indicates vesicle disruption (Figure 5B) but leakage induced by apoA-I in LUV of either composition, while compatible with disordering of surface lipids, does not suggest much, if any, vesicle disruption (Figures 5B-C).
Figure 5. In vitro studies of the interaction of apoA-I and the amphipathic peptide 4F with PL vesicles.
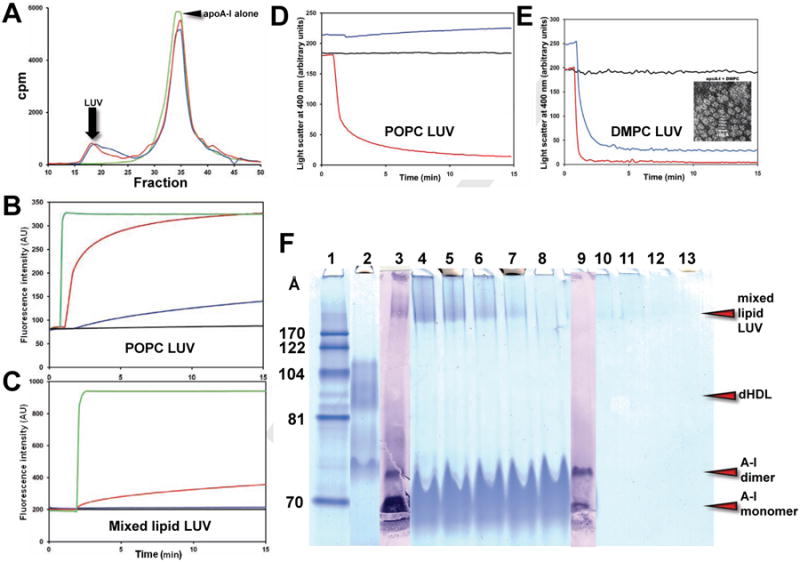
(A) Size exclusion column chromatography of the products of interaction of POPC and mixed lipid LUV after 24 hr incubation with apoA-I at 37°C. ApoA-I alone, green; apoA-I plus POPC LUV, red; apoA-I plus mixed lipid LUV, blue. (B) Kinetics of carboxyfluorescein leakage from POPC LUV. LUV alone, black; LUV plus 4F, red; LUV plus apoA-I, blue; TX-100, green. (C) Kinetics of carboxyfluorescein leakage from mixed lipid LUV. LUV alone, black; LUV plus 4F, red; LUV plus apoA-I, blue; TX-100, green. (D) Kinetics of disruption of POPC LUV. LUV alone, black; LUV plus 4F, red; LUV plus apoA-I, blue. (E) Kinetics of disruption of DMPC LUV. LUV alone, black; LUV + 4F, red; LUV plus apoA-I, blue. Insert, negative stain EM of DMPC:apoA-I discs. (F) NDGGE of apoA-I incubated with mixed lipid LUV for 24 hr at 37°C showing association but little or no free dHDL particle formation: Lane #1. MW markers; 2. dHDL in the 80-110 Å diameter range; 3. ApoA-I western blot of lane #4; 4. Lipid:apoA-I = 3:1, w:w;. 5. Lipid:apoA-I = 2:1, w:w; 6. Lipid:apoA-I = 1:1, w:w; 7. Lipid:apoA-I = 0.5:1, w:w; 8. ApoA-I alone; 9. ApoA-I western blot of lane #9; 10. Lipid of lane #4; 11. Lipid of lane #5; 10. Lipid of lane #6; 10. Lipid of lane #7. All lanes except #3 and #9 are stained using a Colloidal Blue Staining kit.
As shown in Figure 5D, incubation of 4F with POPC LUV disrupts the vesicles within 15 min, showing that this peptide, with a higher surface exclusion pressure (Datta et al., 2001), provides an intrinsic driving force sufficient for disc formation. However, apoA-I, with a lower surface exclusion pressure (Palgunachari et al., 1996), does not disrupt POPC; in fact, a slight increase in light scattering over 15 min suggests that apoA-I may actually induce slow fusion in POPC LUV (Figure 5D). Finally, both 4F and apoA-I disrupt DMPC LUV to form discs (Figure 5E); 4F acts much more rapidly. These results demonstrate an intrinsic driving force for disc formation by apoA-I, with a lower surface exclusion pressure, which is substantially less than that of 4F, with a higher surface exclusion pressure.
The NDGGE analyses in Figure 5F shows that 24 hr incubation of apoA-I with mixed lipid LUV forms little or no free dHDL particles. However, since the synthetic amphipathic α helical peptide, 4F, with a higher monolayer surface exclusion pressure than apoA-I disrupts POPC vesicles to form dHDL particles (Anantharamaiah et al., 1985), 4F is able to produce an amphipathic helix-induced driving force sufficient for vesicle disruption and disc formation, compatible with our contention that surface density is the driving force for bilayer disruption and disc formation.
Even though apoA-l clearly interacts with POPC vesicles (Figure 5), after 30 min the protein has little observable effect on POPC small unilamellar vesicles (SUV) imaged by negative stain EM; no free dHDL are visible in either the control (Figure 6A) or the 30 min sample (data not shown). At 3 hr, however, uniform 100 Å diameter circular densities appear on the surface of the SUV (Figure 6B2). A few free dHDL particles are also seen. After 24 hr incubation at the higher apoA-l concentration, the shapes of the SUV have been dramatically changed and sizes increased, suggesting fusion (Figure 6B3).
POPC and mixed lipid SUV were incubated with apoA-l for 3 h at 37°C and examined by cryonic EM (cryoEM). Several representative cryoEM images in Figure 6C2 shows what appear to be discoidal particles approximately 100 Å in diameter extruded from POPC SUV surfaces. CryoEM tomography images of mixed lipid SUV (Figures 6D- E), showing discoidal particles apparently attached to the surface of vesicles after incubation with apoA-l, support this interpretation.
Figure 6. EM studies of effects of incubation at 37°C of apoA-I with POPC SUV.
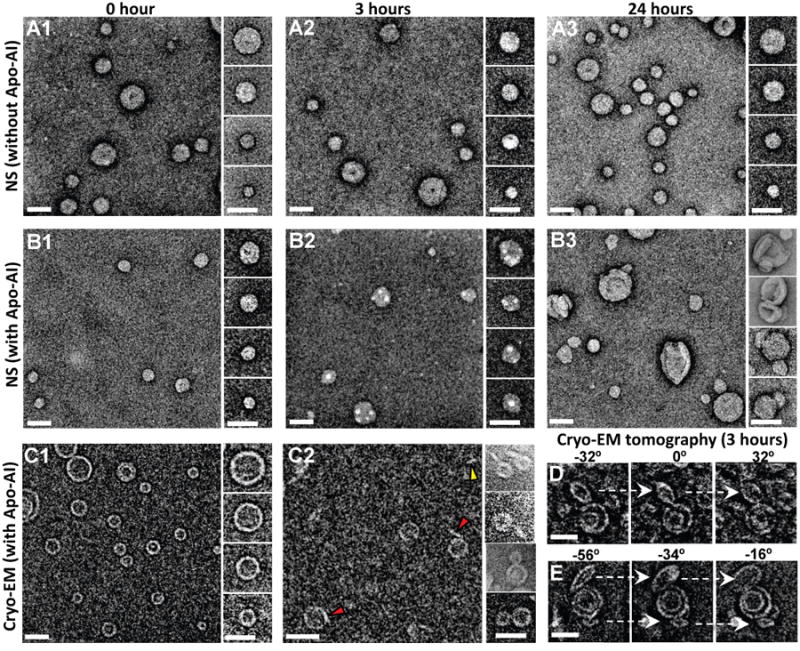
(A-B) Negative stain EM images of POPC SUV. (A1-A3) Survey views and selected particles of POPC SUV incubated at 37˚ C for 0, 3, and 24 hours. (B1-B3) Survey views and selected particles of POPC SUV incubated with apoA-I at 37° C for 0, 3, and 24 hours (molar ratio of POPC SUV:apoA-I is 1:5). (C-E) CryoEM images (inverted contrast) of POPC SUV. (C1, C2) Survey views and selected particles of POPC SUV incubated with apoA-I at 37° C for 0 and 3 hours (molar ratio of POPC SUV:apoA-I is 1:5). Red arrowheads indicate discoidal structures attached to SUV, yellow arrowhead indicates a free discoidal structure. (D, E) Two represented liposome particles attached with discoidal particles were shown by selected tilting images of cryoEM tomography of the same 3-hour sample (tilt angles shown above). Scale bars, 50 nm.
CGMD of amphipathic helix-induced increased surface density on SUV
To address the driving force for the circular densities induced by incubation of apoA-l with SUV, we created 200 Å diameter models of DMPC and POPC SUV by CGMD. Since the synthetic amphipathic helical peptide 4F disrupts both POPC and DMPC vesicles, we added 135 4F to the surface of both types of SUV; Figure 7A shows the starting structure for the DMPC vesicle. CGMD (10 (μs) resulted in formation of discoidal bilayer pleats, Figures 7B-C, which originated entirely from the OM of both types of SUV; these pleats were lined on their exposed edge by multiple molecules of 4F. The DMPC vesicle (Figure 7C) formed a considerably larger disc than the POPC vesicle (Figure 7B); the sizes of these vesicle-attached discs relate to the lower and higher hydrophobicity of the acyl chains of DMPC and POPC, respectively, not to the final size of detached discs. Thus a higher concentration of 4F on the POPC vesicle surface would be required to produce a disc with a size comparable to that on the DMPC vesicle. The 4F-lined disc was attached to the DMPC SUV surface by a stalk, while the smaller 4F-lined disc attached to the POPC SUV as a hemi-disc. These results show that increased surface density induced by insertion of 4F between surface headgroups associating with the surface of lipid vesicles results in extrusion of surface monolayer lipids to form discoidal bilayer pleats. We then added multiple copies of 4F to the surface of a periodic DMPC bilayer. CGMD for 17 μs resulted in formation of peptide-stalk-attached discoidal bilayer pleats (Movie S2) resembling the apoA-l protrusion in Figure 4D.
Figure 7. CGMD of the interaction of apoA-I and 4F with the surface of SUV.
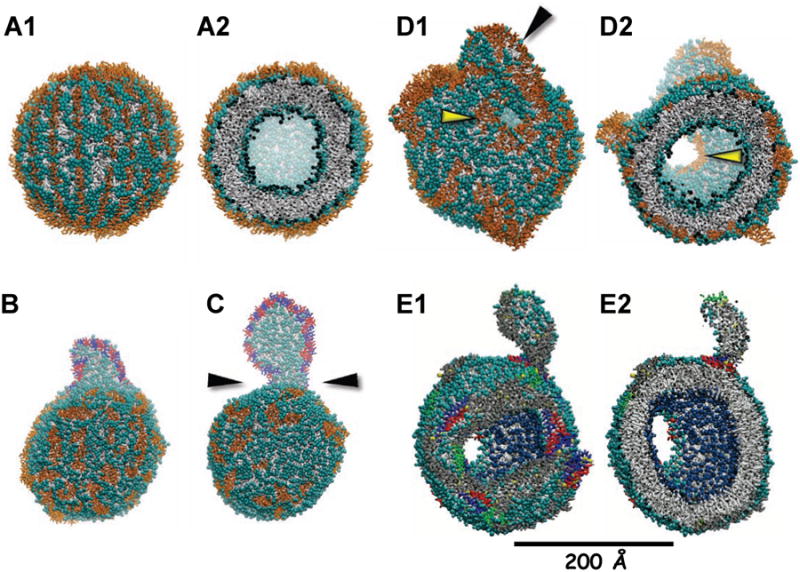
(A) Starting DMPC SUV with 2422 DMPC and 135 4F associated with the surface. (A1) Surface view. (A2) Cross-sectional view. (B) POPC SUV with 2422 POPC and 201 4F at 10 μs. (C) DMPC SUV with 2422 DMPC and 201 4F at 20 μs. (D) DMPC SUV with 2422 DMPC and 268 4F at 18 μs. (D1) Surface view. (D2) Cross-sectional view. Yellow arrowheads show bilayer holes lined by 4F, black arrowhead shows a discoidal pleat. (E) DMPC SUV with 2422 DMPC and 7 apoA-I dimers at 20 μs. (E1) Surface view. (E2) Cross-sectional view. Polar moieties of POPC and DMPC are shown as cyan spheres and the hydrocarbon tails as gray licorice; water and ions are not shown. 4F shown as gold licorice, except (B-C) where 4F associated with the discoidal pleats are red and blue licorice. ApoA-I are shown as gray licorice except residues 1-43, blue; repeat 5 (121-142), green; C-terminal repeat 10 (220-243), red. See also Movie S2.
To increase the surface density, we increased the number of 4F on the DMPC surface from 135 to 268 and performed CGMD for 18 μs. The results in Figure 7D show that the SUV is greatly distorted: pores lined by peptide have been created in the bilayer and multiple hemi-bilayer disc pleats are formed. We then placed 7 apoA-l antiparallel double belt dimers of apoA-l on a DMPC SUV, subjected it to CGMD for 20 μs and the results are shown in Figure 7E. Under these conditions, apoA-l is able to extensively distort the DMPC SUV, creating bilayer pores and pleats, producing results similar to the distortions seen by negative stain EM (Figure 6D) when apoA-l at high concentration is incubated with SUV for 24 hrs. The kinetics of interaction of apoA-l with POPC SUV was too slow to show comparable changes (data not shown).
Discussion
Structural effects of TBDG
Phillips and colleagues (Vedhachalam et al., 2007) suggested that ABCA1-induced lipid surface pressure (density) increase is the basis for driving HDL assembly through evagination of surface membranes to form vesicles containing lipid from both inner and outer monolayers that, because of their high surface curvature, are susceptible to interaction with and disruption by apoA-I to form dHDL particles.
Using CGMD we tested the Phillips model and show that increased OM surface density, whether driven by a PL pump such as ABCA1 or by surface insertion of amphipathic α helical proteins, initially induces membrane protuberances and undulations that ultimately buckle and fold through a metastable intermediate to form a discoidal bilayer pleat extending from the OM. In the presence of amphipathic helical proteins, the disc edges are covered by the protein; in their absence, as the hemi-discoidal bilayer pleat grows it finally curls over to form membrane-attached vesicles.
Supporting our findings, similar results have been reported by Baoukina, et al (Baoukina et al., 2008) for CGMD of PL monolayers at air-water interfaces. These authors conclude that, although not seen in their simulations, separation of hemi-vesicles from the monolayer is favorable for larger vesicles, but can be kinetically hindered. Similar events occur in cells overexpressing ABCA1: in the presence of apoA-I, nascent dHDL are released; in the absence of apoA-I, lipid vesicles are released (Duong et al., 2006).
Although not explicitly addressed in our study of bilayers, temperature, acyl chain length and degree of unsaturation and head group size are known to affect the ability of a monolayer to pleat (Baoukina et al., 2008). We do find that mixed lipid bilayers with the approximate composition of nascent dHDL particles created by cells over-expressing ABCA1 increase the δ required for pleating of POPC and apoA-I from 1.18 to 1.33. Since we show that UC increases the δ required for pleating of POPC vesicles (Figure 3), part of the requirement for an increased δ for mixed lipid bilayers must be due to the presence of UC.
Further supporting our findings, when cholesterol-loaded fibroblasts are incubated with apoA-I, followed by incubation with immunogold-labeled anti-apoA-I, thin-section EM show cell-surface associated electron-opaque ovoid structures 100–300 Å in diameter that protrude from the plasma membrane and are positive for apoA-I (Figures 8A-B). These structures are similar in diameter to apoA-I-lined bilayer pleats—particularly when dimeric ABCA1 is included (Figure 8C)—produced when apoA-I is simulated with bilayers possessing a significant TBDG (Figure 8C). The cholesterol-loaded fibroblasts also show occasional apoA-I positive protrusions that are 500 Å or so in diameter and more vesicle-like (Figure S5A). These larger structures resemble the membrane-attached vesicles produced, in the absence of apoA-I, by our MD of a bilayer with an increased TBDG (Figure S5B).
Figure 8. Proposed mechanism for creation of a TBDG by the ABCA1 lipid pump.
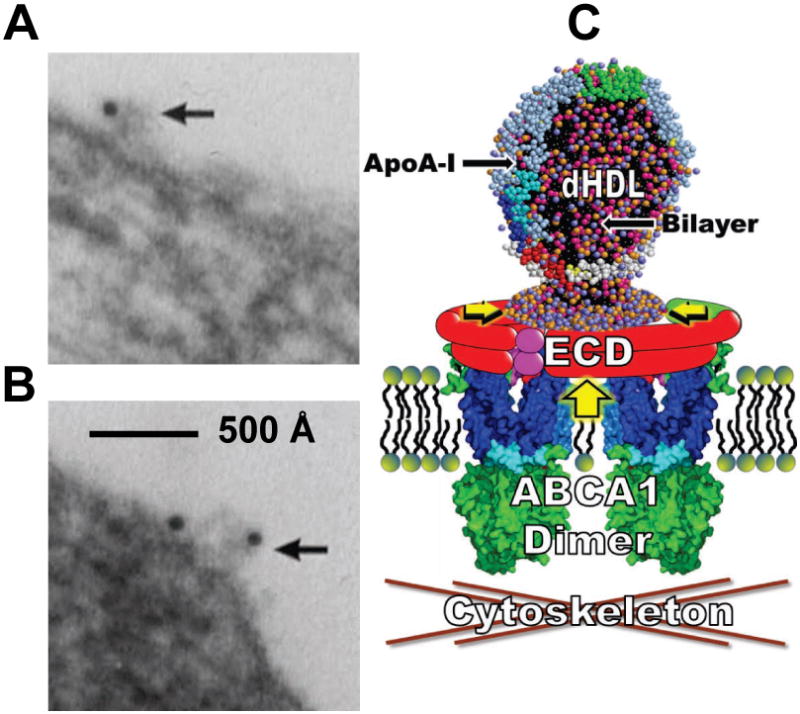
(A-B) Representative morphology of predominant form of apoA-I binding to fibroblasts. (C) Diagram of the dimeric ABCA1 lipid reservoir hypothesis of Nagata, et al (Nagata et al., 2013) modified to show the lipid reservoir in the presence of apoA-I containing a pleated dHDL particle. Large vertical yellow arrow, direction of PL gradient pump; horizontal smaller yellow arrowheads, direction of OM surface density gradient within reservoir. ECD, extracellular domains—different colors illustrate the fact that each ABCA1 monomer contains at least three ECD. See also Figures S5 and S6.
Although, in the absence of amphipathic helical proteins or peptides, membrane-attached vesicles result from TBDG, our results differ from the Phillips hypothesis (Vedhachalam et al., 2007) since we show that a sufficient increase in surface density induces bilayer pleating—or vesicle formation in the absence of amphipathic helical acceptors—that is derived entirely from the outer membrane monolayer (Duong et al., 2006).
Using three in vitro approaches—column chromatography, lipid vesicle leakage and NDGGE—and three EM imaging methods—negative stain, cryoEM and thin sectioning—we show that apoA-I interacts with both POPC and mixed lipid SUV to modify the surface structure of the vesicles and induce leakage, experimental results that tend to validate the MARTINI force field used for our CGMD. These interactions with apoA-I are associated with only a small amount of vesicle disruption to form unattached dHDL particles, even when incubated for as long as 24 hrs. The predominant effect is that apoA-I insertion into the surface of POPC vesicles forms surface-attached pleats or hemi-discs within 3 hr of incubation. This process is kinetically trapped since only a few discs are released into the aqueous phase after 24 hr, suggesting that a driving force would be required for release of apoA-I discs created by ABCA1 in a biologically relevant time frame. The kinetic issue is compounded by membranes composed of mixed lipids.
Mechanisms whereby ABCA1 can create a TBDG
We propose that ABCA1 accelerates nascent HDL formation via active transport of PL from the inner to the outer half of the membrane bilayer to create a transbilayer surface density or pressure gradient highest on the outer membrane surface. For this to occur, however, there should be some mechanism to entrap the lipid pumped to the outside. Otherwise, the concentration gradient seemingly would dissipate into the surrounding membrane.
Since ABCA1 has been shown to reside between lipid rafts (Landry et al., 2006), one possibility is that the surrounding rafts entrap the pumped lipid. However, our CGMD suggest a potential problem with this mechanism of surface density entrapment. When UC, present in plasma membranes, is included in our simulations of surface density asymmetry, UC flips from the high to the low density monolayers, to dampen or uncouple the density gradient (Figure 3I and Table 1). The presence of UC in coupled monolayers subjected to a TBDG by PL pumping will thus slow or inhibit the formation of OM pleats, making any mechanism involving coupled bilayers, including the Phillips model (Vedhachalam et al., 2007), unlikely. However, in spite of the fact that UC flipping occurs in our simulations of mixed lipids, it is nevertheless possible that certain biological lipids, such as SM in high concentrations, might counteract the uncoupling effects of UC.
To the best of our knowledge, our report represents the first direct confirmation of the proposal (Choubey et al., 2013) that UC acts as a surface area buffer by flipping from one side of the bilayer to the other. These dampening effects of UC on formation of a TBDG are a novel argument for disc formation by an entrapped and decoupled OM; the monolayer MD studies by Baoukina, et al (Baoukina et al., 2008) show that monolayers under increased surface pressure (density) are able to form pleats. The possibility that ABCA1 itself provides the mechanism for lipid entrapment via formation of a lipid reservoir has the advantage that it could result in an pressurized OM decoupled from the density dampening of UC.
On the basis of the results presented here and by Nagata, et al (Nagata et al., 2013), we propose the model shown in cartoon form in Figure 8C for the mechanism whereby ABCA1 and apoA-I interact to drive formation of nascent dHDL particles. We suggest that the large extracellular domains of ABCA1, totaling about 2000 residues in the dimeric form—and containing numerous amphipathic helices with high lipid affinity (Figure S6)—create a reservoir enclosing a growing lipid monolayer. As the PL monolayer expands via ABCA1 lipid translocation and the surface pressure increases (Figure 8C), the pressurized lipid monolayer collapses to form a discoidal bilayer pleat. ApoA-I, if present, binds to the extracellular domains in a position to accelerate and stabilize the discoidal pleat (Figure S5C), thus creating nascent dHDL particles (Figure 8C). If apoA-I is not present, PL pumping by ABCA1 creates pleats that fold into vesicles that then are shed into the media (Figure S5B). This model avoids the potentially significant problem of uncoupling of TBDG by the flipping of UC between coupled monolayers of a membrane bilayer.
This model of a decoupled entrapped monolayer compressed by the ABCA1 PL pump begs the question of the origin of UC in nascent dHDL. Since there is no evidence that ABCA1 pumps UC, it seems likely that, during ABCA1 dimerization, UC is entrapped from the surrounding lipid monolayer as the lipid reservoir assembles, likely aided by the pumping of SM into the reservoir.
The presence of UC in biological membranes has been shown to have one additional effect; it increases the bending rigidity of the bilayers (Figures 3F-G). The effect of an increased bending rigidity is also shown in our MD where the size of the pleat is inversely proportional to the UC concentration (Figures 3A-D and Table 1). Thus the concentration of UC in the lipid monolayer reservoir shown in Figure 8C presumably would affect the rate of formation and size of nascent dHDL, a process that could have consequences for RCT and inhibition of atherosclerosis.
The calculated energetics of lipid stalks (Kozlovsky and Kozlov, 2002) and the persistence of apoA-I dHDL particles attached to POPC vesicles (Figures 4-6), presumably by a lipid stalk (Figures 4 and 6), suggest that, as has been shown for membrane fission (Bashkirov et al., 2008), protein-driven scission of the residual lipid stalk of the θ model, especially the larger one produced by mixed lipid bilayers (Figure 4E), may be required to accelerate the release of attached discs from the cell membrane to a biologically relevant rate. Even the θ model run at higher δ forms a thicker lipid stalk but does not fission (Figure S4H). Clearly, the kinetics of this process is slow since dHDL particles form on the membrane surface but are released only slowly into the aqueous phase after 24 hr incubation of apoA-I with vesicles containing a membrane-like composition. Even the highly amphipathic peptide, 4F, takes almost 15 min to form free discoidal particles from POPC SUV and is even less effective against mixed lipid vesicles (Figure 4C). This suggests that a driving force would be required both for creation of a TBDG and for release of apoA-I discs from cell membranes within a biologically relevant time-frame. Besides driving an increase in surface density, ABCA1 itself might drive scission of any residual lipid stalks. Although we performed MD on a series of bilayers containing lipids promoting both positive (PS) and/or negative (PE) membrane curvature without observing scission of the residual lipid stalk during the time-frame of the simulations (data not shown), we cannot rule out the possibility that fission promoting lipids present in biological membranes, such as free fatty acids or lysoPC, would provide the driving force for disc fission.
Conclusions
The current study for the first time, via a combination of computational and in vitro methods, provides a model for the physical forces driving HDL biogenesis. Our proposal that pleating of a decoupled OM of a membrane enclosed by the extracellular lipid reservoir of ABCA1 is the basic physical force driving HDL assembly is relevant to ongoing efforts by pharmaceutical companies to develop drugs that accelerate HDL biogenesis. Our pleating model for the mechanism whereby ABCA1 drives nascent dHDL formation is testable by molecular modeling, site-directed mutagenesis and cryoEM tomography.
Experimental Procedures
MD of Periodic Bilayers
For CGMD we used Gromacs version 4.0 (Hess et al., 2008) and the MARTINI force field for lipids (Marrink et al., 2007) and proteins (Monticelli et al., 2008). Non-bonded van der Waals and electrostatics interactions were truncated at 12 Å. The PME treatment of long range electrostatic interactions was employed. The temperature and pressure were stabilized at 310 K and 1 atm, respectively, using Berendsen's baths (Berendsen et al., 1984). Trajectories were updated every 10 ps. Analysis and imagery were performed with Visual Molecular Dynamics (Humphrey et al., 1996).
For CGMD bilayers without protein, we used a periodic bilayer of 3840 POPC (Jones et al., 2009). To create an increased OM surface pressure by increasing the molecular density, POPC from the IM as well as solvent were removed (Figure S2). The remaining fractions of the POPC in the IM and the solvent were calculated as follows: the volume of the roughly cubical periodic box is approximately equal to the area of one square side raised to the 3/2 power. The lateral density of the IM quickly condensed to one atm while that of the OM increased.
The apoA-I double belt was inserted into the bilayer with the linear protein axes approximately 8.6 Å above the average height of the OM, measured by the z coordinates of the PO4 beads. This orientation satisfied two conditions: i) no lipid beads were displaced; ii) upon hydration, no water beads were inserted between protein and the bilayer surface. For apoA-I experiments, the first 32 residues of the N-terminus were rotated to orient their hydrophobic face towards the lipid surface. For the θ model, the N and C termini were linked by pseudo-bonds.
The secondary structure used for apoA-I CGMD was determined as described (Gu et al., 2010) with all Pro set to turn, all Ser set to bend, the first two N- and C-terminal residues set to coil, and Gly-Gly-Ala (185-187) set to coil-coil-helix. This increased protein flexibility, since MARTINI requires fixed secondary structure. For bilayers containing UC, POPC was mutated to UC and simulated for 320 ns before deletions.
MD of Vesicles
2000 steps of energy minimization were performed to remove steric clashes. Periodic Boundary Conditions using a solvation box with 40 Å of water (Hess et al., 2008) was employed. The total number of CG beads reached a maximum of approximately 520,000 (in the system with 7 apoA-I dimers). SUV were created by CGMD of bilayer discs containing 2422 POPC or DMPC (Markvoort et al., 2009).
Generation of the DMPC SUV with apoA-I dimers
The initial system containing 2422 DMPC molecules was subjected to the docking of 2 apoA-I dimers. We inserted more apoA-I dimers to generate vesicles with an increased apoA-I molar concentration.
Vesicles with apoA-I and the apoA-I mimetic peptide, 4F, and protein secondary structure assignments
CG 4F terminal residues were assigned as random coil and the remaining sixteen as α helical (Catte et al., 2008). 4F were manually inserted into the OM of the DMPC and POPC SUV or a periodic bilayer containing 2500 DMPC. The starting structures were subjected to CGMD as described (Jones et al., 2011).
To sample the effects of protein flexibility on the interaction of apoA-I dimers with DMPC, we employed two protein secondary structure assignments. First, the majority of protein residues were α-helical and the remaining residues were β-turn and random coil (Jones et al., 2010). Second, the more flexible assignment as described above (more residues in β-turn and random coil) was employed (Gu et al., 2010).
Vesicle Clearance and Leakage
POPC, DMPC and mixed lipid (POPC:POPS:POPE:UC:SM = 60:10:10:10:10) films were made by dissolving appropriate amounts of lipids in chloroform and dried under a nitrogen stream to deposit the lipid as a thin film on the test tube wall. The films were hydrated with PBS, vortexed extensively and the mixture sonicated for five 30 min cycles in a bath sonicator (Aquasonic 50T, VWR Scientific Products). The homogeneous lipid suspensions were then passed five times through two stacks of 0.1 μm or 0.05μm polycarbonate filters (Nucleopore Filtration Products, Pleasanton, CA) in a barrel extruder (Lipiex Biomembranes, Vancouver, Canada) to obtain LUVs and SUVs respectively.
4F was synthesized as described (Datta et al., 2001). The disruption of POPC and DMPC 1000 Å diameter LUV by apoA-I and 4F at 1:10 protein:lipid molar ratios was studied as described (Datta et al., 2001). Carboxyfluorescein was entrapped in POPC LUV and leakage monitored at λem/ex = 492nm/517nm as described (Epand et al., 1989).
To study the interaction of apoA-I and LUVs, these components were incubated overnight at 37°C and were separated on a single Superose 6 column by size exclusion chromatography using a Bio-Logic fast protein liquid chromatography system (Bio Rad). The protein in the eluent was detected by spectrophotometry at 280 nm. An aliquot of the reaction mixtures were also analyzed by NDGGE. Association of apoA-I and 4F with DMPC, POPC and mixed lipids vesicles was determined by following the clearance of the respective LUVs by right angle light scattering using a Cary Eclipse fluorescence spectrophotometer (Varion Inc., Palo Alto, CA).
Negative Stain EM
POPC SUV (from Encapsula NanoSciences, Nashville, TN) containing 0.5 mg/ml POPC with 500 Å average diameter were produced and isolated in a 20 mM Tris-Cl, 154 mM NaCl, pH 7.4 buffer. Human recombinant lipid-free apoA-I (from ProSci Inc, Poway, CA) was expressed in Escherichia coli and isolated by high performance liquid chromatography. ApoA-I and POPC-SUV mixtures were prepared at molar ratios of 0:1, 1:1 and 5:1 (apoA-I:vesicle), then incubated at 37°C for up to 24 hr. Aliquots were collected at 0 and 30 min and 3 and 24 hr and were prepared as negative-staining EM specimens with 1% uranyl formate as described (Zhang et al., 2010). Each sample was examined and imaged with a FEI Tecnai T12 transmission electron microscope (Philips Electron Optics/FEI, Eindhoven, The Netherlands) operating at 120 kV. Micrographs were acquired with a Gatan UltraScan 4K × 4K CCD camera at 42,000× magnification with each micrograph pixel corresponding to 0.266 nm.
CryoEM and CryoET
CryoEM specimens were prepared on lacey carbon film coated copper grid (Cu-200LC, Pacific Grid-Tech, San Francisco, CA). CryoEM data of specimens were acquired with a high-sensitivity 4K × 4K pixel Gatan Ultrascan CCD camera at 20K magnification by the Zeiss Libra 120 TEM (Carl Zeiss NTS GmbH, Oberkochen, Germany; each pixel of the micrograph corresponded to 5.9 Å in the specimens). Total dose for un-tilt 2D micrographs is about 10 to 40 e-/Å2. For cryoET data acquisition, the specimens mounted on a Gatan 915 high-tilt cryoEM holder were tilted at angles ranging from -60° to 60° in steps of 1.5°. The total dose of electron illumination was up to ∼120 e-/Å2 or slightly higher. The tilt series of tomographic data was controlled and imaged by manual operation and by Gatan tomography software (Zeiss Libra 120 TEM) that was preinstalled in the microscope.
Thin-section EM
Immunogold-labeled apoA-I studies were performed as described (Lin and Oram, 2000).
Supplementary Material
Highlights.
A transbilayer surface density gradient can pleat the higher density monolayer.
Amphipathic helices accelerate and stabilize monolayer pleating.
Without stabilization by amphipathic helices, pleats collapse to lipid vesicles.
Transbilayer cholesterol movement decouples transbilayer density gradients.
Acknowledgments
Thanks to UAB Information Technology and Department of Mechanical Engineering for use of the cluster Cheaha. A.C. thanks Manjula Chaddha, G.M. Anantharamaiah and Vinod Mishra for helpful discussions. Thanks also to J. C. Gumbart of the Georgia Institute of Technology School of Physics for helpful discussions about lipid pressure profiles. Work supported by NIH grants P01HL34343 and R01HL102515 to JPS. Work at the Molecular Foundry was supported by the Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Footnotes
Author Contributions: JPS designed experiments, analyzed data and wrote the manuscript. MKJ conducted computational research, analyzed data and helped write the manuscript. AC designed and conducted computational experiments and analyzed data. MM performed vesicle clearance and leakage experiments. GD performed vesicle clearance and leakage experiments. LZ performed cryoEM and cryoET experiments. RZ performed NSEM experiments. LL and JCP helped design experiments. MNP synthesized 4F. JFO designed and analyzed thin section EM experiments. GR designed EM experiments, analyzed EM data and helped write the manuscript.
There are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anantharamaiah GM, Jones JL, Brouillette CG, Schmidt CF, Chung BH, Hughes TA, Bhown AS, Segrest JP. Studies of synthetic peptide analogs of the amphipathic helix. Structure of complexes with dimyristoyl phosphatidylcholine. J Biol Chem. 1985;260:10248–10255. [PubMed] [Google Scholar]
- Atkinson D, Small DM, Shipley GG. X-ray and neutron scattering studies of plasma lipoproteins. Ann N Y Acad Sci. 1980;348:284–298. doi: 10.1111/j.1749-6632.1980.tb21308.x. [DOI] [PubMed] [Google Scholar]
- Baoukina S, Monticelli L, Risselada HJ, Marrink SJ, Tieleman DP. The molecular mechanism of lipid monolayer collapse. Proc Natl Acad Sci U S A. 2008;105:10803–10808. doi: 10.1073/pnas.0711563105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashkirov PV, Akimov SA, Evseev AI, Schmid SL, Zimmerberg J, Frolov VA. GTPase cycle of dynamin is coupled to membrane squeeze and release, leading to spontaneous fission. Cell. 2008;135:1276–1286. doi: 10.1016/j.cell.2008.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. The Journal of Chemical Physics. 1984;81:3684–3690. [Google Scholar]
- Bhat S, Sorci-Thomas MG, Tuladhar R, Samuel MP, Thomas MJ. Conformational adaptation of apolipoprotein A-I to discretely sized phospholipid complexes. Biochemistry. 2007;46:7811–7821. doi: 10.1021/bi700384t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodzioch M, Orso E, Klucken J, Langmann T, Bottcher A, Diederich W, Drobnik W, Barlage S, Buchler C, Porsch-Ozcurumez M, et al. The gene encoding ATP-binding cassette transporter 1 is mutated in Tangier disease. Nat Genet. 1999;22:347–351. doi: 10.1038/11914. see comments. [DOI] [PubMed] [Google Scholar]
- Borhani DW, Rogers DP, Engler JA, Brouillette CG. Crystal structure of truncated human apolipoprotein A-I suggests a lipid-bound conformation. Proc Natl Acad Sci U S A. 1997;94:12291–12296. doi: 10.1073/pnas.94.23.12291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks-Wilson A, Marcil M, Clee SM, Zhang LH, Roomp K, van Dam M, Yu L, Brewer C, Collins JA, Molhuizen HO, et al. Mutations in ABC1 in Tangier disease and familial high-density lipoprotein deficiency. Nat Genet. 1999;22:336–345. doi: 10.1038/11905. see comments. [DOI] [PubMed] [Google Scholar]
- Catte A, Patterson JC, Bashtovyy D, Jones MK, Gu F, Li L, Rampioni A, Sengupta D, Vuorela T, Niemela P, et al. Structure of spheroidal HDL particles revealed by combined atomistic and coarse-grained simulations. Biophys J. 2008;94:2306–2319. doi: 10.1529/biophysj.107.115857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choubey A, Kalia RK, Malmstadt N, Nakano A, Vashishta P. Cholesterol translocation in a phospholipid membrane. Biophys J. 2013;104:2429–2436. doi: 10.1016/j.bpj.2013.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta G, Chaddha M, Hama S, Navab M, Fogelman AM, Garber DW, Mishra VK, Epand RM, Epand RF, Lund-Katz S, et al. Effects of increasing hydrophobicity on the physical-chemical and biological properties of a class A amphipathic helical peptide. J Lipid Res. 2001;42:1096–1104. [PubMed] [Google Scholar]
- Davidson WS, Thompson TB. The structure of apolipoprotein A-I in high density lipoproteins. J Biol Chem. 2007;282:22249–22253. doi: 10.1074/jbc.R700014200. [DOI] [PubMed] [Google Scholar]
- Duong PT, Collins HL, Nickel M, Lund-Katz S, Rothblat GH, Phillips MC. Characterization of nascent HDL particles and microparticles formed by ABCA1-mediated efflux of cellular lipids to apoA-I. J Lipid Res. 2006;47:832–843. doi: 10.1194/jlr.M500531-JLR200. [DOI] [PubMed] [Google Scholar]
- Epand RM, Surewicz WK, Hughes DW, Mantsch H, Segrest JP, Allen TM, Anantharamaiah GM. Properties of lipid complexes with amphipathic helix-forming peptides. Role of distribution of peptide charges. J Biol Chem. 1989;264:4628–4635. [PubMed] [Google Scholar]
- Forte T, Norum KR, Glomset JA, Nichols AV. Plasma lipoproteins in familial lecithin: cholesterol acyltransferase deficiency: structure of low and high density lipoproteins as revealed by elctron microscopy. J Clin Invest. 1971;50:1141–1148. doi: 10.1172/JCI106586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis GA, Knopp RH, Oram JF. Defective removal of cellular cholesterol and phospholipids by apolipoprotein A-I in Tangier Disease. J Clin Invest. 1995;96:78–87. doi: 10.1172/JCI118082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu F, Jones MK, Chen J, Patterson JC, Catte A, Jerome WG, Li L, Segrest JP. Structures of discoidal high density lipoproteins: a combined computational-experimental approach. J Biol Chem. 2010;285:4652–4665. doi: 10.1074/jbc.M109.069914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess B, Kutzner C, van der Spoel D, Lindahl E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. Journal of Chemical Theory and Computation. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–38. 27–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Jones MK, Catte A, Li L, Segrest JP. Dynamics of activation of lecithin: cholesterol acyltransferase by apolipoprotein A-I. Biochemistry. 2009;48:11196–11210. doi: 10.1021/bi901242k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MK, Gu F, Catte A, Li L, Segrest JP. “Sticky” and “promiscuous”, the yin and yang of apolipoprotein A-I termini in discoidal high density lipoproteins: a combined computational-experimental approach. Biochemistry. 2011;50:2249–2263. doi: 10.1021/bi101301g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MK, Zhang L, Catte A, Li L, Oda MN, Ren G, Segrest JP. Assessment of the validity of the double superhelix model for reconstituted high density lipoproteins: a combined computational-experimental approach. J Biol Chem. 2010;285:41161–41171. doi: 10.1074/jbc.M110.187799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane JP, Malloy MJ. Prebeta-1 HDL and coronary heart disease. Curr Opin Lipidol. 2012;23:367–371. doi: 10.1097/MOL.0b013e328353eef1. [DOI] [PubMed] [Google Scholar]
- Koppaka V, Silvestro L, Engler JA, Brouillette CG, Axelsen PH. The structure of human lipoprotein A-I. Evidence for the “belt” model. J Biol Chem. 1999;274:14541–14544. doi: 10.1074/jbc.274.21.14541. [DOI] [PubMed] [Google Scholar]
- Kozlovsky Y, Kozlov MM. Stalk model of membrane fusion: solution of energy crisis. Biophys J. 2002;82:882–895. doi: 10.1016/S0006-3495(02)75450-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landry YD, Denis M, Nandi S, Bell S, Vaughan AM, Zha X. ATP-binding cassette transporter A1 expression disrupts raft membrane microdomains through its ATPase-related functions. J Biol Chem. 2006;281:36091–36101. doi: 10.1074/jbc.M602247200. [DOI] [PubMed] [Google Scholar]
- Lawn RM, Wade DP, Garvin MR, Wang X, Schwartz K, Porter JG, Seilhamer JJ, Vaughan AM, Oram JF. The Tangier disease gene product ABC1 controls the cellular apolipoprotein-mediated lipid removal pathway. J Clin Invest. 1999;104:R25–31. doi: 10.1172/JCI8119. see comments. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Chen J, Mishra VK, Kurtz JA, Cao D, Klon AE, Harvey SC, Anantharamaiah GM, Segrest JP. Double belt structure of discoidal high density lipoproteins: molecular basis for size heterogeneity. J Mol Biol. 2004;343:1293–1311. doi: 10.1016/j.jmb.2004.09.017. [DOI] [PubMed] [Google Scholar]
- Lin G, Oram JF. Apolipoprotein binding to protruding membrane domains during removal of excess cellular cholesterol. Atherosclerosis. 2000;149:359–370. doi: 10.1016/s0021-9150(99)00503-1. [DOI] [PubMed] [Google Scholar]
- Markvoort AJ, Spijker P, Smeijers AF, Pieterse K, van Santen RA, Hilbers PA. Vesicle deformation by draining: geometrical and topological shape changes. J Phys Chem B. 2009;113:8731–8737. doi: 10.1021/jp901277h. [DOI] [PubMed] [Google Scholar]
- Marrink SJ, Risselada HJ, Yefimov S, Tieleman DP, de Vries AH. The MARTINI force field: coarse grained model for biomolecular simulations. J Phys Chem B. 2007;111:7812–7824. doi: 10.1021/jp071097f. [DOI] [PubMed] [Google Scholar]
- Martin DD, Budamagunta MS, Ryan RO, Voss JC, Oda MN. Apolipoprotein A-I assumes a “looped belt” conformation on reconstituted high density lipoprotein. Journal of Biological Chemistry. 2006;281:20418–20426. doi: 10.1074/jbc.M602077200. [DOI] [PubMed] [Google Scholar]
- Monticelli L, Kandasamy SK, Periole X, Larson RG, Tieleman DP, Marrink SJ. The MARTINI coarse-grained force field: Extension to proteins. Journal of Chemical Theory and Computation. 2008;4:819–834. doi: 10.1021/ct700324x. [DOI] [PubMed] [Google Scholar]
- Nagata KO, Nakada C, Kasai RS, Kusumi A, Ueda K. ABCA1 dimer-monomer interconversion during HDL generation revealed by single-molecule imaging. Proc Natl Acad Sci U S A. 2013;110:5034–5039. doi: 10.1073/pnas.1220703110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palgunachari MN, Mishra VK, Lund-Katz S, Phillips MC, Adeyeye SO, Alluri S, Anantharamaiah GM, Segrest JP. Only the two end helixes of eight tandem amphipathic helical domains of human apo A-I have significant lipid affinity. Implications for HDL assembly. Arterioscler Thromb Vasc Biol. 1996;16:328–338. doi: 10.1161/01.atv.16.2.328. [DOI] [PubMed] [Google Scholar]
- Quazi F, Molday RS. Differential phospholipid substrates and directional transport by ATP-binding cassette proteins ABCA1, ABCA7, and ABCA4 and disease-causing mutants. J Biol Chem. 2013;288:34414–34426. doi: 10.1074/jbc.M113.508812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rader DJ, Tall AR. The not-so-simple HDL story: Is it time to revise the HDL cholesterol hypothesis? Nat Med. 2012;18:1344–1346. doi: 10.1038/nm.2937. [DOI] [PubMed] [Google Scholar]
- Rust S, Rosier M, Funke H, Real J, Amoura Z, Piette JC, Deleuze JF, Brewer HB, Duverger N, Denefle P, et al. Tangier disease is caused by mutations in the gene encoding ATP-binding cassette transporter 1. Nat Genet. 1999;22:352–355. doi: 10.1038/11921. see comments. [DOI] [PubMed] [Google Scholar]
- Sampaio JL, Gerl MJ, Klose C, Ejsing CS, Beug H, Simons K, Shevchenko A. Membrane lipidome of an epithelial cell line. Proc Natl Acad Sci U S A. 2011;108:1903–1907. doi: 10.1073/pnas.1019267108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segrest JP. Molecular packing of high density lipoproteins: a postulated functional role. FEBS Lett. 1976;69:111–115. doi: 10.1016/0014-5793(76)80665-5. [DOI] [PubMed] [Google Scholar]
- Segrest JP, Jones MK, Klon AE, Sheldahl CJ, Hellinger M, De Loof H, Harvey SC. A detailed molecular belt model for apolipoprotein A-I in discoidal high density lipoprotein. J Biol Chem. 1999;274:31755–31758. doi: 10.1074/jbc.274.45.31755. [DOI] [PubMed] [Google Scholar]
- Sloop CH, Dory L, Hamilton R, Krause BR, Roheim PS. Characterization of dog peripheral lymph lipoproteins: the presence of a disc-shaped “nascent” high density lipoprotein. J Lipid Res. 1983;24:1429–1440. [PubMed] [Google Scholar]
- Tricerri MA, Behling Agree AK, Sanchez SA, Bronski J, Jonas A. Arrangement of apolipoprotein a-i in reconstituted high-density lipoprotein disks: an alternative model based on fluorescence resonance energy transfer experiments. Biochemistry. 2001;40:5065–5074. doi: 10.1021/bi002815q. [DOI] [PubMed] [Google Scholar]
- Vedhachalam C, Duong PT, Nickel M, Nguyen D, Dhanasekaran P, Saito H, Rothblat GH, Lund-Katz S, Phillips MC. Mechanism of ATP-binding cassette transporter A1-mediated cellular lipid efflux to apolipoprotein A-I and formation of high density lipoprotein particles. J Biol Chem. 2007;282:25123–25130. doi: 10.1074/jbc.M704590200. [DOI] [PubMed] [Google Scholar]
- Voight BF, Peloso GM, Orho-Melander M, Frikke-Schmidt R, Barbalic M, Jensen MK, Hindy G, Holm H, Ding EL, Johnson T, et al. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet. 2012 doi: 10.1016/S0140-6736(12)60312-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson PW, Abbott RD, Castelli WP. High density lipoprotein cholesterol and mortality. The Framingham Heart Study. Arteriosclerosis. 1988;8:737–741. doi: 10.1161/01.atv.8.6.737. [DOI] [PubMed] [Google Scholar]
- Wlodawer A, Segrest JP, Chung BH, Chiovetti R, Jr, Weinstein JN. High-density lipoprotein recombinants: evidence for a bicycle tire micelle structure obtained by neutron scattering and electron microscopy. FEBS Lett. 1979;104:231–235. doi: 10.1016/0014-5793(79)80821-2. [DOI] [PubMed] [Google Scholar]
- Zhang L, Song J, Cavigiolio G, Ishida BY, Zhang S, Kane JP, Weisgraber KH, Oda MN, Rye KA, Pownall HJ, et al. The morphology and structure of lipoprotein revealed by optimized negative staining. J Lipid Res. 2010 doi: 10.1194/jlr.D010959. Epub date 10/28. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.