

Abstract
Myosin light chain-2 (MYL2, also called MLC-2) is an ∼19 kDa sarcomeric protein that belongs to the EF-hand calcium binding protein superfamily and exists as three major isoforms encoded by three distinct genes in mammalian striated muscle. Each of the three different MLC-2 genes (MLC-2f; fast twitch skeletal isoform, MLC-2v; cardiac ventricular and slow twitch skeletal isoform, MLC-2a; cardiac atrial isoform) has a distinct developmental expression pattern in mammals. Genetic loss-of-function studies in mice demonstrated an essential role for cardiac isoforms of MLC-2, MLC-2v and MLC-2a, in cardiac contractile function during early embryogenesis. In the adult heart, MLC-2v function is regulated by phosphorylation, which displays a specific expression pattern (high in epicardium and low in endocardium) across the heart. These data along with new data from computational models, genetic mouse models, and human studies have revealed a direct role for MLC-2v phosphorylation in cross-bridge cycling kinetics, calcium-dependent cardiac muscle contraction, cardiac torsion, cardiac function and various cardiac diseases. This review focuses on the regulatory functions of MLC-2 in the embryonic and adult heart, with an emphasis on phosphorylation-driven actions of MLC-2v in adult cardiac muscle, which provide new insights into mechanisms regulating myosin cycling kinetics and human cardiac diseases.
Keywords: myosin light chain-2 (MYL2/MLC-2), cardiac muscle, cardiac disease, contraction, sarcomere, ventricular myosin light chain-2, heart, myosin light chain kinase, cardiac torsion, cardiac function, phosphorylation
1. Myosin Light Chain-2: Isoforms and Expression Patterns In Striated Muscle
Sarcomeres, the functional units of striated muscle, are composed of a multitude of proteins, precisely organized in a very strict crystalline architecture. Despite the immense similarity in sarcomeric ultrastructure among different vertebrate striated muscles (fast versus slow skeletal muscle and atrial versus ventricular cardiac muscle), there is a very high degree of molecular variability due to the existence of multiple isoforms of each sarcomeric component. The distinct function of each striated muscle type is largely due to the differential expression of sarcomeric protein isoforms (Schiaffino and Reggiani, 1996). Myosin light chain-2 (MYL2/MLC-2) is a major sarcomeric protein in mammalian striated muscle of approximately 19 kDa and 166 amino acids in length and is encoded in human and mouse by the MYL2 and Myl2 genes, respectively. Gene structures of MYL2/Myl2 are publicly available for both human (http://www.ncbi.nlm.nih.gov/gene/4633) and mouse (http://www.ncbi.nlm.nih.gov/gene/17906), which are found on chromosomes 12 and 5, respectively. Protein structures of human and mouse MYL2/MLC-2 are shown in Figure 1. MYL2/MLC-2 belongs to a family of a number of homologous EF-hand Ca2+ binding proteins (Grabarek, 2006). The evolutionary lineage of homologous proteins in the EF-hand Ca2+ binding superfamily, which includes MYL2/MLC-2 has been previously described and include at least 66 subfamilies that are grouped into two major categories: canonical EF-hands and pseudo EF-hands (Zhou et al., 2006). MYL2/MLC-2 is a member of the CTER (calmoduin, troponin C, essential and regulatory light chains of myosin) subfamily, which encompasses canonical EF-hands that arose from a common four domain precursor, and thus, includes two pairs (forms a globular domain that encompasses side-to-side pairs; Figure 1) of EF-hand calcium binding domains (Kawasaki and Kretsinger, 2014). MYL2/MLC-2 exists as three major isoforms, each encoded by a distinct gene: The fast twitch skeletal isoform designated as MLC-2f, (Myl2f; Nudel, et al., 1984), the cardiac ventricular and slow twitch skeletal isoform designated as MLC-2v, (Myl2; Henderson, et al., 1988; Kumar, et al., 1986) and the cardiac atrial isoform is designated as MLC-2a, (Myl2a; Kubalak, et al., 1994). These three genes are highly homologous with 73% identity and 93% similarity between the primary amino acid sequence of MLC-2v and MLC-2f (Gulick, et al., 1997). A 56% identity and 86% similarity was also observed in the primary amino acid sequence between MLC-2a and MLC-2v (Gulick, et al., 1997).
Figure 1.
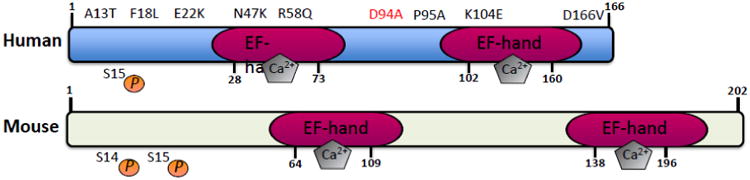
Human and mouse MLC2v protein structures illustrating the location of EF-hand, calcium binding motifs and the sites of dominant missense mutations found in patients with dilated (D94A; red) and hypertrophic cardiomyopathy (A13T, F18L, E22K, N47K, R58Q, P95A and D166V; black). The location of key phosphorylation sites (S14 and S15) implicated in cardiac function and disease are also denoted.
Despite their high degree of homology, each MLC-2 isoform displays a distinct expression pattern during development. MLC-2f expression is thought to be restricted to skeletal muscles of neonatal and adult mice and rats, although expression in myotomal and extramyotomal muscle-forming regions could also be detected during early embryogenesis (Einat et al., 1987; Faerman and Shani, 1993; Katcoff et al., 1980). On the other hand, MLC-2v is considered one of the earliest markers of ventricular specification during mammalian cardiogenesis (O'Brien, et al., 1993). At E7.5-8.0, MLC-2v is expressed in a restricted zone of the cardiogenic crescent prior to fusion of the bilaterally symmetric progenitors at the mid-line. At E8.0, expression of MLC-2v is restricted within the ventricular segment of the linear heart tube and remains restricted to the ventricular chamber throughout embryonic development into adulthood (O'Brien, et al., 1993; Ross, et al., 1996). Unlike MLC-2v, which is expressed in both adult ventricular and slow-twitch skeletal muscle, MLC-2a is only expressed in atria (Chen et al., 1998a). During embryogenesis, MLC-2a is expressed somewhat earlier (E7.5) than MLC-2v and is uniformly expressed throughout the heart tube. Its expression is selectively downregulated in the ventricular chamber beginning at E11 and is undetectable in ventricular chambers by E13.5 (Kubalak, et al., 1994). As a result, the disappearance of MLC-2a is considered to be a marker of ventricular maturation (Dyson, et al., 1995). The early ventricular chamber specificity of MLC2v has lead to the creation of a number of novel genetic tools to help researchers drive gene expression to the mouse ventricles in vivo (Chen et al., 1998b) as well as used as a selection based method to create human ventricular specific disease models using induced pluripotent stem cell derived cardiomyocytes in vitro (Bizy, et al., 2013).
Transgenic approaches have been utilized to dissect the functional differences between the highly homologous MLC-2 isoforms: MLC-2f, MLC-2v, and MLC-2a. Cardiac-specific expression of MLC-2f under the α-myosin heavy chain (MHC) promoter resulted in 100% replacement of MLC-2a protein in the adult atria but only a 53% replacement of MLC-2v protein in adult ventricles (Gulick, et al., 1997). Interestingly, endogenous levels of MLC-2a and MLC-2v mRNA were unaffected in these transgenic lines, indicating that there is no regulatory cross talk between the three MLC-2 isoforms at the mRNA level (Gulick, et al., 1997). Mice with complete replacement of MLC-2a and partial replacement of MLC-2v protein by MLC-2f exhibited a reduction in adult left ventricular contractility and relaxation, as examined in working and non-working isolated heart preparations (Gulick, et al., 1997), demonstrating the non-redundant nature of MLC-2 isoforms since replacement of the skeletal isoform of MLC-2 in the heart contributed to major functional defects in the heart. A limitation to these approaches is the incomplete replacement of isoforms in the adult ventricles presumably due to the existence of endogenous MLC-2v. More importantly, the α-MHC promoter is expressed in both atria and ventricles, thereby making it difficult to interpret the physiological results. Thus, future studies directed at utilizing genetic knock-in strategies to endogenously replace MLC-2v in the MLC-2a locus in mice and vice versa, may help provide further insights into their specialized roles in the heart.
2. Myosin Light Chain-2: The Contractile Protein
Striated muscle contraction occurs when actin-containing thin filaments move past interdigitating myosin-containing thick filaments. The motor in this system is myosin, which transduces chemical energy generated by the hydrolysis of adenosine triphosphate (ATP) into mechanical movement (Huxley and Simmons, 1971). Muscle myosin contains one pair of heavy chains and two pairs of light chains, the essential myosin light chains (MLC-1 or MLC-3) and the regulatory light chain (MLC-2), which is referred to as ventricular myosin light chain-2 (MLC-2v) in cardiac muscle. The three-dimensional structure of myosin indicates that the light chains are arranged in tandem, with MLC-1 and MLC-3 in the amino-terminal half of the neck and MLC-2 in the neck/tail region (Rayment et al., 1993a; Rayment et al., 1993b). Identification of specific interactions between MLC-2 and the cardiac-specific domain of myosin binding protein C further suggested a role for MLC-2 in the myosin tail region, which has exposed binding sites for myosin binding protein C (Ratti et al., 2011). The association between regulatory proteins associated with cardiac myosin, which include a central role for MLC-2, has been detailed elsewhere (for review see Sheikh et al., 2014).
Genetic loss-of-function studies in mice were amongst the first to demonstrate an essential role for the cardiac isoforms of MLC-2 (MLC-2v and MLC-2a) in cardiac contractile function during early embryogenesis (Table 1). Conventional ablation of MLC-2v in mice resulted in embryonic lethality at E12.5, which was associated with ultrastructural defects in ventricular sarcomere assembly, that included (i) disruptions and disorganization of the normal parallel alignment of the thick and thin filaments, (ii) narrower fiber widths and (iii) larger distances between Z discs and misalignment of Z-band between sarcomeres (Chen et al., 1998a). Global loss of MLC-2a in mice also caused embryonic lethality at E10.5-11.5 and resulted in selective ultrastructural defects in atrial sarcomeric assembly that included (i) absence of myofibrillar organization and (ii) lack of parallel alignment of thick and thin filaments and (iii) lack of appropriate Z disc formation (Huang et al., 2003). Both models also exhibited severe cardiac functional deficits. MLC-2v null hearts manifested into an embryonic form of dilated cardiomyopathy resulting in heart failure with reduced ventricular ejection fraction (Chen et al., 1998a). Interestingly, a compensatory increase in MLC-2a protein levels was observed in MLC-2v deficient ventricles that appeared to incorporate into the thick filament (Chen et al., 1998a). These studies revealed an essential early role for MLC-2v in cardiac contractility as well as ventricular chamber development during cardiogenesis (Chen et al., 1998a). In terms of MLC-2a null mice, the atrial ultrastructural defects manifested into defects in atrial contraction (Huang et al., 2003). Interestingly, striking secondary defects in cardiac morphogenesis (looping) and angiogenesis (extraembryonic and intraembryonic) were also observed in MLC-2a deficient embryos, highlighting that selective loss of embryonic atrial function can have a dramatic impact on cardiac and vasculature development, with cardiac function affecting form (Huang et al., 2003). These studies further shed light on mechanisms (alterations in blood flow) that may impact congenital heart diseases (Huang et al., 2003). More recent studies focused on tell tale heart, a mutation identified in zebrafish that encodes the cardiac myosin light chain-2 gene has also revealed a requirement for cardiac MLC-2 in thick filament stabilization (absence of thick filament organization) and contractility in the embryonic zebrafish heart (Rottbauer et al., 2006). Interestingly, zebrafish only contain a single cardiac-specific isoform of MLC-2 and no compensatory increase in another MLC-2 was observed in zMLC-2 deficient settings, demonstrating the requirement of cardiac MLC-2 as a critical contractile protein in the embryonic zebrafish heart (Rottbauer et al., 2006).
Table 1. Summary of Genetic Models Demonstrating An Essential Role for Myosin Light Chain-2 in the Embryonic and Adult Heart.
| Genetic Model | Phenotype | Reference |
|---|---|---|
| Conventional ablation of mouse Ventricular Myosin Light Chain-2 | Embryonic lethality at E12.5 associated with ultrastructural defects in ventricular sarcomere assembly resulting in a dilated cardiomyopathy and heart failure associated with reduced ejection fraction. | Chen et al., 1998b |
| Conventional ablation of mouse Atrial Myosin Light Chain-2 | Embryonic lethality at E10.5-11.5 associated with ultrastructural defects in atrial sarcomere assembly resulting in reduced atrial contraction and leading to secondary effects in cardiac morphogenesis (looping) and vasculature (angiogenesis) development | Huang et al., 2003 |
| Mutation in a zebrafish Myosin Light Chain-2 (tell tale heart) | Embryonic lethality associated with absence of thick and thin filament assembly as well as contractile defects (silent atria and ventricles, loss of fractional shortening) resulting in pericardial edema | Rottbauer et al., 2006 |
| Ventricular Myosin Light Chain-2 Phosphorylation Mutant (Ser14Ala/Ser15Ala) knock-in mouse model | Adult lethality associated with early defects in cardiac twitch relaxation and torsion leading to dilated cardiomyopathy, heart failure and premature death | Sheikh et al., 2012 |
| Cardiac myosin light chain kinase hypomorphic mouse | Adult phenotype with exaggerated cardiac hypertrophy, necrosis, fibrosis and decreased cardiac function | Ding et al., 2010 |
| Conventional ablation of mouse cardiac Myosin Light Chain Kinase | Adult phenotype associated with early defects in cardiac torsion leading to dilated cardiomyopathy | Warren et al., 2012 |
| Cardiac specific overexpression of mouse protein phosphatase 1-b | Adult phenotype associated with dilated cardiomyopathy | Mizutani et al., 2012 |
3. Ventricular Myosin Light Chain-2: Critical Phosphorylation Sites and Regulators
Phosphorylation of MLC-2 was first discovered in rabbit skeletal muscle (Perrie, et al., 1973; Sweeney et al., 1993) and subsequently in cardiac muscle (Frearson and Perry, 1975; Olsson et al., 2004). In cardiac muscle, the critical phosphorylation sites important for endogenous regulation of MLC-2v phosphorylation in vivo are Ser14/Ser15 in the mouse heart and Ser15 in the human heart (Scruggs et al., 2010; Figure 1). Interestingly, compensatory mechanisms exist in the mouse heart to compensate for the loss of one phosphoryation site, that highlight that both Ser14/Ser15 are required to regulate the levels and functions of endogenous MLC-2v phosphorylation in the heart in vivo (Sheikh et al., 2012). The path towards identifying the critical kinase important for phosphorylating MLC-2v has been a long one. Myosin light chain kinase was first purified from rabbit skeletal muscle (Pires and Perry, 1977). The enzyme that phosphorylates MLC-2 is a Ca2+/calmodulin dependent myosin light chain kinase, MLCK (Stull, et al., 1990). In rat ventricle, the basal level of phosphorylated MLC-2v is approximately 30% and increases to about 50% after treadmill exercise (Fitzsimons, et al., 1989). In addition, in skinned cardiac muscle fibers, as in skinned fast skeletal muscle, MLC-2 phosphorylation increased force production at submaximal levels of Ca2+ activation (Sweeney and Stull, 1986). It was not until 2007 and 2008 that cardiac myosin light chain kinase (Mylk3) was identified as the major regulator of MLC-2v phosphorylation (Chan et al., 2008; Seguchi et al., 2007) as genetic mouse models deficient in cardiac MLCK were sufficient to ablate endogenous MLC-2v phosphorylation levels in the heart in vivo (Ding et al., 2010; Warren et al., 2012). Limited studies have focused on identifying the key phosphatases important in regulating MLC-2v dephosphorylation; however, recent studies highlight a contributory role for protein phosphatase 1β (Mizutani et al., 2010). Interestingly, cardiac-specific overexpression of this phosphatase in mice only resulted in a 15% reduction in MLC-2v phosphorylation levels in hearts in vivo (Mizutani et al., 2010), suggesting that the main phosphatase and upstream pathways responsible for dephosphorylating MLC-2v remain to be identified. Data from proteomic studies suggest that other post-translational modifications could affect cardiac MYL2/MLC-2 as well as interfere with the phosphorylated states of MYL2/MLC-2, since MLC-2 Ser 15 is also impacted by O-GlcNAacylation (Ramirez-Correa GA et al., 2008). However, further studies are needed to establish the functional role of these modifications on MLC-2v phosphorylation and function.
3.1. Ventricular Myosin Light Chain-2 Phosphorylation: Effects on Cross-Bridge Cycling Kinetics and Cardiac Muscle Contraction
Phosphorylation is a key player in driving a role for MLC-2 in crossbridge cycling kinetics and calcium-dependent muscle contraction. In vertebrate smooth muscle, the regulation of contraction by Ca2+ is through the myosin-containing thick filament. Phosphorylation of a single serine residue (Ser19) of MLC-2 causes a conformational change in myosin, triggering interaction of myosin with actin, and activating myosin ATPase activity to result in myofilament movement (Sweeney et al, 1993). In contrast, in vertebrate striated muscle, the regulation of actin-myosin interactions by Ca2+ is performed by Ca2+ binding to the thin filament regulatory system involving the troponin-tropomyosin complex. In this setting, MLC-2 was thought to have a modulatory effect on muscle contraction (Diffee et al., 2003; Lowey et al., 1993; Sweeney et al., 1993; Szczesna et al., 1996). However, recent studies using computational models and a non-phosphorylatable (Ser14Ala/Ser15Ala) MLC2v knock-in mouse model have revealed a prominent role for MLC-2v in cross-bridge cycling kinetics and cardiac muscle contraction (Sheikh et al., 2012). The mechanisms driving a role for MCL-2v phosphorylation (Ser14/Ser15) in cross-bridge cycling kinetics in cardiac muscle, included increasing myosin lever arm stiffness (results in greater force production by myosin power stroke) and promoting myosin head diffusion (causing myosin heads to move away from the myosin filament backbone and move closer to actin filaments) (Sheikh et al., 2012). These events altogether slow down myosin kinetics and prolong duty cycle resulting in accumulated myosins being cooperatively recruited to actin binding sites to sustain thin filament activation as a means to fine-tune myofilament Ca2+ sensitivity to force (Sheikh et al., 2012). Computational models harboring these mechanisms were sufficient to reproduce data associated with the classic effects of MLC-2v phosphoryation (via MLCK) in adult skinned cardiac myofilaments in vitro (Olsson et al., 2004; Stelzer et al., 2006). In addition, computation models harboring loss of these mechanisms were able to reproduce the twitch relaxation defects observed in ex-vivo preparations of papillary muscles from non-phosphorylatable (Ser14Ala/Ser15Ala) MLC2v knock-in mice, which were observed in the absence of changes in Ca2+ transients (Sheikh et al., 2012). Thus, these studies further bring to light an important role for MLC-2v phosphorylation in myosin cycling kinetics and a previously unappreciated relationship between muscle tension and Ca2+ that is adaptable (Sheikh et al., 2012).
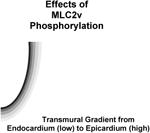
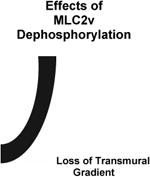
3.2. Ventricular Myosin Light Chain-2 Phosphorylation: Effects on Adult Cardiac Torsion, Function and Disease
Phosphorylation is also a key player in driving a role for MLC-2v in adult cardiac torsion and function (Table 1). In the adult heart, all of the functions of MLC-2v stem from its regulation by phosphorylation, which is uniquely expressed as a spatial gradient (high in epicardium and low in endocardium) across the heart, (Davis et al., 2001; Hidalgo et al., 2006; Sheikh et al., 2012). These expression patterns directly correlated with the expression pattern of its kinase, cardiac myosin light kinase (Warren et al., 2012) and inversely correlated with the expression of its phosphatase, protein phosphatase 1 (Rajashree et al., 2005). The functional relevance of these spatial gradients was only recently revealed as primary defects in cardiac torsion and workload distribution (increased workload in the endocardium versus epicardium) were evident in hearts from a genetic mouse model harboring loss of ML2v phosphorylation (Ser14Ala/Ser15Ala) as well as a genetic mouse model harboring loss of cardiac MLCK at a stage when overt disease was not observed in these models (Sheikh et al., 2012; Warren et al., 2012). Again, computational models harboring loss of myosin cycling mechanisms driving MLC2v phosphorylation were sufficient to reproduce the cardiac torsion defects observed in hearts from non-phosphorylatable (Ser14Ala/Ser15Ala) MLC-2v knock-in mice (Sheikh et al., 2012). These studies further show that spatial differences in MLC-2v phosphorylation and myosin cycling kinetics across the heart not only drive differential mechanics, but that these mechanics impart differential workload distributions across the heart, which are important for driving torsion (Sheikh et al., 2012). A critical role for MLC-2v phosphorylation in adult cardiac function stems from studies performed in a number of genetic mouse models that target loss of MLC-2v phosphorylation and cardiac MLCK as well as overexpression of the myosin phosphatase, protein phosphatase 1, which have been previously comprehensively discussed and summarized elsewhere (Sheikh et al., 2014). Critical to these findings are that MLC-2v dephosphorylation in mice resulted in cardiac dilatation and dysfunction associated with features reminiscent of dilated cardiomyopathy leading to heart failure and premature death (Sheikh et al., 2012; Warren et al., 2012; Mizutani et al., 2012). Interestingly, a cardiac myosin light chain kinase hypomorphic mouse model that resulted in incomplete loss of MLC2v phosphorylation exhibited cardiac hypertrophy, necrosis, fibrosis and cardiac dysfunction (Ding et al. 2010). These findings altogether highlighted a role for MLC2v phosphorylation as an important contractile protein in the adult heart. These studies further reveal that torsion may be an early manifestation of the loss of MLC2v phosphorylation-mediated mechanisms that drive dilated cardiomyopathy (Sheikh et al., 2012). Support for a role for MLC2v dephosphorylation in dilated cardiomyopathy and heart failure also comes from human studies that have reported MLC2v dephosphorylation in hearts obtained from human patients exhibiting dilated (non-ischemic) cardiomyopathy and heart failure (Morano, 1992; van der Velden et al., 2001; van der Velden et al., 2003a; van der Velden et al., 2003b; Scruggs et al., 2010). In addition, some cases of idiopathic dilated cardiomyopathy in humans are also associated with reduction of MLC2v protein levels in the ventricles due to a specific proteinase mediated cleavage of this light chain (Margossian, et al., 1992). It has been shown by several groups that MLC-2v phosphorylation is also dependent on heart rate and stimulation frequency, suggesting a role for MLC-2v in frequency dependent activation (Lamberts et al, 2007; Varian and Janssen, 2007). In addition, MLC2v phosphorylation has also been shown to increase with stretch in cardiac muscle and recent studies have demonstrated its role in length-dependent activation (Monsasky et al., 2010; Monasky et al, 2013), highlighting a role for MLC-2v phosphorylation in regulatory aspects of cardiac output. Future studies investigating these regulatory mechanisms may also give further insights into the mechanisms regulating the altered myocardial growth response to pressure overload and decline of cardiac function in MLC-2v phosphorylation mutant mice.
MLC2v dephosphorylation has also been reported in human patients carrying a rare form of familial hypertrophic cardiomyopathy (FHC) that impacts cardiac papillary muscle and adjacent ventricular muscle, based on specific MLC2v and MLCK mutations (Szczesna et al., 2001; Davis et al., 2001; Jacques et al., 2008). In addition, other MLC-2v mutations associated with this form of FHC are reported likely to be in close proximity to the Ser15 phosphorylation site (Poetter, et al., 1996). FHC is an autosomal dominantly transmitted disease characterized by ventricular wall thickening (concentric hypertrophy) with myofibrillar disarray (Spirito, et al 1997). These findings are in contrast to findings in genetic mouse models that harbor loss of MLC-2v phosphorylation as the cardiac phenotypes are associated with ventricular wall thinning associated with dilated cardiomyopathy as well as absence of markers associated with FHC such as fibrosis and upregulation of fetal gene markers (Sheikh et al., 2012; Warren et al., 2012; Mizutani et al., 2012). In addition, increased MLC2v phosphorylation is thought to have a cardioprotective role in settings of hypertrophic stress (Huang et al., 2008; Warren et al., 2012), while MLC2v dephosphorylation in mice was shown to result in loss of concentric hypertrophy and an exaggerated switch to eccentric hypertrophy resulting in heart failure in settings of cardiac (pathological and physiological) hypertrophic stress (Sheikh et al., 2012; Warren et al., 2012). Mutations in the MYL2 gene have been associated with cardiomyopathies and been extensively studied (Figure 1). These mutations range from the recently association with dilated cardiomyopathy (D94A) to the rare form of familial hypertrophic cardiomyopathy (A13T, F18L, E22K, N47K, R58Q, P95A, K104E, D166V) (Abraham et al., 2009; Farman et al., 2014; Greenberg et al., 2009; Huang et al., 2014; Huang et al., 2015; Muthu et al., 2012; Szczesna-Cordary et al., 2004). Interestingly, ex-vivo expression of MLCK (and restoration of MLC2v phosphorylation) could rescue the cardiac contractile defects but not force deficits in hearts of a transgenic mouse model harboring the FHC-associated MLC-2v D166V mutation (impacts Ca2+ binding affinity) (Muthu et al., 2012), demonstrating the impact of restoring myosin cycling kinetics in the setting of FHC. Recent studies have also revealed novel mechanisms that are influenced by MLCK-mediated pathways, such as ubiquitin-mediated protein degradation pathways (Warren et al., 2012), which may also contribute to the effects of MLC-2v dephosphorylation in the heart, that have yet to be fully explored in FHC settings. Thus, future studies focused on unlocking the mechanisms driving the effects of MLC2v phosphorylation in hypertrophic stretch activated settings may also help explain the variable and rare phenotypes observed in FHC.
4. Conclusions
From its initial discovery in 1969 (Weeds, 1969), MLC-2 has emerged as an established early cardiac developmental marker, with data from genetic mouse models cementing its role as an essential contractile protein in both the embryonic and adult heart. Although previously thought to have a modulatory role in cardiac muscle, new data from computational models and genetic mouse models deficient in the phosphorylatable form of MLC-2v have also elucidated mechanisms driving its role in myosin cycling kinetics and calcium-dependent contraction in cardiac muscle (Table 2). Importantly, recent studies have also demonstrated that the transmural gradient of MLC-2v phosphorylation plays a critical role in adult cardiac torsion, function and diseases, including dilated cardiomyopathy and heart failure (Table 2). However, mechanisms by which MLC-2v mutations found in a rare form of FHC result in disease manifestation are not clear, but may involve the role of MLC-2v phosphorylation in the hypertrophic stretch response. Greater understanding of the role of MLC-2v phosphorylation in cardiac muscle will help fuel therapeutic approaches directed at restoring MLC2v phosphorylation, which appear to have a cardioprotective role in the setting of hypertrophic stress.
Table 2. Summary of the Impact of MLC2v phosphorylation status on cardiac muscle biology and physiology.
|
|
|
|---|---|---|
| Cross-Bridge Cycling Kinetics |
|
|
| Skinned Cardiac Muscle |
|
|
| Whole Cardiac Muscle |
|
|
| Whole Heart |
|
|
Highlights.
MYL2 is sarcomeric protein and member of the EF-hand calcium-binding protein family
MYL2 plays an important role in embryonic heart muscle structure and function.
In adults, MYL2 phosphorylation regulates cardiac myosin cycling kinetics, torsion and function.
Critical phosphorylation sites include: Ser14/Ser15 in mice and Ser15 in humans.
MLC-2v dephosphorylation has been implicated in dilated cardiomyopathy and heart failure.
Acknowledgments
This review and the corresponding Gene Wiki article are written as part of the Cardiac Gene Wiki Review series-- a series resulting from a collaboration between the journal GENE, the Gene Wiki Initiative, and the BD2K initiative. The Cardiac Gene Wiki Initiative is supported by National Institutes of Health (GM083924 and GM114833). Additional support for Gene Wiki Reviews is provided by Elsevier, the publisher of GENE. R.C.L is supported by a NIH T32 training grant. F.S. is supported by NIH/NHLBI (HL09780) and American Heart Association (GRNT22940045) grants. J.C is supported by NIH/NHLBI grants and is an American Heart Association endowed chair in cardiovascular research.
Abbreviations
- ATP
Adenosine triphosphate
- Ca2+
Calcium
- CTER
Calmoduin, troponin C, essential and regulatory light chains of myosin
- E
Embryonic day
- FHC
Familial hypertrophic cardiomyopathy
- MHC
Myosin heavy chain
- MLC-1
Myosin light chain-1
- MYL2/MLC-2
Myosin light chain-2
- MLC-2a
Atrial myosin light chain-2
- MLC-2f
Fast-twitch skeletal myosin light chain-2
- MLC-2v
Ventricular myosin light chain-2 and slow twitch skeletal myosin light chain-2
- MLC-3
Myosin light chain-3
- MLCK
Myosin light chain kinase
- Ser15
Serine 15 phosphorylation site
- Ser14/Ser15
Serine 14 and 15 phosphorylation sites
- Ser14Ala/Ser15Ala
Serine 14 and 15 phosphorylation mutations to Alanine 14 and 15
- Ser19
Serine 19 phosphorylation site
- zMLC-2
Zebrafish myosin light chain-2
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abraham TP, Jones M, Kazmierczak K, Liang HY, Pinheiro AC, Wagg CS, Lopaschuk GD, Szczesna-Cordary D. Diastolic dysfunction in familial hypertrophic cardiomyopathy transgenic model mice. Cardiovasc Res. 2009;82:84–92. doi: 10.1093/cvr/cvp016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bizy A, Guerrero-Serna G, Hu B, Ponce-Balbuena D, Willis BC, Zarzoso M, Ramirez RJ, Sener MF, Mundada LV, Klos M, Devaney EJ, Vikstrom KL, Herron TJ, Jalife J. Myosin light chain 2-based selection of human iPSC-derived early ventricular cardiac myocytes. Stem Cell Res. 2013;11:1335–1347. doi: 10.1016/j.scr.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan JY, Takeda M, Briggs LE, Graham ML, Lu JT, Horikoshi N, Weinberg EO, Aoki H, Sato N, Chien KR, Kasahara H. Identification of cardiac-specific myosin light chain kinase. Circ Res. 2008;102:571–80. doi: 10.1161/CIRCRESAHA.107.161687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Kubalak SW, Minamisawa S, Price RL, Becker KD, Hickey R, Ross J, Jr, Chien KR. Selective requirement of myosin light chain 2v in embryonic heart function. J Biol Chem. 1998a;273:1252–1256. doi: 10.1074/jbc.273.2.1252. [DOI] [PubMed] [Google Scholar]
- Chen J, Kubalak SW, Chien KR. Ventricular muscle-restricted targeting of the RXRa gene reveals a non-cell-autonomous requirement in cardiac chamber morphogenesis. Development. 1998b;125:1943–1949. doi: 10.1242/dev.125.10.1943. [DOI] [PubMed] [Google Scholar]
- Davis JS, Hassanzadeh S, Winitsky S, Lin H, Satorius C, Vemuri R, Aletras Ah, Wen H, Epstein ND. The overall pattern of cardiac contraction depends on a spatial gradient of myosin regulatory light chain phosphorylation. Cell. 2001;107:631–41. doi: 10.1016/s0092-8674(01)00586-4. [DOI] [PubMed] [Google Scholar]
- Diffee GM, Patel JR, Reinach FC, Greaser ML, Moss RL. Altered kinetics of contraction in skeletal muscle fibers containing a mutant myosin regulatory light chain with reduced divalent cation binding. Biophys J. 1996;71:341–350. doi: 10.1016/S0006-3495(96)79231-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding P, Huang J, Battiprolu PK, Hill JA, Kamm KE, Stull J. Cardiac myosin light chain kinase in necessary for myosin regulatory light chain phosphorylation and cardiac performance in vivo. J Biol Chem. 2010;285:40819–40829. doi: 10.1074/jbc.M110.160499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyson E, Sucov HM, Kubalak SW, Schmid-Schonbein GW, DeLano FA, Evans RM, Ross J, Jr, Chien KR. Atrial-like phenotype is associated with embryonic ventricular failure in retinoid X receptor alpha –/– mice. Proc Natl Acad Sci USA. 1995;92:7386–7390. doi: 10.1073/pnas.92.16.7386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einat P, Bergman Y, Yaffe D, Shani M. Expression in transgenic mice of two genes of different tissue specificity integrated into a single chromosomal site. Genes Dev. 1987;1:1075–1084. doi: 10.1101/gad.1.10.1075. [DOI] [PubMed] [Google Scholar]
- Faerman A, Shani M. The expression of the regulatory myosin light chain 2 gene during mouse embryogenesis. Development. 1993;118:919–929. doi: 10.1242/dev.118.3.919. [DOI] [PubMed] [Google Scholar]
- Farman GP, Muthu P, Kazmierczak K, Szczesna-Cordary D, Moore JR. Impact of familial hypertrophic cardiomyopathy-linked mutations in the NH2 terminus of the RLC on β-myosin cross-bridge mechanics. J Appl Physiol. 2014;117:1471–1477. doi: 10.1152/japplphysiol.00798.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzsimons DP, Bodell PW, Baldwin KM. Phosphorylation of rodent cardiac myosin light chain-2: effects of exercise. J Appl Physiol. 1989;67:2447–2453. doi: 10.1152/jappl.1989.67.6.2447. [DOI] [PubMed] [Google Scholar]
- Frearson N, Perry SV. Phosphorylation of the light-chain components of myosin from cardiac and red skeletal muscles. Biochem J. 1975;151:99–107. doi: 10.1042/bj1510099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabarek Z. Structural basis for diversity of the EF-hand calcium binding proteins. J Mol Biol. 2006;359:509–525. doi: 10.1016/j.jmb.2006.03.066. [DOI] [PubMed] [Google Scholar]
- Greenberg MJ, Watt JD, Jones M, Kazmierczak K, Szczesna-Cordary D, Moore JR. Regulatory light chain mutations associated with cardiomyopathy affect myosin mechanics and kinetics. J Mol Cell Cardiol. 2009;46:108–115. doi: 10.1016/j.yjmcc.2008.09.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulick J, Hewett TE, Klevitsky R, Buck SH, Moss RL, Robbins J. Transgenic remodeling of the regulatory myosin light chains in the mammalian heart. Circ Res. 1997;80:655–664. doi: 10.1161/01.res.80.5.655. [DOI] [PubMed] [Google Scholar]
- Henderson SA, Xu YC, Chien KR. Nucleotide sequence of full length cDNAs encoding rat cardiac myosin light chain-2. Nucleic Acids Res. 1988;16:4722. doi: 10.1093/nar/16.10.4722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidalgo C, Wu Y, Peng J, Siems WF, Campbell KB, Granzier H. Effect of diastolic pressure on MLC2v phosphorylation in the rat left ventricle. Arch Biochem Biophys. 2006;456:216–223. doi: 10.1016/j.abb.2006.06.026. [DOI] [PubMed] [Google Scholar]
- Huang C, Sheikh F, Hollander M, Cai C, Becker D, Chu PH, Evans S, Chen J. Embryonic atrial function is essential for mouse embryogenesis, cardiac morphogenesis and angiogenesis. Development. 2003;130:6111–6119. doi: 10.1242/dev.00831. [DOI] [PubMed] [Google Scholar]
- Huang J, Shelton JM, Richardson JA, Kamm KE, Stull JT. Myosin regulatory light chain phosphorylation attenuates cardiac hypertrophy. J Biol Chem. 2008;283:19748–56. doi: 10.1074/jbc.M802605200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Liang J, Yuan CC, Kazmierczak K, Zhou Z, Morales A, McBride KL, Fitzgerald-Butt SM, Hershberger RE, Szczesna-Cordary D. Novel familial dilated cardiomyopathy mutation in MYL2 affects the structure and function of myosin regulatory light chain. FEBS J. 2015 doi: 10.1111/febs.13286. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Liang J, Kazmierczak K, Muthu P, Duggal D, Farman GP, Sorensen L, Pozios I, Abraham TP, Moore JR, Borejdo J, Szczesna-Cordary D. Hypertrophic cardiomyopathy associated Lys104Glu mutation in the myosin regulatory light chain causes diastolic disturbance in mice. J Mol Cell Cardiol. 2014;74:318–329. doi: 10.1016/j.yjmcc.2014.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huxley AF, Simmons RM. Mechanical properties of the cross-bridges of frog striated muscle. J Physiol. 1971;(218) Suppl:59P–60P. [PubMed] [Google Scholar]
- Jacques AM, Briceno N, Messer AE, Gallon CE, Jalilzadeh S, Garcia E, Kikonda-Kanda G, Goddard J, Harding SE, Watkins H, Esteban MT, Tsang VT, McKenna WJ, Marston SB. The molecular phenotype of human cardiac myosin associated with hypertrophic obstructive cardiomyopathy. Cardiovasc Res. 2008;79:481–491. doi: 10.1093/cvr/cvn094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katcoff D, Nudel U, Zevin-Sonkin D, Carmon Y, Shani M, Lehrach H, Frischauf AM, Yaffe D. Construction of recombinant plasmids containing rat muscle actin and myosin light chain DNA sequences. Proc Natl Acad Sci USA. 1980;77:960–964. doi: 10.1073/pnas.77.2.960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaski H, Kretsinger RH. Structural differences among subfamilies of EF-hand proteins—A view from the pseudo two-fold symmetry axis. Proteins. 2014;82:2915–2924. doi: 10.1002/prot.24562. [DOI] [PubMed] [Google Scholar]
- Kubalak SW, Miller-Hance WC, O'Brien TX, Dyson E, Chien KR. Chamber specification of atrial myosin light chain-2 expression precedes septation during murine cardiogenesis. J Biol Chem. 1994;269:16961–16970. [PubMed] [Google Scholar]
- Kumar CC, Cribbs L, Delaney P, Chien KR, Siddiqui MA. Heart myosin light chain 2 gene. Nucleotide sequence of full length cDNA and expression in normal and hypertensive rat. J Biol Chem. 1986;261:2866–2872. [PubMed] [Google Scholar]
- Lamberts RR, Hamdani N, Soekhoe TW, Boontje NM, Zaremba R, Walker LA, de Tombe PP, van der Velden J, Stienen GJ. Frequency-dependent myofilament Ca2+ desensitization in failing rat myocardium. J Physiol. 2007;582:695–709. doi: 10.1113/jphysiol.2007.134486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowey S, Waller GS, Trybus KM. Skeletal muscle myosin light chains are essential for physiological speeds of shortening. Nature. 1993;365:454–456. doi: 10.1038/365454a0. [DOI] [PubMed] [Google Scholar]
- Margossian SS, White HD, Caulfield JB, Norton P, Taylor S, Slayter HS. Light chain 2 profile and activity of human ventricular myosin during dilated cardiomyopathy. Identification of a causal agent for impaired myocardial function. Circulation. 1992;85:1720–1733. doi: 10.1161/01.cir.85.5.1720. [DOI] [PubMed] [Google Scholar]
- Monasky MM, Biesiadecki BJ, Janssen PM. Increased phosphorylation of tropomyosin, troponin I, and myosin light chain-2 after stretch in rabbit ventricular myocardium under physiological conditions. J Mol Cell Cardiol. 2010;48:1023–1028. doi: 10.1016/j.yjmcc.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monasky MM, Taglieri DM, Jacobson AK, Haizlip KM, Solaro RJ, Janssen PM. Post-translational modifications of myofilament proteins involved in length-dependent prolongation of relaxation in rabbit right ventricular myocardium. Arch Biochem Biophys. 2013;535:22–29. doi: 10.1016/j.abb.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morano I. Effects of different expression and posttranslational modifications of myosin light chains on contractility of skinned human cardiac fibers. Basic Res Cardiol. 1992;87:129–141. doi: 10.1007/978-3-642-72474-9_11. [DOI] [PubMed] [Google Scholar]
- Mizutani H, Okamoto R, Moriki N, Konishi K, Taniguchi M, Fujita S, Dohi K, Onishi K, Suzuki N, Satoh S, Makino N, Itoh T, Hartshorne DJ, Ito M. Overexpression of myosin phosphatase reduces Ca(2+) sensitivity of contraction and impairs cardiac function. Circ J. 2010;74:120–28. doi: 10.1253/circj.cj-09-0462. [DOI] [PubMed] [Google Scholar]
- Muthu P, Kazmierczak K, Jones M, Szczesna-Cordary D. The effect of myosin RLC phosphorylation in normal and cardiomyopathic mouse hearts. J Cell Mol Med. 2012;16:911–919. doi: 10.1111/j.1582-4934.2011.01371.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nudel U, Calvo JM, Shani M, Levy Z. The nucleotide sequence of a rat myosin light chain 2 gene. Nucleic Acids Res. 1984;12:7175–7186. doi: 10.1093/nar/12.18.7175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien TX, Lee KJ, Chien KR. Positional specification of ventricular myosin light chain 2 expression in the primitive murine heart tube. Proc Natl Acad Sci USA. 1993;90:5157–5161. doi: 10.1073/pnas.90.11.5157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson M, Patel J, Fitzsimons D, Walker J, Moss RL. Basal myosin light chain phosphorylation is a determinant of Ca2+ sensitivity of force and activation dependence of the kinetics of myocardial force development. Am J Physiol Heart Circ Physiol. 2004;287:H2712–H2718. doi: 10.1152/ajpheart.01067.2003. [DOI] [PubMed] [Google Scholar]
- Pires EM, Perry SV. Purification and properties of myosin light-chain kinase from fast skeletal muscle. Biochem J. 1977;167:137–146. doi: 10.1042/bj1670137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrie WT, Smillie LB, Perry SB. A phosphorylted light-chain component of myosin from skeletal muscle. Biochem J. 1973;135:151–164. doi: 10.1042/bj1350151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poetter K, Jiang H, Hassandeh S, Master SR, Chang A, Dalakas MC, Rayment I, Sellers JR, Fananapazir L, Epstein ND. Mutations in either the essential or regulatory light chains of myosin are associated with a rare myopathy in human heart and skeletal muscle. Nat Genet. 1996;13:63–69. doi: 10.1038/ng0596-63. [DOI] [PubMed] [Google Scholar]
- Rajashree R, Blunt BC, Hofmann PA. Modulation of myosin phosphatase targeting subunit and protein phosphatase 1 in the heart. Am J Physiol Heart Circ Physiol. 2005;289:H1736–H1743. doi: 10.1152/ajpheart.00318.2004. [DOI] [PubMed] [Google Scholar]
- Ramirez-Correa GA, Jin W, Wang Z, Zhong X, Gao WD, Dias WB, Vecoli C, Hart GW, Murphy AM. O-linked GlcNAc modification of cardiac myofilament proteins: a novel regulator of myocardial contractile function. Circ Res. 2008;103:1354–1358. doi: 10.1161/CIRCRESAHA.108.184978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratti J, Rostkova E, Gautel M, Pfuhl M. Structure and interactions of myosin binding protein C domain C0: cardiac-specific regulation of myosin at its neck? J Biol Chem. 2011;286:12650–12658. doi: 10.1074/jbc.M110.156646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayment I, Holden HM, Whittaker M, Yohn CB, Lorenz M, Holmes KC, Milligan RA. Structure of the actin-myosin complex and its implications for muscle contraction. Science. 1993a;261:58–65. doi: 10.1126/science.8316858. [DOI] [PubMed] [Google Scholar]
- Rayment I, Rypniewski WR, Schmidt-Base K, Smith R, Tomchick DR, Benning MM, Winkelmann DA, Wesenberg G, Holden HM. Three-dimensional structure of myosin subfragment-1: a molecular motor. Science. 1993b;261:50–58. doi: 10.1126/science.8316857. [DOI] [PubMed] [Google Scholar]
- Ross RS, Navankasattusas S, Harvey RP, Chien KR. An HF-1a/HF-1b/MEF-2 combinatorial element confers cardiac ventricular specificity and established an anterior-posterior gradient of expression. Development. 1996;122:1799–1809. doi: 10.1242/dev.122.6.1799. [DOI] [PubMed] [Google Scholar]
- Rottbauer W, Wessels G, Dahme T, Just S, Trano N, Hassel D, Burns CG, Katus HA, Fishman MC. Cardiac myosin light chain-2: a novel essential component of thick-myofilament assembly of the heart. Circ Res. 2006;99:323–331. doi: 10.1161/01.RES.0000234807.16034.fe. [DOI] [PubMed] [Google Scholar]
- Scruggs SB, Reisdorph R, Armstrong ML, Warren CM, Reisdorph N, Solaro RJ, Buttrick PM. A novel, in-solution separation of endogenous cardiac sarcomeric proteins and identification of distinct charged variants of regulatory light chain. Mol Cell Proteomics. 2010;9:1804–18. doi: 10.1074/mcp.M110.000075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiaffino S, Reggiani C. Molecular diversity of myofibrillar proteins: gene regulation and functional significance. Physiol Rev. 1996;76:371–423. doi: 10.1152/physrev.1996.76.2.371. [DOI] [PubMed] [Google Scholar]
- Seguchi O, Takashima S, Yamazaki S, Asakura M, Asano Y, Shintani Y, Wakeno M, Minamino T, Kondo H, Furukawa H, Nakamura K, Naito A, Takahashi T, Ohtsuka T, Kawakami K, Isomura T, Kitamura S, Tomoike H, Mochizuki N, Kitakaze M. A cardiac myosin light chain kinase regulates sarcomere assembly in the vertebrate heart. J Clin Invest. 2007;117:2812–24. doi: 10.1172/JCI30804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheikh F, Lyon RC, Chen J. Getting the skinny on thick filament regulation in cardiac muscle biology and disease. Trends Cardiovasc Med. 2014;24:133–141. doi: 10.1016/j.tcm.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheikh F, Ouyang K, Campbell SG, Lyon RC, Chuang J, Fitzsimons D, Tangney J, Hidalgo CG, Chung CS, Cheng H, Dalton ND, Gu Y, Kasahara H, Ghassemian M, Omens JH, Peterson KL, Granzier HL, Moss RL, McCulloch AD, Chen J. Mouse and computational models link Mlc2v dephosphorylation to altered myosin kinetics in early cardiac disease. J Clin Invest. 2012;122:1209–21. doi: 10.1172/JCI61134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spirito P, Seidman CE, McKenna WJ, Maron BJ. The management of hypertrophic cardiomyopathy. N Engl J Med. 1997;336:775–785. doi: 10.1056/NEJM199703133361107. [DOI] [PubMed] [Google Scholar]
- Stelzer JE, Patel JR, Moss RL. Acceleration of stretch activation in murine myocardium due to phosphorylation of myosin regulatory light chain. J Gen Physiol. 2006;128:261–72. doi: 10.1085/jgp.200609547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stull JT, Hsu LC, Tansey MG, Kamm KE. Myosin light chain kinase phosphorylation in tracheal smooth muscle. J Biol Chem. 1990;265:16683–16690. [PubMed] [Google Scholar]
- Sweeney HL, Stull JT. Phosphorylation of myosin in permeabilized mammalian cardiac and skeletal muscle cells. Am J Physiol. 1986;250:C657–C660. doi: 10.1152/ajpcell.1986.250.4.C657. [DOI] [PubMed] [Google Scholar]
- Sweeney HL, Bowman BF, Stull JT. Myosin light chain phosphorylation in vertebrate striated muscle: regulation and function. Am J Physiol. 1993;264:C1085–C1095. doi: 10.1152/ajpcell.1993.264.5.C1085. [DOI] [PubMed] [Google Scholar]
- Szczesna D, Ghosh D, Li Q, Gomes AV, Guzman G, Arana C, Zhi G, Stull JT, Potter JD. Familial hypertrophic cardiomyopathy mutations in the regulatory light chains of myosin affect their structure, Ca2+ binding, and phosphorylation. J Biol Chem. 2001;276:7086–7092. doi: 10.1074/jbc.M009823200. [DOI] [PubMed] [Google Scholar]
- Szczesna-Cordary D, Guzman G, Ng SS, Zhao J. Familial hypertrophic cardiomyopathy-linked alterations in Ca2+ binding of human cardiac myosin regulatory light chain affect cardiac muscle contraction. J Biol Chem. 2004;279:3535–3542. doi: 10.1074/jbc.M307092200. [DOI] [PubMed] [Google Scholar]
- Szczesna D, Zhao J, Potter JD. The regulatory light chains of myosin modulate cross-bridge cycling in skeletal muscle. J Biol Chem. 1996;271:5246–5250. doi: 10.1074/jbc.271.9.5246. [DOI] [PubMed] [Google Scholar]
- van der Velden J, Klein LJ, Zaremba R, Boontje NM, Huybregts MA, Stooker W, Eijsman L, de Jong JW, Visser CA, Visser FC, Stienen GJ. Effects of calcium, inorganic phosphate, and pH on isometric force in single skinned cardiomyocytes from donor and failing human hearts. Circulation. 2001;104:1140–1146. doi: 10.1161/hc3501.095485. [DOI] [PubMed] [Google Scholar]
- van der Velden J, Papp Z, Zaremba R, Boontje NM, de Jong JW, Owen VJ, Burton PB, Goldmann P, Jaquet K, Stienen GJ. Increased Ca2+ sensitivity of the contractile apparatus in end-stage human heart failure results from altered phosphorylation of contractile proteins. Cardiovasc Res. 2003a;57:37–47. doi: 10.1016/s0008-6363(02)00606-5. [DOI] [PubMed] [Google Scholar]
- van der Velden J, Papp Z, Boontje NM, Zaremba R, de Jong JW, Janssen PM, Hasenfuss G, Stienen GJ. The effect of myosin light chain 2 dephosphorylation on Ca2+ -sensitivity of force is enhanced in failing hearts. Cardiovasc Res. 2003b;57:505–514. doi: 10.1016/s0008-6363(02)00662-4. [DOI] [PubMed] [Google Scholar]
- Varian KD, Janssen PM. Frequency-dependent acceleration of relaxation involves decreased myofilament calcium sensitivity. Am J Physiol Heart Circ Physiol. 2007;292:H2212–H2219. doi: 10.1152/ajpheart.00778.2006. [DOI] [PubMed] [Google Scholar]
- Warren SA, Briggs LE, Zeng H, Chuang J, Chang EI, Terada R, Li M, Swanson MS, Lecker SH, Willis MS, Spinale FG, Maupin-Furlowe J, McMullen JR, Moss RL, Kasahara H. Myosin light chain phosphorylation is critical for adaptation to cardiac stress. Circulation. 2012;126:2575–2588. doi: 10.1161/CIRCULATIONAHA.112.116202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeds AG. Light chains of myosin. Nature. 1969;223:1362–1364. doi: 10.1038/2231362a0. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Yang W, Kirberger M, Lee HW, Ayalasomayajula G, Yang JJ. Prediction of EF-hand calcium-binding proteins and analysis of bacterial EF-hand proteins. Proteins. 2006;65:643–655. doi: 10.1002/prot.21139. [DOI] [PubMed] [Google Scholar]