

Abstract
Targeting brown adipose tissue (BAT) content or activity has therapeutic potential for treating obesity and the metabolic syndrome by increasing energy expenditure. Both inter- and intra-individual differences contribute to heterogeneity in human BAT and potentially to differential thermogenic capacity in human populations. Here, we demonstrated the generated clones of brown and white preadipocytes from human neck fat of four individuals and characterized their adipogenic differentiation and thermogenic function. Combining an uncoupling protein 1(UCP1) reporter system and expression profiling, we defined novel sets of gene signatures in human preadipocytes that could predict the thermogenic potential of the cells once they were maturated in culture. Knocking out the positive UCP1 regulators identified by this approach, PREX1 and EDNRB in brown preadipocytes using CRISPR/Cas9 markedly abolished the high level of UCP1 in brown adipocytes differentiated from the preadipocytes. Finally, we were able to prospectively isolate adipose progenitors with great thermogenic potential using cell surface marker CD29. These data provide new insights into the cellular heterogeneity in human fat and offer the identification of possible biomarkers of thermogenically competent preadipocytes.
Obesity is a pandemic and major contributor to metabolic disorders. Increased adiposity is the main characteristic of obesity. In mammals, there are two functionally distinct types of fat: white adipose tissue (WAT), which is specialized for energy storage, and brown adipose tissue (BAT), which dissipates energy for thermogenesis1,2 via the activity of uncoupling protein 1 (UCP1). In addition to the classical brown adipocytes, UCP1-positive “beige” or “brite” adipocytes can be recruited within WAT upon chronic cold or β3-adrenergic stimulation3–6.
Owing to the immense capacity of BAT to combust fuels for heat production7,8 and the presence of BAT in adult humans9–14, increasing the amount or activity of brown or beige fat has been considered as an appealing approach for the treatment or prevention of obesity and related metabolic disorders. Indeed, in rodents activation of brown or beige fat can promote increased energy expenditure and protects from diet-induced obesity5,6,15. In humans, BAT mass or activity is inversely correlated to body mass index and percent body fat10–12. Cold exposure in humans can elevate BAT volume and activity and increase energy expenditure, pointing towards a therapeutic potential of BAT in humans for the treatment of obesity and metabolic disease16–18.
Recent data indicate that the neck, supraclavicular and spinal cord regions of adult humans contain substantial deposits of UCP1-positive adipocytes19–22. The presence of brown, beige, and white adipocytes as well as perhaps other unidentified adipose cell types highlights the heterogeneity of adipose tissue depots, which potentially links to their diverse functions in energy metabolism. Both inter-subject differences and various cellular compositions within a given fat tissue contribute to the heterogeneity of human BAT and affect thermogenic potential. In rodents, lineage tracing and cell sorting analyses demonstrate that the various types of fat cells arise from discrete pools of progenitors, which express distinct molecular markers19,23–26. However, whether these markers identified in mouse cells can unambiguously define different types of human adipose progenitors is currently unknown.
A key impediment for these studies is the lack of human-derived brown and white fat progenitor cell models. In order to investigate the heterogeneous nature of the progenitor cell population in human BAT and WAT, we have generated clonal cell lines from human neck fat and characterized their adipogenic differentiation and metabolic function in vitro and in vivo after transplantation into immune deficient nude mice. Using clonal analysis and gene expression profiling, we have defined unique sets of gene signatures in human preadipocytes that could predict the thermogenic potential of these cells once matured in culture into adipocytes. These data highlight the cellular heterogeneity in human BAT and WAT and provide novel gene targets that may be targeted or selected for to prime preadipocytes for strong thermogenic differentiation.
Results
Generation and characterization of human fat progenitors
We have previously reported that adult human BAT and WAT are present in defined neck locations20, and found that deeper human neck fat was predominantly brown as these depots express significantly higher levels of the brown fat-specific marker UCP1 compared with expression detected in the superficial neck fat. To define molecular and functional characteristics of specific adipose progenitors, we generated human preadipocyte pooled cell populations derived from a total of four human subjects by isolating cells from the stromal vascular fraction (SVF) of human neck fat and immortalizing them via stable expression of human telomere reverse transcriptase (hTert)27 (Supplementary Fig. 1a). Pairs of immortalized progenitors for human BAT (hBAT-SVF, isolated from deep neck fat) and human WAT (hWAT-SVF, isolated from superficial neck fat) of the same individuals were established from each of the four individuals for proper comparisons (Supplementary Table 1a). The immortalized cells could be passaged in culture for more than 90 days and have been followed for at least 20 population doublings (Supplementary Fig. 1b).
After immortalization the cells from both WAT and BAT depots of the four human subjects maintained a fibroblast-like morphology and following induction with a standard adipogenic differentiation protocol all precursors became lipid-laden cells expressing a high level of the mature adipocyte marker fatty acid synthase (FASN) (Fig. 1a and Supplementary Fig. 1c). Notably, in differentiated hBAT-SVF cells (referred as human brown adipocytes, hBA), expression of the brown fat marker UCP1 was up to 200-fold higher than in differentiated hWAT-SVF cells (human white adipocytes, hWA) (Fig. 1b), and was accompanied by robust induction of UCP1 protein (Fig. 1c and Supplementary Fig. 1d). A comparable pattern of expression was observed for other brown fat markers such as deiodinase 2 (DIO2) and peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PPARGC1A)(Supplementary Fig. 1c). LEP, a marker of WAT, was selectively expressed in hWA compared to hBA in all subjects (Fig. 1d). Importantly, the immortalized cells retained differentiation characteristics of primary cells (Supplementary Fig. 2).
Figure 1.
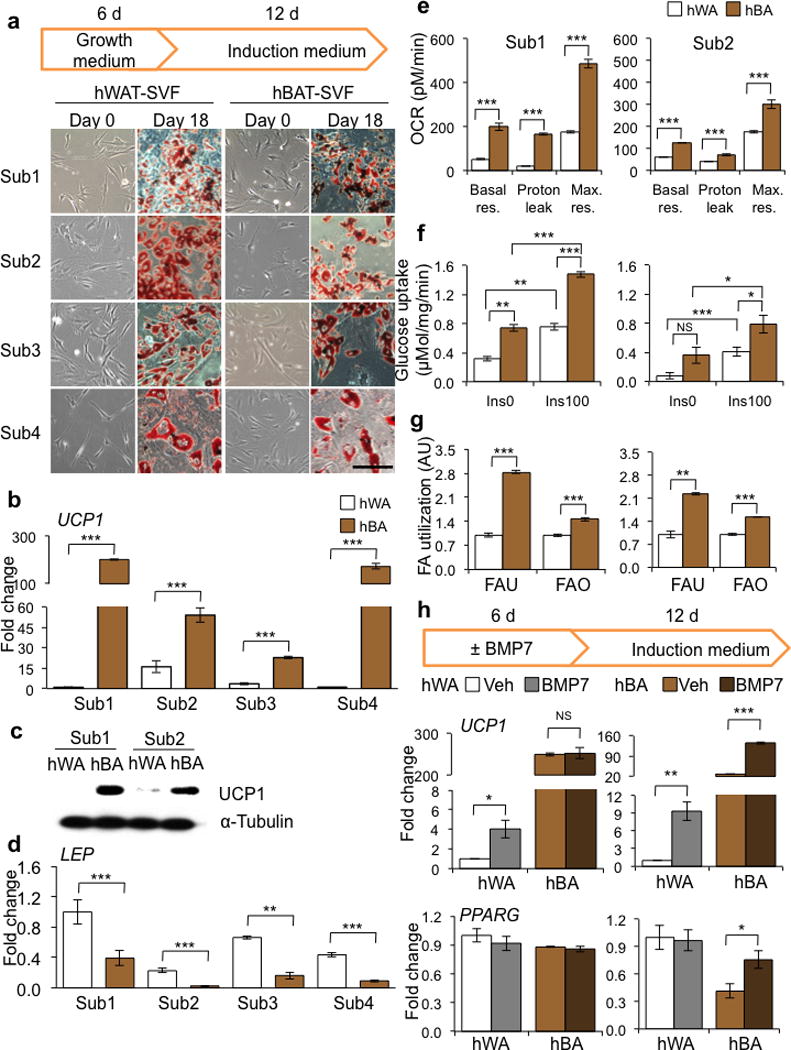
Generation and characterization of immortalized human brown and white fat progenitors. (a) Light microscopic images of immortalized human WAT progenitors (hWAT-SVF) and human BAT progenitors (hBAT-SVF) at day 0 and 18 (stained with Oil Red O) from 4 subjects. Scale bar, 100 μm. (b,d) Q-RT-PCR analysis for UCP1 and LEP mRNA expression in differentiated adipocytes from hWAT-SVF and hBAT-SVF of 4 subjects. Data are presented as fold changes relative to Sub1 hWA (mean ± s.e.m., n=3). (c) Western blot analysis of UCP1 protein level in hWA and hBA differentiated from progenitors of Sub1 and Sub2. α-Tubulin serves as a loading control. (e) Oxygen consumption rate (OCR) was measured in the absence (Basal respiration, Basal Res.) and presence of oligomycin (Proton Leak) or FCCP (Maximal respiration, Max. Res.) in hWA and hBA from Sub1 (Left) and Sub2 (Right). Data are presented as mean ± s.e.m. (n=10; hWA vs hBA). (f) Glucose uptake was measured using 3H-2-deoxy-glucose in hWA and hBA stimulated with (Ins100) or without (Ins0) 100 nM insulin from Sub1 (Left) and Sub2 (Right). Data are presented as mean ± s.e.m. (n=3). (g) Fatty acid uptake (FAU) and fatty acid oxidation (FAO) were measured using 14C-palmitic acid in hWA and hBA from Sub1 (Left) and Sub2 (Right). Data are presented as a fold change compared to hWA (mean ± s.e.m., n=3). (h) Q-RT-PCR analysis for UCP1 and PPARG mRNA expression in hWA and hBA from Sub1 (Left) and Sub2 (Right). Data are presented as fold changes compared to vehicle-hWA for each subject (mean ± s.e.m., n=3; NS, not significant; Veh vs BMP7). Two-tailed Student’s t-test was used to determine P values (* P < 0.05, ** P < 0.01, *** P < 0.001).
To determine whether the differentiated cells possessed metabolic capacity, we evaluated cellular respiration and fuel utilization in hBA and hWA of Subject 1 and 2. Consistent with differences in gene expression, the levels of basal and maximal respiration as well as proton leak in hBA were higher compared with hWA (Fig. 1e). Glucose uptake in both the basal and insulin-stimulated states was also notably higher in hBA than hWA as was both fatty acid uptake and oxidation rate (Fig. 1f,g). Differentiated brown, but not white, adipocytes were able to respond to forskolin, a chemical mimic of β-adrenergic stimulation, by increasing the oxygen consumption rate (Supplementary Fig. 3a). In addition, hBA from Subject 2 could respond to stimulations of norepinephrine and other browning agents by increasing the levels of UCP1 and DIO2 (Supplementary Fig. 3b and Supplementary Fig. 4), suggesting that the mature human brown adipocytes are responsive to both physiological and pharmacological adrenergic stimuli.
Recent studies using mouse systems have demonstrated that adipose progenitors can respond to inductive signals and increase their thermogenic capacity in mature adipocytes4,23,28. To determine whether human-derived progenitors can respond to browning agents, we pretreated the aforementioned precursors from Subject1 and Subject2 with BMP723,29 for 6 days, followed by adipogenic induction. Pre-exposure to BMP7 of hWAT-SVF from both subjects and hBAT-SVF from Subject2 led to increased UCP1 expression, mitochondrial activity and fuel utilization in mature adipocytes (Fig. 1h and Supplementary Fig. 5a–f), suggesting a fraction of these progenitors are inducible. Pretreatment with BMP7 also augmented peroxisome proliferator-activated receptor gamma (PPARG) expression in brown adipocytes from Subject2 only. BMP8, another browning agent28, exerted similar effects (Supplementary Fig. 5a).
The fact that hBAT-SVF and hBA derived from Subject2, but not Subject1, consistently responded to browning agents suggest that the cells derived from Subject2 are more inducible while cells derived from Subject1 may represent the classical brown fat cells which possess a very high level of UCP1 at basal. The distinction of classical versus inducible hBA between Subject1 and Subject2 was further supported by the differential expression levels of the classical BAT maker ZIC14,30,31 (Supplementary Fig. 5g).
These data support the previous characterization of the tissue from human neck BAT and WAT20 and demonstrate that the progenitor cell populations we have generated recapitulate adipogenic differentiation and thermogenic expression profiles in vitro. Further, inter-subject differences not only exist in whole adipose tissue as previously noted20, but also exist in adipose progenitors and their derived adipocytes in culture. Despite the inter-subject variations, human brown adipocytes clearly possess high levels of UCP1 and great bioenergetic capacity.
Creation of a reporter system for monitoring UCP1 expression
To allow direct assessment of the thermogenic potential of differentiated cells, we introduced a transgenic reporter construct into the white and brown fat precursors to measure UCP1 gene expression by coupling a bicistronic luciferase and green fluorescent protein (GFP) reporter system to a 4.1-Kb human UCP1 promoter fragment (Fig. 2a). In mature adipocytes that stably expressed the reporter construct, luciferase activity was strongly correlated with endogenous UCP1 gene expression and only detected in mature brown adipocytes but not in undifferentiated cells (Fig. 2b). We monitored differentiating cells using time-lapse microscopy and could detect activation of the GFP reporter as early as day 9 in differentiating BAT cells (Fig. 2c and Supplementary video). To determine if these cells were capable of differentiation in vivo, we transplanted progenitor cells into immune-deficient nude mice and used in vivo bioluminescent imaging to measure UCP1 reporter activity. Luciferase activity was high in mice implanted with hBAT progenitors, and could be further induced by BMP7 pretreatment of progenitors (Fig. 2d). Conversely, mice receiving transplanted hWAT progenitors displayed almost no detectable luciferase activity. While both grafts expressed similar level of FABP4, fat grafts from hBAT-SVF displayed at least 100-fold increase in UCP1 mRNA compared to hWAT-SVF-derived fat pads which is consistent with luciferase activity. LEP was selectively expressed in fat grafts from hWAT-SVF (Fig. 2e). These data demonstrate that the UCP1 reporter system accurately indicates differentiation into mature brown adipocytes in both in vivo and in vitro settings.
Figure 2.
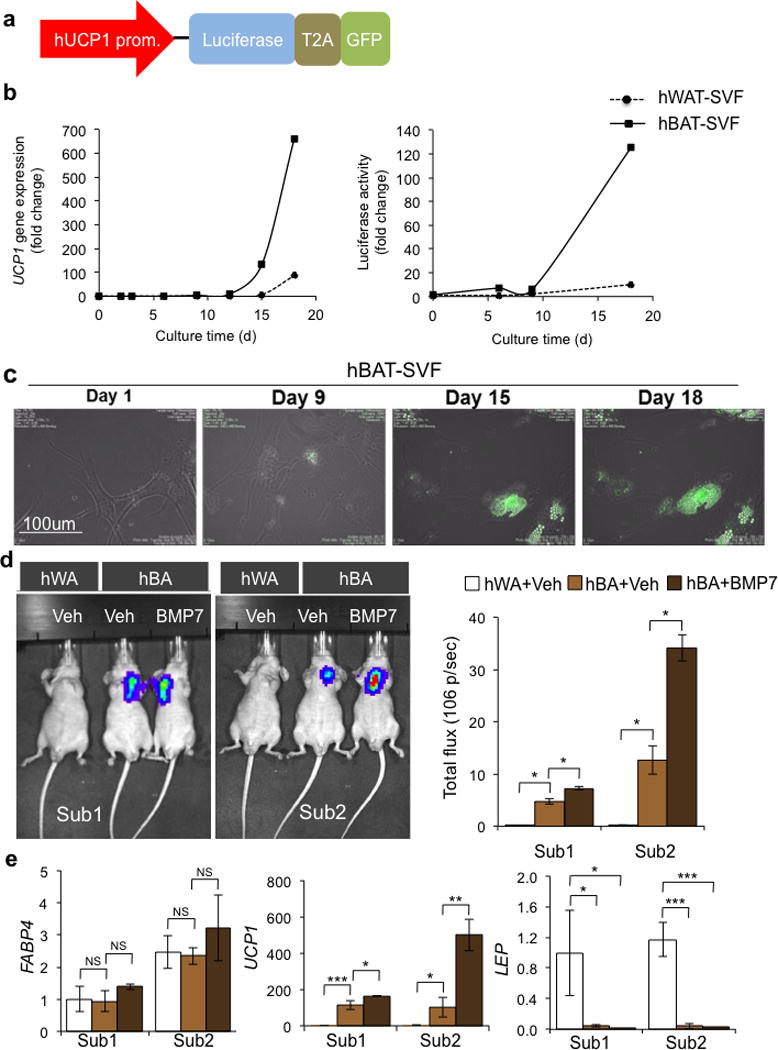
Utilization of a UCP1 reporter system for in vitro and in vivo monitoring of UCP1 expression. (a) Schematic structure of the hUCP1 promoter reporter system. 4148 bp of human UCP1 promoter drives the expression of bicistronic luciferase and GFP. T2A is the internal ribosomal entry site. (b) In hBAT-SVF and hWAT-SVF stably expressed the reporter construct, luciferase activity (Right) was strongly correlated with endogenous UCP1 gene expression (Left) during the course of differentiation (see Fig. 1a and Method). Data are presented as fold changes compared to hWAT-SVF on day 0 (mean ± s.e.m., n=3). A representative experiment from a total of two independent studies is shown. (c) Monitoring UCP1 expression by GFP in vitro using a time lapse imaging system during differentiation of hBAT-SVF from Sub1. (d) Representative IVIS images of nude mice after 22 days of transplantation of hWAT-SVF and hBAT-SVF are shown on the left panel. Quantifications of luciferase activity by total flux are shown on the right panel (mean ± s.e.m.). The experiments have been repeated twice (n=2 for hWAT-SVF group; n=3 for hBAT-SVF group). (e) Q-RT-PCR analysis for expression of FABP4, UCP1 and LEP in fat pads developed from the transplanted cells. Data are presented as fold changes compared to fat pads developed from hWAT-SVF with vehicle treatment (mean ± s.e.m.). Two-tailed Student’s t-test was used to determine P values (* P < 0.05, ** P < 0.01, *** P < 0.001).
Clonal analysis of human brown and white fat progenitors
In order to study homogenous cell populations derived from human adipose precursor cells, we isolated a total of 280 clonal preadipocyte cell lines (152 hWAT-SVF clones and 128 hBAT-SVF clones) from the immortalized pool populations of all four subjects. Of these lines, 44% (67 out of 152) of hWAT-SVF and 70% (90 out of 128) of hBAT-SVF clones robustly differentiated into mature adipocytes (Fig. 3 and Supplementary Fig. 6). To determine UCP1 induction in the differentiated state, we measured luciferase reporter activity in each clonal line after 18-days of adipogenic differentiation (Supplementary Fig. 7). Importantly, the data revealed that up to 96% of the hWAT-SVF clones were UCP1 negative, while more than 94% of the hBAT-SVF clones displayed differential levels of UCP1-luciferase activities (Fig. 3).
Figure 3.
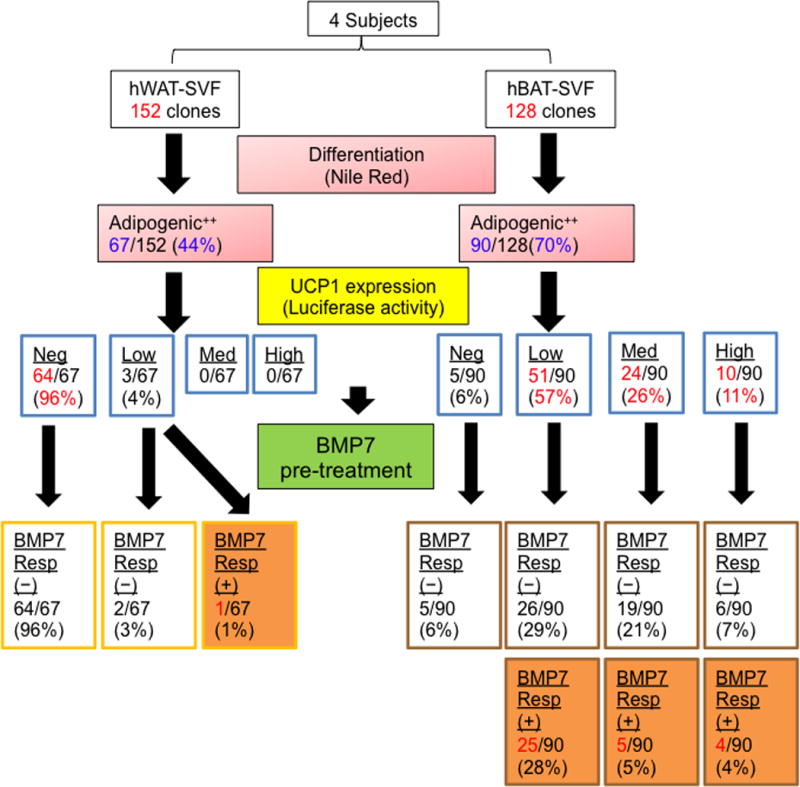
Clonal analysis of human brown and white fat progenitors. The strategy of clonal analysis of hWAT-SVF and hBAT-SVF progenitors is shown as a dendrogram. 152 clones from hWAT-SVF and 128 clones from hBAT-SVF were derived by limiting dilution from 4 subjects. Adipogenic capacity was determined by Nile red staining and UCP1 level was determined by luciferase activity on day 18. Detailed selection criteria are described in Supplementary Figures 6 and 7. Selected highly adipogenic clones (adipogenic ++) were pre-treated with 3.3 nM BMP7 for 6 days and then differentiated into mature adipocytes in a 96-well plate. Luciferase activity was measured on day 18 and divided into different levels (negative, Neg; low; medium, Med; high) after normalized to protein content. The positive response (+) to BMP7 pretreatment was defined by more than 1.5-fold increase of luciferase activity between BMP7-pretreated and vehicle groups.
As shown in Figure 1h, certain subpopulations of hWAT or hBAT precursors could respond to inductive signals, such as BMP7, to further increase their thermogenic capacity. To identify the precursor clones that could respond to stimulation, we pretreated undifferentiated cells with BMP7 and determined reporter expression in mature cells. While only 1% of the highly adipogenic hWAT-SVF clones could respond to BMP7 pretreatment, a substantial number of hBAT-SVF clones (up to 37%) could be induced by BMP7 pretreatment (Fig. 3). Further analyzing the clones in terms of their human subject origins revealed that more than 60% of the hBAT-SVF clones from Subject2 and Subject3 could respond to BMP7 stimulation while the majority of the hBAT-SVF clones from Subject1 and Subject4 were not responsive to BMP7 pretreatment (Supplementary Table 2). These data not only support the analysis of the pooled progenitor populations described above, but also suggest that the thermogenic features of mature adipocytes are regulated by the anatomical location of the tissue they originate from in addition to genetic influences of the individual human subjects. They also highlight the heterogeneity of the human adipose clones, even among cell lines isolated from a common subject and a common tissue.
Gene signatures predict thermogenic potential of adipocytes
We aimed to identify molecular markers of thermogenically competent cells. To this end, we took advantage of the UCP1 reporter system in each clonal cell line to select a set of clones from all four subjects that represented a wide range of luciferase activity after adipogenic differentiation for further analysis (Fig. 4a). We assayed these clones’ gene expression in the preadipocyte state with microarrays, and correlated this with UCP1 expression in the differentiated state. After applying the stringent threshold of P < 0.001, which is associated with a false discovery rate (FDR) of 0.03, we prioritized 581 genes that displayed significant positive correlation and 454 genes that displayed significant negative correlation (Fig. 4b). A subset of these genes is shown along with UCP1 in Figure 4c, illustrating the association between gene expression in the preadipocyte state and UCP1 expression in the differentiated state. Interestingly, several of previously identified brown and white fat makers are among the list of positive or negative predictors (Supplementary Table 3).
Figure 4.
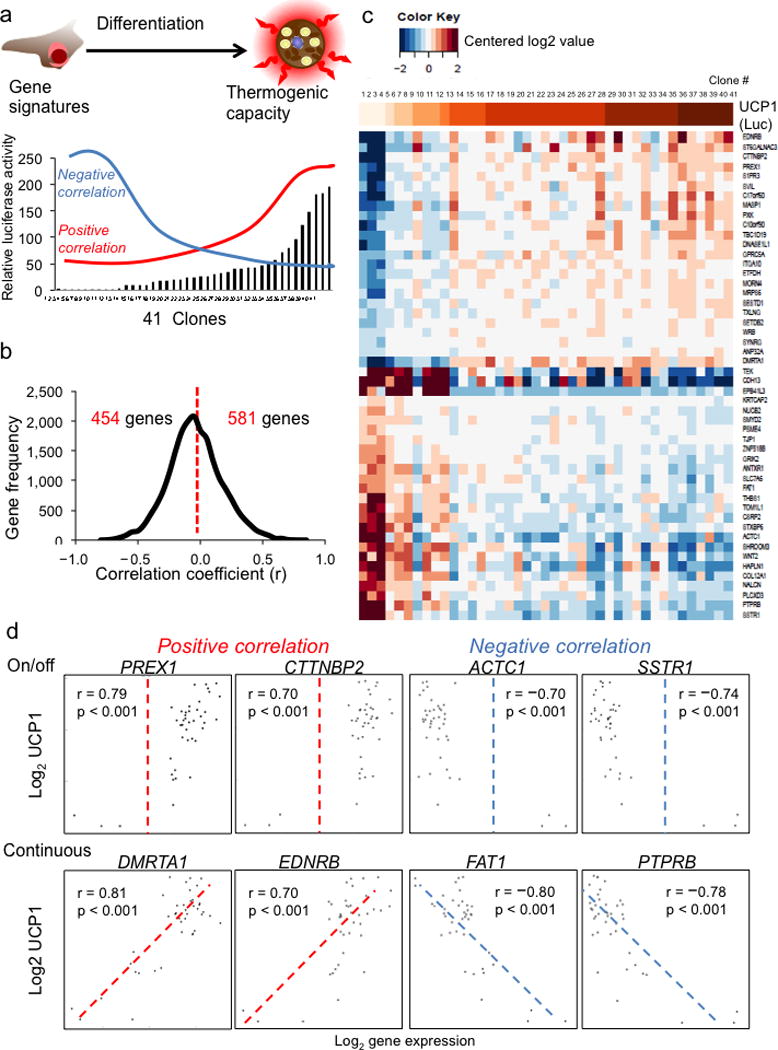
Gene expression profiles in adipose progenitors predict the thermogenic capacity of mature adipocytes. (a) A schematic presentation outlining the strategy utilized to identify the genes in preadipocytes with positive or negative correlation with UCP1 levels in mature adipocytes. Microarray analyses were done in 41 selected highly adipogenic clones from 4 subjects (8 clones from hWAT-SVF and 33 clones from hBAT-SVF). (b) Histogram showing the distribution of genes that are positively and negatively correlated with UCP1 levels (determined by luciferase activities). We used P-value < 0.001 as the cutoff to prioritize candidate genes (two-tailed alternative with function cor.test). The correlation coefficient (R) is shown in the X-axis and gene frequency is shown in the Y-axis. (c) Log2 gene expression data from 50 genes most associated with UCP1 were centered to have mean zero and restricted to the interval [−2,2] and are shown in a heatmap, along with a color bar representing UCP1 at top, where darker indicates higher UCP1. UCP1 levels were determined by luciferase activities in the reporter clones. (d) Scatter plots showing the positive and negative correlations between the UCP1-luciferase levels and expression levels of candidate genes from microarrays. The log2 gene expression level of progenitor (day 0) is shown in the X-axis. The Y-axis represents the log2 of UCP1 luciferase level of mature adipocyte (day 18).
Scatter plot analysis revealed two general categories of genes in preadipocytes that may regulate thermogenic program turned on during late stage of differentiation (Fig. 4d). The first category of genes acted as binary on and off switches to determine cell fate. Positive regulators in this category are likely required for thermogenic differentiation, while negative regulators would be completely suppressed to allow UCP1 expression of any level. Representatives of this category included phosphatidylinositol-3,4,5-trisphosphate-dependent Rac exchange factor 1 (PREX1)32, cortactin binding protein 2 (CTTNBP2)33, cardiac actin 1(ACTC1)34, and somatostatin receptor 1 (SSTR1)35. The second category of genes acted as genetic rheostats to suppress or enhance thermogenic capacity incrementally as their expression level changed. Positive regulators in this category, such as doublesex and mab-3-related transcription factor-like family A1 (DMRTA1)36 and endothelin receptor type B (EDNRB)37, might support thermogenic differentiation in proportion to their expression levels; while negative regulators, such as FAT atypical cadherin 1 (FAT1)38 and protein tyrosine phosphatase, receptor type B (PTPRB)39, might further suppress thermogenic potential as they are more highly expressed. Interestingly, most of these candidate genes have never been directly implicated in adipocyte differentiation or thermogenic regulation.
Essential role of PREX1 and EDNRB in thermogenic competency
To select promising candidate genes for further analyses, we applied the following three criteria. First, the primary selection criterion was based on their correlation coefficients, P-values and FDR values (Fig. 4c and Supplementary Table 4). Second, the top-ranking candidate genes were further verified by Q-RT-PCR assay in a set of 10 independent single cell clones derived from the same four subjects (but not included in the original microarray analysis) for positive or negative correlations between the expression levels of selected candidate genes in preadipocytes and UCP1 mRNA levels in mature adipocytes (Fig. 5a). Third, they were also validated in 7 pairs of the human neck brown and white adipose tissues (Supplementary Fig. 8).
Figure 5.
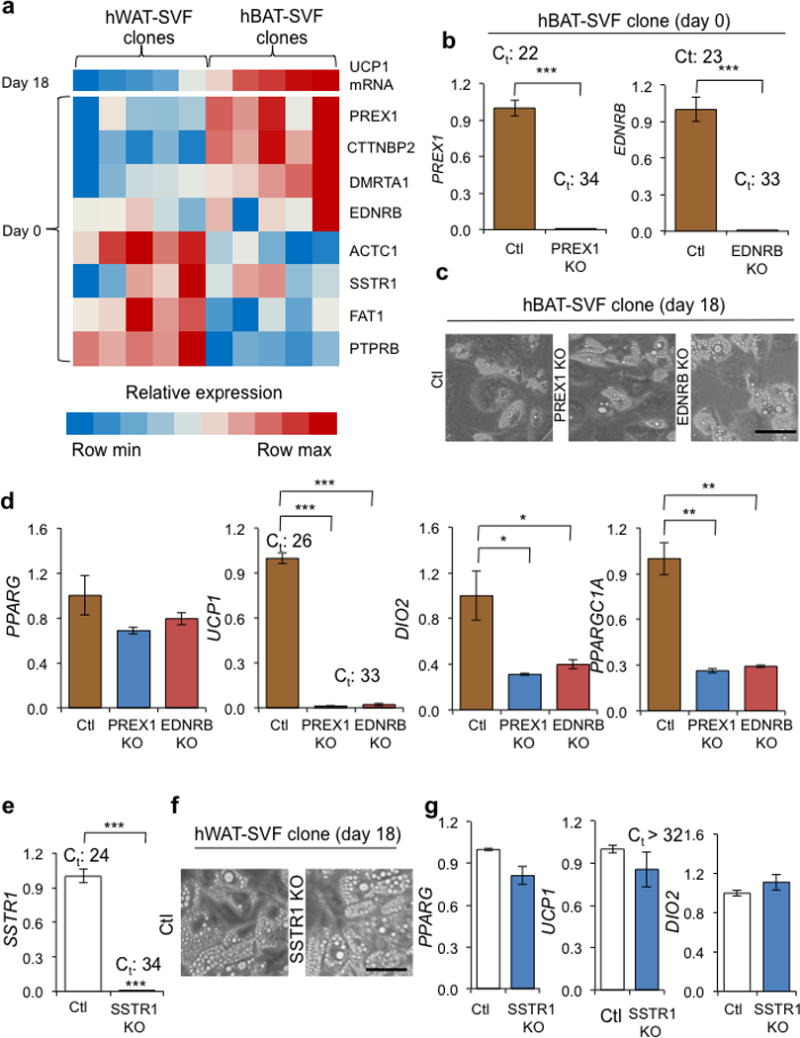
PREX1 and EDNRB are required for determining thermogenic competency. (a) A Heatmap displaying correlations between UCP1 mRNA levels on day 18 (top row) and expression levels of candidate genes on day 0. Data were obtained from 10 independent hWAT-SVF and hBAT-SVF clones derived from the same 4 subjects that were not included in microarray analyses. Values were normalized within each row using a linear color scale. (b) Levels of PREX1 and EDNRB mRNA were measured by Q-RT-PCR in PREX1 (PREX1 KO) and EDNRB (EDNRB KO) knockout hBAT-SVF clone using CRISPR/Cas9. The experiments were verified in another progenitor clone. (c) Microscopic views of differentiated PREX1 KO and EDNRB KO hBAT-SVF cells. Scale bar, 100 μm. (d) Q-RT-PCR analysis for PPARG and brown-fat-specific markers (UCP1, DIO2 and PPARGC1A) in differentiated PREX1 KO and EDNRB KO hBAT-SVF cells. (e) SSTR1 level was detected by Q-RT-PCR in a SSTR1 knockout (SSTR1 KO) hWAT-SVF clone using CRISPR/Cas9. The experiments were verified in another progenitor clone. (f) Microscope views of differentiated SSTR1 KO hWAT-SVF clone. Scale bar, 100 μm. (g) Q-RT-PCR analysis for PPARG and brown-fat-specific markers (UCP1 and DIO2) in differentiated SSTR1 KO clone. Q-RT-PCR data are presented as fold changes compared to control vector transfected cells (Ctl) (mean ± s.e.m., n=3; two-tailed Student’s t-test; * P < 0.05, ** P < 0.01, *** P < 0.001). The Ct values (Ct) are indicated to reflect the actual levels of gene expression.
To validate the role of some of these identified biomarkers in thermogenic capacity, we used CRISPR/Cas9 to knockout the positive UCP1 regulators PREX1 and EDNRB in an hBAT-SVF clone (Fig. 5b). Gene ablation had no effect on the differentiation of precursor cells into lipid-laden adipocytes that expressed normal levels of PPARG (Fig. 5c,d). Expression of the thermogenic markers UCP1, DIO2 and PPARGC1A, however, were markedly decreased in both knockout cell lines (Fig. 5d). Consequently, basal respiration, proton leak, and maximal respiration capacity were significantly reduced in PREX1 knockout cells (P = 0.03835, 0.02884, and 0.00932, respectively). Similarly, EDNRB knockout cells displayed a significant reduction of maximal respiration (P = 0.03351) and a trend of lower levels of basal respiration and proton leak (Supplementary Fig. 9a–c). To test the effects of a negative regulator of UCP1 capacity, we knocked out SSTR1 in an hWAT-SVF clone (Fig. 5e). Similar to the positive regulators, gene deletion had no effect on adipogenic differentiation; yet thermogenic gene expression remained repressed in white adipocytes (Fig. 5f,g). These data demonstrate the predictive value of the genes that we identified with our microarray analysis and suggest the gene expression network positively regulating thermogenic competency may be more sensitive to perturbation while there is at least some redundancy in the negative regulatory network of genes.
Isolation of thermogenically competent progenitors by CD29
To identify surface markers that can be used to isolate precursors with thermogenic competency, we focused on genes encoding cell surface proteins that had expression patterns positively correlated with UCP1 reporter activity (Fig. 4). Two members of the integrin family, integrin α10 (ITGA10) (P < 0.001) and integrin β1 (ITGB1, also known as CD29) (P < 0.001) exhibited significantly positive correlation with UCP1 level (Fig. 6a,b). Integrins are heterodimeric trans-membrane receptors that mediate various biological functions, such as cell proliferation, differentiation, and migration40,41. Using fluorescence-activated-cell-sorting (FACS) with an antibody against CD29, we were able to separate subpopulations of cells from pooled hWAT-SVF and hBAT-SVF based on the abundance of CD29 on the cell surface. Interestingly, hWAT-SVF contained 22.2% CD29low, 68.5% CD29med and 9.3% CD29high cells, while hBAT-SVF had almost equal proportions of CD29med and CD29high cells (50.2% and 49.7%, respectively), and very few CD29low cells (0.01%) (Fig. 6c). The ability of CD29-positive hWAT-SVF cells to differentiate into lipid-laden cells appeared to be positively correlated with CD29 levels (Fig. 6d). Importantly, CD29high hBAT-SVF cells could effectively differentiate into brown adipocytes that expressed the highest level of UCP1 among all the groups (Fig. 6e,f). These data suggest the exciting potential of using an antibody against CD29 to prospectively isolate human adipose progenitors that can give rise to mature adipocytes with great thermogenic capability.
Figure 6.
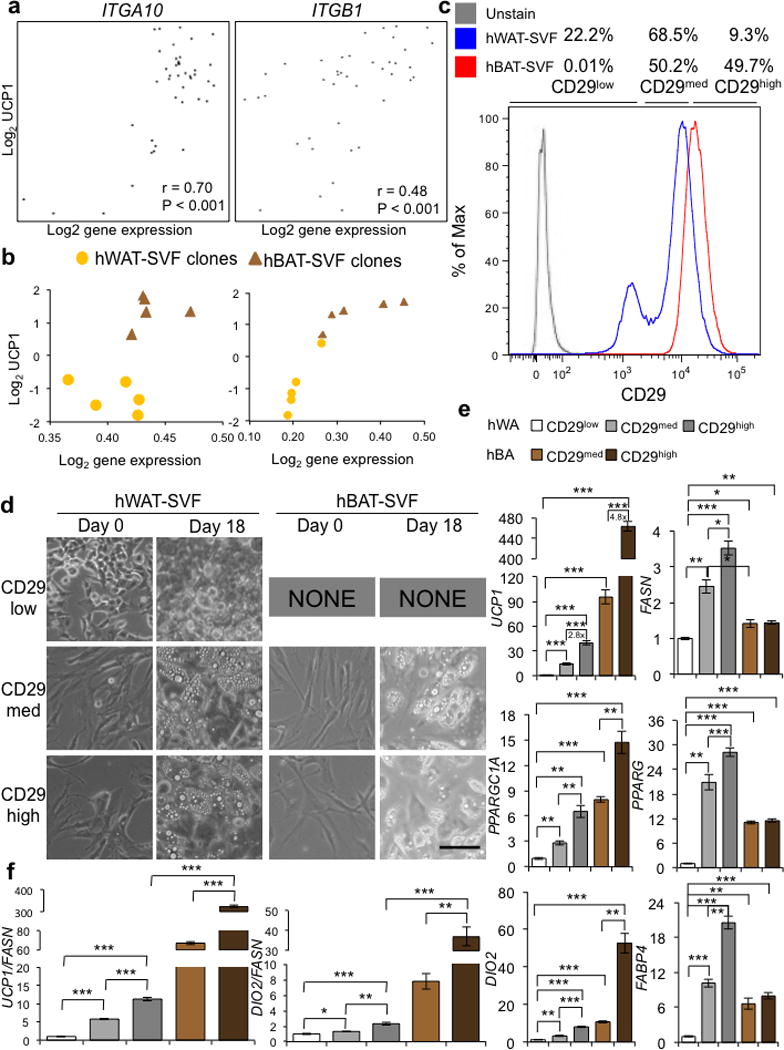
Isolation of progenitors possessing thermogenic potential using a cell surface marker. (a) Scatter plots showing positive correlation between the UCP1-luciferase levels (shown as log2 level in the Y-axis) on day 18 and expression levels of ITGA10 and ITGB1 (shown as log2 level in the X-axis) on day 0 from microarray analyses. (b) Correlation between the mRNA levels of ITGA10 and ITGB1 (shown as log2 level in the X-axis) on day 0 and UCP1 mRNA levels (shown as log2 level in the Y-axis) on day 18, in 10 independent hWAT-SVF and hBAT-SVF clones as described in Figure 5a. (c) Histogram displaying subpopulations with differential levels of CD29 from pooled hWAT-SVF (Blue) and hBAT-SVF (Red) using fluorescence-activated cell sorting. Gray line represents unstained cells. (d) Microscopic views of sorted subpopulation with different level of CD29 (CD29low, CD29med and CD29high) on day 0 and 18 are shown. Note that we couldn’t sort enough numbers of CD29low from pooled hBAT-SVF, and thus results from this subpopulation are not shown. Scale bar, 100 μm. A representative experiment from two independent studies is shown. (e) Q-RT-PCR analysis for the adipocyte markers (FASN, PPARG and FABP4) and brown-fat-specific markers (UCP1, PPARGC1A and DIO2) on the differentiated indicated populations. (f) To correct for the different degrees of adipogenesis shown in (e), expression levels of UCP1 and DIO2 were normalized to the level of mature adipocyte marker, FASN. Data are presented as mean ± s.e.m. (n=3; two-tailed Student’s t-test; * P < 0.05, ** P < 0.01, *** P < 0.001).
Discussion
In this study we have generated cell lines that serve as models to tease apart the heterogeneity of human brown and white adipose tissue, and will allow investigations into the function of these types of cells. The cell lines we have generated robustly differentiate into cells and tissue that recapitulate the gene expression signature and metabolic capacity of BAT. Interestingly, differentiated hBA display greater ability in glucose and fatty acid uptake and fatty acids oxidation compared with differentiated hWA, indicating human brown adipocytes are capable of utilizing both glucose and fatty acids as fuels. In addition, we have generated cells lines stably expressing a UCP1 reporter construct. These cells offer great opportunities for high-throughput screens aimed at identifying targets that enhance thermogenic differentiation or activate mature cells by measuring UCP1 reporter activity longitudinally. Further, the UCP1 reporter allows for the generation of human xenograft models wherein human BAT and WAT can be dynamically assayed for induction of UCP1 in vivo. Our proof of concept experiment using BMP7 pretreatment of hBAT preadipocytes demonstrates the exciting prospect of using mice with human-derived BAT and WAT to screen for novel activators of thermogenesis in an in vivo setting.
Microarray analysis in adipose clones reveals two general classifications of genes that regulate thermogenic differentiation: the binary on and off and the continuous categories. These two categories of genes suggest a distinct commitment step in brown adipogenesis, followed by differentiation. Both categories of genes present interesting opportunities for modulating cellular energy balance. By activating on switches and deactivating off switches, it may be possible to activate pools of precursor cells to differentiate into brown adipocytes. This approach may need to be combined with a second strategy targeting the genes that act as genetic rheostats with the goal of fine-tuning regulators to increase UCP1 expression in adipocytes.
Indeed, using CRISPR/Cas9 to knockout the positive UCP1 regulators PREX1 and EDNRB in brown preadipocytes, we can almost completely abolish the high level of UCP1 in mature brown fat cells. However, ablation of the negative regulator SSTR1 in white fat precursors failed to turn on the thermogenic program. These findings suggest that these positive regulators play an essential role in determining thermogenic competency in brown preadipocytes. By contrast, it may take a combined approach to remove multiple negative regulators and perhaps turn on positive modulators in the human white preadipocytes in order to switch on their thermogenic program.
PREX1 is a guanine-nucleotide exchange factor for the Rho small GTP-binding proteins. It has been shown to promote Glut4 trafficking in 3T3-L1 adipocytes42, and SNPs near PREX1 are linked to susceptibility of type 2 diabetes through its potential effect on adiposity43. Endothelin, the ligand of EDNRB, can modulates intracellular calcium and cAMP levels, stimulate glucose uptake and activate lipolysis in adipocytes44,45. Thus, it is possible that PREX1 and EDNRB might regulate pathways controlling fuel utilization in preadipocytes, which poise the cells to become thermogenically competent in mature adipocytes. The role of these markers in human obesity warrants future investigation.
Recently, Shinoda et al. identify two human BAT markers, namely KCNK3 and MTUS146. Interestingly, while the expression of these genes in our preadipocyte clones does not show significant correlation with UCP1 expression in adipocytes (Supplementary Table 3), they are indeed expressed at higher levels in the differentiated hBA compared with hWA (Supplementary Fig. 9e). Given the differences in anatomical locations where the cells are derived from and the approaches employed in these two studies, it is conceivable that distinct markers would be identified. Additionally, these findings actually complement each other because the markers identified by Shinoda et al. serve as mature hBAT markers, whereas gene signatures identified in the current study represent the predicting markers in human preadipocytes for thermogenic capacity.
Single cell clonal analysis reveals a large number of clones that differentiate and accurately recapitulate BAT and WAT behavior. These cells represent a broad range of thermogenic competency but importantly demonstrate the consistent increase in thermogenic capacity in BAT cells compared to WAT cells. However, the influence of each subject’s characteristics is still apparent. These include genetic background, gender, body mass index, as well as the anatomic locations where the fat depots are collected. These factors may contribute to the differential responses to stimuli to become thermogenically active cells, and need to be investigated by the generation and characterization of adipose precursors from more individuals of representing populations.
In this study, we have shown the utility of a CD29 antibody to prospectively isolate human preadipocytes with high thermogenic potential suggesting the promising prospect of using this approach to profile the thermogenic potential of different patient populations. CD29 positive cells from adipose tissue possess great adipogenic differentiation potential47. Our data also shows a similar result wherein the degrees of lipid accumulation positively correlate with the levels of CD29 in hWAT-SVF. CD29 is involved in the formation of the transmembrane linkage between the extracellular matrix and the microfilaments which in turn control48,49, suggesting that regulatory events that affect cell adhesion and cell shape during adipocyte differentiation might play a role in determining thermogenic potential.
Understanding of the cellular identity of human adipose tissue is key to formulating therapeutic interventions to treat obesity and its sequelae. The cell models reported here not only provide a promising opportunity to study adipogenesis and the gene patterns that regulate thermogenic competency, but also offer potential avenues for developing effective therapies for obesity and its many associated metabolic diseases.
On-Line Methods
Materials
Recombinant human BMP7 was kindly provided by Stryker Regenerative Medicine (Hopkinton, MA), recombinant human BMP8 was purchased from R&D Systems (Minneapolis, MN). Antibody sources are as follows: anti-UCP1 was from Abcam (Cambridge, MA, ab155117) and AnaSpec (Fremont, CA, Cat # 53936); anti-α-tubulin was from Sigma-Aldrich (Dallas, TX, T6074); anti-CD29 was from eBioscience (San Diego, CA, clone TS2/16). All other chemicals were purchased from Sigma-Aldrich (Dallas, TX), unless otherwise specified.
Human subject
This study followed the institutional guidelines of and was approved by the Human Studies Institutional Review Boards of Beth Israel Deaconess Medical Center and Joslin Diabetes Center. Details on procedures of human subject collection were described previously20. There were two independent human subject cohorts: for isolation and immortalization of fat progenitors, human neck fat from 4 subjects was analyzed(clinical characteristics of subjects are provided in Supplementary Table 1a); for gene expression verification, neck fat from 7 different people was studied (clinical characteristics of subjects are provided in Supplementary Table 1b). All subjects gave written informed consent before taking part in the study.
Isolation and culture of primary human white and brown fat progenitors
Isolation of primary stromal-vascular fraction (SVF) from human neck fat was described previously20. Due to the limited amount of human brown fat tissue collected from surgeries, we pooled fat depots of the same subject in order to obtain sufficient numbers of SVF cells for primary culture as well as immortalization. Specifically, subcutaneous and subplatysmal neck fat depots were pooled to generate hWAT-SVF and deep neck fat depots collected from the carotid sheath, longus colli and prevertebral regions were combined for generation of hBAT-SVF. Freshly resected fat depots were collected, minced and digested using collagenase 1 (2 mg/mL in PBS with the addition of 3.5% BSA; Worthington Biochemical Corporation, Lakewood, NJ), and the SVF was isolated. SVF cells were plated and grown in high glucose Dulbecco’s modified Eagle’s medium (DMEM/H) supplemented with 10% (v/v) fetal bovine serum (FBS) and 1% penicillin/streptomycin. For adipocyte differentiation, cells were grown to confluent for 6 days (referred as day 6) and then exposed to adipogenic induction mixture in DMEM/H medium containing isobutylmethylxanthine (0.5 mM), dexamethasone (0.1 μM), human insulin (0.5 μM; Roche Applied Science, Indianapolis, IN), T3 (2 nM), indomethacin (30 μM), pantothenate (17 μM), biotin (33 μM) and 2% FBS for another 12 days (referred as day 18). Induction medium was changed every 3 days until collected.
Generation of immortalized human brown and white fat progenitors
Primary SVF cells were immortalized with human telomere reverse transcriptase (hTERT) as described27. Primary SVF isolated from 4 subjects that had undergone 4–5 population doublings were separately infected with a retrovirus containing the plasmid, pBABE-hTERT-Hygro (Addgene #1773, Cambridge, MA) that expresses hTERT driven by long terminal repeat promoter. Phoenix-A cells (ATCC) were transfected with hTERT-Hygro DNA using PolyJet DNA in vitro transfection reagent (SignaGen Laboratories, Rockville, MD). Culture supernatants containing virus were collected every 24 h after transfection and filtered through a 0.45 μm filter (Fisher Scientific, Pittsburgh, PA). Primary SVF cells from human white and brown fat at 80% confluence were infected with supernatants in the presence of 4 μg/mL Polybrene every day until cells reached 90% confluence. Cells were then treated with hygromycin (concentrations ranging from 100 μg/mL to 400 μg/mL depending on cell conditions) in DMEM/H medium containing 10% FBS and antibiotics. Once drug selection was finished, the cells were maintained in culture medium with 50 μg/mL hygromycin for 2 weeks.
Culture and differentiation of immortalized human white and brown fat progenitors
Immortalized progenitor cells were plated and grown in DMEM/H medium supplemented with 10% FBS (referred as day 0). For adipocyte differentiation, cell were grown for 6 days until reaching confluence (day 6), and then treated with the adipogenic induction medium as described above for 12 days (day 18). To further stimulate thermogenic program, fully differentiated cells were incubated with 10μM forskolin or 1 μM norepinephrine for 4 h. For BMPs and FGF21 pre-treatment, recombinant BMP7 (3.3 nM), BMP8 (3.3 nM), or FGF21 (50 nM) were added to undifferentiated cells in medium containing insulin (0.5 μM), T3 (2 nM) and 2% FBS for 6 days followed by adipogenic induction for 12 days. For BMPs and FGF21 post-treatment, fully differentiated adipocytes at day 18 were treated with recombinant BMP7 (3.3 nM), BMP8 (3.3 nM), or FGF21 (50 nM) in medium containing insulin (0.5 μM), T3 (2 nM) and 2% FBS for 2 days. We routinely check for mycoplasma contamination and all the cells used in this study are free of mycoplasma.
Oil Red O staining
Cells were washed twice with PBS and fixed with 10% buffered formalin for 30 min at room temperature. Cells were then stained for 4 h at room temperature with a filtered Oil Red O solution (0.5% Oil Red O in isopropyl alcohol), washed twice with distilled water, and visualized.
Quantitative RT-PCR
RNA extraction, cDNA synthesis, and quantitative real-time PCR (Q-RT-PCR) were performed as described before20, 29. Q-RT-PCR assays were run in duplicates and quantified in the ABI Prism 7900 sequence-detection system using SYBR (Roche Applied Science, Indianapolis, IN). Relative mRNA expression was determined by the delta-Ct method and the values were normalized to the expression of 18S ribosomal RNA (18s). The sequences of primers used in this study are provided in Supplementary Table 5.
Western blotting
Protein detection by western blotting was performed as described before29. Primary antibodies were incubated overnight at 4°C: UCP1 (1:500, rabbit polyclonal; Abcam, Cambridge, MA) and α-tubulin (1:4,000, mouse monoclonal). HRP-coupled secondary antibodies (Cell Signaling Technologies, Beverly, MA) were used at 1:2,000 dilutions at room temperature for 2 h followed by detection using the ECL system.
Seahorse bioenergetic profiling
To assess mitochondrial respiration, a Seahorse Extracellular Flux Analyzer (Seahorse Bioscience Inc., North Billerica, MA) was used to quantify oxygen consumption rates (OCR) of differentiated human white and brown adipocytes. Progenitor cells were seeded on 24-well format plates and allowed to adhere overnight. After 6 days, adipogenesis was induced as described above. After adipogenic induction for 12 days, OCR was analyzed. To measure OCR independent of oxidative phosphorylation, 0.5 μM oligomycin (EMD Chemicals Inc., Gibbstown, NJ) was added to cells. Subsequently, 0.8 μM FCCP (carbonyl cyanide-p-trifluoromethoxyphenylhydrazone) and 1 μM respiratory chain inhibitors (rotenone) were added to measure maximal respiration and basal rates of nonmitochondrial respiration. For cAMP-induced respiration, fully differentiated adipocytes were incubated with 10 μM forskolin for 4 hours. For BMP7 pretreatment-induced respiration, recombinant BMP7 (3.3 nM) was added to the undifferentiated cells for 6 days and then adipogenic induction mixture medium was added to the confluent cells for 12 days, followed by measurement of cellular respiration. All data were average of four-time points with 10 wells per time point by quantified in bar plots, and error bars are standard error of the mean (s.e.m.). Statistical comparisons were done by Student’s t-test.
Glucose uptake assay
After serum starvation in DMEM/H medium containing 1% of BSA for 2–3 h, differentiated human white and brown adipocytes were washed with HEPES buffer. Then they were incubated with or without 100 nM insulin for 30 min in DMEM/H medium containing 1% of BSA. Glucose transport was determined by the addition of 2-deoxy-[3H]glucose (0.1 mM, 0.5 μCi/mL; PerkinElmer Life and Analytical Science, Waltham, MA). After 5 min of incubation, the reaction was stopped by ice-cold PBS and cells were washed twice with ice-cold PBS. Cells were then lysed in 0.1% SDS, and glucose uptake was assessed in 4 mL of scintillant using Beckman LS6500 scintillation counter (Beckman Coulter, Indianapolis IN). Nonspecific 2-deoxy-[3H]glucose uptake was measured in the presence of cytochalasin B (20 μM) and was subtracted from the total uptake to get specific glucose uptake. Results were expressed as the mean ± s.e.m. of the indicated number of experiments. The protein content was determined by the Bradford method.
Fatty acid uptake and fatty acid oxidation assays
Fatty acid uptake and oxidation were determined by measuring both 14C-labeled palmitic acid uptake and conversion of 14C-labeled palmitic acid into CO2. Briefly, the culture medium was removed, and cells were incubated with DMEM/H containing 4% fatty acid free BSA, 0.5 mM palmitic acid, and 0.2 μCi/mL [1-14C]-palmitic acid (PerkinElmer Life and Analytical Science, Waltham, MA) for 1 h. The incubation medium was transferred to a vial containing 1 M acetic acid, capped quickly, and allowed to sit 1 h for CO2 gas to be released. 14CO2 released was absorbed by hyamine hydroxide, and activity was counted. Fatty acid oxidation was calculated from CO2 generated. To measure fatty acid uptake, cells were rinsed twice with PBS and lysed after incubation with [1-14C]-palmitic acid. Lipids were extracted using a chloroform-methanol mixture (2:1), and 14C counts were determined in the organic phase. Fatty acid uptake was calculated as the total of 14C lipids in the cells and 14CO2 generated.
Generation of cells with a hUCP1 reporter system
Immortalized human fat progenitor cells were infected with a lentivirus containing the plasmid, pLV.ExBi.P/Puro-hUCP1promoter-Luc(firefly)-T2A-hrGFP that expresses luciferase and GFP driven by human UCP1 promoter. 4148bp human UCP1 promoter was cloned from pLightSwitch_hUCP1-Prom (S723122; Switch Gear Genomics, Carlsbad, CA) and was then sub-cloned into a lentiviral plasmid to generate plasmid containing a UCP1 reporter system (Cyagen Biosciences Inc., Santa Clara, CA). 293T cells (ATCC) were transfected with hUCP1promoter-Luc-T2A-GFP, pMD2.G and psPAX2 DNA using PolyJet DNA in vitro transfection reagent (SignaGen Laboratories, Rockville, MD). Culture supernatants containing virus were collected every 24 h after infection and filtered through a 0.45 μm filter (Fisher, Scientific, Pittsburgh, PA). Immortalized human white and brown fat progenitors at 80% confluence were infected with viral supernatants in the presence of 4 μg/mL Polybrene every day until cells reached 90% confluence. Then cells were treated with 1 μg/mL puromycin in DMEM/H medium containing 10% FBS and antibiotics. Once drug selection was finished, the cells were maintained in culture medium with 0.2 μg/mL puromycin for 2 weeks.
Luciferase reporter assay
In vitro luciferase assays were performed using luciferase assay kits (Promega, Madison, WI) according to the manufacturer’s instructions. Remove culture medium from differentiated adipocytes and wash cells in PBS. Dispense an appropriate volume of 1X lysis reagent (Passive Lysis Buffer) into each culture well. Scrape attached cells from the wells, and transfer the cell lysates into white 96-well plate (Corning Inc., Tewksbury, MA) for detection of the bioluminescence signal using luminometer plate reader (BioTek Instruments, Inc., Winooski, VT). Use a reagent injector to dispense 100 μL of Luciferase assay buffer with substrate and 100 μL of Stop & Glo Reagent. And perform a 2-second pre-measurement delay, followed by a 10-second measurement period for each reporter assay. Luciferase activity data were normalized to protein content.
Time lapse imaging system
Human immortalized cells with hUCP1-promoter-Luc/GFP were plated on a Hi-Q4 culture dish (Nikon, Tokyo, Japan) and cultured in a Nikon BioStation IM-Q (Nikon, Tokyo, Japan), which is a compact cell incubator and monitoring system that allows for live cell imaging. Cells were maintained in BioStation IM-Q at 37°C in 5% CO2 environment. Adipogenesis was induced as described. Bright field and fluorescent images were obtained every hour over the course every three days between medium changes for a total of 18 days.
Cell transplantations and IVIS imaging system
Human immortalized white and brown fat progenitors with hUCP1-promoter-Luc/GFP were grown in the presence and absence of 3.3 nM BMP7 for 6 days to reach confluence. Cells were washed, trypsinzed, and resuspended in growth medium with an equal amount of Matrigel Matrix (BD Biosciences, San Diego, CA). Then, 1.0 × 107 cells in 0.3 mL volume were injected into the thoracic/sternum region of 6-week-old male BALB/c athymic nude mice (n=2 mice for white fat progenitors transplantation group, n=3 mice for brown fat progenitors transplantation group; Harlan laboratories, Indianapolis, IN) using an 18-gauge needle, according to the methods described previously23,29. No statistical method was used to predetermine sample size and experiments were not randomized. For an acquisition of the bioluminescence images, the mice were sedated with 2% isoflurane in 100% O2 in the chamber. D-Luciferin (PerkinElmer Life and Analytical Science, Waltham, MA) was diluted to 3 mg/100 μL in normal saline and 0.6 mg of D-Luciferin was administrated intraperitoneally into mice. An IVIS-Spectrum CT imaging system equipped with a CCD camera (Caliper, PerkinElmer Life and Analytical Science, Hopkinton, MA) was used for in vivo bioluminescence imaging. The luminescence intensity in regions of interest from each image was quantified to examine the viability of the implanted cells. Mice were scanned by IVIS each week after transplantation. After 6 weeks of transplantation, mice were sacrificed, and adipose tissue derived from implanted cells was excised and processed for Q-RT-PCR analysis. There was no blinding during animal experiments. The animal experiment was performed according to procedures approved by the Joslin Diabetes Center Institutional Animal Care and Use Committee (IACUC).
Generation of immortalized clonal cell lines
To derive subclones of immortalized human white and brown progenitors with hUCP1-promoter-Luc/GFP reporter, limiting dilution of cells into 96-well plates was performed as previously described27. Briefly, cells were plated at 50 cells/96-well plate in DMEM/H containing 10% FBS. After 2 weeks, colonies were evident. Cells at approximately 80% confluence were trypsinized and further propagated in 48-well, then 12-well and finally 6-well plate. 152 clonal lines originated from neck superficial fat depot and 128 clonal lines from deep fat were selected for adipogenic potential after induction. The highly adipogenic clonal lines were selected for further analysis (67 clonal white fat progenitor lines; 90 clonal brown fat progenitor lines).
Microarray analysis
Analysis of gene expression using GeneChip® PrimeView (Affymetrix, Santa Clara, CA) was performed on 42 highly adipogenic clonal white and brown cell lines. RNA was isolated from clonal cell lines using Direct-zol RNA MiniPrep kit (Zymo Research, Irvine, CA) according to the manufacturer’s instructions. The quality of total RNA was evaluated by A260/A280 ratio, which was within the value of 1.9 to 2.0 defined as high quality total RNA. Biotin-labeled cRNA was synthesized, purified and fragmented using GeneChip 3′IVT Express Kit (Affymetrix, Santa Clara, CA). Integrity and fragmented cRNA was assessed by running aliquots on the Agilent 2100 Bioanalyzer prior to proceeding further. The high quality cRNA meets the following criteria: the A260/A280 ratio should fall within the value of 1.9 to 2.0; The 28S/18S RNA bands (from the gel) should be crisp and the intensity of the 28S band should be roughly twice the intensity of the 18S band. As one clone from hWAT-SVF had poor cRNA quality, this clone was excluded from analysis. Array hybridization and scanning were performed by the Advanced Genomics and Genetics Core of Joslin Diabetes Center according to established methods. Microarray data were normalized using robust multi-array average (RMA)50, which placed it on a log-2 scale, and were deposited to the Gene Expression Omnibus (GEO) repository (accession number GSE68544). The log transformation is particularly helpful for making gene expression data approximately normally distributed, so (although we did not explicitly test for normality) the normalized data for each probeset were correlated to log2 (UCP1) using Pearson correlation with a two-sided alternative (with function cor.test), which yielded correlation coefficients and P-values. P-values were adjusted for multiple testing using the False Discovery Rate (FDR)51 with function p.adjust. The 50 probesets from unique genes most strongly associated with log2 (UCP1) were plotted in a heatmap along with log2 (UCP1) using the heatmap.2 function in the gplots package and color palettes from the RColorBrewer package. The probesets’ values were centered to have mean zero and restricted to the interval [−,2] to aid visualization, and a color bar representing UCP1 was added at top, where darker indicates higher UCP1. All microarray analyses were done in the R programming language (www.r-project.org).
Engineering target gene knockout cell clones using CRISPR/Cas9 system
Cas9 vectors express the Cas9 nuclease and guide RNA (gRNA) were obtained from Horizon Discovery Group plc (Cambridge, United Kingdom). 5 gRNAs of each target gene were designed using Horizon’s proprietary gUIDEbook software and cloned into a Case9 expressing plasmid on behalf of Horizon by DNA2.0. The plasmid carrying Cas9, gRNA and a GFP selection marker was introduced into immortalized cell clone by transfection using PolyJet DNA in vitro transfection reagent (SignaGen Laboratories, Rockville, MD). Next day, the cells were replaced with fresh medium supplemented with 10% FBS, and re-seed when the cells became confluent. To derive subclones with plasmid insertion, we selected and expanded the clones with GFP signal, followed by extraction of total RNA for Q-RT-PCR using standard methods.
Cell sorting
CD29 positive progenitors were sorted by FACS, as previously described before23. Briefly cells were trypsinized, centrifuged, and resuspended in HBSS (Invitrogen, Life Technologies, Carlsbad, CA) with 2% FBS. CD29 antibody (β1-integrin, 1:200, APC conjugate, clone TS2/16; eBioscience, San Diego, CA) incubation was performed for 20 min on ice in HBSS containing 2% FBS. For cell sorting, Cytomation Moflo (Cytomation Inc.,) instrument was used. FACS data were collected using Summit software (Cytomation Inc.,) and analyzed offline using FlowJo software (Tree Star, Inc., Macintosh version 8.1.1).
Immunofluorescence staining
Cells were grown and differentiation on a chamber covered with a glass slide (Lab-Tek II chamber CC2 glass slide cover; Thermo Scientific, Waltham, MA). Fully differentiated cells were fixed in 4% paraformaldehyde for 10 min, washed three times with PBS. Cells were permeabilized with 0.1% Triton X-100 for 30 min and incubated with primary antibodies overnight at 4°C: UCP1 (1:50, rabbit polyclonal; AnaSpec, Fremont, CA). After primary antibody incubation, cells were washed and incubated with appropriate secondary antibody (Alexa Fluor-488 (green); Invitrogen, Life Technologies, Carlsbad, CA) at a 1:200 dilution for 10 min in the dark. After secondary antibody incubation, cells were washed with distilled water for DAPI staining (0.1 μg/mL in water for 5 min in the dark), and mounted. Cells were kept in the dark after mounting and analyzed by a fluorescence microscope (Olympus BX60F-3; Olympus Corporation, Tokyo, Japan). Identical conditions and settings were used for picture acquisition and analysis. A threshold was set for each image to eliminate background and to create a binary mode image. For every sections and cells, images from three representative areas were analyzed.
Nile Red staining
To detect intracellular lipid droplet in live cells, Nile Red staining was performed in differentiated adipocytes. Cells were washed twice in PBS and then incubated in PBS containing 3 μM Nile Red (Life Technologies, Carlsbad, CA) for 60 min at 37°C. To remove the Nile Red working solution from the cells, wash the cells with PBS. Monitor the fluorescence change at Ex/Em = 552/636 nm with a fluorescence microscope or fluorescent plate reader (BioTek Instruments, Inc., Winooski, VT).
Statistics
All results were expressed as mean ± s.e.m.. All statistical analyses were performed using the programs Excel (Microsoft) and Statview (SAS Institute). Two-tailed Student’s t-test was used to determine P values. Statistical significance was defined as P < 0.05. Gene expression level in human WAT versus BAT was analyzed by using Wilcoxon matched-pairs signed-rank test. No statistical method was used to predetermine sample size. The experiments were not randomized. All experiments were not blinded. Exclusion criteria were mentioned under Microarray Analysis.
Supplementary Material
Acknowledgments
This work was supported in part by US National Institutes of Health (NIH) grants R01DK077097 (to Y.-H.T.), R01DK099511 (to L.J.G.), K23DK081604 (to A.M.C) and P30DK036836 (to Joslin Diabetes Center’s Diabetes Research Center, DRC) from the National Institute of Diabetes and Digestive and Kidney Diseases, a sponsored research grant from Chugai Pharmaceutical Co., Ltd (to Y.-H.T and A.M.C), a research grant from the American Diabetes Association (ADA 7-12-BS-191, to Y.-H.T.) and by funding from the Harvard Stem Cell Institute (to Y.-H.T.). M.D.L was supported by NIH fellowships (T32DK007260 and F32DK102320). We thank M.-E. Patti and K. Hughes of the Advanced Genomics and Genetics Core of Joslin’s DRC for advice and expert technical assistance. The authors thank Stryker Regenerative Medicine (Hopkinton, MA) for the generous gift of recombinant BMP7.
Footnotes
AUTHOR CONTRIBUTIONS
The study was designed by Y.-H.T., R.X., M.D.L. and A.M.C. The manuscript was written by Y.-H.T., M.D.L., R.X. and J.D.. R.X. performed the majority of the experiments. M.D.L. did the time-lapse imaging, IVIS scanning and FACS. J.D. analyzed microarray data. F.S performed bioenergetics analyses in knockout cells. T.J.S. and H.Z. established the method of isolation, immortalization and differentiation of human fat progenitors. T.L.H did the human cell implantation and gene expression microarrays. K.L.T. provided assistance with the Seahorse bioanalyzer. Y.L. provided research assistance. H.Z. and L.J.G helped with fuel utilization experiments. A.M.C., L.S.W. and A.P.W. collected human fat samples. M.S.L. and L.L.R. helped with the time-lapse imaging. All authors contributed to editing the manuscript.
References
- 1.Cannon B, Nedergaard J. Brown adipose tissue: function and physiological significance. Physiol Rev. 2004;84:277–359. doi: 10.1152/physrev.00015.2003. [DOI] [PubMed] [Google Scholar]
- 2.Schulz TJ, Tseng YH. Brown adipose tissue: development, metabolism and beyond. Biochem J. 2013;453:167–178. doi: 10.1042/BJ20130457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guerra C, Koza RA, Yamashita H, Walsh K, Kozak LP. Emergence of brown adipocytes in white fat in mice is under genetic control. Effects on body weight and adiposity. J Clin Invest. 1998;102:412–420. doi: 10.1172/JCI3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Petrovic N, et al. Chronic peroxisome proliferator-activated receptor gamma (PPARgamma) activation of epididymally derived white adipocyte cultures reveals a population of thermogenically competent, UCP1-containing adipocytes molecularly distinct from classic brown adipocytes. J Biol Chem. 2010;285:7153–7164. doi: 10.1074/jbc.M109.053942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harms M, Seale P. Brown and beige fat: development, function and therapeutic potential. Nat Med. 2013;19:1252–1263. doi: 10.1038/nm.3361. [DOI] [PubMed] [Google Scholar]
- 6.Nedergaard J, Cannon B. The browning of white adipose tissue: some burning issues. Cell Metab. 2014;20:396–407. doi: 10.1016/j.cmet.2014.07.005. [DOI] [PubMed] [Google Scholar]
- 7.Bartelt A, et al. Brown adipose tissue activity controls triglyceride clearance. Nat Med. 2011;17:200–205. doi: 10.1038/nm.2297. [DOI] [PubMed] [Google Scholar]
- 8.Stanford KI, et al. Brown adipose tissue regulates glucose homeostasis and insulin sensitivity. J Clin Invest. 2013;123:215–223. doi: 10.1172/JCI62308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nedergaard J, Bengtsson T, Cannon B. Unexpected evidence for active brown adipose tissue in adult humans. Am J Physiol Endocrinol Metab. 2007;293:E444–E452. doi: 10.1152/ajpendo.00691.2006. [DOI] [PubMed] [Google Scholar]
- 10.Cypess AM, et al. Identification and importance of brown adipose tissue in adult humans. N Engl J Med. 2009;360:1509–1517. doi: 10.1056/NEJMoa0810780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Marken LW, et al. Cold-activated brown adipose tissue in healthy men. N Engl J Med. 2009;360:1500–1508. doi: 10.1056/NEJMoa0808718. [DOI] [PubMed] [Google Scholar]
- 12.Virtanen KA, et al. Functional brown adipose tissue in healthy adults. N Engl J Med. 2009;360:1518–1525. doi: 10.1056/NEJMoa0808949. [DOI] [PubMed] [Google Scholar]
- 13.Saito M, et al. High incidence of metabolically active brown adipose tissue in healthy adult humans: effects of cold exposure and adiposity. Diabetes. 2009;58:1526–1531. doi: 10.2337/db09-0530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zingaretti MC, et al. The presence of UCP1 demonstrates that metabolically active adipose tissue in the neck of adult humans truly represents brown adipose tissue. FASEB J. 2009;23:3113–3120. doi: 10.1096/fj.09-133546. [DOI] [PubMed] [Google Scholar]
- 15.Himms-Hagen J, et al. Effect of CL-316,243, a thermogenic beta 3-agonist, on energy balance and brown and white adipose tissues in rats. Am J Physiol. 1994;266:R1371–R1382. doi: 10.1152/ajpregu.1994.266.4.R1371. [DOI] [PubMed] [Google Scholar]
- 16.Yoneshiro T, et al. Recruited brown adipose tissue as an antiobesity agent in humans. J Clin Invest. 2013;123:3404–3408. doi: 10.1172/JCI67803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van der Lans AA, et al. Cold acclimation recruits human brown fat and increases nonshivering thermogenesis. J Clin Invest. 2013;123:3395–3403. doi: 10.1172/JCI68993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ouellet V, et al. Brown adipose tissue oxidative metabolism contributes to energy expenditure during acute cold exposure in humans. J Clin Invest. 2012;122:545–552. doi: 10.1172/JCI60433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu J, et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell. 2012;150:366–376. doi: 10.1016/j.cell.2012.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cypess AM, et al. Anatomical localization, gene expression profiling and functional characterization of adult human neck brown fat. Nat Med. 2013;19:635–639. doi: 10.1038/nm.3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lidell ME, et al. Evidence for two types of brown adipose tissue in humans. Nat Med. 2013;19:631–634. doi: 10.1038/nm.3017. [DOI] [PubMed] [Google Scholar]
- 22.Jespersen NZ, et al. A classical brown adipose tissue mRNA signature partly overlaps with brite in the supraclavicular region of adult humans. Cell Metab. 2013;17:798–805. doi: 10.1016/j.cmet.2013.04.011. [DOI] [PubMed] [Google Scholar]
- 23.Schulz TJ, et al. Identification of inducible brown adipocyte progenitors residing in skeletal muscle and white fat. Proc Natl Acad Sci USA. 2011;108:143–148. doi: 10.1073/pnas.1010929108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee YH, Petkova AP, Mottillo EP, Granneman JG. In vivo identification of bipotential adipocyte progenitors recruited by beta3-adrenoceptor activation and high-fat feeding. Cell Metab. 2012;15:480–491. doi: 10.1016/j.cmet.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berry R, Rodeheffer MS. Characterization of the adipocyte cellular lineage in vivo. Nat Cell Biol. 2013;15:302–308. doi: 10.1038/ncb2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang W, et al. Ebf2 is a selective marker of brown and beige adipogenic precursor cells. Proc Natl Acad Sci USA. 2014;111:14466–14471. doi: 10.1073/pnas.1412685111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tchkonia T, et al. Fat depot-specific characteristics are retained in strains derived from single human preadipocytes. Diabetes. 2006;55:2571–2578. doi: 10.2337/db06-0540. [DOI] [PubMed] [Google Scholar]
- 28.Whittle AJ, et al. BMP8B increases brown adipose tissue thermogenesis through both central and peripheral actions. Cell. 2012;149:871–885. doi: 10.1016/j.cell.2012.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tseng YH, et al. New role of bone morphogenetic protein 7 in brown adipogenesis and energy expenditure. Nature. 2008;454:1000–1004. doi: 10.1038/nature07221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seale P, et al. Transcriptional control of brown fat determination by PRDM16. Cell Metab. 2007;6:38–54. doi: 10.1016/j.cmet.2007.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Timmons JA, et al. Myogenic gene expression signature establishes that brown and white adipocytes originate from distinct cell lineages. Proc Natl Acad Sci USA. 2007;104:4401–4406. doi: 10.1073/pnas.0610615104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Welch HC, et al. P-Rex1, a PtdIns(3,4,5)P3- and Gbetagamma-regulated guanine-nucleotide exchange factor for Rac. Cell. 2002;108:809–821. doi: 10.1016/s0092-8674(02)00663-3. [DOI] [PubMed] [Google Scholar]
- 33.Cheung J, et al. Identification of the human cortactin-binding protein-2 gene from the autism candidate region at 7q31. Genomics. 2001;78:7–11. doi: 10.1006/geno.2001.6651. [DOI] [PubMed] [Google Scholar]
- 34.Zhang SX, et al. Identification of direct serum-response factor gene targets during Me2SO-induced P19 cardiac cell differentiation. J Biol Chem. 2005;280:19115–19126. doi: 10.1074/jbc.M413793200. [DOI] [PubMed] [Google Scholar]
- 35.Yamada Y, et al. Cloning and functional characterization of a family of human and mouse somatostatin receptors expressed in brain, gastrointestinal tract, and kidney. Proc Natl Acad Sci USA. 1992;89:251–255. doi: 10.1073/pnas.89.1.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kikkawa T, et al. Dmrta1 regulates proneural gene expression downstream of Pax6 in the mammalian telencephalon. Genes Cells. 2013;18:636–649. doi: 10.1111/gtc.12061. [DOI] [PubMed] [Google Scholar]
- 37.Garciafigueroa DY, Klei LR, Ambrosio F, Barchowsky A. Arsenic-stimulated lipolysis and adipose remodeling is mediated by G-protein-coupled receptors. Toxicol Sci. 2013;134:335–344. doi: 10.1093/toxsci/kft108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen TY, et al. Endogenous n-3 polyunsaturated fatty acids (PUFAs) mitigate ovariectomy-induced bone loss by attenuating bone marrow adipogenesis in FAT1 transgenic mice. Drug Des Devel Ther. 2013;7:545–552. doi: 10.2147/DDDT.S45263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Behjati S, et al. Recurrent PTPRB and PLCG1 mutations in angiosarcoma. Nat Genet. 2014;46:376–379. doi: 10.1038/ng.2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Takada Y, Ye X, Simon S. The integrins. Genome Biol. 2007;8:215. doi: 10.1186/gb-2007-8-5-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Margadant C, Monsuur HN, Norman JC, Sonnenberg A. Mechanisms of integrin activation and trafficking. Curr Opin Cell Biol. 2011;23:607–614. doi: 10.1016/j.ceb.2011.08.005. [DOI] [PubMed] [Google Scholar]
- 42.Balamatsias D, et al. Identification of P-Rex1 as a novel Rac1-guanine nucleotide exchange factor (GEF) that promotes actin remodeling and GLUT4 protein trafficking in adipocytes. J Biol Chem. 2011;286:43229–43240. doi: 10.1074/jbc.M111.306621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lewis JP, et al. Analysis of candidate genes on chromosome 20q12-13.1 reveals evidence for BMI mediated association of PREX1 with type 2 diabetes in European Americans. Genomics. 2010;96:211–219. doi: 10.1016/j.ygeno.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu-Wong JR, Berg CE, Dayton BD. Endothelin-stimulated glucose uptake: effects of intracellular Ca(2+), cAMP and glucosamine. Clin Sci (Lond) 2002;103(Suppl 48):418S–423S. doi: 10.1042/CS103S418S. [DOI] [PubMed] [Google Scholar]
- 45.Juan CC, et al. Effect of endothelin-1 on lipolysis in rat adipocytes. Obesity (Silver Spring) 2006;14:398–404. doi: 10.1038/oby.2006.53. [DOI] [PubMed] [Google Scholar]
- 46.Shinoda K, et al. Genetic and functional characterization of clonally derived adult human brown adipocytes. Nat Med. 2015;21:389–394. doi: 10.1038/nm.3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gierloff M, et al. Adipogenic differentiation potential of rat adipose tissue-derived subpopulations of stromal cells. J Plast Reconstr Aesthet Surg. 2014;67:1427–1435. doi: 10.1016/j.bjps.2014.05.042. [DOI] [PubMed] [Google Scholar]
- 48.Farnier C, et al. The signaling pathway for beta1-integrin/ERKs is involved in the adaptation of adipocyte functions to cell size. Ann N Y Acad Sci. 2002;973:594–597. doi: 10.1111/j.1749-6632.2002.tb04706.x. [DOI] [PubMed] [Google Scholar]
- 49.Kawaguchi N, et al. ADAM12 induces actin cytoskeleton and extracellular matrix reorganization during early adipocyte differentiation by regulating beta1 integrin function. J Cell Sci. 2003;116:3893–3904. doi: 10.1242/jcs.00699. [DOI] [PubMed] [Google Scholar]
- 50.Irizarry RA, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 51.Benjamin Y, et al. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J Roy Statist Soc Ser B (Methodological) 1995;57:289–300. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.