

Abstract
Brain peroxisome proliferator-activated receptor gamma (PPARγ), a member of the nuclear receptor superfamily of ligand-dependent transcription factors, is involved in neuroprotection. It is activated by the drug rosiglitazone, which then can increase the prosurvival protein BCL-2, to mediate neuroprotection. However, the mechanism underlying this molecular cascade is unknown. Here we show that the neuroprotective protein Neurotrophic Factor-α1 (NF-α1), which also induces the expression of BCL-2, has a promoter that contains PPARγ-binding sites that are activated by rosiglitazone. Treatment of Neuro2a cells and primary hippocampal neurons with rosiglitazone increased endogenous NF-α1 expression and prevented H2O2-induced cytotoxicity. Concomitant with the increase of NF-α1, BCL-2 was also increased in these cells. When siRNA against NF-α1 was used, the induction of BCL-2 by rosiglitazone was prevented, and the neuroprotective effect of rosiglitazone was reduced. These results demonstrate that rosiglitazone-activated PPARγ directly induces the transcription of NF-α1, contributing to neuroprotection in neurons.
Keywords: Neurotrophic Factor-α1(NF-α1), Rosiglitazone, neuroprotection, BCL-2, hippocampus
Introduction
Peroxisome proliferator-activated receptor gamma (PPARγ), a nuclear receptor and transcription factor, can be activated by the high-affinity agonists thiazolidinediones (TZDs), a class of drugs that have been used for the treatment of type 2 diabetes, of which rosiglitazone is a member (Lehmann et al. 1995). In the central nervous system (CNS), PPARγ activation plays a role in neuroprotection against neurodegenerative diseases (Heneka & Landreth 2007) and, more recently, in the central regulation of energy balance and hyperphagia, resulting in obesity (Ryan et al. 2011, Lu et al. 2011). Numerous studies have indicated positive effects of PPARγ activation by the agonist rosiglitazone in traumatic brain injuries (Yi et al. 2008, Qi et al. 2010), cerebral ischemia (Luo et al. 2006, Giaginis et al. 2008) and neurodegenerative disorders such as Alzheimer's and Parkinson's disease (Chen et al. 2012). PPARγ activation was also shown to prevent neuronal cell death resulting from NMDA-induced excitotoxicity in primary cortical neurons (Zhao et al. 2006), and protected an immortalized hippocampal cell line from glutamate or H2O2-induced oxidative stress (Aoun et al. 2003). In addition, in a pilot study, rosiglitazone has been shown to successfully treat bipolar disorder in patients with insulin resistance (Rasgon et al. 2010) and to reduce depressive-like behavior in mice and rats (Eissa Ahmed et al. 2009, Sharma et al. 2012), although the mechanism of action in these cases is unknown. These reports, nonetheless, suggest that PPARγ activation plays significant beneficial roles in the CNS.
Carboxypeptidase E (CPE), an exopeptidase responsible for the maturation of numerous neuropeptides (Fricker 1988), has been shown to be involved in many physiological processes (see (Cawley et al. 2012) for review). Several studies have shown a correlation between an increase in the expression of CPE and neuronal survival in the ischemic brain (Jin et al. 2001, Zhou et al. 2004) and after chronic restraint stress in mice (Murthy et al. 2013). In addition, CPE has recently been identified as a neuroprotective protein, now also known as neurotrophic factor-α1 (henceforth referred to as NF-α1 in this work), which is secreted from neurons and acts extra-cellularly in an autocrine or paracrine manner (Woronowicz et al. 2008, Koshimizu et al. 2009, Cheng et al. 2013).
Due to the neuroprotective effect of rosiglitazone in ischemic brain injury and the correlation of increased expression of NF-α1 in mesenchymal stem cells stably over-expressing PPARγ (Shockley et al. 2009), we hypothesized that activation of PPARγ may contribute to neuroprotection by inducing NF-α1 expression. Using a combination of bioinformatics, luciferase reporter and cytotoxicity assays, we show that rosiglitazone up-regulated the expression of BCL-2 in an NF-α1-dependent manner, in Neuro2A cells and hippocampal neurons and protected the cells against cell death under conditions of oxidative stress.
Material and Methods
Animals
Pregnant rats were purchased from Taconic Farms, Inc., (Hudson, NY). All animal treatment and care protocols conformed to NICHD ACUC Guidelines.
Transfection and treatment of Neuro2A cells, hippocampal and cortical neurons
Neuro2A cells were purchased from ATCC (Manassas, VA) and cultured as previously described (Lou et al. 2013). Hippocampal and cortical neurons were prepared from rat embryos (E18) as previously described (Cheng et al. 2013). Cells were treated with 1 μM rosiglitazone (Cayman) or vehicle (0.1% DMSO) for 24 or 48 h. Where noted, the cells were transiently transfected with 30 nM Stealth siRNA oligonucleotides directed against NF-α1 (5′-GGUUUGUCCGUGACCUUCAGGGUAA-3′) or a scrambled control sequence (5′-UUAAACGUCCGGAACACUCAGGACC-3′) (Life Technologies, Grand Island, NY) for 24 h using Lipofectamine RNAiMAX (Invitrogen), prior to drug treatment. In other experiments, cells were incubated with 100 μM H2O2 to induce oxidative stress or with GW9662 (Cayman), a selective PPARγ inhibitor, to inhibit the protective effect of rosiglitazone.
Bioinformatics analysis of NF-α1 promoter and luciferase constructs and assays
Mouse, rat and human NF-α1-promoter sequences were analyzed using the Genomatix software suite (GmbH, Munich, Germany). Based on this analysis, a 1944 bp fragment of the mouse NF-α1 promoter comprising 2 PPARγ binding sites (PPREs) (position -1187 and -1086), was custom cloned (GenScript, Piscataway, NJ) into the pGL3-basic luciferase vector (Promega, Madison, WI) to generate the PPRE reporter construct. A slightly shorter fragment of this NF-α1 promoter lacking PPREs was used to generate the control reporter construct. Hippocampal or cortical neurons were transfected with the luciferase reporter constructs using the Amaxa Rat Neuron Nucleofector Kit (Lonza). A pGL4.73 vector (Renilla luciferase reporter vector) was used as a control for transfection efficiency. Luminescence was measured using the TD-20/20 Glomax luminometer (Promega). Results were expressed as percentage of luciferase activity compared to the controls. These experiments were performed three times (N=3) with n=12 replicates for the hippocampal neurons and n=4-5 replicates for the cortical neurons, per experiment.
LDH release assay for cell cytotoxicity
The release of lactate dehydrogenase (LDH) was used to measure cell cytotoxicity. This was done with a CytoTox 96 Non-Radioactive Cytotoxicity Assay kit (Promega, Madison, WI).
Real Time Quantitative-PCR
Complimentary DNA was made (Improm-II Reverse Transcription System, Promega) from 500 ng of RNA extracted (RNeasy Mini Kit, Qiagen) from cells and used for qPCR on an ABI 7500 real time PCR system with Power SYBR green I (Applied Biosystems). The cycling conditions were: 10 min denaturation at 95 °C and 40 cycles of DNA synthesis at 95 °C for 15 s and 60 °C for 1 min. Primer sequences were: rat NF-α1 fwd: 5′-GCAACTGCTTTGAGATCACT-3′, rev: 5′-TTGATGAGGGAGTTTTTGTT-3′; rat Bcl2 fwd: 5′-AAGCTGTCACAGAGGGGCTA-3′, rev: 5′-CAGGCTGGAAGGAGAAGATG-3′ (300 nM); for 18 S fwd: 5′-CTCTTAGCTGAGTGTCCCGC-3′, rev: 5′-CTGATCGTCTTCGAACCTCC-3′ (100 nM). All reactions were performed in duplicate and relative amounts of target mRNA were normalized to 18S rRNA.
Western blot analysis
Soluble protein extracts from cells and tissue were prepared by homogenizing with T-per or M-per extraction reagent (Pierce, Rockford) supplemented with protease inhibitors (Roche) and analyzed by Western blotting as described previously (Cheng et al. 2013). NF-α1 and BCL-2 proteins were detected using mouse anti-NF-α1 (1:5000) (BD Biosciences, San Jose, CA) and mouse anti-BCL-2 (1:2000) (Cell Signaling, Danvers, MA) monoclonal antibodies. Detection was by the Odyssey infrared imaging system as described previously (Cheng et al. 2013), using fluorophore conjugated anti-mouse secondary antibodies (Amersham). Expression levels were normalized to β-actin and expressed in arbitrary units as the mean ± standard error of the mean.
Statistical analysis
Data were analyzed by Student's t-test or the nonparametric Mann-Whitney U test where noted and one-way analysis of variance (ANOVA) followed by Tukey post-hoc multiple comparisons tests. Correlation studies were performed with the Spearman's nonparametric test. Significance was set at p < 0.05. Data were analyzed using the Prism program (GraphPad Software, Inc, San Diego, CA).
Results
Rosiglitazone-activated PPARγ binds to NF-α1 promoter to activate gene transcription
Bioinformatic analysis of mouse, rat and human NF-α1 promoter sequences revealed the presence of several potential PPARγ-binding sites (PPRE), the positions and sequences of which are indicated in Fig. 1A. For mouse, 2 PPREs were found at position -1200/-1150 (plus strand) and -1100/-1050 (minus strand) relative to the transcriptional start site (Fig. 1A). Luciferase reporter studies (Fig. 1C, D) using mouse NF-α1 promoter sequences (Fig. 1B), showed that a 24 h treatment of primary cortical (C) or hippocampal (D) neurons, transfected with the PPRE reporter construct, with 1 μM rosiglitazone resulted in a significant increase in luciferase activity, whereas no increase was observed when neurons were transfected with the control reporter construct.
Figure 1.
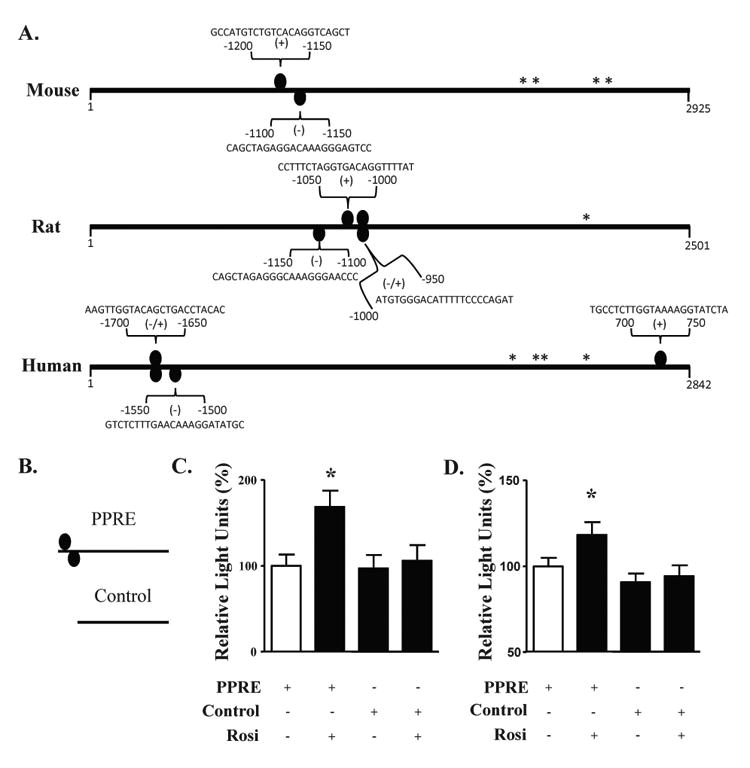
The mouse NF-α1 promoter contains PPARγ binding sites that are functional in neurons. (A) PPARγ binding sites (PPRE, ovals) are found in mouse, rat and human NF-α1 promoters. * indicate transcription start sites and the numbers represent the base pair position relative to the first transcription start site. Sequences are the PPARγ binding sites as determined by the Genomatix software. (B) Schematic diagram of the luciferase constructs containing a fragment of NF-α1 promoter without (control) or with the mouse PPREs. (C, D) Bar graphs showing luciferase activity from cortical (C) and hippocampal neurons (D) treated with and without rosiglitazone and transfected with a PPRE-containing construct, compared to neurons transfected with the control construct containing no PPRE. In both cell types, an increase in promoter activity was found when the cells were treated with Rosi. n=13/group for cortical neurons and n=36/group for hippocampal neurons; values are mean ±SEM, *p<0.05, one way ANOVA.
Neuroprotective effect of rosiglitazone-activated PPARγ is dependent on NF-α1 expression in Neuro2A cells
In Neuro2A cells treated with rosiglitazone for 24 h, NF-α1 and BCL-2 increased by ∼50 % and ∼100 %, respectively (Fig. 2A-C). To elucidate the role of increased expression of NF-α1 in neuroprotection, we used siRNA. In Neuro2A cells transfected with scrambled siRNA, cytotoxicity caused by H2O2 was prevented by 24 h rosiglitazone pre-treatment (Fig. 2D, left). However, in cells transfected with siNF-α1, which inhibited NF-α1 expression by >70 %, (siScr, 101.5 ± 9.7 versus siNF-α1, 24.9 ± 4.2), the significant protective effect of rosiglitazone in H2O2 treated cells was lost (Fig. 2D, right). The increased levels of NF-α1 and BCL-2 after 24 h treatment with rosiglitazone were maintained after subsequent treatment with H2O2 for 24 h (Fig. 3 A, B). Under these conditions, Neuro2a cells pre-treated with rosiglitazone exhibited reduced cytotoxicity caused by the H2O2 (Fig. 3C) and this effect of rosiglitazone was lost when the Neuro2a cells were co-incubated with the PPARγ inhibitor GW9662 (Fig. 3C), suggesting rosiglitazone activates PPARγ to protect the Neuro2a cells.
Figure 2.
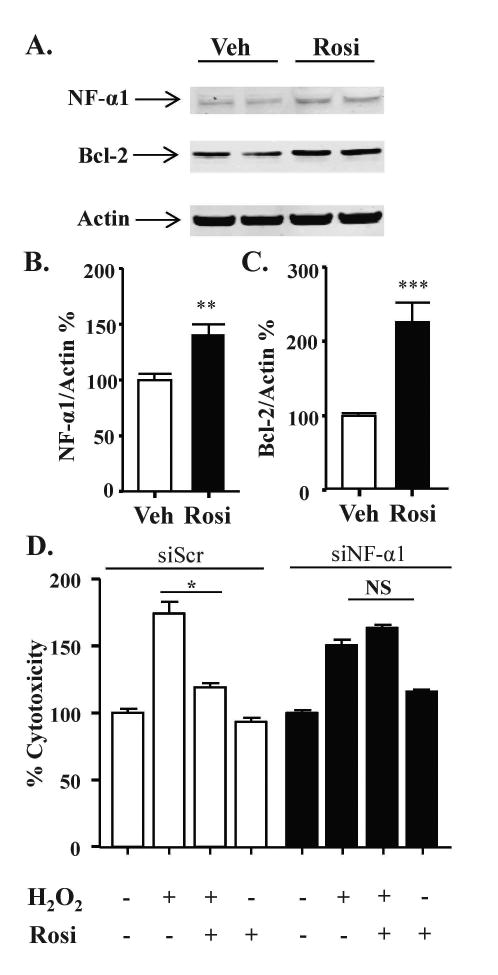
Rosiglitazone protects Neuro2A cells against oxidative stress in an NF-α1-dependent manner. (A) Representative Western blot and (B, C) quantification showing increased NF-α1 (A, B) and BCL-2 (A, C) protein levels in Neuro2a cells treated with vehicle (Veh) or 1 μM rosiglitazone (Rosi) for 24 h. (n = 6/group; values are mean ±SEM, **p<0.01, ***p<0.001, t-test). (D) Neuro2A cells were transfected with siScr or siNF-α1 for 24 h and then treated with 1 μM Rosi or vehicle for 24 h, then 100 μM H2O2 or vehicle was added to the cells for an additional 24 h. The LDH release assay showed Rosi inhibited H2O2 induced cell cytotoxicity in siScr treated Neuro2A cells but not in siNF-α1 treated cells. n=5/group; values are mean ±SEM, one way ANOVA for siScr [treatment effect: F(3, 16)=60.11, p<0.001]; for siCPE [treatment effect: F(3, 16)=121.9, p<0.001], followed by Tukey test. *p <0.05, H2O2 group compared to Rosi + H2O2 group.
Figure 3.
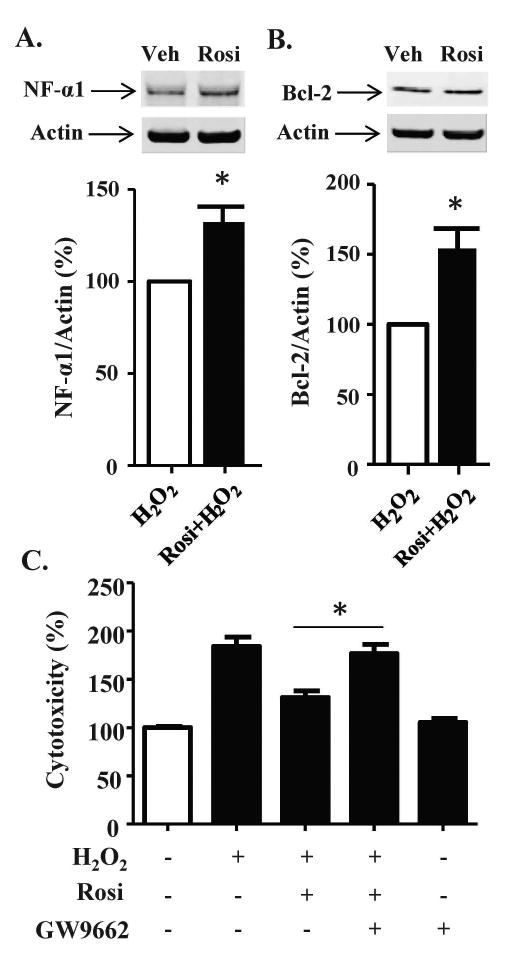
PPARγ mediates the protective effect of Rosi in Neuro2a cells. (A, B) Neuro2a cells were incubated with vehicle or Rosi for 24 h, then the cells were treated with 100 μM H2O2 for 24 h. Western Blots showed (A) NF-α1 and (B) BCL-2 protein levels increased with Rosi treatment (see Fig. 2) and remained elevated after 100 μM H2O2 treatment in Neuro2a cells. n = 3/group; values are mean ±SEM, p<0.05, t-test. (C) Neuro2a cells were treated with 1 μM Rosi and/or 1 μM GW9662 for 24 h and then the cells were treated with 100 μM H2O2 for 24 h. The LDH release assay showed that GW9662 inhibited the protective effect of Rosi against oxidative stress in Neuro2a cells. n=5/ group; values are mean ±SEM, one way ANOVA for treatment effect: F(4, 20)=93.69, p<0.001, followed by Tukey test. *p <0.05, GW9662 + Rosi + H2O2 group compared to Rosi + H2O2 group.
Rosiglitazone protects hippocampal neurons in a NF-α1 dependent manner
Treatment of hippocampal neurons with 1 μM rosiglitazone for 24 h and 48 h increased NF-α1 mRNA (Fig. 4C) and protein (Fig. 4A, B) levels, respectively by ∼50 %. In hippocampal neurons transfected with scrambled siRNA, cytotoxicity caused by H2O2 was inhibited by rosiglitazone treatment (Fig. 4D, left). However, in cells transfected with siNF-α1, the protective effect of rosiglitazone in H2O2 treated neurons was reduced (Fig. 4D, right).
Figure 4.
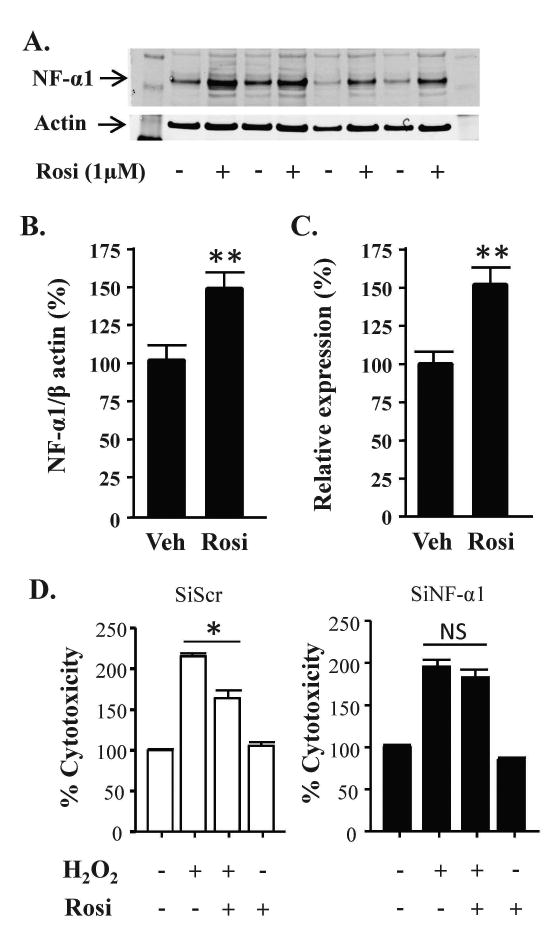
Rosiglitazone protects hippocampal neurons in an NF-α1-dependent manner. (A) Representative Western blots and (B) quantification of NF-α1 protein levels in hippocampal neurons treated with vehicle or 1 μM rosiglitazone (Rosi) for 48 h. Actin served as an internal control. n = 8/group; values are mean ±SEM, **p<0.01, Mann-Whitney test. (C) Bar graphs showing NF-α1 mRNA levels in hippocampal neurons treated with vehicle (Veh) or 1 μM rosiglitazone (Rosi) for 24 h. n = 6/group; values are mean ±SEM, **p < 0.01, Mann-Whitney test. (D) Primary cultured hippocampal neurons were transfected with siScr or siNF-α1 for 24 h and then treated with 1 μM Rosi or vehicle for 24 h, then 50 μM H2O2 was added to the cells for a further 24 h. The LDH release assay showed Rosi inhibited H2O2 induced cell cytotoxicity in siScr treated neurons but not in siNF-α1 treated neurons. n=5/ group; values are mean ±SEM, one way ANOVA for siScr [treatment effect: F(3, 16)=93.69, p<0.001]; for siNF-α1 [treatment effect: F(3, 16)=78.54, p<0.001], followed by Tukey test. *p <0.05, H2O2 group compared to Rosi + H2O2 group.
Rosiglitazone up-regulates BCL-2 expression in a NF-α1-dependent manner
Bcl-2 mRNA levels increased ∼2-fold with a 24 h treatment of rosiglitazone in primary hippocampal neurons (Fig. 5A). Bcl-2 expression levels were strongly and significantly correlated with NF-α1 mRNA levels (Fig. 5B). In hippocampal neurons transfected with scrambled siRNA, treatment with rosiglitazone significantly increased BCL-2 protein levels by ∼2-fold (Fig. 5C, D). However, this increase was inhibited when neurons were transfected with NF-α1 siRNA (Fig. 5C, D).
Figure 5.
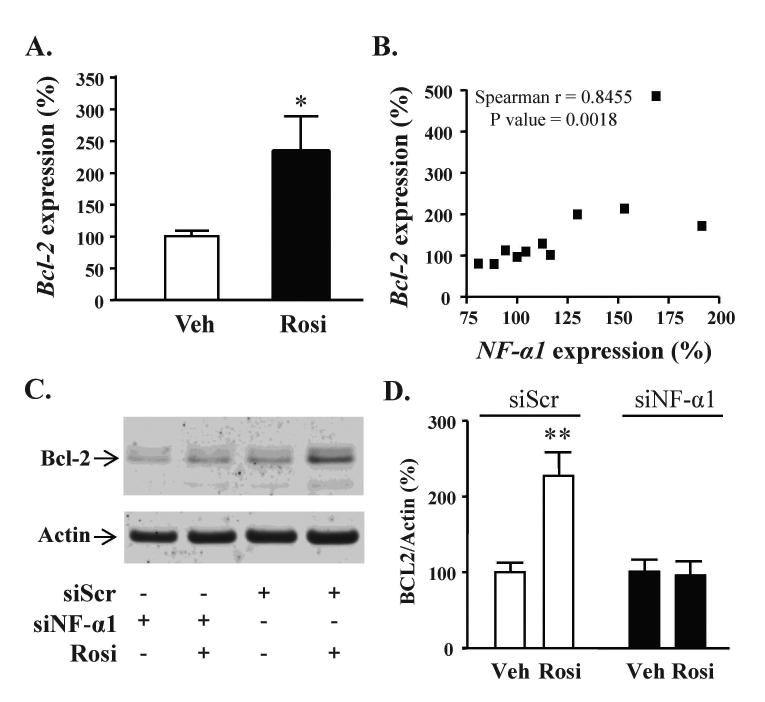
NF-α1 mediates the neuprotective effect of rosiglitazone by enhancing Bcl-2 expression. (A) Bar graphs showing Bcl-2 mRNA levels in hippocampal neurons treated with vehicle (Veh) or 1 μM rosiglitazone (Rosi) for 24 h. Rosiglitazone significantly increased Bcl-2 mRNA levels in hippocampal neurons, n = 5/group; values are mean ±SEM, *p < 0.05, Mann-Whitney test. (B) Bcl-2 mRNA levels were strongly and significantly correlated with NF-α1 mRNA levels. (C) Representative Western blots and (D) Bar graphs show BCL-2 protein levels in hippocampal neurons transfected with scrambled siRNA or siNF-α1. In hippocampal neurons transfected with scrambled siRNA and treated for 24 h with rosiglitazone, BCL-2 protein levels increased significantly, n=7/group; values are mean ±SEM, **p<0.01, Mann-Whitney test. In contrast, BCL-2 protein did not increase with rosiglitazone treatment when NF-α1 expression was inhibited by siNF-α1 (n= 7/group).
Discussion
Our previous work using a chronic restraint stress paradigm in male mice demonstrated that NF-α1 increased in the hippocampus in response to this stress (Murthy et al. 2013). At that time our bioinformatic analysis identified glucocorticoid binding sites on the NF-α1 promoter to suggest that glucocorticoids released from the adrenal cortex during this stress may have induced the expression of NF-α1 under these conditions. Further analysis using the Genomatix software identified several potential PPARγ binding sites on the NF-α1 promoters of rat, mouse and human (Fig. 1A); binding sites that we have now demonstrated by luciferase reporter assays to be functional in cortical and hippocampal neurons (Fig. 1B, C). Hence, rosiglitazone-activated PPARγ may directly regulate NF-α1 expression in neurons through binding to the NF-α1 promoter. This is important in light of the new finding that NF-α1 has neuroprotective properties (Cheng et al. 2013) and may contribute to a mechanism for some of the neuroprotective properties of rosiglitazone. Furthermore, a correlation between increased NF-α1 expression and over-expression of PPARγ in mesenchymal stem cells (Shockley et al. 2009) suggests an inherent connection between NF-α1 expression and PPARγ and its agonists.
Indeed, treatment of Neuro2A cells (Figs. 2 and 3), a mouse neuro-endocrine cell line, and rat hippocampal neurons in culture (Fig. 4), with rosiglitazone, specifically increased the endogenous levels of NF-α1 in these cells and is consistent with previous in vivo studies showing a correlation between oral rosiglitazone administration in mice and an increase in NF-α1 mRNA expression in the mouse hippocampus (Cheng et al. 2014). Of particular significance in our experiments was the demonstration that the hydrogen peroxide-induced cell cytotoxicity, which was prevented by rosiglitazone pre-treatment in Neuro2a cells and hippocampal neurons transfected with a scrambled siRNA (Figs. 2D and 4D, left panel), was eliminated when siNF-α1 was used (Figs. 2D and 4D, right panel). These results demonstrate that the neuroprotective effect of rosiglitazone-activated PPARγ in these cells is dependent on NF-α1 expression.
In the CNS, numerous studies, both in vivo and ex vivo using animals and neuronal cell lines or primary neurons, have provided evidence that rosiglitazone protects neurons against oxidative stress-induced cell death under various conditions, such as ischemia (Giaginis et al. 2008) or traumatic brain injuries (Yi et al. 2008, Qi et al. 2010). In primary hippocampal neurons and NGF-differentiated PC12 cells, rosiglitazone was shown to protect cells against hydrogen peroxide and/or Amyloid-beta-induced cell stress through the pro-survival mitochondria protein BCL-2-mediated pathway (Fuenzalida et al. 2007, Abraki et al. 2012), a finding we were able to replicate in Neuro2A cells (Fig. 2C, 3B) and hippocampal neurons (Fig. 5C, D). While these previous studies showed enhanced expression of BCL-2 upon rosiglitazone treatment, leading to neuroprotection, the mechanism leading to this event was not fully understood, since bioinformatic analysis revealed no putative PPARγ binding sites in the Bcl-2 promoter (our unpublished study), suggesting that up-regulation of the transcription of this protein is indirect. BCL-2 is a pro-survival protein involved in mitochondrial membrane integrity and previously we showed that NF-α1 can enhance BCL-2 expression through activation of the ERK and AKT signaling pathways in hippocampal neurons (Cheng et al. 2013). In our current experiments on hippocampal neurons, the rosiglitazone-dependent increase of BCL-2 was eliminated when the neurons were transfected with siNF-α1 but not when scrambled siRNA was used (Fig. 4C-E), indicating that rosiglitazone increases BCL-2 levels in neurons in a NF-α1-dependent manner, leading to neuroprotection. We also analyzed the levels of BCL-xl and BAX, other members of the Bcl family of mitochondrial proteins, and found them to be unchanged when Neuro2a cells were treated with rosiglitazone (data not shown), indicating that induction of BCL-2 in these cells appears specific for rosiglitazone through NF-α1.
An extensive number of studies have investigated the mechanisms by which PPARγ activation functions to protect neurons (for review see (Kapadia et al. 2008)). Primary among them appears to be the prevention of an inflammatory response to the stressor by reducing pro-inflammatory cytokines and transcription factors and inducing the anti-inflammatory response, including components of the anti-oxidant pathway. In our experiments, we used oxidative stress to damage the neurons; however, we did not find a difference in expression levels of several anti-oxidant enzymes, including superoxide dismutase 1 and 2, glutathione peroxidase-1 and catalase (data not shown), suggesting that the neuroprotection we observed under these conditions in vitro was not through anti-oxidant enzymes. While the mechanisms of action of rosiglitazone are complex, our specific assay identifies NF-α1 as a significant molecule in rosiglitazone-mediated neuroprotection. It would be premature to conclude, however, that NF-α1 is solely responsible for neuroprotection in vivo since many molecules are involved in the complex mechanism (Kapadia et al. 2008) and our in vitro assays are specific to dose, time and mode of stressor. Nevertheless, there is evidence that there is a connection between NF-α1, rosiglitazone and BCL2 in vivo since we have demonstrated an increase in NF-α1 expression in the hippocampus of rosiglitazone-fed mice (Cheng et al. 2014) and that NF-α1 levels are elevated after chronic restraint stress which then drives the increase in BCL-2 expression in the hippocampus (Murthy et al. 2013); additionally, BCL2 has previously been demonstrated to mediate neuroprotection by rosiglitazone (Fuenzalida et al. 2007, Qin et al. 2014).
In conclusion, we have demonstrated that rosiglitazone-activated PPARγ directly induces the expression of NF-α1 in neurons. This increase in NF-α1 signals the activation of the ERK and AKT pathways (Cheng et al. 2013), leading to enhanced BCL-2 expression. BCL-2 is known to function as a pro-survival protein by regulating mitochondrial energetics and inhibition of caspase 3, resulting in neuroprotection (Fig. 6) and suggesting that PPARγ agonists such the TZDs that enhance the expression of NF-α1 may be useful for treatment of neurodegenerative diseases.
Figure 6.
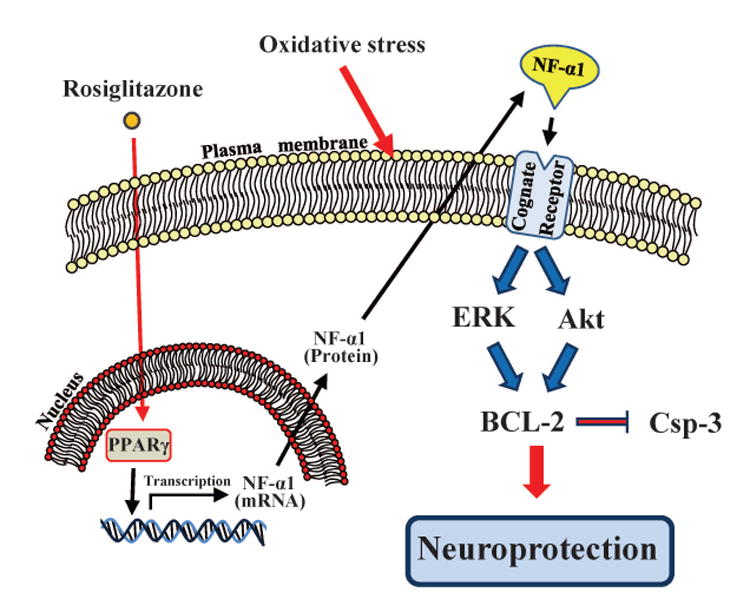
Schematic mechanism of action of rosiglitazone-activated PPARγ in neuroprotection. Rosiglitazone enters the nucleus and binds PPARγ. The activated PPARγ binds the NF-α1 promoter to activate transcription. The NF-α1 mRNA is consequently translated and the protein is secreted. It then binds a cognate receptor and activates the ERK and AKT pathways leading to an increase in BCL-2 expression which mediate neuroprotection by inhibition of caspases.
Acknowledgments
This research was supported by the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD), National Institutes of Health, USA.
Abbreviations
- CPE
carboxypeptidase E
- CNS
Central Nervous System
- PPARγ
Peroxisome proliferator-activated receptor gamma
- PPRE
PPARγ-binding site
- NF-α1
Neurotrophic Factor-α1
- siRNA
small interfering ribonucleic acid
- TZD
thiazolidinediones
- H2O2
hydrogen peroxide
- DMSO
dimethyl sulfoxide
Footnotes
The authors declare no conflicts of interest.
References
- Abraki SB, Khalaj L, Shaerzadeh F, Khodagholi F. Simultaneous inhibition of COX-2 and activation of PPAR-gamma resulted in the same level and pattern of neuroprotection as they were targeted separately. J Mol Neurosci. 2012;49:116–129. doi: 10.1007/s12031-012-9903-5. [DOI] [PubMed] [Google Scholar]
- Aoun P, Watson DG, Simpkins JW. Neuroprotective effects of PPARgamma agonists against oxidative insults in HT-22 cells. European journal of pharmacology. 2003;472:65–71. doi: 10.1016/s0014-2999(03)01867-3. [DOI] [PubMed] [Google Scholar]
- Cawley NX, Wetsel WC, Murthy SR, Park JJ, Pacak K, Loh YP. New roles of carboxypeptidase E in endocrine and neural function and cancer. Endocrine reviews. 2012;33:216–253. doi: 10.1210/er.2011-1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YC, Wu JS, Tsai HD, Huang CY, Chen JJ, Sun GY, Lin TN. Peroxisome proliferator-activated receptor gamma (PPAR-gamma) and neurodegenerative disorders. Molecular neurobiology. 2012;46:114–124. doi: 10.1007/s12035-012-8259-8. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Cawley NX, Loh YP. Carboxypeptidase E/NFalpha1: a new neurotrophic factor against oxidative stress-induced apoptotic cell death mediated by ERK and PI3-K/AKT pathways. PloS one. 2013;8:e71578. doi: 10.1371/journal.pone.0071578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Rodriguiz RM, Murthy SR, et al. Neurotrophic factor-alpha1 prevents stress-induced depression through enhancement of neurogenesis and is activated by rosiglitazone. Molecular psychiatry. 2014 doi: 10.1038/mp.2014.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eissa Ahmed AA, Al-Rasheed NM, Al-Rasheed NM. Antidepressant-like effects of rosiglitazone, a PPARgamma agonist, in the rat forced swim and mouse tail suspension tests. Behavioural pharmacology. 2009;20:635–642. doi: 10.1097/FBP.0b013e328331b9bf. [DOI] [PubMed] [Google Scholar]
- Fricker LD. Carboxypeptidase E. Annual review of physiology. 1988;50:309–321. doi: 10.1146/annurev.ph.50.030188.001521. [DOI] [PubMed] [Google Scholar]
- Fuenzalida K, Quintanilla R, Ramos P, Piderit D, Fuentealba RA, Martinez G, Inestrosa NC, Bronfman M. Peroxisome proliferator-activated receptor gamma up-regulates the Bcl-2 anti-apoptotic protein in neurons and induces mitochondrial stabilization and protection against oxidative stress and apoptosis. J Biol Chem. 2007;282:37006–37015. doi: 10.1074/jbc.M700447200. [DOI] [PubMed] [Google Scholar]
- Giaginis C, Tsourouflis G, Theocharis S. Peroxisome proliferator-activated receptor-gamma (PPAR-gamma) ligands: novel pharmacological agents in the treatment of ischemia reperfusion injury. Current molecular medicine. 2008;8:562–579. doi: 10.2174/156652408785748022. [DOI] [PubMed] [Google Scholar]
- Heneka MT, Landreth GE. PPARs in the brain. Biochimica et biophysica acta. 2007;1771:1031–1045. doi: 10.1016/j.bbalip.2007.04.016. [DOI] [PubMed] [Google Scholar]
- Jin K, Graham SH, Nagayama T, Goldsmith PC, Greenberg DA, Zhou A, Simon RP. Altered expression of the neuropeptide-processing enzyme carboxypeptidase E in the rat brain after global ischemia. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2001;21:1422–1429. doi: 10.1097/00004647-200112000-00006. [DOI] [PubMed] [Google Scholar]
- Kapadia R, Yi JH, Vemuganti R. Mechanisms of anti-inflammatory and neuroprotective actions of PPAR-gamma agonists. Frontiers in bioscience : a journal and virtual library. 2008;13:1813–1826. doi: 10.2741/2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshimizu H, Senatorov V, Loh YP, Gozes I. Neuroprotective protein and carboxypeptidase E. J Mol Neurosci. 2009;39:1–8. doi: 10.1007/s12031-008-9164-5. [DOI] [PubMed] [Google Scholar]
- Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma) J Biol Chem. 1995;270:12953–12956. doi: 10.1074/jbc.270.22.12953. [DOI] [PubMed] [Google Scholar]
- Lou H, Park JJ, Phillips A, Loh YP. gamma-Adducin promotes process outgrowth and secretory protein exit from the Golgi apparatus. J Mol Neurosci. 2013;49:1–10. doi: 10.1007/s12031-012-9827-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M, Sarruf DA, Talukdar S, et al. Brain PPAR-gamma promotes obesity and is required for the insulin-sensitizing effect of thiazolidinediones. Nature medicine. 2011;17:618–622. doi: 10.1038/nm.2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, Yin W, Signore AP, Zhang F, Hong Z, Wang S, Graham SH, Chen J. Neuroprotection against focal ischemic brain injury by the peroxisome proliferator-activated receptor-gamma agonist rosiglitazone. Journal of neurochemistry. 2006;97:435–448. doi: 10.1111/j.1471-4159.2006.03758.x. [DOI] [PubMed] [Google Scholar]
- Murthy SR, Thouennon E, Li WS, Cheng Y, Bhupatkar J, Cawley NX, Lane M, Merchenthaler I, Loh YP. Carboxypeptidase E protects hippocampal neurons during stress in male mice by up-regulating prosurvival BCL2 protein expression. Endocrinology. 2013;154:3284–3293. doi: 10.1210/en.2013-1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi L, Jacob A, Wang P, Wu R. Peroxisome proliferator activated receptor-gamma and traumatic brain injury. International journal of clinical and experimental medicine. 2010;3:283–292. [PMC free article] [PubMed] [Google Scholar]
- Qin H, Tan W, Zhang Z, Bao L, Shen H, Wang F, Xu F, Wang Z. 15d-Prostaglandin J Protects Cortical Neurons Against Oxygen-Glucose Deprivation/Reoxygenation Injury: Involvement of Inhibiting Autophagy Through Upregulation of Bcl-2. Cellular and molecular neurobiology. 2014 doi: 10.1007/s10571-014-0125-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasgon NL, Kenna HA, Williams KE, Powers B, Wroolie T, Schatzberg AF. Rosiglitazone add-on in treatment of depressed patients with insulin resistance: a pilot study. TheScientificWorldJournal. 2010;10:321–328. doi: 10.1100/tsw.2010.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan KK, Li B, Grayson BE, Matter EK, Woods SC, Seeley RJ. A role for central nervous system PPAR-gamma in the regulation of energy balance. Nature medicine. 2011;17:623–626. doi: 10.1038/nm.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma AN, Elased KM, Lucot JB. Rosiglitazone treatment reversed depression- but not psychosis-like behavior of db/db diabetic mice. Journal of psychopharmacology. 2012;26:724–732. doi: 10.1177/0269881111434620. [DOI] [PubMed] [Google Scholar]
- Shockley KR, Lazarenko OP, Czernik PJ, Rosen CJ, Churchill GA, Lecka-Czernik B. PPARgamma2 nuclear receptor controls multiple regulatory pathways of osteoblast differentiation from marrow mesenchymal stem cells. Journal of cellular biochemistry. 2009;106:232–246. doi: 10.1002/jcb.21994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woronowicz A, Koshimizu H, Chang SY, et al. Absence of carboxypeptidase E leads to adult hippocampal neuronal degeneration and memory deficits. Hippocampus. 2008;18:1051–1063. doi: 10.1002/hipo.20462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi JH, Park SW, Brooks N, Lang BT, Vemuganti R. PPARgamma agonist rosiglitazone is neuroprotective after traumatic brain injury via anti-inflammatory and anti-oxidative mechanisms. Brain research. 2008;1244:164–172. doi: 10.1016/j.brainres.2008.09.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Ou Z, Grotta JC, Waxham N, Aronowski J. Peroxisome-proliferator-activated receptor-gamma (PPARgamma) activation protects neurons from NMDA excitotoxicity. Brain research. 2006;1073-1074:460–469. doi: 10.1016/j.brainres.2005.12.061. [DOI] [PubMed] [Google Scholar]
- Zhou A, Minami M, Zhu X, Bae S, Minthorne J, Lan J, Xiong ZG, Simon RP. Altered biosynthesis of neuropeptide processing enzyme carboxypeptidase E after brain ischemia: molecular mechanism and implication. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2004;24:612–622. doi: 10.1097/01.WCB.0000118959.03453.17. [DOI] [PubMed] [Google Scholar]