

Abstract
The chaperone-usher (CU) pathway assembles organelles termed pili or fimbriae in Gram-negative bacteria. Type 1 pili expressed by uropathogenic Escherichia coli are prototypical structures assembled by the CU pathway. Biogenesis of pili by the CU pathway requires a periplasmic chaperone and an outer membrane protein termed the usher (FimD). We show that the FimD C-terminal domains provide the high-affinity substrate binding site, but that these domains are masked in the resting usher. Domain masking requires the FimD plug domain, which serves as a switch controlling usher activation. We demonstrate that usher molecules can act in trans for pilus biogenesis, providing conclusive evidence for a functional usher oligomer. These results reveal mechanisms by which molecular machines such as the usher regulate and harness protein-protein interactions, and suggest that ushers may interact in a cooperative manner during pilus assembly in bacteria.
INTRODUCTION
The chaperone-usher (CU) pathway is a conserved secretion system dedicated to the assembly of virulence-associated organelles termed pili or fimbriae in Gram-negative bacteria1-4. The type 1 and P pili expressed by uropathogenic Escherichia coli are prototypical structures assembled by the CU pathway5,6. CU pili are linear polymers composed of multiple different subunit proteins (pilins). The assembled pilus adopts a composite architecture, consisting of a rigid helical rod that is anchored to the outer membrane (OM) and a flexible tip fiber that contains the adhesive subunit (adhesin). The type 1 pilus rod contains more than 1,000 copies of the FimA major pilin; the type 1 pilus tip contains the FimH adhesin at its distal end, followed by single copies of the FimG and FimF adaptor subunits (Fig. 1a)7,8. FimH binds to mannosylated proteins present on the bladder epithelium, leading to bacterial invasion and the development of cystitis5.
Figure 1. Models for type 1 pilus biogenesis and usher domain architecture.

(a) Assembly of type 1 pili by the CU pathway. Pilus subunits traverse the inner membrane (IM) via the Sec translocon. Upon entering the periplasm, the subunits form binary complexes with the FimC chaperone (yellow). The chaperone enables proper folding of pilus subunits via the DSC mechanism (see also Supplementary Fig. 1a). Chaperone-subunit complexes next interact with the FimD usher. The usher is depicted as a monomer, with its ß-barrel channel domain in the OM and its N, plug, C1, and C2 domains indicated. Binding of a chaperone-adhesin complex (FimC-FimH) to the N domain activates the usher for pilus biogenesis. The plug is expelled from the usher channel to accommodate the FimH adhesin, and the FimC-FimH complex is handed off from the N to the C domains. The N domain is now free to recruit additional chaperone-subunit complexes, which undergo DSE with the last-incorporated subunit bound at the C domains (see also Supplementary Fig. 1b). Repeated rounds of subunit recruitment and DSE result in assembly of the pilus fiber in a top-down manner and secretion through the usher channel to the bacterial surface. (b) Cartoon representations of WT FimD and the domain-deletion mutants used in this study. The N, plug (P), C1, and C2 domains are indicated.
The CU pathway assembles and secretes pili in a highly regulated manner (Fig. 1a). Nascent pilins enter the periplasm via the Sec translocon9, and then form binary complexes with the periplasmic chaperone in a process termed donor-strand complementation (DSC)10,11. In DSC, the chaperone donates a ß-strand to complete the incomplete immunoglobulin (Ig)-like fold of the subunit (Supplementary Fig. 1a)10-12. For assembly of subunits into a pilus fiber and secretion to the cell surface, chaperone-subunit complexes must interact with the OM usher. The usher catalyzes the exchange of chaperone-subunit for subunit-subunit interactions13. Subunit-subunit interactions form by a mechanism termed donor strand exchange (DSE)12,14, in which the N-terminal extension (Nte) of an incoming subunit replaces the donated chaperone ß-strand from the preceding subunit (Supplementary Fig. 1b). Type 1 pili are assembled starting with the FimH adhesin, and the pilus extends by step-wise addition of new chaperone-subunit complexes to the base of the fiber (Fig. 1a). Each subunit specifically interacts with its appropriate neighbor in the pilus, with the specificity of binding determined by the DSE reaction15,16. In addition, the usher aids ordered pilus assembly by differentially recognizing chaperone-subunit complexes16-19.
Ushers are large, integral OM proteins composed of five domains20-23: a periplasmic N-terminal (N) domain, a transmembrane ß-barrel channel domain, a plug domain located within the ß-barrel region, and two periplasmic C-terminal domains (C1 and C2) (Fig. 1 and Supplementary Fig. 1). The N domain provides the initial binding site for chaperone-subunit complexes (Figs. 1a and 2a)21,24-26. The C1 and C2 domains provide a second binding site and anchor the growing pilus fiber (Figs. 1a and 2b)23,27,28. In the resting apo-FimD usher, the plug domain occludes the lumen of the ß-barrel channel (Supplementary Fig. 1c)20,22,23. The usher must be activated for pilus biogenesis by binding of a FimC-FimH complex to the N domain13,18,29. Activation results in displacement of the plug to the periplasm, insertion of the FimH adhesin into the channel lumen, and transfer of FimC-FimH from the usher N domain to the C domains (Fig. 2b)23. The mechanism and specific sequence of events driving usher activation and handoff of chaperone-subunit complexes from the N to the C domains is not understood. The usher N and C domains bind to the same surface of the chaperone, and handoff requires rotation of the chaperone-subunit complex, concomitant with translocation of the pilus fiber through the usher channel toward the cell surface21,23,26,27. The usher exists in the OM as an oligomer20,28,30,31. However, the pilus fiber is secreted through only one protomer of the usher oligomer, and the usher monomer appears to be sufficient for pilus biogenesis20,23,27,32. Therefore, whether and how the additional usher molecules contribute to the catalysis of pilus assembly in vivo is a subject of debate.
Figure 2. In vivo detection of FimC-FimH binding to the FimD usher.

(a) Structure of the FimD N domain (blue) bound to a FimC-FimH pilin domain complex (yellow and green, respectively) (PDB ID: 1ZE3, ref. 21). FimD residue Phe4 is depicted in red in stick representation. Phe4 is in close proximity to the FimC chaperone (see also Supplementary Fig. 2a). (b) Structure of the FimD-FimC-FimH complex (PDB ID: 3RFZ, ref. 23). FimH is in green, FimC is in yellow, and the FimD domains are colored as in Figure 1. Residues Tyr704 (C1), Thr717 (C1), and Tyr788 (C2) are depicted in red in stick representation. Tyr704 is in close proximity to the FimH adhesin domain, whereas Thr717 and Tyr788 are in proximity to the chaperone (see also Supplementary Fig. 2b). (c) SDS-PAGE and immunoblot analysis of in vivo, site-directed photocrosslinking. Samples are purified His-tagged WT FimD or FimD Phe4 (N domain), Tyr704 (C1 domain), Thr717 (C1 domain), or Tyr788 (C2 domain) amber mutants and associated crosslinked products. Additional controls are shown in Supplementary Figure 2. The position of the FimD monomer is indicated on the right for each panel. The asterisks (*) mark FimD crosslinked products. Uncropped images are shown in Supplementary Data Set 2.
In this study, we sought to understand how the usher controls and coordinates protein-protein interactions during pilus biogenesis. We used site-directed photocrosslinking to confirm the usher N, C1, and C2 domains as specific binding sites during pilus assembly in vivo. Using a fluorescence-based affinity assay to compare binding of FimC-FimH to wild-type (WT) and domain deleted FimD ushers (Fig. 1b), we show that the FimD C domains provide the high-affinity binding site, suggesting that handoff of chaperone-subunit complexes from the N to the C domains is driven by differential affinity. We provide evidence that the C domains are masked in apo-FimD through interaction with the plug domain, explaining why FimC-FimH must first bind to the N domain to activate the usher. We show that the plug domain is essential for fiber polymerization and acts as a switch controlling usher activation. Finally, by using a plug deletion mutant to pre-activate the usher, we demonstrate that the usher is functional as an oligomer in vivo.
RESULTS
FimD N and C domains are in vivo binding sites for FimC-FimH
We used site-directed photocrosslinking via unnatural amino acid mutagenesis to map points of contact between chaperone-subunit complexes and the usher, as predicted by crystal structures of FimC-FimH bound to the FimD N domain or the complete FimD usher (Fig. 2a and b)21,23. Amber stop codon (TAG) substitutions were constructed for residues in the N, C1 and C2 domains of FimD. Each FimD amber mutant was transformed together with a FimC-FimH expression plasmid into an E. coli strain containing plasmid pEVOL-pBpF, allowing incorporation of the photoreactive phenylalanine derivative p-benzoyl-phenylalanine (pBpa) at the position of the amber stop codon33. Bacteria grown in the presence of pBpa were exposed to UV light to promote reaction of the carbonyl oxygen of pBpa with nearby carbon-hydrogen bonds, forming stable crosslinks33,34.
We constructed and analyzed nine different FimD amber mutants, obtaining crosslinks between the usher and the FimC chaperone or FimH adhesin for each mutant except one (Supplementary Fig. 2a). Each of these FimD amber mutants formed a stable usher in the OM in the presence of pBpa, and each was functional for pilus assembly (data not shown). We obtained the most efficient crosslinking when pBpa was located at FimD positions Phe4 in the N domain, Tyr704 and Thr717 in the C1 domain, and Tyr788 in the C2 domain (Fig. 2 and Supplementary Figs. 2 and 3). Crosslinked products that reacted with anti-FimC-FimH antibody were visible for each of these FimD amber mutants (Fig. 2c). The anti-FimC-FimH antibody cross-reacts with the His-tag epitope, and thus also detects the His-tagged FimD usher. Immunoblotting with anti-His-tag antibody verified the presence of the usher in the crosslinked products, and analysis of a strain expressing a Strep-tagged FimD (which does not cross-react with the anti-FimC-FimH antibody) confirmed the presence of the chaperone or adhesin (Supplementary Fig. 2b and c). In addition, the crosslinked bands for the FimD Phe4, Thr717, and Tyr788 mutants, but not the Tyr704 mutant, reacted with anti-FimC-FimG antibody, which recognizes the FimC chaperone but not the FimH adhesin (Fig. 2c). Taken together, these results confirm the predicted interactions of FimD residues Phe4, Thr717, and Tyr788 with FimC, and FimD residue Tyr704 with FimH. More broadly, these results validate the N, C1, and C2 domains of the usher as specific binding sites for chaperone-subunit complexes during pilus biogenesis in vivo.
The bands obtained for the FimD Phe4 and Tyr788 amber mutants migrated at the expected size for a crosslinked FimD-FimC product (114 kDa; mature FimD and FimC are 91 and 23 kDa, respectively) (Fig. 2c). A doublet was obtained for the FimD Phe4 mutant, with the lower band reacting with both the anti-FimC-FimH and anti-FimC-FimG antibodies, identifying it as the usher-chaperone complex (Fig. 2c). Mass spectrometry analysis confirmed the presence of FimD and FimC in the lower FimD Phe4 band, as well as in the FimD Tyr788 crosslinked band (Supplementary Data Set 1). Mass spectrometry analysis of the upper band of the FimD Phe4 doublet suggested that this was a crosslink with the abundant OM protein OmpA (Supplementary Data Set 1). This is consistent with the usher having a dynamic and flexible N domain, able to sample the periplasm for chaperone-subunit complexes. An ompA mutant strain assembled type 1 pili similarly to the parental WT strain, as determined by hemagglutination (HA) assay (data not shown), indicating that that OmpA does not have a direct role in pilus biogenesis. In contrast to the FimD Phe4 and Tyr788 crosslinked products, the bands obtained for the FimD Tyr704 and Thr717 mutants migrated with slower relative mobility than expected for either a FimD-FimH complex (120 kDa; mature FimH is 29 kDa) or a FimDFimC complex, respectively (Fig. 2c). Mass spectrometry confirmed the presence of FimH and FimC in the crosslinked bands, and did not identify other crosslinked partners for the FimD Tyr704 and Thr717 mutants (Supplementary Data Set 1). Therefore, although we do not know the basis for the altered mobilities, the Tyr704 and Thr717 crosslinked products represent FimD interactions with FimH or FimC, respectively.
FimD C1 and C2 domains are the high-affinity binding site
Having validated the usher N and C domains as binding sites for chaperone-subunit complexes, we next sought to determine the relative contributions of each domain to affinity for FimC-FimH. Affinity (Kd) was measured using a fluorescence-based assay17,35. We first measured binding affinity of WT FimD for FimCFimH, where FimC was labeled with the thiol-reactive probe coumarin maleimide (CPM) at Q19C, T51C, or N86C single cysteine substitution mutations. These labeling sites were chosen because of their close proximity to the usher when the chaperone-adhesin complex is bound at either the N or C domains (Fig. 3 and Supplementary Fig. 4)21,23. Each of the FimC substitution mutants expressed stably and functioned similarly to WT FimC for pilus assembly in bacteria (data not shown). Binding assays using these FimC constructs yielded Kd of 9.50–12.6 nM (Fig. 3c and Supplementary Fig. 5). The measured affinities were not significantly different (P = 0.18), indicating agreement among the different labeling sites. Moreover, these values correspond well with a previously reported Kd of 9.1 nM, determined by surface plasmon resonance18. We chose the FimCQ19C CPM labeling site (Kd = 12.6 nM) for subsequent affinity measurements.
Figure 3. Binding affinities of FimC-FimH chaperone-adhesin complexes for WT and domain-deleted FimD ushers.

(a,b) Structures of the FimD N domain bound to a FimC-FimH pilin domain complex (a), and the FimD-FimC-FimH complex (b). The structures and colors are as in Figure 2. The FimC cysteine substitution sites Q19, T51, and N86 are shown in red in stick representation. These sites are in close proximity to the usher when FimC-FimH is bound at either the N or C domains (see also Supplementary Fig. 4). (c) Binding curve of FimCQ19C- FimH for WT FimD. The graph represents normalized changes in total fluorescence emission intensity plotted as a function of FimD concentration, where the total change in intensity was 20%. The data points represent means ± SEM of three independent experiments, with three replicates per experiment. See also Supplementary Figure 5.
(d) Affinities of FimC-FimH for FimD WT and domain deletion mutants. Affinities were calculated as in c. Additional binding curves of FimC-FimH for WT FimD are shown in Supplementary Figure 6. *P < 0.005 compared to WT FimD by two-tailed t test.
To examine the contribution of the usher C domains to affinity for chaperone-subunit complexes, we measured the binding of FimDΔC2 and FimDΔC1ΔC2 domain deletion mutants to FimC-FimH. Both of these, and all other usher deletion mutants used in this study, expressed stably and folded properly in the bacterial OM (data not shown). We obtained Kd values of 213 and 389 nM for the FimDΔC2 and FimDΔC1ΔC2 mutants, respectively (Fig. 3d and Supplementary Fig. 5). The decreased affinities obtained for these mutants compared to WT FimD identify the C domains as the high-affinity binding site on the usher for chaperone-subunit complexes.
These results also reveal that the usher N domain, which remains available for binding in the FimDΔC2 and FimDΔC1ΔC2 mutants (Fig. 1b), has lower affinity for FimC-FimH. In the P pilus system, the isolated usher plug domain was shown to interact with chaperone-subunit complexes19,36. To determine if the plug contributes to affinity for FimC-FimH in the context of the full-length usher, we examined a FimDΔplug mutant. The affinity of FimDΔplug for FimC-FimH (12.5 nM; Fig. 3d and Supplementary Fig. 5) was similar to WT FimD. This indicates no direct role for the plug domain, at least for binding to the initiating chaperone-adhesin complex.
The plug domain masks the C domains in the inactive usher
Given that the C domains provide the high-affinity binding site, it is not clear why the N domain is required for the initial binding of chaperone-subunit complexes to the usher. To address this question, we measured the affinity of a FimDΔN mutant for FimC-FimH. Despite the presence of the C domains, there was no appreciable binding of FimC-FimH to FimDΔN (Kd > 1200 nM; Fig. 3d and Supplementary Fig. 5). This suggests that the high-affinity C domains are unavailable for binding in the absence of the N domain. In its apo state, the usher plug domain resides within the lumen of the ß-barrel channel (Supplementary Fig. 1c)20,22,23. We reasoned that in this position, the plug could interact with the C1 and C2 domains, keeping the C domains inaccessible until activation of the usher by binding of a chaperone-adhesin complex to the N domain and expulsion of the plug to the periplasm. To test this, we constructed a FimD usher deleted for both the N and plug domains (FimDΔNΔplug) (Fig. 1b). The affinity of FimDΔNΔplug for FimC-FimH was 40.8 nM (Fig. 3d and Supplementary Fig. 5), which is dramatically increased compared to the FimDΔN mutant and close to the affinity observed for WT FimD. This result indicates that the high-affinity C1 and C2 domains become accessible to chaperone-subunit complexes in the absence of the plug domain, supporting our hypothesis that the plug functions to mask the C domains in the inactive usher.
The plug domain is required for higher-order pilus assembly
Previous studies demonstrated that the plug domain is essential for pilus assembly by the usher22,37,38. Our results indicate that the plug functions to maintain the usher in the inactive state by masking the C domains, but this does not explain why the plug is necessary for pilus biogenesis. One possibility is that, in the absence of the plug domain, chaperone-subunit complexes no longer bind to the N domain. We used site-directed photocrosslinking to detect binding of FimC-FimH to the N and C domains of the FimDΔplug mutant. As shown in Figure 4, we obtained a similar pattern of crosslinks for the FimDΔplug mutant as for WT FimD. Note that the level of FimDΔplug in the OM is lower than for WT FimD, explaining the weaker appearance of the crosslinked bands for the Δplug mutant. Thus, chaperone-subunit complexes still interact with both the N and C domains in the absence of the plug domain.
Figure 4. FimC-FimH binds to the N, C1, and C2 domains of the FimDΔplug usher in vivo.
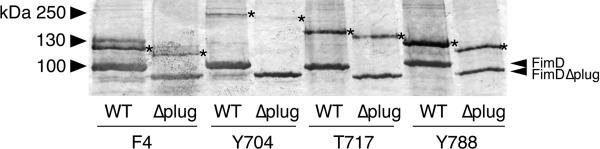
Anti-FimC-FimH immunoblot analysis of in vivo, site-directed photocrosslinking. Samples are purified His-tagged WT or Δplug FimD ushers containing amber mutations at positions Phe4 (N domain), Tyr704 (C1 domain), Thr717 (C1 domain), or Tyr788 (C2 domain) and associated crosslinked products. The positions of the WT and Δplug FimD monomers are indicated on the right, and the asterisks (*) mark the crosslinked products. The uncropped image is shown in Supplementary Data Set 2.
To further investigate the role of the plug in pilus assembly, we expressed His-tagged WT FimD or the FimDΔplug mutant in bacteria together with the FimC chaperone and FimH and FimG pilus tip subunits. These experimental conditions allow testing of the ability of the usher to polymerize pilus fibers (consisting of FimH followed by multiple copies of FimG), using a co-purification assay39. FimC, FimG, and FimH co-purified with both WT FimD and the FimDΔplug mutant, as revealed by immunoblotting with anti-FimC-FimG or anti-FimC-FimH antibodies (Fig. 5). Subunit-subunit, but not chaperone-subunit, interactions are stable to SDS at low temperatures39. Analysis of the WT FimD samples incubated at 25°C revealed a ladder of higher molecular mass species, indicating polymerization of FimG into a pilus fiber with FimH at its tip (Fig 5). In contrast, examination of the FimDΔplug samples incubated at 25°C demonstrated that the mutant was greatly impaired in its ability to promote fiber polymerization (Fig. 5). Consistent with this, bacteria expressing the FimDΔplug mutant were unable to assemble pilus fibers of sufficient length on the bacterial surface to agglutinate red blood cells (Supplementary Table 1). Taking these findings together, we conclude that the plug domain is dispensable for the binding of chaperone-subunit complexes to the usher and initiation of pilus assembly, but essential for efficient polymerization of the pilus fiber.
Figure 5. Assembly of type 1 pilus tip fibers by the WT and Δplug FimD ushers.

(a,b) anti-FimC-FimG (a) or anti-FimC-FimH (b) immunoblots. Samples are purified His-tagged WT or Δplug FimD ushers and associated pilus assembly intermediates incubated at 25 or 95°C in SDS sample buffer. The identities of the pilus proteins and assembly intermediates are indicated on the right using single letters (C, FimC; G, FimG; H, FimH; D, FimD). Uncropped images are shown in Supplementary Data Set 2.
Plug deletion reveals a functional usher oligomer
The usher exists as an oligomer in the OM; however, only one usher protomer is involved in secretion of the pilus fiber, and the function of the oligomer is not known20,23,30-32. One possibility is that the N domains of the nontranslocating ushers recruit chaperone-subunit complexes to the OM assembly platform, and these complexes are then transferred to the C domains of the actively translocating usher. If true, then co-expression of FimDΔC1ΔC2 and FimDΔNΔplug usher mutants (N and C domains available, respectively; Fig. 6a) should allow reconstitution of pilus biogenesis. Indeed, co-expression of the ΔNΔplug and ΔC1ΔC2 FimD constructs resulted in assembly of functional type 1 pili, as measured by the HA assay (Table 1). Note that the ΔNΔplug and ΔC1ΔC2 mutants did not assemble pili when expressed individually (Table 1). Pilus biogenesis on the bacterial surface by the strain co-expressing FimDΔC1ΔC2 and FimDΔNΔplug was confirmed by electron microscopy (EM), which revealed levels of pilus fibers comparable to the strain expressing WT FimD (Fig. 6). Consistent with our finding that that the the plug domain masks the C domains in the inactive usher, co-expression of a FimDΔN mutant (plug domain intact) with FimDΔC1ΔC2 did not result in pilus assembly (Table 1). In additional experiments, we found that co-expression of FimDΔNΔplug with a FimDΔplugΔC1ΔC2 mutant did not restore pilus assembly, and neither did co-expression of FimDΔN with a FimDΔplugΔC1ΔC2 mutant (Table 1). This indicates that a plug domain must be present for successful complementation, but the plug cannot be located together with the C domains. These data demonstrate that individual usher molecules are capable of functioning in trans for pilus biogenesis in bacteria, and provide confirmation that the plug domain masks the C domains in the inactive usher.
Figure 6. Co-expression of FimDΔNΔplug and FimDΔC1ΔC2 ushers results in pilus assembly on the bacterial surface.

(a) Cartoon representations of the FimDΔC1ΔC2 (usher 1) and FimDΔNΔplug (usher 2) deletion mutants co-expressed in (c). The N, plug (P), C1, and C2 domains present in each usher construct are indicated. (b,c,d) whole-bacteria, negative-stain transmission EM of E. coli strain MM294ΔfimD expressing WT FimD (b), FimDΔNΔplug together with FimDΔC1ΔC2 (c), or vector only (d). The FimD expression plasmids used were as listed in Table 1. Scale bars = 500 nm.
Table 1.
Assembly of adhesive pili on the bacterial surface by co-expression of WT or domain-deleted FimD ushers
| FimD | Plasmids | HA titera |
|---|---|---|
| WT + WT | pNH382 + pNH213 | 128 |
| ΔN + ΔC1ΔC2 | pNH383 + pNH295 | 0 |
| ΔNΔplug + ΔC1ΔC2 | pGW217 + pNH295 | 32 |
| ΔNΔplug + vector | pGW217 + pMMB66 | 0 |
| vector + ΔC1ΔC2 | pTRYC + pNH295 | 0 |
| ΔN + ΔplugΔC1ΔC2 | pNH383 + pNH423 | 0 |
| ΔNΔplug + ΔplugΔClΔC2 | pGW217 + pNH423 | 0 |
Hemagglutination (HA) titer is the maximum fold dilution of bacteria (strain MM294ΔfimD expressing the indicated FimD constructs) able to agglutinate guinea pig red blood cells.
DISCUSSION
The usher is a remarkable molecular machine that catalyzes ordered polymerization of the pilus fiber and provides the channel for secretion of the fiber to the cell surface. The usher performs its functions in the absence of an external energy source such as ATP, relying instead on harnessing protein-protein interactions40. Our findings reveal mechanisms by which the usher controls access to its domains and show how these domains act in concert to ensure the assembly of adhesive organelles. We also demonstrate that individual usher molecules can act in trans for pilus biogenesis in bacteria, providing conclusive evidence for a functional usher oligomer.
Using site-directed photocrosslinking, we confirmed that the usher N, C1, and C2 domains function as binding sites for chaperone-subunit complexes during pilus biogenesis in bacteria. The crosslinked products obtained for the FimD Tyr704 and Thr717 mutants, located in the C1 domain, migrated with slower-than-expected relative mobility. Other potential binding partners were not identified by mass spectrometry and only the usher is capable of forming a crosslink in our experimental system. One possibility for the slower mobility is that these crosslinked products migrate aberrantly during electrophoresis. A second possibility is that these complexes represent higher-order assembly intermediates comprising FimD-FimD as well as FimD-FimH or FimD-FimC contacts, consistent with the usher oligomer forming an actively engaged assembly unit in vivo.
Our comparison of full-length and domain-deleted FimD ushers revealed that the C domains provide the high-affinity binding site for FimC-FimH chaperone-adhesin complexes. Based on our results, we conclude that the Kd measured for WT FimD (12.6 nM) reflects the contribution of the C domains to affinity for FimC-FimH, and the Kd measured for FimDΔC1ΔC2 (389 nM) reflects the contribution of the N domain to affinity for FimC-FimH. In contrast to our results, Volkan et al. found that the C2 domain of the P pilus usher PapC had lower affinity for chaperone-subunit complexes compared to the N domain19. This difference likely reflects the fact that the PapC C2 domain was studied in isolation, rather than in its native orientation in the context of the C1 domain and the rest of the usher protein. Chaperone-subunit complexes bind first to the usher N domain and then transfer to the C domains, through an unknown mechanism21,23-25,27,28. Our results suggest that this handoff is driven by differential affinity, with the high affinity C domains outcompeting the lower affinity N domain for the common binding site on the chaperone. Handoff may also be facilitated by allosteric destabilization of the N domain-chaperone-subunit complex by the C2 domain or through interactions with the plug, as suggested by studies in the P pilus system19,36.
Our finding that the C domains have higher affinity compared to the N domain for FimC-FimH raised the question as to why chaperone-subunit complexes first bind to the N domain of the apo usher. We demonstrated that the C domains are not available for binding in the absence of the N domain, but become available in the absence of the plug. Based on these results, we propose that the apo usher employs a domain masking strategy to keep the C domains inaccessible, dependent on interaction of the C domains with the plug (Supplementary Fig. 6). Only FimC-FimH chaperone-adhesin complexes are able to activate the FimD usher13,18,29. Therefore, masking of the C domains would allow the usher to sample chaperone-subunit complexes in the periplasm via its N domain, with only FimC-FimH initiating pilus assembly by triggering release of the plug from the channel and freeing the C domains (Supplementary Fig. 6). Domain masking thus provides a mechanism to ensure assembly of a functional pilus fiber with the adhesin at its tip, poised to bind host cell receptors.
The plug domain is essential for the function of the usher in pilus biogenesis22,37,38. The plug occupies the channel of the apo usher, and we show here that the plug masks the C domains. However, these functions are related to maintenance of the usher in its inactive state. We found that the plug domain is not needed for recruitment of chaperone-subunit complexes to the usher or formation of stable pilus assembly intermediates in vivo. Instead, we found that the plug is required for efficient polymerization of the pilus fiber. The catalytic activity of the usher in fiber polymerization is postulated to be due to optimal positioning of chaperone-subunit complexes to promote the DSE reaction23. In the activated usher, the plug resides in the periplasm, adjacent to the N domain (Supplementary Fig. 1d)23,27. We propose that the plug contributes to catalytic activity by fixing orientation of the N domain relative to C domains. Thus, the location of the plug may act as a central switch that determines the activation state of the usher. In the resting usher, the plug closes the channel and masks the C domains. Expulsion of the plug to the periplasm then activates the usher by (i) opening the channel, (ii) unmasking the C domains, and (iii) ensuring optimal positioning of the N domain to promote subunit-subunit interactions.
The usher exists as an oligomeric complex in the bacterial OM20,23,30-32, but whether the oligomer makes a functional contribution to pilus biogenesis has been a subject of debate. We show here that co-expression of FimDΔNΔplug and FimDΔC1ΔC2 ushers results in assembly of adhesive pili on the bacterial surface. Pilus biogenesis by these ushers necessitates that the N and C domains from different usher molecules cooperatively interact, providing a mechanistic basis for the function of the usher oligomer (Supplementary Fig. 6). Analysis of various combinations of FimD deletion mutants revealed that a plug domain is required for successful complementation, but the plug cannot be present on the same usher as the C domains. The requirement for the plug together with the N domain emphasizes the active role of the plug in the catalytic activity of the usher. The finding that complementation does not work when the plug is present together with the C domains reflects our finding that the plug masks the C domains in the inactive usher.
In a prior study, we found that PapC C-terminal deletion mutants could interact with FimD to drive assembly of P pili on the bacterial surface by the Fim system28. This supports the existence of cooperative interactions between different ushers in bacteria. The FimH adhesin was also required for functional interaction between PapC and FimD in the prior study, suggesting that FimD needed to be activated by binding to the adhesin28. In light of the results from the current study, we can now understand that the C domains of FimD were masked and unavailable to participate in pilus assembly prior to usher activation.
We propose that identical usher molecules act in an asymmetric manner during pilus biogenesis, with multiple ushers serving to recruit chaperone-subunit complexes to the OM, but only one usher providing the active translocation channel (Supplementary Fig. 6). Such an arrangement may enhance the catalytic activity of the usher by increasing the local concentration of chaperone-subunit complexes, and may allow for greater regulatory control of fiber polymerization through usher-usher interactions or changes in the oligomeric state of the usher. Other transporters found in both prokaryotes and eukaryotes also exist as oligomeric complexes41-44. Studies suggest that these complexes may also function in an asymmetric manner, with the oligomeric arrangement providing additional binding sites or allowing regulatory interactions41,44-46. Thus, the use of identical channels in an asymmetric manner may be a common strategy employed by diverse transport systems.
ONLINE METHODS
Strains and plasmids
The bacterial strains and plasmids used in this study are listed in Supplementary Table 2. Unless otherwise noted, bacteria were grown at 37°C with aeration in LB medium. When appropriate, the growth medium was supplemented with antibiotics as follows: 100 μg/ml ampicillin (Amp); 50 μg/ml kanamycin (Kan); 100 μg/ml spectinomycin (Spec); 25 μg/ml chloramphenicol (Clm); 15 μg/ml tetracycline (Tet).
The molecular biology techniques and primers used to construct the plasmids made in this study are listed in Supplementary Table 3. E. coli DH5α was used as the host strain for plasmid manipulations. The FimD amber mutants used for site-directed photocrosslinking were derived from plasmids pNH213 or pNH400 using QuikChange Site-Directed Mutagenesis (Stratagene). Plasmid pNH213 encodes the FimD usher with a C-terminal, thrombin-cleavable, polyhistadine tag (His-tag) under isopropyl-ß-D-thiogalactoside (IPTG)-inducible expression. For pNH400, the His-tag of plasmid pNH213 was switched to a Step-tag using site-directed, ligase-independent mutagenesis (SLIM)47,48. The FimC cysteine mutants for fluorescence labeling were derived from pETS1000 using QuikChange mutagenesis. Plasmid pETS1000 encodes the FimC chaperone with a C-terminal His-tag under arabinose-inducible expression. Plasmid pNH324, encoding FimDΔplug, was derived from pNH213 using SLIM to delete residues 244-323. In addition to deletion of the plug domain, an N243G substitution mutation was created. Similarly, the plug domain was deleted from plasmids pNH295 and pNH296, encoding FimDΔC1ΔC2 and FimDΔN, respectively, to make plasmids pNH423 and pGW117. All constructs generated using SLIM or QuikChange mutagenesis methods were sequenced to verify that the correct mutations were made.
In vivo site-directed photocrosslinking
Strain SF100 was transformed with plasmid pEVOL-pBpF, encoding an arabinose-inducible amber suppressor tRNA and aminoacyl-tRNA synthetase, allowing incorporation of pBpa at amber stop codons (TAG)33. Strain SF100/pEVOL-pBpF was then transformed with plasmid pNH212, encoding IPTG-inducible FimC and FimH proteins. Finally, strain SF100/pEVOL-pBpF + pNH212 was transformed with plasmids for IPTG-inducible expression of His-tagged wild-type (WT) FimD (pNH213) or FimD Phe4 (pNH319), Tyr704 (pNH320), Thr717 (pNH321), or Tyr788 (pNH329) amber codon mutants. Overnight cultures were diluted 1:20 into 30–50 ml fresh LB containing 0.2 mM pBpa (VWR). Cultures were induced at OD600 = 0.6 with 0.1% arabinose and 50 μM IPTG for 1–2 h. Cultures were pelleted and resuspended in 1 ml 20 mM Tris-HCl (pH 8.0), transferred to wells in an untreated six-well culture plate (CytoOne), and exposed to a UV lamp (365 nm, 100 W, Fisher Scientific) for 10 min. Exposed bacteria were then transferred to microcentrifuge tubes and pelleted at maximum speed in a microcentrifuge for 15 min at 4 °C. Pellets were weighed and resuspended in 500 μl BugBuster Master Mix (Novagen) per 0.1 g wet weight. EDTA-free Complete protease inhibitor (Roche) was added, and the samples were rocked for 20 min at room temperature. Samples were then spun at maximum speed in a microcentrifuge for 20 min at 4 ºC, and supernatant fractions were transferred to clean tubes. Imidazole was added to 20 mM, 50 µl of 50% Ni-NTA agarose beads (Qiagen) were added, and samples were rocked for 30 min at room temperature. The beads were washed 3 times with 1 ml 20 mM Tris-HCl (pH 8.0), 0.3 M NaCl, 20 mM imidazole, and then boiled in 60 µl of 2X SDS-PAGE sample buffer. Boiled samples were separated by SDS-PAGE, and analyzed either by staining with Coomassie blue or immunoblotting with anti-His-tag (BioLegend, catalog number MMS-156P, validation profile provided on manufacturer website), anti-FimC-FimH20, or anti-FimC-FimG20 antibodies. The blots were developed with alkaline phosphatase-conjugated secondary anti-mouse (Sigma catalog number A9316) or anti-rabbit (Sigma catalog number A3812) antibodies and BCIP (5-bromo-4-chloro-3-indolylphosphate)-NBT (nitroblue tetrazolium) substrate (KPL).
For some experiments, SF100/pEVOL-pBpF + pNH212 strains expressing Strep-tagged instead of His-tagged FimD were used (plasmids pNH400 through pNH404). For these experiments, following UV exposure, OM fractions were isolated as described24. The OM fractions were separated by SDS-PAGE and analyzed by immunoblotting with anti-His-tag or anti-FimC-FimH antibodies, as above. The expression and folding of the FimD amber mutants in the OM was compared with WT FimD, as described below. SF100/pEVOL-pBpF was used as the host strain for these experiments and the bacteria were grown in the presence of 0.2 mM pBpa. The ability of the FimD amber mutants to assemble adhesive pili on the bacterial surface was compared with WT FimD using the HA assay, as described below. For these assays, MM294ΔfimD/pEVOL-pBpF was used as the host strain and the bacteria were grown in the presence of 0.2 mM pBpa.
Mass spectrometry analysis of crosslinked products
Excised gel pieces were destained, reduced, alkylated and digested with trypsin (Promega Gold, Mass Spectrometry Grade), essentially as described49. The resulting concentrated peptide extract was diluted into a solution of 2% Acetonitrile (ACN), 0.1% Formic Acid (FA) (buffer A) for analysis. The peptide mixture was analyzed by automated microcapillary liquid chromatography-tandem mass spectrometry. Fused-silica capillaries (100 μm i.d.) were pulled using a P-2000 CO2 laser puller (Sutter Instruments) to a < 5 μm i.d. tip, and packed with 10 cm of 5 μm ProntoSil 120-5-C18H material (Agilent) using a pressure bomb. The column was installed in-line with an Eksigent Nano2D High Performance Liquid Chromatography (HPLC) pump running at 300 nl min-1. The column was equilibrated in buffer A, and the peptide mixture was loaded onto the column using an autosampler. The HPLC separation was provided by a gradient between buffer A and buffer B (98% ACN, 0.1% FA). The HPLC gradient was held constant at 100% buffer A for 10 min after peptide loading, followed by a 35-min gradient from 0% buffer B (100% Buffer A) to 40% buffer B. Then, another gradient was performed for 3 min to 80% buffer B, where it was held constant for 2 min. Finally, the gradient was changed from 80% buffer B to 100% buffer A over 1 min, and then held constant at 100% buffer A for 29 more minutes. The application of a 1.8 kV distal voltage electrosprayed the eluted peptides directly into a Thermo Fisher Scientific LTQ XL ion trap mass spectrometer equipped with a custom built nanoLC electrospray ionization source. Full mass spectra (MS) were recorded on the peptides over a 400-2000 m/z range, followed by five tandem mass (MS/MS) events sequentially generated in a data-dependent manner on the first, second, third, fourth and fifth most intense ions selected from the full MS spectrum (at 35% collision energy). Mass spectrometer scan functions and HPLC solvent gradients were controlled by the Xcalibur data system (ThermoFinnigan, San Jose, CA). The resultant MS/MS spectra were extracted from the RAW file with Readw.exe (http://sourceforge.net/projects/sashimi). The resulting mzXML file contains all the data for all MS/MS spectra and can be read by the subsequent analysis software. The MS/MS data were searched using InsPecT50 and GPM X!Tandem against the Ecoli_K12 UniProt database (downloaded 3/19/2013) with optional modifications: +16 on Methionine, +57 on Cysteine, and +80 on Threonine, Serine and Tyrosine. Only peptides with a P value of ≤ 0.01 were analyzed further. Common contaminants (e.g. keratins) were removed from the database. Proteins identified by at least 2 distinct peptides within a sample were considered valid.
Purification of FimD and FimC-FimH for affinity measurements
The WT FimD usher and FimD domain deletion mutants contained C-terminal, thrombin-cleavable His-tags and were purified as described51. Briefly, 6 l cultures of strain Tuner harboring plasmid pNH213 (WT FimD), pNH295 (FimDΔC1ΔC2), pNH317 (FimDΔC2), pNH296 (FimDΔN), pNH324 (FimDΔplug), or pGW117 (FimDΔNΔplug) were induced for usher expression at OD600 = 0.6 with 100 μM IPTG and grown overnight at room temperature. Bacteria were lysed using a French press and the OM fraction was isolated by Sarkosyl extraction and centrifugation. OM fractions were then solubilized in 20 mM Tris-HCl (pH 8), 0.3 M NaCl, 1% dodecyl-maltopyranoside (DDM; Anatrace). Imidazole was added to 20 mM and the samples were loaded onto a cobalt affinity column using an FPLC apparatus (GE Healthcare). The bound FimD protein was eluted using an imidazole step gradient in buffer 20 mM Tris-HCl (pH 8), 0.3 M NaCl, 10 mM lauryl(dimethyl)amine oxide (LDAO; Anatrace). The His-tag was cleaved by digestion with thrombin overnight, and then the sample was passed again over a cobalt affinity column coupled to a benzamidine colum (GE Healthcare). The pure, His-tag-free FimD was collected in the flow-through fraction. The purified usher was dialyzed into 20 mM HEPES (pH 7.5), 150 mM NaCl, 5 mM LDAO, and concentrated using a Millipore Ultrafree centrifugal concentrator (50 kDa molecular weight cutoff). Protein concentrations were determined using the bicinchoninic acid (BCA) protein assay (Pierce).
FimC-FimH complexes were purified from strain Tuner/pHJ20 harboring plasmid pGW1 (FimCT51C), pGW2 (FimCN86C), or pGW3 (FimCQ19C). Plasmid pHJ20 encodes IPTG-inducible FimH, and plasmids pGW1–3 encode arabinose-inducible, His-tagged FimC with the indicated cysteine substitutions. Purification was performed as described51. Briefly, 2 l cultures were grown at 37 °C and induced at OD600 = 0.6 with 0.002% arabinose and 1 mM IPTG for 2 h. Periplasm fractions were isolated by EDTA-lysozyme treatment and dialyzed into 20 mM Tris-HCl (8.0), 0.3 M NaCl. Imidazole was added to 20 mM and samples were loaded onto a nickel affinity column using an FPLC apparatus. Bound FimC-FimH complex was eluted using an imidazole step gradient. Fractions containing FimC-FimH were pooled and dialyzed into 20 mM MES (pH 5.4). The samples were then run on a Resource S column (GE Healthcare) and eluted using a linear NaCl gradient, to separate excess unbound FimC chaperone from FimCFimH chaperone-adhesin complex.
Fluorescence-based affinity assay
Fluorescence labeling reactions and titration experiments were performed as described17,35. FimCQ19C-, FimCT51C-, or FimCN86C-FimH complexes (500 nM) were labeled with the thiol-reactive probe coumarin maleimide (Life Technologies) for 2 h at 4 °C at a 5:1 probe:protein molar ratio. For labeling, the pH of the protein solution was first raised to 8.0 via the addition of K2HPO4. Unbound probe was removed via dialysis against 20 mM HEPES (pH 7.5), 150 mM NaCl, and 5 mM LDAO was added to the final exchanged solution. Labeling efficiency was calculated using Beer's Law, and was typically > 80%.
Fluorescence measurements were performed using a PC1 photon-counting spectrofluorometer (ISS), as described17. Coumarin-labeled chaperone-subunit complexes were diluted to 25 nM and 120 μl was transferred to a 3 mm microcuvette. Purified FimD was then titrated into the FimC-FimH solution. The fluorophore was excited at 384 nM, and its emission spectrum was measured from 420-520 nm with a step size of 2 nm. Variability in lamp intensity was accounted for using Vinci (ISS) data acquisition software. The integral of the curve was calculated, providing the total emission intensity. Buffer measurements were also performed, and background emissions were subtracted. Data were normalized to account for dilution during titration, and set to a scale of 0 (starting value) to 1 (the final value). Apparent equilibrium bimolecular dissociation constants (Kd) were obtained by fitting the data using a sigmoidal curve function in Prism (GraphPad) and solving for the inflection point. Each titration curve shown is the result of at least three independent experiments with three replicates per experiment. All of our observed Kd's were independent of starting FimC-FimH concentrations (below the dissociation constant), and thus were dependent only on the mass action of the titrant, FimD. Statistical comparison of Kd's for FimD mutants with WT FimD was performed using a two-tailed t test in Prism (GraphPad). Comparison of the different FimC cysteine substitution mutants was performed using one-way analysis of variance and Tukey's multiple-comparison post test. P values < 0.05 were considered significant.
Analysis of usher expression and folding in the OM
The expression levels and folding of the FimD mutants in the OM were compared to WT FimD, as described39. Briefly, OM fractions were isolated by French press disruption and Sarkosyl extraction, and proper folding of the ushers was determined by heat-modifiable mobility on SDS-PAGE. Strain SF100 was used as the host strain for these studies.
Hemagglutination (HA) assay
HA assays were performed by serial dilution in microtiter plates, as described39. HA titers were determined visually and are reported as the greatest fold dilution of bacteria able to agglutinate guinea pig red blood cells (Colorado Serum Company). For each HA assay, at least three independent experiments were performed, with three replicates per experiment. Analysis of the FimC WT and cysteine substitution mutants was performed in strain MM294ΔfimC, which contains a fimC deletion in the chromosomal fim operon. Analysis of the FimD WT and mutant ushers was performed in strain MM294ΔfimD, which contains a fimD deletion in the chromosomal fim operon. The experiments in which FimD WT or domain deletion mutant ushers were co-expressed were also done in strain MM294ΔfimD. Bacteria harboring appropriate FimD or FimC plasmids were grown statically for 24–48 h to induce the chromosomal fim genes, and then FimD or FimC expression was induced with 50 μM IPTG or 0.15% arabinose, respectively, for an additional 3 h with shaking at 100 rpm. To test the role of OmpA in type 1 pilus biogenesis, HA titers were determined for strain JF568 and its isogenic ompA– derivative JF699. The strains were grown statically for 24–48 h to induce the chromosomal fim genes. For analysis of assembly of FimG-FimH type 1 pilus tips, strain AAEC185/pNH222 (encoding FimC, FimG, and FimH) was transformed with the FimD expression plasmids pNH213 (WT FimD) or pNH324 (FimDΔplug). HA titers were determined from strains grown with aeration and induced at OD600 = 0.6 with 50 μM IPTG and 0.1% arabinose for 1 h.
Electron microscopy (EM)
Whole bacteria, negative-stain transmission EM was performed as described24. Aliquots (1 ml) of cultures grown for the HA assay were washed with PBS and resuspended in 1.5 ml PBS. Bacteria were fixed with 1% gluteraldehyde in PBS, washed with PBS followed by water, and then stained for 20 s with phosphotungstic acid. Grids were examined on a TECNAI 12 BioTwin G02 microscope (FEI) and representative images were acquired with an XR-60 CCD digital camera system (Advanced Microscopy Techniques).
Co-purification of type 1 pilus assembly intermediates with WT and Δplug FimD ushers
Co-purification assays were performed as previously described39. Briefly, OM fractions were isolated by French press disruption and Sarkosyl extraction from strain AAEC185/pNH222 harboring plasmids pNH213 or pNH324, grown as described for the HA assay. OM fractions were solubilized with the non-denaturing detergent DDM, and the His-tagged FimD was purified by cobalt affinity chromatography. FimD-containing fractions from the column were incubated for 10 min at 25 or 95°C in SDS-PAGE sample buffer, separated by SDS-PAGE, and immunoblotted with anti-FimC-FimH20 or anti-FimC-FimG20 antibodies to detect pilus assembly intermediates that co-purified with the usher. Immunoblots were developed with alkaline phosphatase-conjugated secondary anti-rabbit (Sigma catalog number A3812) antibody and BCIP-NBT substrate (KPL).
Original images of gels and blots used in this study can be found in Supplementary Data Set 2.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the Schultz laboratory (Scripps Research Institute) for providing plasmid pEVOL-pBpF. We thank S. Van Horn of the Stony Brook University Central Microscopy Imaging Center and V. Sampath (Stony Brook University) for assistance with electron microscopy. We thank J. Haley, D. Martin and R. Rieger of the Stony Brook Proteomics Center for performing the mass spectrometry analysis and for helpful discussions. We thank S. Scarlata (Stony Brook University), A. W. Karzai (Stony Brook University), K. W. Dodson (Washington University), and A. H. Delcour (University of Houston) for helpful discussions and critical reading of the manuscript.
This study was supported by US National Institutes of Health (NIH) grants R01GM062987 (to D.G.T. and H.L.) and R01AI029549 (to S.J.H.). G.T.W. was supported by Medical Scientist Training Program award T32GM008444 and National Research Service Award F30AI112252 from the NIH. The Stony Brook Proteomics Center receives support from NIH award S10RR023680.
Footnotes
AUTHOR CONTRIBUTIONS
G.T.W., N.S.H., S.J.H., H.L. and D.G.T. designed the experiments. G.T.W., N.S.H., E.B.P and S.S. performed the experiments. All authors were involved data interpretation and discussion. G.T.W. and D.G.T wrote the manuscript with contributions from all other authors.
REFERENCES
- 1.Nuccio SP, Baumler AJ. Evolution of the chaperone/usher assembly pathway: fimbrial classification goes Greek. Microbiol. Mol. Biol. Rev. 2007;71:551–575. doi: 10.1128/MMBR.00014-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zav'yalov V, Zavialov A, Zav'yalova G, Korpela T. Adhesive organelles of Gram-negative pathogens assembled with the classical chaperone/usher machinery: structure and function from a clinical standpoint. FEMS Microbiol. Rev. 2010;34:317–378. doi: 10.1111/j.1574-6976.2009.00201.x. [DOI] [PubMed] [Google Scholar]
- 3.Geibel S, Waksman G. The molecular dissection of the chaperone-usher pathway. Biochim. Biophys. Acta. 2014;1843:1559–1567. doi: 10.1016/j.bbamcr.2013.09.023. [DOI] [PubMed] [Google Scholar]
- 4.Thanassi DG, Bliska JB, Christie PJ. Surface organelles assembled by secretion systems of Gram-negative bacteria: diversity in structure and function. FEMS Microbiol. Rev. 2012;36:1046–1082. doi: 10.1111/j.1574-6976.2012.00342.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mulvey MA, et al. Induction and evasion of host defenses by type 1-piliated uropathogenic Escherichia coli. Science. 1998;282:1494–1497. doi: 10.1126/science.282.5393.1494. [DOI] [PubMed] [Google Scholar]
- 6.Roberts JA, et al. The Gal(alpha1-4)Gal-specific tip adhesin of Escherichia coli P-fimbriae is needed for pyelonephritis to occur in the normal urinary tract. Proc. Natl. Acad. Sci. USA. 1994;91:11889–11893. doi: 10.1073/pnas.91.25.11889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones CH, et al. FimH Adhesin of Type-1 Pili Is Assembled into a Fibrillar Tip Structure in the Enterobacteriaceae. Proc. Natl. Acad. Sci. U S A. 1995;92:2081–2085. doi: 10.1073/pnas.92.6.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hahn E, et al. Exploring the 3D molecular architecture of Escherichia coli type 1 pili. J. Mol. Biol. 2002;323:845–857. doi: 10.1016/s0022-2836(02)01005-7. [DOI] [PubMed] [Google Scholar]
- 9.Lycklama ANJA, Driessen AJ. The bacterial Sec-translocase: structure and mechanism. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2012;367:1016–1028. doi: 10.1098/rstb.2011.0201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choudhury D, et al. X-ray structure of the FimC-FimH chaperone-adhesin complex from uropathogenic Escherichia coli. Science. 1999;285:1061–1066. doi: 10.1126/science.285.5430.1061. [DOI] [PubMed] [Google Scholar]
- 11.Sauer FG, et al. Structural basis of chaperone function and pilus biogenesis. Science. 1999;285:1058–1061. doi: 10.1126/science.285.5430.1058. [DOI] [PubMed] [Google Scholar]
- 12.Zavialov AV, et al. Structure and biogenesis of the capsular F1 antigen from Yersinia pestis: preserved folding energy drives fiber formation. Cell. 2003;113:587–596. doi: 10.1016/s0092-8674(03)00351-9. [DOI] [PubMed] [Google Scholar]
- 13.Nishiyama M, Ishikawa T, Rechsteiner H, Glockshuber R. Reconstitution of pilus assembly reveals a bacterial outer membrane catalyst. Science. 2008;320:376–379. doi: 10.1126/science.1154994. [DOI] [PubMed] [Google Scholar]
- 14.Sauer FG, Pinkner JS, Waksman G, Hultgren SJ. Chaperone priming of pilus subunits facilitates a topological transition that drives fiber formation. Cell. 2002;111:543–551. doi: 10.1016/s0092-8674(02)01050-4. [DOI] [PubMed] [Google Scholar]
- 15.Rose RJ, et al. Unraveling the molecular basis of subunit specificity in P pilus assembly by mass spectrometry. Proc. Natl. Acad. Sci. U S A. 2008;105:12873–12878. doi: 10.1073/pnas.0802177105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nishiyama M, Glockshuber R. The outer membrane usher guarantees the formation of functional pili by selectively catalyzing donor-strand exchange between subunits that are adjacent in the mature pilus. J. Mol. Biol. 2010;396:1–8. doi: 10.1016/j.jmb.2009.12.005. [DOI] [PubMed] [Google Scholar]
- 17.Li Q, et al. The differential affinity of the usher for chaperone-subunit complexes is required for assembly of complete pili. Mol. Microbiol. 2010;76:159–172. doi: 10.1111/j.1365-2958.2010.07089.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saulino ET, Thanassi DG, Pinkner JS, Hultgren SJ. Ramifications of kinetic partitioning on usher-mediated pilus biogenesis. EMBO J. 1998;17:2177–2185. doi: 10.1093/emboj/17.8.2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Volkan E, et al. Domain activities of PapC usher reveal the mechanism of action of an Escherichia coli molecular machine. Proc. Natl. Acad. Sci. U S A. 2012;109:9563–9568. doi: 10.1073/pnas.1207085109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Remaut H, et al. Fiber formation across the bacterial outer membrane by the chaperone/usher pathway. Cell. 2008;133:640–652. doi: 10.1016/j.cell.2008.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nishiyama M, et al. Structural basis of chaperone-subunit complex recognition by the type 1 pilus assembly platform FimD. EMBO J. 2005;24:2075–2086. doi: 10.1038/sj.emboj.7600693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang Y, Smith BS, Chen LX, Baxter RH, Deisenhofer J. Insights into pilus assembly and secretion from the structure and functional characterization of usher PapC. Proc. Natl. Acad. Sci. U S A. 2009;106:7403–7407. doi: 10.1073/pnas.0902789106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Phan G, et al. Crystal structure of the FimD usher bound to its cognate FimC-FimH substrate. Nature. 2011;474:49–53. doi: 10.1038/nature10109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ng TW, Akman L, Osisami M, Thanassi DG. The usher N terminus is the initial targeting site for chaperone-subunit complexes and participates in subsequent pilus biogenesis events. J. Bacteriol. 2004;186:5321–5331. doi: 10.1128/JB.186.16.5321-5331.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dubnovitsky AP, et al. Conserved hydrophobic clusters on the surface of the Caf1A usher C-terminal domain are important for F1 antigen assembly. J. Mol. Biol. 2010;403:243–259. doi: 10.1016/j.jmb.2010.08.034. [DOI] [PubMed] [Google Scholar]
- 26.Eidam O, Dworkowski FS, Glockshuber R, Grutter MG, Capitani G. Crystal structure of the ternary FimC-FimF(t)-FimD(N) complex indicates conserved pilus chaperone-subunit complex recognition by the usher FimD. FEBS Lett. 2008;582:651–655. doi: 10.1016/j.febslet.2008.01.030. [DOI] [PubMed] [Google Scholar]
- 27.Geibel S, Procko E, Hultgren SJ, Baker D, Waksman G. Structural and energetic basis of folded-protein transport by the FimD usher. Nature. 2013;496:243–246. doi: 10.1038/nature12007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shu Kin So S, Thanassi DG. Analysis of the requirements for pilus biogenesis at the outer membrane usher and the function of the usher C-terminus. Mol. Microbiol. 2006;60:364–375. doi: 10.1111/j.1365-2958.2006.05111.x. [DOI] [PubMed] [Google Scholar]
- 29.Munera D, Hultgren S, Fernandez LA. Recognition of the N-terminal lectin domain of FimH adhesin by the usher FimD is required for type 1 pilus biogenesis. Mol. Microbiol. 2007;64:333–346. doi: 10.1111/j.1365-2958.2007.05657.x. [DOI] [PubMed] [Google Scholar]
- 30.Thanassi DG, et al. The PapC usher forms an oligomeric channel: implications for pilus biogenesis across the outer membrane. Proc. Natl. Acad. Sci. U S A. 1998;95:3146–3151. doi: 10.1073/pnas.95.6.3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li H, et al. The outer membrane usher forms a twin-pore secretion complex. J. Mol. Biol. 2004;344:1397–1407. doi: 10.1016/j.jmb.2004.10.008. [DOI] [PubMed] [Google Scholar]
- 32.Allen WJ, Phan G, Hultgren SJ, Waksman G. Dissection of pilus tip assembly by the FimD usher monomer. J. Mol. Biol. 2013;425:958–967. doi: 10.1016/j.jmb.2012.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Young TS, Ahmad I, Yin JA, Schultz PG. An enhanced system for unnatural amino acid mutagenesis in E. coli. J. Mol. Biol. 2010;395:361–374. doi: 10.1016/j.jmb.2009.10.030. [DOI] [PubMed] [Google Scholar]
- 34.Chin JW, Martin AB, King DS, Wang L, Schultz PG. Addition of a photocrosslinking amino acid to the genetic code of Escherichia coli. Proc. Natl. Acad. Sci. U S A. 2002;99:11020–11024. doi: 10.1073/pnas.172226299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Royer CA, Scarlata SF. Fluorescence approaches to quantifying biomolecular interactions. Methods Enzymol. 2008;450:79–106. doi: 10.1016/S0076-6879(08)03405-8. [DOI] [PubMed] [Google Scholar]
- 36.Morrissey B, et al. The role of chaperone-subunit usher domain interactions in the mechanism of bacterial pilus biogenesis revealed by ESI-MS. Mol. Cell. Proteomics. 2012;11:M111.015289. doi: 10.1074/mcp.M111.015289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mapingire OS, Henderson NS, Duret G, Thanassi DG, Delcour AH. Modulating effects of the plug, helix and N- and C-terminal domains on channel properties of the PapC usher. J. Biol. Chem. 2009;284:36324–36333. doi: 10.1074/jbc.M109.055798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu X, et al. Caf1A usher possesses a Caf1 subunit-like domain that is crucial for Caf1 fibre secretion. Biochem. J. 2009;418:541–551. doi: 10.1042/BJ20080992. [DOI] [PubMed] [Google Scholar]
- 39.Henderson NS, Ng TW, Talukder I, Thanassi DG. Function of the usher N-terminus in catalysing pilus assembly. Mol. Microbiol. 2011;79:954–967. doi: 10.1111/j.1365-2958.2010.07505.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thanassi DG, Stathopoulos C, Karkal A, Li H. Protein secretion in the absence of ATP: the autotransporter, two-partner secretion, and chaperone/usher pathways of Gram-negative bacteria. Molec. Membr. Biol. 2005;22:63–72. doi: 10.1080/09687860500063290. [DOI] [PubMed] [Google Scholar]
- 41.Deville K, et al. The oligomeric state and arrangement of the active bacterial translocon. J. Biol. Chem. 2011;286:4659–4669. doi: 10.1074/jbc.M110.175638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rehling P, et al. Protein insertion into the mitochondrial inner membrane by a twin-pore translocase. Science. 2003;299:1747–1751. doi: 10.1126/science.1080945. [DOI] [PubMed] [Google Scholar]
- 43.Ahting U, et al. Tom40, the pore-forming component of the protein-conducting TOM channel in the outer membrane of mitochondria. J. Cell Biol. 2001;153:1151–1160. doi: 10.1083/jcb.153.6.1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reichow SL, et al. Allosteric mechanism of water-channel gating by Ca2+-calmodulin. Nat. Struct. Mol. Biol. 2013;20:1085–1092. doi: 10.1038/nsmb.2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dalal K, Chan CS, Sligar SG, Duong F. Two copies of the SecY channel and acidic lipids are necessary to activate the SecA translocation ATPase. Proc. Natl. Acad. Sci. U S A. 2012;109:4104–4109. doi: 10.1073/pnas.1117783109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mao C, et al. Stoichiometry of SecYEG in the active translocase of Escherichia coli varies with precursor species. Proc. Natl. Acad. Sci. U S A. 2013;110:11815–11820. doi: 10.1073/pnas.1303289110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chiu J, March PE, Lee R, Tillett D. Site-directed, Ligase-Independent Mutagenesis (SLIM): a single-tube methodology approaching 100% efficiency in 4 h. Nucleic Acids Res. 2004;32:e174. doi: 10.1093/nar/gnh172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chiu J, Tillett D, Dawes IW, March PE. Site-directed, Ligase-Independent Mutagenesis (SLIM) for highly efficient mutagenesis of plasmids greater than 8kb. J. Microbiol. Methods. 2008;73:195–198. doi: 10.1016/j.mimet.2008.02.013. [DOI] [PubMed] [Google Scholar]
- 49.Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 1996;68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 50.Tanner S, et al. InsPecT: identification of posttranslationally modified peptides from tandem mass spectra. Anal. Chem. 2005;77:4626–4639. doi: 10.1021/ac050102d. [DOI] [PubMed] [Google Scholar]
- 51.Henderson NS, Thanassi DG. Purification of the outer membrane usher protein and periplasmic chaperone-subunit complexes from the P and type 1 pilus systems. Methods Mol. Biol. 2013;966:37–52. doi: 10.1007/978-1-62703-245-2_3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.