

Summary
Hemophilia B is an X‐chromosome‐linked inherited bleeding disorder primarily affecting males, but those carrier females with reduced factor IX activity (FIX:C) levels may also experience some bleeding. Genetic analysis has been undertaken for hemophilia B since the mid‐1980s, through linkage analysis to track inheritance of an affected allele, and to enable determination of the familial mutation. Mutation analysis using PCR and Sanger sequencing along with dosage analysis for detection of large deletions/duplications enables mutation detection in > 97% of patients with hemophilia B. The risk of the development of inhibitory antibodies, which are reported in ~ 2% of patients with hemophilia B, can be predicted, especially in patients with large deletions, and these individuals are also at risk of anaphylaxis, and nephrotic syndrome if they receive immune tolerance induction. Inhibitors also occur in patients with nonsense mutations, occasionally in patients with small insertions/deletions or splice mutations, and rarely in patients with missense mutations (p.Gln237Lys and p.Gln241His). Hemophilia B results from several different mechanisms, and those associated with hemophilia B Leyden, ribosome readthrough of nonsense mutations and apparently ‘silent’ changes that do not alter amino acid coding are explored. Large databases of genetic variants in healthy individuals and patients with a range of disorders, including hemophilia B, are yielding useful information on sequence variant frequency to help establish possible variant pathogenicity, and a growing range of algorithms are available to help predict pathogenicity for previously unreported variants.
Keywords: DNA Mutational Analysis, factor IX, genetic carrier detection, hemophilia B, prenatal diagnosis
Introduction
Hemophilia B is a coagulation factor deficiency resulting from reduced levels or an absence of factor IX. Symptoms of recurrent prolonged bleeding result from reduced levels or an absence of plasma FIX, whose function is to cleave and activate FX within the coagulation cascade. FIX is a vitamin K‐dependent plasma protease that participates in the intrinsic blood coagulation pathway. FIX circulates as a zymogen, and is activated to activated FIX (FIXa) by sequential cleavage at p.Arg191‐Ala192 and p.Arg226‐Val227 by activated FXI or tissue factor–activated FVII. With activated FVIII as a cofactor providing correct orientation, FIXa cleaves FX at p.Arg182–Ser183 and p.Arg234–Ile235, resulting in its activation.
The disorder affects approximately one in 30 000 males worldwide. It was discriminated from hemophilia A around 1950, and named Christmas disease by Biggs et al. 1; Christmas was the family name of their first affected individual examined in detail. Patients are classified as having severe (< 1 IU dL−1), moderate (1–5 IU dL−1) or mild (5–40 IU dL−1) hemophilia B, according to residual FIX activity (FIX:C). The US Universal Data Collection (UDC) database and the UK Haemophilia Centre Doctors’ Organization (UKHCDO) annual report for 2013–2014 describe 3785 (UDC) and 1205 (UKHCDO) patients as having mild (26–45%), moderate (21–38%) and severe (34–36%) hemophilia B 2, 3.
The gene encoding FIX (F9) was cloned in 1982–1983, and its full sequence was published in 1985 4. Molecular genetic analysis using linkage analysis to follow the affected allele in hemophilia B families and of mutations responsible for the disorder to facilitate carrier status determination and prenatal diagnosis (PND) became available from this time. F9 comprises eight exons, with a 2780‐bp transcript encoding a 461‐residue pre‐pro‐protein, which is subsequently cleaved to the 421‐residue mature FIX serine protease.
Although the prevalence of hemophilia B is generally stated as being one in 30 000 males, reported rates in different countries vary widely, and probably partly depend on the level of healthcare provision. In a survey using World Federation for Hemophilia data, the prevalence of hemophilia B per 100 000 males in 103 countries ranged from 0.01 (Nigeria) to 8.07 (Ireland) 5.
The recently revised F9 locus‐specific mutation database (F9db) lists > 3000 pathogenic mutations and neutral polymorphisms, with mutations affecting 73% of the 461 residues 6, 7. The addition of FIX molecular models for missense changes adds useful information about specific substitutions.
The aim of this review is to summarize the current understanding of hemophilia B symptoms in males and carrier females, and its mutation detection, molecular pathology and pathogenicity assessment for diagnostic and prognostic use.
Nomenclature
Gene names and nucleotide and protein changes all follow standard formats. Gene names are defined by the Human Genome Organization (HUGO) Gene Nomenclature Committee (HGNC) 8, according to which the gene encoding FIX is designated F9; nucleotide and protein variants are described according to Human Genome Variation Society (HGVS) nomenclature 9. Nucleotides and proteins are numbered from the protein initiator methionine; c. denotes cDNA numbering, and p. denotes protein numbering. The HGVS website provides examples highlighting the reporting of different mutation types. The F9 mutation database (F9db) uses HGVS nomenclature plus legacy numbering to facilitate reference to historically reported mutations 6, 7. Legacy protein numbering was from the start of mature FIX (HGVS codon 47), and is presented in this article in parentheses. Stable reference sequences known as locus reference genomic (LRG), which should not change over time, are available for F9; transcript ID LRG_556t1, and protein ID LRG_556p1 10.
F9db
The first version of the F9db was published in 1990 by Giannelli et al. 11. It described 216 mutations spanning from c.‐49T>A (legacy –20) to c.1369A>T (legacy 31 352). Following several annual publications, an internet database was initiated in 1997, but it was last updated in 2004 12, and was recently replaced by the current database 6, 7. Mutation data were extracted from the F9db with query tools, or were kindly provided by P.M. Rallapalli (personal communication, 25 November 2014) for 3656 or 3713 patients.
Hemophilia phenotype
Phenotype in affected males
Hemophilia B symptoms include prolonged bleeding and easy bruising. Bleeding into joints and sometimes muscles can occur, accompanied by pain and swelling. Bleeding can be from the nose, can result from cuts, tooth extraction, surgery, or trauma, may continue for a prolonged period, and can restart after stopping. Bleeding can affect the gastrointestinal and urinary tracts. Spontaneous bleeding generally only affects severely/moderately affected patients, who will often experience their first bleed during their first 2 years, with the first bleed sometimes occurring after circumcision. In contrast, mildly affected males may not be diagnosed until later in life.
Hemophilia B severity
There has been some speculation that the symptoms of hemophilia B may be less severe than those of hemophilia A 13. A recent study of children with severe and moderate hemophilia A (n = 679) and hemophilia B (n = 123) showed that there was no significant difference in bleeding between patients with the two disease types during their first 75 exposure days 14. The factors compared included age at first bleed, age at diagnosis, and age at first exposure to factor concentrate. A study of older individuals examining the rate of joint arthroplasty 15 found that the risk of arthroplasty was higher among patients with severe hemophilia A than among those with severe hemophilia B, with a hazard ratio of 2.65, and a 95% confidence interval of 1.62–4.33 (P < 0.001). Results were adjusted for age, hepatitis C virus, human immunodeficiency virus and inhibitor status in a Cox regression model. Schulman 16 used a hemophilia severity score combining annual joint bleed incidence, orthopedic joint score and concentrate consumption, and collected data between 1990 and 1999 on 100 adult and adolescent patients with hemophilia of all severities without an inhibitor history to document bleeding. Scores from 0 to 0.94 were higher in patients with severe hemophilia A than in those with severe hemophilia B (medians of 0.50 and 0.24; P = 0.031). The higher prevalence of missense mutations in hemophilia B provides biological evidence for it having lower severity than hemophilia A, although clinical evidence is limited.
Phenotype in female carriers
Plug et al. reported postal survey data on females from hemophilia families in The Netherlands who were carriers or non‐carriers of hemophilia A or hemophilia B 17. The median FIX:C was 60 IU dL−1 (range, 5–219 IU dL−1) in carriers, and 102 IU dL−1 (range, 45–328 IU dL−1) in non‐carriers. For those with FIX:C levels of > 60 IU dL−1, 41–60 IU dL−1, and < 40 IU dL−1, a significant trend in number of events when bleeding occurred was observed for bleeding from small wounds and following surgery or tooth extraction.
The relative bleeding risk demonstrated that bleeding following dental extraction or tonsillectomy/adenoidectomy was elevated in carriers as compared with non‐carriers, with relative risks of 23.1 (3.1–169) for dental extraction and 9.9 (1.3–76.3) following tonsillectomy/adenoidectomy.
Sharathkumar et al. reported on phenotypic variability in hemophilia B carriers from a large Amish pedigree with a p.Thr342Met mutation (male levels in the F9db of < 1–16 IU dL−1). Among up to 64 females, menorrhagia (47%), bruising (31%) and epistaxis (17%) were the most common symptoms. FIX:C levels ranged between 13 IU dL−1 and 112 IU dL−1. Those with lower FIX:C levels (mean, ~ 35–45 IU dL−1) had significantly more subcutaneous hematoma, bleeding following dental extraction, postpartum hemorrhage and surgical bleeding than those with higher levels (mean, ~ 53–64 IU dL−1) 18.
Mutation detection
Several organizations recommend that mutation analysis should be performed for all patients with hemophilia, highlighting any excess risk of inhibitor development associated with specific mutations, and enabling carrier testing for female relatives and prenatal or possibly preimplantation genetic diagnosis. The first hemophilia B mutation was reported in 1985 19. Mutation analysis generally uses PCR DNA amplification followed by Sanger sequencing of the eight exons, the 5′‐untranslated region, the 3′‐untranslated region and splice boundaries of F9. In some laboratories, mutation scanning is used to highlight the amplicon containing a candidate mutation prior to its sequence analysis 20. Next‐generation DNA sequencing (NGS) is also being used to determine the F9 sequence 21. Variants comprise point mutations, deletions, insertions, duplications, insertions and deletions, complex changes, and neutral polymorphisms; the proportions currently in the F9db 7 are shown in Fig. 1. The largest proportion of patients with hemophilia B have missense mutations (65%).
Figure 1.
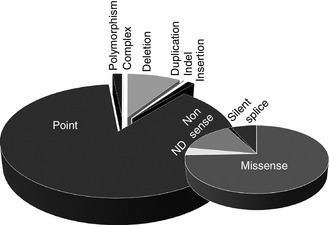
Factor IX mutation type distribution in patients with hemophilia B on the F9 mutation database. Three thousand six hundred and fifty‐six mutations (P.M. Rallapalli, personal communication) are broken down by type in the larger pie chart. Point mutations are broken down by type in the smaller pie chart. ND, mutation type not determined.
Large deletions of an exon or more constitute ~ 2% of all unique mutations. They are readily detected in affected males through lack of amplification of one or more exons. Large duplication of an exon or more has only been reported in one hemophilia B case 22, and, although such mutations are likely to be rare, they may be under‐reported, as a technique that is sensitive to the number of F9 copies (dosage analysis), such as multiplex ligation‐dependent probe amplification, is required for their identification, but is not in common use. Dosage analysis is required to determine female carrier status for large deletion mutations, and in males and females to identify large duplications 23, 24, 25 (Fig. 2).
Figure 2.
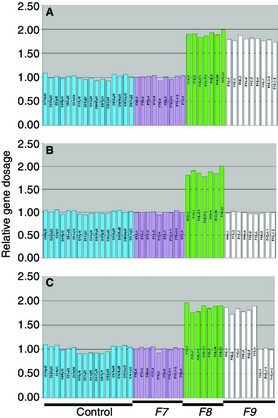
F9 gene dosage analysis using multiplex ligation‐dependent probe amplification (MLPA). MLPA probes include those for normalization across the genome (control) and for F7, F8, and F9. (A) Dosage in a female lacking a large deletion. (B) Dosage in a female with complete F9 deletion. (C) Dosage in a female with a partial F9 deletion of exons 7–8.
Complex mutations include large deletion and insertion mutations, e.g. a deletion of 4.4 kb from exons 4–6, and its replacement by a 47‐bp sequence 26, and interruption of the coding sequence by insertion of the mobile genetic element Alu into exon 5 in two different patients 27, 28, and by a long interspersed nuclear element insertion into exon 7 29.
Splice site mutations constitute 212 of 3196 (6.6%) database entries (P.M. Rallapalli, personal communication). Most are within ± 7 nucleotides of the splice site, and much smaller numbers are within ± 25 bp. However, a handful of F9db mutations lie deeper in introns, with c.392‐569C>A in intron 4 having been reported in severe hemophilia B (unpublished); however, these variants have not been validated as causing the disorder. Deep intronic mutations are rarely reported in hemophilia B 30, but are not routinely sought.
Chaivali et al. described hemophilia B as being ‘a quasi‐quantitative condition with certain mutations showing phenotypic plasticity’ 31. By the use of unsupervised learning computational analysis of 201 missense mutations in the F9db with two or more reports, mutations were divided into three clusters. Mutations in clusters 1 and 2 showed FIX:C level variation, whereas cluster 3 mutations had a uniform phenotype. Mutations associated with variable phenotypes were located at residues that were less conserved across species, with fully conserved residues being present in 27% of cluster 1 and 30% of cluster 2, but in 61% of cluster 3.
Analysis of F9 mRNA
F9 cDNA reverse‐transcribed from mRNA could provide a good source of material for understanding the effects of splicing defects, and also for the small group of patients with hemophilia B in whom mutations have not been identified by F9 genomic DNA examination. In contrast to hemophilia A, few studies have been published on F9 cDNA analysis. It appears challenging in some instances to obtain full‐length F9 cDNA from peripheral leukocytes. Sarkar et al. analyzed F9 mRNA by using single‐strand conformation polymorphism, but did not highlight difficulties in obtaining mRNA 32, and van der Water et al. 33 sequenced leukocyte exon 2–4 transcript in wild‐type and mutant to identify an intron 2 splice mutation. Green et al. could only obtain a partial transcript of exons 7–8 from leukocytes, in contrast to liver‐derived F9 mRNA, where the entire transcript was present 34; similarly, Cutler et al. 35 reported leukocyte transcripts from exons 1–2 and 7–8, but not from central F9 exons. Cao et al. recently reported using mRNA analysis to help identify an intron 3 (c.278‐3A>G) splice defect, and obtained the sequence of the additional intron 3 nucleotides incorporated into the transcript by the mutation 36. It is possible that transcripts expressed ectopically in cells not from the main site of FIX synthesis may produce mRNA lacking certain exons. The genome browser Ensembl 10 lists three F9 transcripts with between four and eight exons. It may be worth revisiting the analysis of F9 leukocyte mRNA for patients in whom mutations have not yet been detected, or to apply NGS to the entire F9 locus, as undertaken for F8 37, 38, 39.
Mutation detection rate
In 21 studies undertaken from 1993 to 2014 with 20 or more unrelated patients with hemophilia B, F9 mutation detection rates varied from 83% to 100% in genetic analysis using PCR and Sanger sequencing with occasional dosage analysis. The mean mutation detection rate in the 1811 individuals in these studies was 97.4% (Table S1). Additionally, the UKHCDO Genetic Testing Network collected mutation detection rates of 724 apparently unrelated patients for analysis undertaken to 2010 20. Mutations were identified in 704 cases (97.2%), but sensitivity varied by FIX:C level. Mutations were detected in 99.5% of severe cases and in 98.2% of patients with FIX:C of < 40 IU dL−1; individuals with higher levels included most of those with no mutation identified.
De novo F9 mutations
Eleven of the above studies examined whether there were one or more reported hemophilic patients among the families analyzed, to estimate sporadic case frequency. A study on sporadic mutation prevalence was included 40. Of 773 patients, 380 were classed as having familial hemophilia B (49%; range, 31–67%), the lowest prevalence being in Ireland 41 and the highest in the USA 42 and Iran 43.
Causes of reduced FIX levels in symptomatic females
Approximately 10–15% of hemophilia carriers experience bleeding symptoms. Skewed X‐chromosome inactivation (Lyonization) is implicated in some cases, but chromosome translocation, compound heterozygosity for two different mutations and Turner syndrome (XO) can also result in bleeding. Orstavik et al. examined 15 hemophilia B carriers; there was no tendency for there to be skewed X‐chromosome inactivation in those with high or low FIX:C levels 44. Di Michele et al. reported on bleeding symptoms and phenotype in US hemophilic females, identified through the UDC database. Of six affected females with hemophilia B (one severe; five moderate), each with a missense mutation, three had skewed X‐chromosome inactivation, and one had a de novo X‐chromosome deletion; the reason for hemophilia B was not characterized in two individuals 45.
Mosaicism
Somatic and gonadal mosaicism for hemophilia B (the existence of two or more cell lines in an individual with different genotypes) has been reported occasionally. Sommer et al. reported on 414 hemophilia B families among whom hemophilia B origin was identified in 49 families by sequencing and haplotype analysis. No ‘mutation origin’ individuals passed on the affected allele to more than one child, but, in four individuals, the mutant sequence was visible on sequence traces (sensitivity down to ~ 10%), 11% demonstrating somatic mosaicism, suggesting that this is not uncommon 46. Additionally, the maternal grandmother of a patient had an approximately 50 : 50 ratio of mutant/wild‐type sequence in her leukocyte DNA, and transmitted the same allele to five daughters, only one of whom carried the F9 mutation 47. Green et al. 48 sought evidence of gonadal mosaicism in 47 families, but detected no ovarian mosaicism, suggesting that the risk of a ‘non‐carrier’ mother transmitting the mutation to a further child was < 0.062. The origin of mosaic conditions in hemophilia has been reviewed by Kasper et al. 42, and that of mosaicism in general by Biesecker et al. 49.
Prenatal diagnosis
PND became available for hemophilia B in the mid‐1980s, when single‐nucleotide variants (SNVs) in F9 were identified, and became more useful as further genetic markers throughout F9 were found 50, 51, 52. From the early 1990s, DNA sequencing was increasingly used to determine the familial mutation, and the majority of carrier status determination and PND is now undertaken with this analysis. However, a mix of linkage and sequence analysis can be used to provide a cost‐effective approach to carrier analysis and PND 53. Early PND at 11–14 weeks of gestation, amniocentesis at 16–18 weeks, and late gestation analysis at ≥ 32 weeks, also using amniocentesis, are all available, the last to inform delivery management for known carriers 54. Examination of free fetal DNA in the maternal circulation is being used increasingly for early fetal sex determination, and can be utilized from ~ 10 weeks of gestation to avoid invasive PND analysis 55.
The Italian hemophilia B database records mutations in 373 unrelated patients, and this information has identified 274 carriers of childbearing age. Of 66 PNDs undertaken for 52 hemophilia B carriers (mean age, 34 years; range, 22–44 years), 44 (85%) had received genetic counseling prior to pregnancy, and eight presented once they were pregnant (15%). Chorionic villus samples were obtained in 52 cases, amniocentesis in 12 cases, and free fetal DNA in two cases. Most had a single PND, but nine had two, and one each had three or four. Hemophilia B in PND families was severe in 35 (67%), moderate in four, mild in five, and unknown in others 56.
F9 sequence variants
SNVs
The Exome Aggregation Consortium (ExAC) investigator coalition collected exome sequencing data from large sequencing projects on 60 706 unrelated individuals, and made the data freely available. These, along with the Database of Single Nucleotide Polymorphisms (dbSNP) and Exome Sequencing Project (ESP) frequency data, provide a useful indication of whether a variant is seen frequently in a population, and is thus more likely to be a neutral than a pathogenic variant 57, 58, 59. The ExAC F9 missense variant frequency is shown in Table S2.
Mutation sex ratio
The origin of different mutation types was investigated by Montandon et al., who identified different ratios of mutations of male or female origin (sex ratio of germline mutation) for different F9 mutation types among the Swedish hemophilia B population; of 77 families, 23 had a single affected male 60. Mothers of 15 males were available, and of these, 13 had parents available. Three patients and 10 mothers had de novo mutations, and extrapolation to the Swedish population of 8.52 × 106 individuals enabled the calculation that eight males and 98 females would harbor de novo F9 mutations (~ 10% of hemophilia B males). The mutation rate was estimated to be as follows: overall, 4.1 × 10−6; male, 2.1 × 10−5; and female, 1.9 × 10−6; the ratio of male/female mutation rates was 11 : 1.
Mutation mechanisms
Historically, FIX mutations were divided into classes including cross‐reacting material‐positive (reduction in activity, but not antigen, type II), reduced, and negative (parallel antigen and activity reduction, type I). Mutations can also be described as having discrepant or equivalent activity and antigen levels. Equivalent mutations reduce FIX mRNA or protein synthesis, stability, or secretion, or combinations of these properties, whereas those with discrepant hemophilia B have qualitative defects, with mutations (mostly missense) impairing functional interactions 7, 61. Sixty‐four unique mild hemophilia B mutations in mature FIX can be divided into predominantly equivalent types (23%) and predominantly discrepant types (64%). These mutations and their FIX location have been presented in detail by Rallapalli et al., and are not further discussed here 7.
‘Silent’ mutations
‘Silent’ (synonymous) mutations can be classified as those that alter a nucleotide but not the encoded amino acid. Many have no discernible pathogenic effect, but some influence protein production, and this can occur through disruption of different processes. Splice mutations may change nucleotides at the end of exons, altering splice‐site recognition, and may also result in splice‐site creation within an exon/intron. Additionally, nucleotide changes may alter splicing regulatory protein recognition sites. Nucleotide substitution can alter mRNA stability, changing the quantity of protein produced. Codon usage alterations can influence the protein translation rate when a common tRNA anticodon is replaced by one having fewer copies, leading to protein misfolding, as illustrated by p.Val153=, which results in mild hemophilia B 62. These mechanisms are reviewed by Sauna et al. 63. Silent variants at FIX amino acids are listed in Table 1.
Table 1.
‘Silent’ variants at factor IX residues
| Nucleotide change | Amino acid position (legacy numbering) | No. of patients | FIX:C (%) | Pathogenic mode | rs No. | ESP MAF | ExAC MAF | ExAC allele prevalence | References |
|---|---|---|---|---|---|---|---|---|---|
| c.87A>G | p.Thr29 = (–18) | 7 | < 1–10 | Splice donor | – | – | – | – |
Thompson et al. 92
Li et al. 93 |
| c.396A>G | p.Val132 = (86) | 1 | NA | Insufficient data | – | – | – | – | Mahajan et al. 94 |
| c.459G>A | p.Val153 = (107) | 6 | 12–20 | Codon usage change | rs144314232 | EurAm A = 0.01% | 0.00001141 | 1/87 612 |
Thompson et al. 92
Knobe et al. 62 |
| c.484C>A | p.Arg162 = (116) | 5 | 3–6 | Possible new splice site | – | – | – | – | Saad et al. 95 |
| c.519A>C | p.Ala173 = (127) | 2 | 2 | Splice donor | – | – | – | – | Unpublished |
| c.519A>G | p.Ala173 = (127) | 5 | NA | Splice donor | – | – | – | – | Wulff et al. 96 |
| c.519A>T | p.Ala173 = (127) | 1 | NA | Splice donor | – | – | – | – | Unpublished |
| c.579G>A | p.Glu193 = (147) | 1 | NA | Insufficient data | – | – | – | – | McGraw et al. 97 |
| c.711A>G | p.Gln237 = (191) | 2 | < 1–3 | Possible new splice site | – | – | – | – | Thompson et al. 98 |
| c.723G>A | p.Gln241 = (195) | 4 | < 1–4 | Splice acceptor | – | – | – | – |
Chen et al. 92
Li et al. 93 |
| c.738T>C | p.Gly246 = (200) | 1 | NA | Insufficient data | – | – | – | – | Mahajan et al. 94 |
| c.819T>C | p.Val273 = (227) | 7 | < 1–37* | Neutral variant | rs1800455 | EurAm C = 0.16% | 0.001436 | 126/87 723 | Koeberl et al. 99 |
| c.1029C>T | p.Asn343 = (297) | 1 | NA | Neutral variant | rs145026483 | EurAm T = 0.64% | 0.00003422 | 3/87 672 | Bottema et al. 100 |
| c.1077C>A | p.Val359 = (313) | 1 | NA | Insufficient data | – | – | – | – | Mahajan et al. 94 |
| c.1110G>A | p.Gln370 = (324) | 1 | NA | Neutral variant | rs34698779 | EurAm A = 0.04% AfAm = 3.26% | 0.002998 | 263/87 724 | Unpublished |
| c.1248T>C | p.Val416 = (370) | 1 | NA | Insufficient data | – | – | – | – | Mahajan et al. 94 |
ESP, Exome Sequencing Project; ExAC, Exome Aggregation Consortium; FIX:C, FIX activity; MAF, minor allele frequency; NA, no data on FIX levels. Data from the F9 mutation database, accessed 25 January 2015. *10–37% in six cases. ‘=’ following the amino acid position indicates no amino acid change.
Ribosome readthrough of nonsense mutations
Pinotti et al. analyzed spontaneous ribosome readthrough, i.e. misrecognition of premature termination codons resulting in protein production, for four F9 nonsense mutations 64. In vitro expression demonstrated detectable FIX antigen (≤ 5% of wild‐type levels) for three of four common nonsense mutations investigated, but not for p.Arg103*. Ribosome readthrough may enable low‐level protein production for some nonsense mutations, impacting both on disease severity and on the likelihood of inhibitor development, and sequence context appears to influence the process. Rates of inhibitor formation associated with nonsense and other mutation types are shown in Table 2.
Table 2.
Association of inhibitors with mutation type in patients listed in the F9 mutation database
| Mutation type | No. of patients with inhibitors | Total no. of patients with the same mutation | Patients with inhibitors (%) |
|---|---|---|---|
| Large deletion | 30 | 69 | 43 |
| Nonsense | 23 | 229 | 10 |
| Frameshift | 5 | 5 | –* |
| Splice | 2 | 6 | –* |
| Insertion/deletion | 1 | 1 | –* |
| Missense | 1 | 1 | –* |
| Total | 62 | 311 | 20 |
Mutation details are shown in Table S3. Mutation data are from 6. For large deletions, breakpoints may not have been identified, and deletions of the same exons may have different intronic sequence breakpoints. *Numbers too small for percentage to be relevant.
Hemophilia B Leyden
In 1970, a new form of hemophilia B was recognized 65, in which FIX:C levels rise rapidly (up to 5 IU dL−1 per year) from puberty onwards, reaching the lower normal range in many cases. Depending on the mutation, some individuals are initially affected by severe hemophilia B, whereas others have a milder phenotype 66. Patients may become clotting factor‐independent by early adulthood 67. The phenotype is seen in 1.9% of patients in the F9db (P.M. Rallapalli, personal communication). A short region of the proximal promoter (HGVS c.‐50 to c.‐18, legacy −27 to +13) contains binding sites for three transcription factors: hepatic nuclear factor 4α from c.‐49 (−20), CCAAT enhancer‐binding protein from c.‐19 (+10), and the recently identified ONECUT1 (also called HNF6) and the related ONECUT2, which binds less well, with binding centered around c.‐35_34 (−5/−6) 67, 68. Disruption of these sites by point mutation can reduce transcription factor binding during infancy/childhood, but the postpubertal FIX level rise induced by growth hormone production still occurs as in other pubertal children 69. Mutations are listed in Table 3.
Table 3.
Hemophilia B Leyden mutations
| HGVS nucleotide no. | Legacy nucleotide no. | FIX:C (%)* | No. of patients |
|---|---|---|---|
| c.‐50T>G | −21 | 0–10 | 3 |
| c.‐49T>A | −20 | 0–60 | 3 |
| c.‐48G>C | −19 | 3 | 2 |
| c.‐35G>A | −6 | 0–25, 13–70 | 31 |
| c.‐35G>C | −6 | 1–30 | 5 |
| c.‐34A>T | −5 | 3 | 3 |
| c.‐24T>A | 6 | 0–8, 0–37 | 4 |
| c.‐22T>C | 8 | 0–60 | 4 |
| c.‐22delT | 8 | 5 | 1 |
| c.‐21C>G | 9 | NA | 1 |
| c.‐18A>G | 12 | 2.5–28, 0–60 | 11 |
| c.‐18delA | 12 | 0–60 | 1 |
FIX:C, FIX activity; HGVS, Human Genome Variation Society; NA, not available. Mutation data from the F9 mutation database 6. *Initial and most recent FIX:C level.
Early reports suggested that females did not experience the same FIX level rise, but a recent report documented symptomatic hemophilia B carrier sisters who had elevated postpubertal levels 70. Two male relatives had childhood FIX levels of 1 IU dL−1 and 2 IU dL−1, which rose to 24 IU dL−1 at 21 years in one individual, and to 45 IU dL−1 in an uncle only analyzed in adulthood. Prepubertal carrier levels (11 IU dL−1 and 17 IU dL−1) rose to 21 IU dL−1 (16 years) and 35 IU dL (14 years). Their elevated levels are consistent with work in mice 69 demonstrating that an increased FIX level is associated with rising postpubertal growth hormone levels rather than testosterone, as previously postulated.
Recurrent mutations (RMs) or identity by descent (IBD)?
When little treatment was available for hemophilia B, it was estimated that mutations causing severe disease would last for only two to four generations before being lost through the patient's death prior to reproduction. De novo mutations are more common in severe than in mild hemophilia B, but may involve the same residues, leading to RMs that are not associated with specific haplotypes or ethnic groups. As patients with moderate/mild disease often lived to reproduce, mutations were transmitted through the population, and sometimes resulted in a founder effect, whereby several individuals shared the same mutation through IBD; this is seen particularly in isolated populations.
Mukherjee et al. analyzed mutations occurring at the CpG dinucleotide mutation hotspot in individuals reported in the F9db, and identified several different haplotypes for many RMs 71. For example, in 2003, p.Thr342Met had 106 F9db entries on 15 apparently different haplotypes. Bottema et al. studied 38 patients with F9 CpG dinucleotide mutations, and, using six SNVs, found that patients with severe hemophilia B were more likely to have RMs than IBD 72. More recently, Hallden et al. investigated IBD in the entire Swedish hemophilia B population, and found that, that among all 86 families, substitution mutations identified in more than one family were present in 47 of 77 families (61%). Haplotyping using 74 markers in and flanking F9 showed that 24 families had IBD mutations, the majority of which had mild disease. Mutation ages were estimated at 2–23 generations. The remaining 23 families appeared to have RMs 73.
Analysis of the F9 mutation rate suggests that the introduction of modern chemicals does not appear to have altered the rate 74, and that the mutation rate is comparable among different population groups 75, 76, 77. Germline F9 mutations therefore largely result from endogenous processes rather than exogenous mutagens.
More than one mutation
In a review of the F9db (3656 patients), 49 were reported to have multiple mutations (1.3%; P.M. Rallapalli, personal communication). Forty‐six had two candidate mutations, two had three, and one had four. Among these variants, 11 individuals had silent changes at six different positions (22%), seven had c.391+7A>G, six had c.839‐20dupA (reported as c.839‐20_839‐19insA), and two had the common exon 6 variant c.580A>G (p.Ala194Thr). Variant frequencies in dbSNP, along with amino acid conservation and splice‐site prediction, suggest that pathogenicity is unlikely (c.391+7A>G, minor allele frequency [MAF] in African Americans, 0.13; c.839‐20dupA, MAF in European Americans, 0.0164; c.580A>G, MAF in European Americans, 0.298), leaving 20 patients with two possible mutations (0.55%). Ghosh et al. examined 103 patients, and identified eight with two mutations (7.8%, including two related individuals) 78. None of the variants was seen in either ExAC or dbSNP 71, 72.
Inhibitors
Inhibitory antibodies are high‐affinity polyclonal antibodies that neutralize the procoagulant activity of a coagulation factor. Inhibitors are much less common in patients with hemophilia B than in those with hemophilia A. They are reported in 2% of patients in the F9db, almost exclusively in patients with severe disease 6, and in 2% of 3785 patients with hemophilia B in the UDC project 2. Most occur in patients with large deletions or nonsense mutations, and they occur less commonly in patients with small deletions/insertion–deletions, splice mutations, and a single F9db missense mutation (p.Gln237Lys) 79, plus another from the USA (p.Gln241His) 80. Table 2 summarizes mutation types in patients with hemophilia B and inhibitors, and Table S3 provides further detail.
In contrast to hemophilia A, where anaphylaxis is rare, anaphylaxis can occur in severe hemophilia B, particularly during early treatment, and several guidelines therefore suggest that the first 20 days of FIX concentrate exposure should take place in hospital. Several factors may contribute to anaphylaxis risk. In comparison with FVIII, FIX is relatively small and readily diffuses into extravascular spaces. Its plasma concentration of 5 mg mL−1 is much higher (FVIII 0.1 mg mL−1), and FIX treatment doses contain more protein than used for hemophilia A. Lack of tolerance to FIX in patients with mutations resulting in complete absence of FIX production, along with a high therapeutic dose and extravascular dispersion, may contribute to hypersensitivity and inhibitor development 81. In contrast to antibodies against FVIII, antibodies against FIX may form circulating immune complexes. These can initiate anaphylaxis when the individual is re‐exposed to FIX concentrate.
The ISTH Scientific and Standardization Committee international FIX inhibitor registry (1997–2006) analyzed 94 patients with severe hemophilia B and an inhibitor 82. Thirty‐two of 94 patients underwent genetic analysis demonstrating that large deletions and nonsense mutations were associated with both inhibitor development and anaphylaxis. Individuals with complete gene deletions had the highest risk; 56 of 94 had experienced anaphylaxis (60%), with the risk being stated as ≥ 26% in individuals with complete gene deletions. Nephrotic syndrome (kidney disorder characterized by proteinuria, hypoalbuminemia, and edema) affected the same group, with 13 patients developing this complication (14%). Nephrotic syndrome developed while patients were receiving immune tolerance induction therapy 82.
Pathogenicity prediction
When mutation analysis is undertaken, previously identified mutations may be found. Documenting their pathogenicity is often straightforward, particularly when the variant has been seen in other patients with similar FIX levels. Where internet searching does not identify the variant in other patients, in silico pathogenicity prediction can help to determine whether the variant is likely to be pathogenic. Guideline documents on assessing predicted pathogenicity have been published by the Association for Clinical Genetic Science 83 and the American College of Medical Genetics 84, and incorporate searching for reports on population frequency; for missense variants, the parameters examined include extent of conservation across a range of species; for possible splice variants, the use of different algorithm types for splice‐site prediction; functional studies, e.g. in vitro expression of a missense variant; and mRNA analysis to determine the effects of splice mutations and cosegregation of the variant with disease. A specific pathogenicity prediction tool has been designed for hemophilia 85, and modeling of hydrogen bond changes suggests that these bonds are less frequently affected in mild than in severe hemophilia B 86. Modeling protein structure can help in understanding the impact of amino acid substitution 6, 7, 87. Table S4 lists suggested resources.
External quality assessment (EQA)
Genetic diagnostic services are often required by their laboratory accreditation to participate in EQA. The UK National External Quality Assessment Survey (UK NEQAS) runs a scheme for Molecular Genetics of Heritable Bleeding Disorders, and arranges two surveys annually on the analysis of hemophilia A, hemophilia B, or von Willebrand disease 88. The analyses resemble a diagnostic referral, posing a specific question regarding the patient, frequently accompanied by a relative. Reports are scored on: (i) relevant referrer and patient details; (ii) identification of correct genotype; and, importantly, (iii) correct interpretation of the results’ significance for the patient and family. General recommendations are returned to all participants, with specific feedback on each laboratory's performance. Two points are awarded for each of the survey areas, and participants must gain at least one point in each area to pass the exercise. The scheme has seen rapid improvement in report format and essential inclusions from new scheme participants 89, 90.
Hemophilia B gene therapy
A successful trial of hemophilia B gene therapy has been undertaken in London, with adeno‐associated virus 8 as an F9 vector. Ten patients were enrolled: an initial six for a phase 1 dose‐escalation study, with two patients each receiving low, intermediate or high doses of vector genome (2 × 1011 kg–1 to 2 × 1012 kg–1); plus four further patients given the high dose. All had extensive clinical and laboratory follow‐up after a single intravenous vector infusion. All 10 patients showed rises in FIX:C level of 1–6% over a median of 3.2 years. The high‐dose group of six patients had FIX:C levels of 5.1% ± 1.7%. FIX concentrate use was significantly reduced for all patients following treatment (median 2613 IU kg−1 per year in the year before vector infusion; median 206 IU kg−1 per year in the year following infusion; P = 0.002). The median number of bleeding episodes for all patients was 15.5 in the year prior to infusion, but only 1.5 in the year following infusion, i.e. a 90% reduction (P = 0.009). Of seven patients previously receiving prophylaxis, four discontinued this treatment. Alanine aminotransferase levels were transiently raised in four of six high‐dose patients, but resolved with prednisolone treatment over a median of 5 days, and no late toxic effects were reported 91. The study provides very encouraging data on the future of gene therapy in hemophilia.
Conclusion
Hemophilia B is one of the most heavily studied of genetic disorders, and many thousands of patients have had their mutations identified. Mutations are detected in > 97% of cases, but remain elusive in a few. Mode of pathogenicity has been explored through in vitro expression for a minority of mutations. Knowledge of the mutation can facilitate inhibitor risk prediction, carrier detection, and PND for affected families.
Disclosure of Conflict of Interests
The author has received honoraria from Novo Nordisk and Octapharma. CSL Behring provides support for the ISTH and EAHAD von Willebrand disease database.
Supporting information
Table S1. Mutation detection rate in 21 studies on patients with hemophilia B.
Table S2. Factor IX missense variant data from the Exome Aggregation Consortium.
Table S3. Mutations associated with inhibitors in hemophilia B.
Table S4. Resources for candidate mutation analysis.
Acknowledgements
The author is grateful to P. Rallapalli for providing data from the F9db, and to D. Hampshire for proofreading the manuscript. A. C. Goodeve receives support from the National Institutes of Health program project grant HL081588 Zimmerman Program for the Molecular and Clinical Biology of VWD.
Goodeve AC. Hemophilia B: molecular pathogenesis and mutation analysis. J Thromb Haemost 2015; 13: 1184–95.
Manuscript handled by: F. R. Rosendaal
Final decision: F. R. Rosendaal, 2 April 2015
References
- 1. Biggs R, Douglas AS, Macfarlane RG, Dacie JV, Pitney WR, Merskey C. Christmas disease – a condition previously mistaken for haemophilia. Br Med J 1952; 2: 1378–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Puetz J, Soucie JM, Kempton CL, Monahan PE, Hemophilia Treatment Center Network Investigators . Prevalent inhibitors in haemophilia B subjects enrolled in the Universal Data Collection database. Haemophilia 2014; 20: 25–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. UKHCDO . UK National Haemophilia Database Bleeding Disorder Statistics for 2013–2014 http://www.ukhcdo.org/annReport.htm. Accessed 2 February 2015.
- 4. Yoshitake S, Schach BG, Foster DC, Davie EW, Kurachi K. Nucleotide sequence of the gene for human factor IX (antihemophilic factor B). Biochemistry 1985; 24: 3736–50. [DOI] [PubMed] [Google Scholar]
- 5. Stonebraker JS, Bolton‐Maggs PH, Michael Soucie J, Walker I, Brooker M. A study of variations in the reported haemophilia B prevalence around the world. Haemophilia 2012; 18: e91–4. [DOI] [PubMed] [Google Scholar]
- 6. Rallapalli PM. Factor IX Variant Database http://www.factorix.org/. Accessed 15 November 2014.
- 7. Rallapalli PM, Kemball‐Cook G, Tuddenham EG, Gomez K, Perkins SJ. An interactive mutation database for human coagulation factor IX provides novel insights into the phenotypes and genetics of hemophilia B. J Thromb Haemost 2013; 11: 1329–40. [DOI] [PubMed] [Google Scholar]
- 8. HGNC . HUGO Gene Nomenclature Committee information homepage http://www.genenames.org/. Accessed 15 November 2014.
- 9. HGVS . Nomenclature for the description of sequence variations homepage http://www.hgvs.org/mutnomen/. Accessed 20 December 2014.
- 10. Ensembl . Ensembl Genome browser homepage http://www.ensembl.org/index.html. Accessed 31 January 2015.
- 11. Giannelli F, Green PM, High KA, Lozier JN, Lillicrap DP, Ludwig M, Olek K, Reitsma PH, Goossens M, Yoshioka A, Sommer S, Brownlee GG. Haemophilia B: database of point mutations and short additions and deletions. Nucleic Acids Res 1990; 18: 4053–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Green P. Haemophilia B Mutation Database http://www.kcl.ac.uk/ip/petergreen/haemBdatabase.html. Accessed 3 December 2010.
- 13. Mannucci PM, Franchini M. Is haemophilia B less severe than haemophilia A? Haemophilia 2013; 19: 499–502. [DOI] [PubMed] [Google Scholar]
- 14. Clausen N, Petrini P, Claeyssens‐Donadel S, Gouw SC, Liesner R, PedNet, Research of Determinants of Inhibitor Development Study Group . Similar bleeding phenotype in young children with haemophilia A or B: a cohort study. Haemophilia 2014; 20: 747–55. [DOI] [PubMed] [Google Scholar]
- 15. Tagariello G, Iorio A, Santagostino E, Morfini M, Bisson R, Innocenti M, Mancuso ME, Mazzucconi MG, Pasta GL, Radossi P, Rodorigo G, Santoro C, Sartori R, Scaraggi A, Solimeno LP, Mannucci PM, Italian Association Hemophilia Centre . Comparison of the rates of joint arthroplasty in patients with severe factor VIII and IX deficiency: an index of different clinical severity of the 2 coagulation disorders. Blood 2009; 114: 779–84. [DOI] [PubMed] [Google Scholar]
- 16. Schulman S, Eelde A, Holmstrom M, Stahlberg G, Odeberg J, Blomback M. Validation of a composite score for clinical severity of hemophilia. J Thromb Haemost 2008; 6: 1113–21. [DOI] [PubMed] [Google Scholar]
- 17. Plug I, Mauser‐Bunschoten EP, Brocker‐Vriends AH, van Amstel HK, van der Bom JG, van Diemen‐Homan JE, Willemse J, Rosendaal FR. Bleeding in carriers of hemophilia. Blood 2006; 108: 52–6. [DOI] [PubMed] [Google Scholar]
- 18. Sharathkumar A, Hardesty B, Greist A, Salter J, Kerlin B, Heiman M, Sulkin M, Shapiro A. Variability in bleeding phenotype in Amish carriers of haemophilia B with the 31008 C–>T mutation. Haemophilia 2009; 15: 91–100. [DOI] [PubMed] [Google Scholar]
- 19. Rees DJ, Rizza CR, Brownlee GG. Haemophilia B caused by a point mutation in a donor splice junction of the human factor IX gene. Nature 1985; 316: 643–5. [DOI] [PubMed] [Google Scholar]
- 20. Mitchell M, Keeney S, Goodeve A. Practice Guidelines for the Molecular Diagnosis of Haemophilia B http://www.acgs.uk.com/media/774631/haemophilia_b_bpg_revision_sept_2011_approved.pdf. Accessed 15 January 2015.
- 21. Josephson NC, Martin B, Nakaya S, Konkle BA, Luk A, Pierce GF, Shendure J, O'Roak B. A next generation sequencing approach for genotyping patients with hemophilia. J Thromb Haemost 2013; 11: 85–289. [Google Scholar]
- 22. Chan V, Au P, Lau P, Chan TK. A novel haemophilia B defect due to partial duplication of the factor IX gene. Br J Haematol 1994; 86: 601–9. [DOI] [PubMed] [Google Scholar]
- 23. Kwon MJ, Yoo KY, Kim HJ, Kim SH. Identification of mutations in the F9 gene including exon deletion by multiplex ligation‐dependent probe amplification in 33 unrelated Korean patients with haemophilia B. Haemophilia 2008; 14: 1069–75. [DOI] [PubMed] [Google Scholar]
- 24. Casana P, Haya S, Cid AR, Oltra S, Martinez F, Cabrera N, Aznar JA. Identification of deletion carriers in hemophilia B: quantitative real‐time polymerase chain reaction or multiple ligation probe amplification. Transl Res 2009; 153: 114–17. [DOI] [PubMed] [Google Scholar]
- 25. Payne AB, Bean CJ, Hooper WC, Miller CH. Utility of multiplex ligation‐dependent probe amplification (MLPA) for hemophilia mutation screening. J Thromb Haemost 2012; 10: 1951–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Solera J, Magallon M, Martin‐Villar J, Coloma A. Factor IXMadrid 2: a deletion/insertion in factor IX gene which abolishes the sequence of the donor junction at the exon IV–intron d splice site. Am J Hum Genet 1992; 50: 434–7. [PMC free article] [PubMed] [Google Scholar]
- 27. Vidaud D, Vidaud M, Bahnak BR, Siguret V, Gispert Sanchez S, Laurian Y, Meyer D, Goossens M, Lavergne JM. Haemophilia B due to a de novo insertion of a human‐specific Alu subfamily member within the coding region of the factor IX gene. Eur J Hum Genet 1993; 1: 30–6. [DOI] [PubMed] [Google Scholar]
- 28. Wulff K, Gazda H, Schroder W, Robicka‐Milewska R, Herrmann FH. Identification of a novel large F9 gene mutation – an insertion of an Alu repeated DNA element in exon e of the factor 9 gene. Hum Mutat 2000; 15: 299. [DOI] [PubMed] [Google Scholar]
- 29. Mukherjee S, Mukhopadhyay A, Banerjee D, Chandak GR, Ray K. Molecular pathology of haemophilia B: identification of five novel mutations including a LINE 1 insertion in Indian patients. Haemophilia 2004; 10: 259–63. [DOI] [PubMed] [Google Scholar]
- 30. Feng J, Liu Q, Drost J, Sommer SS. Deep intronic mutations are rarely a cause of hemophilia B. Hum Mutat 1999; 14: 267–8. [DOI] [PubMed] [Google Scholar]
- 31. Chavali S, Ghosh S, Bharadwaj D. Hemophilia B is a quasi‐quantitative condition with certain mutations showing phenotypic plasticity. Genomics 2009; 94: 433–7. [DOI] [PubMed] [Google Scholar]
- 32. Sarkar G, Yoon HS, Sommer SS. Screening for mutations by RNA single‐strand conformation polymorphism (rSSCP): comparison with DNA‐SSCP. Nucleic Acids Res 1992; 20: 871–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. van de Water NS, Tan T, May S, Browett PJ, Harper P. Factor IX polypyrimidine tract mutation analysis using mRNA from peripheral blood leukocytes. J Thromb Haemost 2004; 2: 2073–5. [DOI] [PubMed] [Google Scholar]
- 34. Green PM, Rowley G, Giannelli F. Unusual expression of the F9 gene in peripheral lymphocytes hinders investigation of F9 mRNA in hemophilia B patients. J Thromb Haemost 2003; 1: 2675–6. [DOI] [PubMed] [Google Scholar]
- 35. Cutler JA, Mitchell MJ, Savidge GF. More on: unusual expression of the F9 gene in peripheral lymphocytes hinders investigation of F9 mRNA in hemophilia B patients. J Thromb Haemost 2004; 2: 1021. [DOI] [PubMed] [Google Scholar]
- 36. Cao DH, Liu XL, Mu K, Ma XW, Sun JL, Bai XZ, Lin CK, Jin CL. Identification and genetic analysis of a factor IX gene intron 3 mutation in a hemophilia B pedigree in China. Turk J Haematol 2014; 31: 226–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pezeshkpoor B, Zimmer N, Marquardt N, Nanda I, Haaf T, Budde U, Oldenburg J, El‐Maarri O. Deep intronic ‘mutations’ cause hemophilia A: application of next generation sequencing in patients without detectable mutation in F8 cDNA. J Thromb Haemost 2013; 11: 1679–87. [DOI] [PubMed] [Google Scholar]
- 38. Castaman G, Giacomelli SH, Mancuso ME, D'Andrea G, Santacroce R, Sanna S, Santagostino E, Mannucci PM, Goodeve A, Rodeghiero F. Deep intronic variations may cause mild hemophilia A. J Thromb Haemost 2011; 9: 1541–8. [DOI] [PubMed] [Google Scholar]
- 39. Pezeshkpoor B, Pavlova A, Oldenburg J, El‐Maarri O. F8 genetic analysis strategies when standard approaches fail. Hamostaseologie 2014; 34: 167–73. [DOI] [PubMed] [Google Scholar]
- 40. Kasper CK, Lin JC. Prevalence of sporadic and familial haemophilia. Haemophilia 2007; 13: 90–2. [DOI] [PubMed] [Google Scholar]
- 41. Jenkins PV, Egan H, Keenan C, O'Shea E, Smith OP, Nolan B, White B, O'Donnell J. Mutation analysis of haemophilia B in the Irish population: increased prevalence caused by founder effect. Haemophilia 2008; 14: 717–22. [DOI] [PubMed] [Google Scholar]
- 42. Kasper CK, Buzin CH. Mosaics and haemophilia. Haemophilia 2009; 15: 1181–6. [DOI] [PubMed] [Google Scholar]
- 43. Karimipoor M, Zeinali S, Nafissi N, Tuddenham EG, Lak M, Safaee R. Identification of factor IX mutations in Iranian haemophilia B patients by SSCP and sequencing. Thromb Res 2007; 120: 135–9. [DOI] [PubMed] [Google Scholar]
- 44. Orstavik KH, Scheibel E, Ingerslev J, Schwartz M. Absence of correlation between X chromosome inactivation pattern and plasma concentration of factor VIII and factor IX in carriers of haemophilia A and B. Thromb Haemost 2000; 83: 433–7. [PubMed] [Google Scholar]
- 45. Di Michele DM, Gibb C, Lefkowitz JM, Ni Q, Gerber LM, Ganguly A. Severe and moderate haemophilia A and B in US females. Haemophilia 2014; 20: e136–43. [DOI] [PubMed] [Google Scholar]
- 46. Knoll A, Ketterling RP, Sommer SS. Absence of somatic mosaicism in 17 families with hemophilia B: an analysis with a sensitivity 10‐ to 1000‐fold greater than that of sequencing gels. Hum Genet 1996; 98: 539–45. [DOI] [PubMed] [Google Scholar]
- 47. Sommer SS, Knoll A, Greenberg CR, Ketterling RP. Germline mosaicism in a female who seemed to be a carrier by sequence analysis. Hum Mol Genet 1995; 4: 2181–2. [DOI] [PubMed] [Google Scholar]
- 48. Green PM, Saad S, Lewis CM, Giannelli F. Mutation rates in humans. I. Overall and sex‐specific rates obtained from a population study of hemophilia B. Am J Hum Genet 1999; 65: 1572–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Biesecker LG, Spinner NB. A genomic view of mosaicism and human disease. Nat Rev Genet 2013; 14: 307–20. [DOI] [PubMed] [Google Scholar]
- 50. Peake IR, Lillicrap DP, Boulyjenkov V, Briet E, Chan V, Ginter EK, Kraus EM, Ljung R, Mannucci PM, Nicolaides K, Tuddenham EGD. Report of a joint WHO/WFH meeting on the control of haemophilia: carrier detection and prenatal diagnosis. Blood Coagul Fibrinolysis 1993; 4: 313–44. [DOI] [PubMed] [Google Scholar]
- 51. Jayandharan GR, Srivastava A, Srivastava A. Role of molecular genetics in hemophilia: from diagnosis to therapy. Semin Thromb Hemost 2012; 38: 64–78. [DOI] [PubMed] [Google Scholar]
- 52. Dai J, Lu Y, Ding Q, Wang H, Xi X, Wang X. The status of carrier and prenatal diagnosis of haemophilia in China. Haemophilia 2012; 18: 235–40. [DOI] [PubMed] [Google Scholar]
- 53. Bors A, Andrikovics H, Illes Z, Jager R, Kardos M, Marosi A, Nemes L, Tordai A. Carrier and prenatal diagnostic strategy and newly identified mutations in Hungarian haemophilia A and B families. Blood Coagul Fibrinolysis 2015; 26: 161–6. [DOI] [PubMed] [Google Scholar]
- 54. Cutler J, Chappell LC, Kyle P, Madan B. Third trimester amniocentesis for diagnosis of inherited bleeding disorders prior to delivery. Haemophilia 2013; 19: 904–7. [DOI] [PubMed] [Google Scholar]
- 55. Peyvandi F, Garagiola I, Mortarino M. Prenatal diagnosis and preimplantation genetic diagnosis: novel technologies and state of the art of PGD in different regions of the world. Haemophilia 2011; 17(Suppl. 1): 14–17. [DOI] [PubMed] [Google Scholar]
- 56. Belvini D, Salviato R, Acquila M, Bicocchi MP, Frusconi S, Garagiola I, Sanna V, Santacroce R, Rocino A, Tagariello G. Prenatal diagnosis of haemophilia B: the Italian experience. Haemophilia 2013; 19: 898–903. [DOI] [PubMed] [Google Scholar]
- 57. Exome Aggregation Consortium . ExAC Browser. http://exac.broadinstitute.org/. Accessed 30 November 2014.
- 58. National Centre for Biotechnology Information . NCBI dbSNP http://www.ncbi.nlm.nih.gov/sites/entrez. Accessed 29 January 2015.
- 59. NHLBI . Exome Sequencing Project (ESP) http://evs.gs.washington.edu/EVS/. Accessed 29 January 2015.
- 60. Montandon AJ, Green PM, Bentley DR, Ljung R, Kling S, Nilsson IM, Giannelli F. Direct estimate of the haemophilia B (factor IX deficiency) mutation rate and of the ratio of the sex‐specific mutation rates in Sweden. Hum Genet 1992; 89: 319–22. [DOI] [PubMed] [Google Scholar]
- 61. Green PM, Bentley DR, Mibashan RS, Nilsson IM, Giannelli F. Molecular pathology of haemophilia B. EMBO J 1989; 8: 1067–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Knobe KE, Sjorin E, Ljung RC. Why does the mutation G17736A/Val107Val (silent) in the F9 gene cause mild haemophilia B in five Swedish families? Haemophilia 2008; 14: 723–8. [DOI] [PubMed] [Google Scholar]
- 63. Sauna ZE, Kimchi‐Sarfaty C. Understanding the contribution of synonymous mutations to human disease. Nat Rev Genet 2011; 12: 683–91. [DOI] [PubMed] [Google Scholar]
- 64. Pinotti M, Caruso P, Canella A, Campioni M, Tagariello G, Castaman G, Giacomelli S, Belvini D, Bernardi F. Ribosome readthrough accounts for secreted full‐length factor IX in hemophilia B patients with nonsense mutations. Hum Mutat 2012; 33: 1373–6. [DOI] [PubMed] [Google Scholar]
- 65. Veltkamp JJ, Meilof J, Remmelts HG, van der Vlerk D, Loeliger EA. Another genetic variant of haemophilia B: haemophilia B Leyden. Scand J Haematol 1970; 7: 82–90. [DOI] [PubMed] [Google Scholar]
- 66. Crossley M, Winship PR, Austen DE, Rizza CR, Brownlee GG. A less severe form of haemophilia B Leyden. Nucleic Acids Res 1990; 18: 4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Funnell AP, Wilson MD, Ballester B, Mak KS, Burdach J, Magan N, Pearson RC, Lemaigre FP, Stowell KM, Odom DT, Flicek P, Crossley M. A CpG mutational hotspot in a ONECUT binding site accounts for the prevalent variant of hemophilia B Leyden. Am J Hum Genet 2013; 92: 460–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Funnell AP, Crossley M. Hemophilia B Leyden and once mysterious cis‐regulatory mutations. Trends Genet 2014; 30: 18–23. [DOI] [PubMed] [Google Scholar]
- 69. Kurachi S, Huo JS, Ameri A, Zhang K, Yoshizawa AC, Kurachi K. An age‐related homeostasis mechanism is essential for spontaneous amelioration of hemophilia B Leyden. Proc Natl Acad Sci USA 2009; 106: 7921–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hildyard C, Keeling D. Effect of age on factor IX levels in symptomatic carriers of haemophila B Leyden. Br J Haematol 2015; 169: 448–9. [DOI] [PubMed] [Google Scholar]
- 71. Mukherjee S, Mukhopadhyay A, Chaudhuri K, Ray K. Analysis of haemophilia B database and strategies for identification of common point mutations in the factor IX gene. Haemophilia 2003; 9: 187–92. [DOI] [PubMed] [Google Scholar]
- 72. Bottema CD, Ketterling RP, Vielhaber E, Yoon HS, Gostout B, Jacobson DP, Shapiro A, Sommer SS. The pattern of spontaneous germ‐line mutation: relative rates of mutation at or near CpG dinucleotides in the factor IX gene. Hum Genet 1993; 91: 496–503. [DOI] [PubMed] [Google Scholar]
- 73. Hallden C, Martensson A, Nilsson D, Sall T, Lind‐Hallden C, Liden AC, Ljung R. Origin of Swedish hemophilia B mutations. J Thromb Haemost 2013; 11: 2001–8. [DOI] [PubMed] [Google Scholar]
- 74. Feng J, Drost JB, Scaringe WA, Liu Q, Sommer SS. Mutations in the factor IX gene (F9) during the past 150 years have relative rates similar to ancient mutations. Hum Mutat 2002; 19: 49–57. [DOI] [PubMed] [Google Scholar]
- 75. Sommer SS, Scaringe WA, Hill KA. Human germline mutation in the factor IX gene. Mutat Res 2001; 487: 1–17. [DOI] [PubMed] [Google Scholar]
- 76. Liu JZ, Li X, Drost J, Thorland EC, Liu Q, Lind T, Roberts S, Wang HY, Sommer SS. The human factor IX gene as germline mutagen test: samples from Mainland China have the putatively endogenous pattern of mutation. Hum Mutat 2000; 16: 31–6. [DOI] [PubMed] [Google Scholar]
- 77. Li X, Drost JB, Roberts S, Kasper C, Sommer SS. Factor IX mutations in South Africans and African Americans are compatible with primarily endogenous influences upon recent germline mutations. Hum Mutat 2000; 16: 371. [DOI] [PubMed] [Google Scholar]
- 78. Ghosh K, Shetty S, Quadros L, Kulkarni B. Double mutations causing haemophilia B: a double whammy! Br J Haematol 2009; 145: 433–5. [DOI] [PubMed] [Google Scholar]
- 79. Chen SH, Zhang M, Lovrien EW, Scott CR, Thompson AR. CG dinucleotide transitions in the factor IX gene account for about half of the point mutations in hemophilia B patients: a Seattle series. Hum Genet 1991; 87: 177–82. [DOI] [PubMed] [Google Scholar]
- 80. Saini S, Hamasaki‐Katagiri N, Pandey GS, Yanover C, Guelcher C, Simhadri VL, Dandekar S, Guerrera MF, Kimchi‐Sarfaty C, Sauna ZE. Genetic determinants of immunogenicity to factor IX during the treatment of haemophilia B. Haemophilia 2015; 21: 210–18. [DOI] [PubMed] [Google Scholar]
- 81. Warrier I, Lusher JM. Development of anaphylactic shock in haemophilia B patients with inhibitors. Blood Coagul Fibrinolysis 1998; 9(Suppl. 1): S125–8. [PubMed] [Google Scholar]
- 82. Chitlur M, Warrier I, Rajpurkar M, Lusher JM. Inhibitors in factor IX deficiency: a report of the ISTH‐SSC international FIX inhibitor registry (1997–2006). Haemophilia 2009; 15: 1027–31. [DOI] [PubMed] [Google Scholar]
- 83. Wallis Y, Payne S, McAnulty C, Bodmer D, Sistermans E, Robertson K, Moore D, Abbs S, Deans Z, Devereau A. Practice Guidelines for the Evaluation of Pathogenicity and the Reporting of Sequence Variants in Clinical Molecular Genetics http://www.acgs.uk.com/media/774853/evaluation_and_reporting_of_sequence_variants_bpgs_june_2013_-_finalpdf.pdf. Accessed 20 December 2014
- 84. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier‐Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17: 405–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Hamasaki‐Katagiri N, Salari R, Simhadri VL, Tseng SC, Needlman E, Edwards NC, Sauna ZE, Grigoryan V, Komar AA, Przytycka TM, Kimchi‐Sarfaty C. Analysis of F9 point mutations and their correlation to severity of haemophilia B disease. Haemophilia 2012; 18: 933–40. [DOI] [PubMed] [Google Scholar]
- 86. Mukherjee S, Saha A, Biswas P, Mandal C, Ray K. Structural analysis of factor IX protein variants to predict functional aberration causing haemophilia B. Haemophilia 2008; 14: 1076–81. [DOI] [PubMed] [Google Scholar]
- 87. Bicocchi MP, Pasino M, Rosano C, Molinari AC, Della Valle E, Lanza T, Bottini F, Acquila M. Insight into molecular changes of the FIX protein in a series of Italian patients with haemophilia B. Haemophilia 2006; 12: 263–70. [DOI] [PubMed] [Google Scholar]
- 88. UK NEQAS . United Kingdom National External Quality Assessment Service for Molecular Genetics homepage www.ukneqasbc.org/. Accessed 31 January 2015.
- 89. Perry DJ, Cumming T, Goodeve A, Hill M, Jennings I, Kitchen S, Walker I. The UK National External Quality Assessment Scheme for heritable bleeding disorders. Semin Thromb Hemost 2014; 40: 261–8. [DOI] [PubMed] [Google Scholar]
- 90. Perry DJ, Goodeve A, Hill M, Jennings I, Kitchen S, Walker I, UK NEQAS for Blood Coagulation . The UK National External Quality Assessment Scheme (UK NEQAS) for molecular genetic testing in haemophilia. Thromb Haemost 2006; 96: 597–601. [PubMed] [Google Scholar]
- 91. Nathwani AC, Reiss UM, Tuddenham EG, Rosales C, Chowdary P, McIntosh J, Della Peruta M, Lheriteau E, Patel N, Raj D, Riddell A, Pie J, Rangarajan S, Bevan D, Recht M, Shen YM, Halka KG, Basner‐Tschakarjan E, Mingozzi F, High KA, et al Long‐term safety and efficacy of factor IX gene therapy in hemophilia B. N Engl J Med 2014; 371: 1994–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Thompson AR, Bajaj SP, Chen SH, MacGillivray RT. ‘Founder’ effect in different families with haemophilia B mutation. Lancet 1990; 335: 418. [DOI] [PubMed] [Google Scholar]
- 93. Li T, Miller CH, Driggers J, Payne AB, Ellingsen D, Hooper WC. Mutation analysis of a cohort of US patients with hemophilia B. Am J Hematol 2014; 89: 375–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Mahajan A, Chavali S, Kabra M, Chowdhury MR, Bharadwaj D. Molecular characterization of hemophilia B in North Indian families: identification of novel and recurrent molecular events in the factor IX gene. Haematologica 2004; 89: 1498–503. [PubMed] [Google Scholar]
- 95. Saad S, Rowley G, Tagliavacca L, Green PM, Giannelli F. First report on UK database of haemophilia B mutations and pedigrees. UK Haemophilia Centres. Thromb Haemost 1994; 71: 563–70. [PubMed] [Google Scholar]
- 96. Wulff K, Schroder W, Wehnert M, Herrmann FH. Twenty‐five novel mutations of the factor IX gene in haemophilia B. Hum Mutat 1995; 6: 346–8. [DOI] [PubMed] [Google Scholar]
- 97. McGraw RA, Davis LM, Lundblad RL, Stafford DW, Roberts HR. Structure and function of factor IX: defects in haemophilia B. Clin Haematol 1985; 14: 359–83. [PubMed] [Google Scholar]
- 98. Thompson AR, Schoof JM, Weinmann AF, Chen SH. Factor IX mutations: rapid, direct screening methods for 20 new families with hemophilia B. Thromb Res 1992; 65: 289–95. [DOI] [PubMed] [Google Scholar]
- 99. Koeberl DD, Bottema CD, Buerstedde JM, Sommer SS. Functionally important regions of the factor IX gene have a low rate of polymorphism and a high rate of mutation in the dinucleotide CpG. Am J Hum Genet 1989; 45: 448–57. [PMC free article] [PubMed] [Google Scholar]
- 100. Bottema CD, Ketterling RP, Yoon HS, Sommer SS. The pattern of factor IX germ‐line mutation in Asians is similar to that of Caucasians. Am J Hum Genet 1990; 47: 835–41. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Mutation detection rate in 21 studies on patients with hemophilia B.
Table S2. Factor IX missense variant data from the Exome Aggregation Consortium.
Table S3. Mutations associated with inhibitors in hemophilia B.
Table S4. Resources for candidate mutation analysis.