

Abstract
Porphyromonas gingivalis is an established pathogen in periodontal disease and an emerging pathogen in serious systemic conditions, including some forms of cancer. We investigated the effect of P. gingivalis on β-catenin signaling, a major pathway in the control of cell proliferation and tumorigenesis. Infection of gingival epithelial cells with P. gingivalis did not influence the phosphorylation status of β-catenin but resulted in proteolytic processing. The use of mutants deficient in gingipain production, along with gingipain-specific inhibitors, revealed that gingipain proteolytic activity was required for β-catenin processing. The β-catenin destruction complex components Axin1, adenomatous polyposis coli (APC), and GSK3β were also proteolytically processed by P. gingivalis gingipains. Cell fractionation and Western blotting demonstrated that β-catenin fragments were translocated to the nucleus. The accumulation of β-catenin in the nucleus following P. gingivalis infection was confirmed by immunofluorescence microscopy. A luciferase reporter assay showed that P. gingivalis increased the activity of the β-catenin-dependent TCF/LEF promoter. P. gingivalis did not increase Wnt3a mRNA levels, a finding consistent with P. gingivalis-induced proteolytic processing causing the increase in TCF/LEF promoter activity. Thus, our data indicate that P. gingivalis can induce the noncanonical activation of β-catenin and disassociation of the β-catenin destruction complex by gingipain-dependent proteolytic processing. β-Catenin activation in epithelial cells by P. gingivalis may contribute to a proliferative phenotype.
INTRODUCTION
Porphyromonas gingivalis, a Gram-negative anaerobe, is a keystone pathogen in chronic and severe manifestations of periodontal disease (1, 2). Among the first host cells encountered by P. gingivalis in the gingival compartment are the epithelial cells that line the crevice and provide both a physical barrier to microbial intrusion and an interactive interface that signals the presence of bacteria to the underlying cells of the immune system. P. gingivalis and gingival epithelial cells engage in an intricate molecular dialogue that facilitates entry of P. gingivalis into the epithelial cell cytoplasm, where internalized P. gingivalis remains viable and can spread to adjacent cells (3–5). P. gingivalis also impinges upon several aspects of innate immunity, creating a dysbiotic host response that is unable to eliminate periodontal bacteria (6–9). Furthermore, the misdirected inflammatory responses to P. gingivalis favor the persistence of the organism by providing a source of nutrients in the form of tissue breakdown products (10).
P. gingivalis is an asaccharolytic organism and requires the action of proteolytic enzymes to provide the nitrogen and carbon sources necessary for growth. A family of cysteine proteases comprising the arginine-specific gingipains RgpA and RgpB and the lysine-specific gingipain Kgp is responsible for the majority of the proteolytic activity of P. gingivalis (11, 12). Gingipains are also major components of P. gingivalis outer membrane vesicles, which may facilitate their penetration of the periodontal tissues (13–15). These enzymes are directed to the bacterial cell surface and secreted through the type IX pathway, which requires a C-terminal secretion signal known as the CTD (12, 16). Gingipains can degrade the structural components of periodontal tissues, immune effector molecules, and host heme-sequestering proteins and thus constitute major virulence factors of P. gingivalis (3, 17).
In addition to demonstrating a well-defined role for P. gingivalis in periodontal disease, epidemiological evidence is accumulating that links P. gingivalis with serious systemic conditions, including pancreatic cancer and oral squamous cell carcinoma (OSCC) (18, 19). Processes that could be relevant to the development of cancer include the ability of P. gingivalis to suppress apoptosis in gingival epithelial cells (20, 21) and accelerate progression through the S phase of the cell cycle (22). Indeed, P. gingivalis alters the expression and activity of a number of proteins involved in cell cycle regulation, including several cyclins and cyclin-dependent kinases (22). Furthermore, P. gingivalis infection differentially regulates the expression of a large percentage of epithelial cell genes and pathways (23), indicating a broadly based subversion of host cell signal transduction and physiological status.
The Wnt/β-catenin pathway is a conserved signaling circuit that plays a role in cell growth, differentiation, and survival (24). In the absence of Wnt ligands, β-catenin is maintained in a destruction complex and targeted for proteasomal degradation. The destruction complex is comprised of scaffold proteins, including tumor suppressor adenomatous polyposis coli (APC) and Axin, on which kinases, such as GSK3β, phosphorylate β-catenin at N-terminal serine and threonine residues. Subsequently, β-catenin is ubiquitinated and degraded by the 26S proteasome (25). Conversely, binding of Wnt glycoproteins to Frizzled (Fzd) receptors leads to phosphorylation of the LRP 5/6 coreceptors and recruitment of Axin and Dishevelled (Dvl) proteins to the plasma membrane (24). Phosphoinactivation of GSK3β on the serine 9 residue occurs, followed by functional disruption of the destruction complex (26). Cytoplasmic β-catenin is stabilized by phosphorylation of serine 552 (27) and is translocated to the nucleus, where it binds the TCF/LEF transcription factors and stimulates expression of Wnt/β-catenin target genes, including Myc and cyclin D1 (28). Activated β-catenin promotes a prosurvival proliferative phenotype and is associated with epithelial-to-mesenchymal transition (29). Accordingly, accumulation and nuclear translocation of β-catenin are observed in a number of human cancers, including OSCC (30, 31).
Fusobacterium nucleatum, which is emerging as an important pathogen in colorectal cancer, binds to E-cadherin and activates β-catenin signaling with oncogenic responses (32). Although P. gingivalis has been shown to affect a number of processes, such as cell survival, that intersect with the Wnt/β-catenin pathway, little is known regarding the impact of P. gingivalis cells on β-catenin signaling. We investigated the influence of P. gingivalis on β-catenin activation. The results indicate that P. gingivalis can proteolytically process and activate β-catenin independent of Wnt and suggest a novel mechanism by which P. gingivalis could contribute to disruption of oral tissue homeostasis.
MATERIALS AND METHODS
Bacterial strains, eukaryotic cells, and growth conditions.
The P. gingivalis strains used in the present study were W83, ATCC 33277 (33277), and the ΔfimA (33), ΔrgpAB, Δkgp, and ΔrgpAB Δkgp (34, 35) isogenic 33277 mutants. Bacteria were cultured in Trypticase soy broth supplemented with yeast extract (1 mg/ml), hemin (5 μg/ml), and menadione (1 μg/ml). Medium was supplemented with antibiotics erythromycin (10 μg/ml), tetracycline (1 μg/ml), or chloramphenicol (20 μg/ml) as appropriate. Streptococcus gordonii strain DL1 was grown in Todd-Hewitt broth. F. nucleatum strain ATCC 25586 was cultured in brain heart infusion broth supplemented with hemin (5 μg/ml) and menadione (1 μg/ml). All bacteria were cultured anaerobically at 37°C. Telomerase immortalized gingival epithelial keratinocytes (TIGKs) derived from a primary gingival epithelial cell line were maintained in DermaLife keratinocyte medium with supplements (Lifeline Cell Technology, Carlsbad, CA) as described previously (36). Cells between passages 10 and 20 were cultured to 80% confluence and infected with P. gingivalis under tissue culture conditions (5% CO2, 37°C). Where indicated, P. gingivalis was pretreated with TLCK (Nα-p-tosyl-l-lysine chloromethyl ketone; a gingipain inhibitor, 100 μM) for 2 h (37), and TIGK cells were pretreated with MG132 (a proteasome inhibitor, 10 μM) for 2 h (38).
Antibodies, chemicals, and protease purification.
All antibodies were obtained from Cell Signaling (Danvers, MA). TLCK was obtained from Sigma-Aldrich (St. Louis, MO). β-Catenin and MG132 were from EMD Millipore (Billerica, MA). Wnt3a was obtained from R&D Systems (Minneapolis, MN). RgpB was purified from P. gingivalis culture supernatants by acetone precipitation, size-exclusion chromatography using Sephadex G-150, and affinity chromatography on arginine-Sepharose as described previously (37).
Western blot analysis.
TIGKs were lysed with cold cell lysis buffer containing PhosSTOP phosphatase inhibitor and protease inhibitor. Cytoplasmic and nuclear fractions were collected with a nuclear extract kit (Active Motif, Carlsbad, CA) according to the manufacturer's instructions. Proteins (20 ng) were separated by SDS–10% PAGE, blotted onto a polyvinylidene difluoride membrane, and blocked using 5% bovine serum albumin in Tris-buffered saline with 0.1% Tween 20. Blots were reacted for 16 h with primary antibody at 4°C and 1 h with horseradish peroxidase-conjugated secondary antibody at room temperature. The membrane was developed using ECL detection, and densitometric analyses were conducted using a ChemiDoc XRS Plus (Bio-Rad, Hercules, CA). GAPDH (glyceraldehyde-3-phosphate dehydrogenase) was used as a loading control.
Transfection and TCF/LEF reporter assay.
TCF/LEF reporter plasmids (Qiagen, Valencia, CA) were transfected into TIGKs using 0.15% Lipofectamine (Life Technologies, Grand Island, NY). At 48 h posttransfection, cells were infected with P. gingivalis or mock treated. The luciferase activity was measured with the Dual-Glo luciferase assay system (Promega, Madison, WI) and normalized to the Renilla internal control.
Immunofluorescence and confocal microscopy.
TIGKs on glass coverslips were infected with P. gingivalis, washed twice in phosphate-buffered saline, and fixed for 10 min in 4% paraformaldehyde. Permeabilization was with 0.3% Triton X-100 for 10 min at room temperature, prior to blocking in 10% goat serum for 1 h. β-Catenin was detected by reacting with primary antibodies at 1:100 for 1 h, followed by fluorescein isothiocyanate-conjugated secondary antibody (1:200) for 1 h in the dark. Nuclear staining was with Hoechst 33342 (Life Technologies) at 1:2,000. Slides were mounted with Vectashield and observed in a Leica SP8 confocal microscope, and z-stacks were obtained (20 layers/stack, 0.7-μm-pore-size intervals) through the z-axis of cells (three z-stacks/coverslip). The percent volume of β-catenin in the nucleus was calculated by using Volocity 3D image analysis software (Perkin-Elmer, Waltham, MA).
qRT-PCR.
Total RNA was isolated from P. gingivalis-infected or mock-treated TIGKs with a Perfect Pure RNA cell kit (5Prime, Gaithersburg, MD) and reverse transcribed using a high-capacity cDNA reverse transcription kit (Applied Biosystems, Grand Island, NY). TaqMan primers were obtained commercially (Applied Biosystems), and quantitative reverse transcriptase PCR (qRT-PCR) was performed on an Applied Biosystems StepOne plus. mRNA levels were normalized with those of GAPDH mRNA using the ΔΔCT method as described previously (39).
RESULTS
P. gingivalis infection results in cleavage of β-catenin in gingival epithelial cells.
The stability and location of β-catenin is controlled by phosphorylation of specific amino acid residues. Since P. gingivalis expresses phosphatases that can target eukaryotic proteins (9, 40), we first assessed the impact of P. gingivalis infection on β-catenin phosphorylation status in TIGKs by Western blotting. As shown in Fig. 1, infection with P. gingivalis had no significant effect on phosphorylation of the S552 residue, which enhances stability of β-catenin (24). However, P. gingivalis infection at multiplicities of infection (MOIs) of 50 and 100 induced a time-dependent partial degradation of β-catenin. Processing of β-catenin also occurred at an MOI of 10, although to a lesser degree than was observed at the higher MOIs, and the production of β-catenin fragments was maximal at 30 min. We also observed a P. gingivalis-dependent partial degradation of GSK3β, indicating disruption of the β-catenin destruction complex. GSK3β is inactivated by phosphorylation of the S9 residue (41); however, the level of S9 phosphorylation of GSK3β was not altered by P. gingivalis, indicating that P. gingivalis does not modulate GSK3β signaling to affect the status of β-catenin. To confirm that the ability of P. gingivalis to cleave β-catenin and GSK3β was not restricted to the 33277 lineage, TIGK cells were also infected with strain W83. Western blotting demonstrated that W83 incited processing of β-catenin and GSK3β to the same extent as 33277 (Fig. 2).
FIG 1.

P. gingivalis induces cleavage of β-catenin and GSK3β. TIGKs were infected with P. gingivalis 33277 at the MOIs and times indicated. Control cells (lanes C) were not infected. Immunoblots of cell lysates were probed with the antibodies shown. Composite images are representative of four biological replicates.
FIG 2.
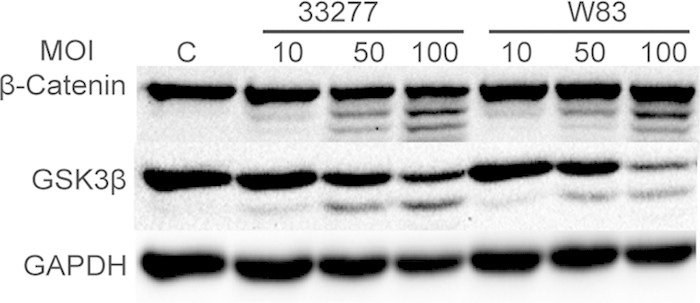
P. gingivalis-induced cleavage of β-catenin is not strain dependent. TIGKs were infected with P. gingivalis strain 33277 or W83 for 2 h at the MOIs indicated. Control cells (lane C) were not infected. Immunoblots of cell lysates were probed with the antibodies shown. Images are representative of three biological replicates.
P. gingivalis gingipains are involved in β-catenin processing.
An increase in the proteolysis of β-catenin could be the result of the direct action of the P. gingivalis proteinases or elevated epithelial cell proteasomal degradation. To distinguish between these possibilities, P. gingivalis was preincubated with TLCK, a gingipain inhibitor, or TIGKs were preincubated with MG132, an inhibitor of the eukaryotic proteasome, prior to infection. Western blotting (Fig. 3) showed that TLCK completely prevented cleavage of β-catenin by P. gingivalis, whereas MG132 had no effect on the integrity of β-catenin. Moreover, proteolytic processing of GSK3β was also inhibited by TLCK. These data suggest that proteolysis of β-catenin and GSK3β is a direct action of the gingipain proteases and not a consequence of increased proteasomal activity in the host cells. To corroborate these data, we investigated the properties of mutants of P. gingivalis deficient in gingipain production (Fig. 4). Loss of the arginine-specific proteases RgpA and RgpB caused a significant reduction in the processing of β-catenin, while in the absence of the lysine-specific Kgp the reduction in β-catenin proteolysis was less pronounced. These results were also reflected in the degradation pattern of GSK3β. In contrast, loss of the structural subunit protein of the major fimbriae (FimA) did not impair P. gingivalis-mediated processing of β-catenin or GSK3β. The FimA fimbriae are required for efficient invasion of epithelial cells by P. gingivalis (33), and hence, collectively, these data indicate that P. gingivalis can cause breakdown of β-catenin from an extracellular location through the action of secreted gingipains, primarily the arginine-specific proteases. The ability of cell-free gingipains to degrade β-catenin was corroborated with the purified RgpB. As shown in Fig. S1A in the supplemental material, RgpB was capable of proteolysis of β-catenin in vitro. Moreover, culture supernatant from P. gingivalis wild type but not from the ΔrgpAB Δkgp mutant caused degradation of β-catenin (see Fig. S1B in the supplemental material). In silico interrogation of the amino acid sequence of human β-catenin (GenBank accession no. CAA61107) revealed 39 arginine and 26 lysine residues in a total of 781 residues; hence, β-catenin has the potential to act as a substrate for both arginine- and lysine-specific gingipains.
FIG 3.
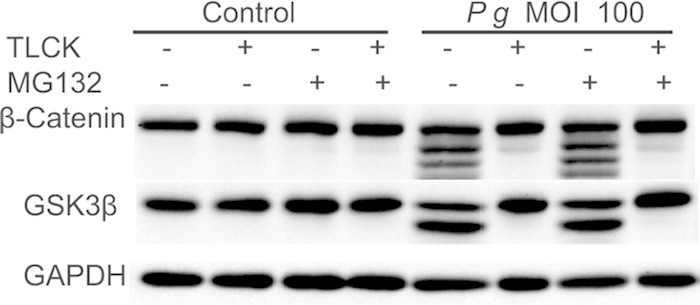
TLCK prevents cleavage of β-catenin and GSK3β by P. gingivalis. Immunoblots of lysates of TIGK cells infected with P. gingivalis 33277 (Pg) for 2 h at an MOI of 100. P. gingivalis was pretreated with/without TLCK (100 μM, gingipain inhibitor), and TIGK cells were pretreated with or without MG132 (10 μM, proteasome inhibitor). Control cells were not infected. Blots were probed with the antibodies shown. The image is representative of three biological replicates.
FIG 4.

Mutation of the gingipain genes reduces P. gingivalis-induced processing of β-catenin and GSK3β. Immunoblots of lysates of TIGK cells infected with P. gingivalis 33277 or isogenic mutants for 2 h at the MOIs indicated are shown. Control cells (lane C) were not infected. Blots were probed with the antibodies shown. Composite images (the ΔfimA mutant results are from a different blot) are representative of three biological replicates.
P. gingivalis induces the degradation of the β-catenin destruction complex.
The cleavage of GSK3β by P. gingivalis prompted us to speculate that P. gingivalis gingipain activity induces degradation and disassociation of the destruction complex. In addition to GSK3, the cytoplasmic β-catenin destruction complex contains the structural proteins Axin and APC. Western blots (Fig. 5) showed that both Axin1 and APC are degraded by P. gingivalis, and inhibition of degradation by TLCK implicates gingipains as the effectors. The loss of structural integrity of the β-catenin destruction complex will disrupt targeting to the proteasome and release β-catenin into the cytoplasmic compartment.
FIG 5.

P. gingivalis infection causes loss of APC and Axin1. Immunoblots of lysates of TIGK cells infected with P. gingivalis 33277 for 2 h at the MOIs indicated are shown. P. gingivalis was treated with or without TLCK (100 μM) for 2 h. Control cells (lanes C) were not infected. Blots were probed with the antibodies shown. The composite image is representative of three biological replicates.
β-Catenin cleavage products translocate to the nucleus.
Although P. gingivalis cleaved β-catenin, complete degradation did not occur. However, the phosphorylation status of β-catenin did not change following P. gingivalis infection, and thus the question remained as to whether the β-catenin fragments were stable and could be translocated to the nucleus or whether they were ultimately degraded by the proteasome. To address this issue, we first prepared nuclear and cytoplasmic fractions of infected TIGKs and probed with β-catenin antibodies by Western blotting. As shown in Fig. 6, β-catenin fragments were identified in the nuclear fraction after P. gingivalis infection, and there was also a significant drop in the level of cytoplasmic β-catenin. To obtain further support for these results, we visualized and quantified the level of β-catenin in the nucleus using confocal microscopy (Fig. 7). At a low MOI, there was no significant increase in detection of β-catenin; however, at an MOI of 100 P. gingivalis induced a significantly higher level of β-catenin in the nuclear area of TIGKs. In contrast, the triple gingipain mutant of P. gingivalis at an MOI of 100 was unable to increase the amount of β-catenin in the nucleus. Hence, P. gingivalis gingipain-processed β-catenin remains capable of recognition by the nuclear transport machinery.
FIG 6.
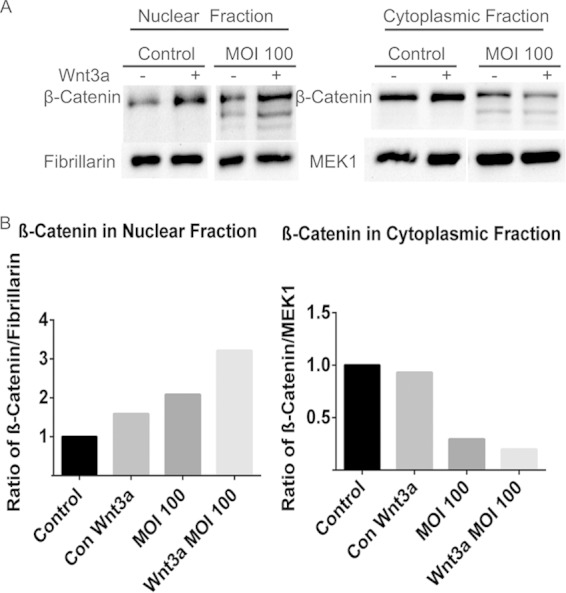
P. gingivalis induces nuclear translocation of β-catenin. (A) TIGK cells pretreated with/without Wnt3a (100 ng/ml, a positive control for nuclear translocation of β-catenin) for 2 h were infected with P. gingivalis 33277 at an MOI of 100 for 2 h. Control cells were not infected. Nuclear and cytoplasmic fractions were prepared and immunoblotted with the antibodies indicated. Fibrillarin is a control for nuclear fraction loading and MEK1 is a control for cytoplasmic fraction loading (74, 75). (B) Quantitative scanning densitometry of the images in panel A. The data are representative of three biological replicates.
FIG 7.

Immunofluorescent staining of nuclear β-catenin. TIGK cells were left uninfected (A and B), were infected with P. gingivalis 33277 (WT) at an MOI of 10 (C and D) or 100 (E and F), or were infected with P. gingivalis ΔrgpAB Δkgp at an MOI of 100 (G and H) for 2 h. The cells were fixed, stained with Hoechst 33342, and probed with β-catenin antibodies. Panels A, C, E, and G show images of β-catenin (green), and panels B, D, F, and H show merged images of β-catenin (green) and nuclei (blue). Cells were imaged at magnification ×63 and are shown as representative confocal projections. (I) Quantitative analysis of confocal image stacks showing β-catenin in the nuclear area of TIGK cells infected with P. gingivalis WT or ΔrgpAB Δkgp strains at the MOIs and times shown or left uninfected (column C). The data are means, and error bars indicate standard deviations (n = 100 cells analyzed under each condition; ***, P < 0.005 by ANOVA with Tukey multiple-comparison test). The data are representative of three biological replicates.
P. gingivalis-processed β-catenin is functionally active.
To assess the functional consequences of β-catenin proteolytic processing following P. gingivalis infection, we performed a promoter-reporter assay using the TCF/LEF-responsive element as the target. Figure 8 shows that P. gingivalis infection of TIGKs induces luciferase promoter activity, indicating that P. gingivalis-processed β-catenin remains capable of activating responsive genes. The role of gingipains in the activation of β-catenin was verified by the finding that the RgpA/B- and Kgp-deficient mutant of P. gingivalis did not activate the promoter, whereas purified RgpB was functionally active. In addition, preincubation of P. gingivalis or RgpB with the gingipain inhibitor TLCK prevented promoter activity. The prevalent oral organisms S. gordonii and F. nucleatum, which do not produce gingipains, did not activate the β-catenin-responsive reporter (Fig. 8), indicating specificity of this response for P. gingivalis.
FIG 8.
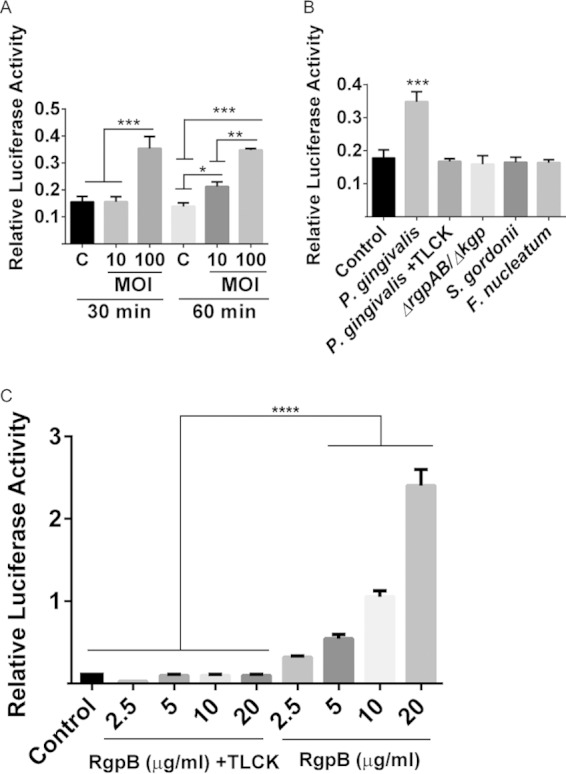
P. gingivalis gingipains upregulate the activity of a β-catenin-responsive promoter. TIGKs were transiently transfected with the TCF/LEF promoter-luciferase reporter plasmid or a constitutively expressing Renilla luciferase reporter. The TCF/LEF luciferase activity was normalized to the level of Renilla luciferase. (A) Transfected cells were infected with P. gingivalis 33277 at the MOIs and times indicated or were left uninfected (column C). (B) Transfected cells were infected for 1 h at an MOI of 100 with P. gingivalis 33277 with or without pretreatment with TLCK (100 μM, 2 h), P. gingivalis ΔrgpAB Δkgp, S. gordonii, or F. nucleatum strains. (C) Transfected cells were reacted with purified RgpB, with or without TLCK (100 μM), at the concentration indicated for 1 h. Control cells received no exogenous protein. The data are means, and error bars indicate the standard deviations (n = 3; *, P < 0.01; **, P < 0.005; ***, P < 0.001 by ANOVA with Tukey multiple-comparison test). The data are representative of three biological replicates.
P. gingivalis does not increase Wnt levels.
To ensure that activation of β-catenin by P. gingivalis is not the result of independent upregulation of Wnt, we tested mRNA and protein levels of Wnt3a in TIGK cells (Fig. 9). No increase in expression of Wnt3a protein or mRNA was observed following P. gingivalis infection. Rather, P. gingivalis modestly suppressed expression of Wnt3a over a 15- to 120-min time period. Collectively, therefore, our results indicate that proteolytic processing of β-catenin, along with disassociation of the destruction complex, allows the release of β-catenin fragments and recognition by the nuclear translocation machinery. Within the nucleus, the processed β-catenin remains functionally active, and transcription of β-catenin-responsive genes will ensue.
FIG 9.
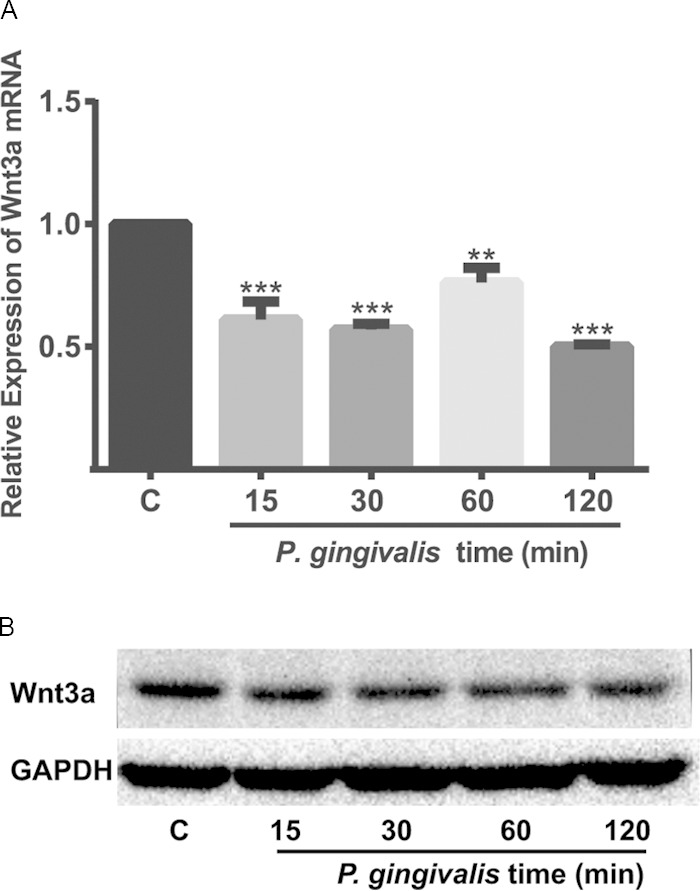
P. gingivalis infection downregulates Wnt3a. TIGK cells were infected with P. gingivalis 33277 at an MOI of 100 for the times indicated. (A) Wnt3a mRNA levels were measured by qRT-PCR. The data were normalized to GAPDH mRNA and are expressed relative to noninfected controls (column C). The results are means, and error bars indicate standard deviations (n = 3; **, P < 0.005; ***, P < 0.001 by ANOVA with Tukey multiple-comparison test). The data are representative of three biological replicates. (B) Western blot of cell lysates probed with Wnt3a or GAPDH antibodies. The image is representative of three biological replicates.
DISCUSSION
The results of the present study suggest a novel noncanonical mechanism of β-catenin activation by proteolytic processing. We show that infection of epithelial cells with P. gingivalis does not significantly impact the phosphorylation status of β-catenin but rather results in its cleavage. The proteolytically processed products of β-catenin are translocated into the nucleus, where they can activate the TCF/LEF promoter element. Although manipulation of Wnt/β-catenin signaling by pathogenic bacteria has been established previously, the mechanisms identified thus far do not involve direct action on the stability of β-catenin. For example, Salmonella strains can impair β-catenin signaling by causing an upregulation of GSK3β-dependent phosphorylation with consequent increased proteasomal degradation (42). Interestingly, Salmonella strains expressing AvrA can activate β-catenin, as the deubiquitinase activity of AvrA prevents ubiquitination and degradation (43, 44). β-Catenin activation is also antagonized by the edema toxin of Bacillus anthracis, which prevents the Wnt-dependent phosphoinactivation of GSK3β (45), and by the Clostridium difficile toxin A (TcdA) through the inactivation of Rho GTPases (46).
In addition to the hierarchical system of posttranslational modifications that regulate β-catenin, localization and activity can also be controlled by proteolytic activity in the cell. Matrilysin (MMP-7) has been demonstrated to release β-catenin from the cell membrane, after which the β-catenin is degraded in the cytosol. However, in the presence of a β-catenin-stabilizing Wnt signal, β-catenin can be translocated in active form to the nucleus (47). P. gingivalis is an asaccharolytic organism and requires the action of proteolytic enzymes to provide nutritional substrates. The arginine-specific gingipains RgpA and RgpB and the lysine-specific gingipain Kgp are secreted by the organism and are the predominant extracellular proteinases of P. gingivalis (11). Gingipains can also enter host epithelial cells, both from the extracellular milieu and packaged in outer membrane vesicles (48, 49). The observations with gingipain mutants of P. gingivalis suggest that the RgpA/B gingipains are more potent than Kgp at processing of β-catenin by P. gingivalis, although the presence of all three enzymes was required for maximal β-catenin breakdown. Although care should be exercised in the interpretation of data from gingipain mutant experiments, since the enzymes are involved in the processing of a number of cell surface proteins of P. gingivalis, the finding that TLCK, a broad-spectrum inhibitor of gingipains, also reduced the level of β-catenin degradation lends support to the central role of RgpA/B. In addition to activation of β-catenin signaling, gingipains potentially play a multimodal role in the disruption of cellular and inflammatory homeostasis. In established OSCC cell lines, P. gingivalis gingipains activate PAR2 and PAR4, leading to the phosphorylation of IκB, the nuclear translocation of NF-κB, and increased production of proMMP9 (50, 51). In addition, gingipains can cleave proMMP9, generating the mature active enzyme (52), which is important for cancer cell invasion and metastasis. Gingipains can also proteolytically process proteins on the epithelial cell surface, causing release and redistribution, with consequent effects on signal transduction and inflammatory responses (53, 54). Intracellularly, P. gingivalis gingipains can degrade mammalian target of rapamycin (mTOR), thus disrupting the mTOR pathway which regulates the cytoskeleton, as well as cleave β-actin directly (55, 56). In trophoblasts, gingipains can degrade P53 and the E3 ubiquitin protein ligase homolog protein (MDM2) and modulate the activity of multiple signaling pathways, resulting in both cell cycle arrest and cell death (57). The capacity of gingipains to regulate the expression of inflammatory mediators at the mRNA level has also been demonstrated. In gingival fibroblasts, gingipains increase TGFβ gene expression, while suppressing the expression of CXCL8 (58).
P. gingivalis is a host-adapted organism that occupies several microenvironments in the oral cavity, including the subgingival biofilm on the root surfaces of the teeth, and the crevicular fluid, as well as in and on the epithelial cells that line the gingival crevice. Several distinct lineages can be recovered in vivo, which can vary in a number of properties, including fimbriation state and capsule production. Strain 33277 is fimbriated, but it does not produce a discrete capsule, whereas strain W83 is afimbriate but encapsulated (59, 60). 33277 and W83 showed similar activities, a finding consistent with a role for proteases in β-catenin processing. Indeed, clinical isolates of P. gingivalis are generally strongly proteolytic (61); hence, the ability to activate β-catenin may be widely conserved in the species. Furthermore, as gingipains are secreted, the organism does not require a close association with epithelial cells to impact β-catenin signaling. This concept is supported by the result with the FimA-deficient mutant, which, while unable to instigate fimbrial attachment and intracellular invasion, was as effective as 33277 in the cleavage of β-catenin.
Purified lipopolysaccharide (LPS) from P. gingivalis has been shown to both increase the phosphoinactivation of GSK3β and attenuate β-catenin activity in human stem cells from the apical papilla (62) and activate β-catenin through decreasing phosphoinactivation of GSK3β in rat bone marrow mesenchymal cells (63). In addition, P. gingivalis LPS can inhibit osteoblast differentiation by promoting the expression of Notch target genes and suppressing canonical Wnt/β-catenin signaling through GSK3β (64). In gingival epithelial cells challenged with whole cells of P. gingivalis we did not observe an effect on GSK3β phosphorylation. However, processing of GSK3β occurred, which was also dependent on the activity of the P. gingivalis gingipains. Although GSK3β partial degradation products can remain catalytically active (65), we postulate that GSK3β proteolysis negatively affected the integrity of the destruction complex. This notion was supported by the finding that P. gingivalis caused degradation of the scaffolding proteins Axin1 and APC. Disassociation of the destruction complex will divert β-catenin from the proteasomal pathway and allow access to the nuclear translocation machinery.
β-Catenin controls the expression of a number of genes involved in cell proliferation (Myc and cyclin D1) and migration (MMP-7) (66). Aberrant β-catenin signaling is associated with the development of malignancies, including OSCC (67–69), and β-catenin activation and nuclear localization are correlated with progression of the severity of OSCC (70, 71). P. gingivalis is a dysbiotic organism that is becoming increasingly associated with cancer, pancreatic cancer and OSCC in particular (18, 19). P. gingivalis can be recovered from OSCC surfaces in significantly higher numbers than from contiguous healthy mucosa (72) and detected by immunohistochemistry in gingival carcinomas (73). Hence, the results of the present study suggest that noncanonical activation of β-catenin signaling by P. gingivalis may be a potential mechanism by which P. gingivalis could contribute to tumorigenesis.
The P. gingivalis-epithelial cell interface is dynamic and multidimensional, and the properties of infected cells will reflect the collective output of the interaction between host signaling pathways and a number of bioactive P. gingivalis molecules, including fimbriae, LPS, and phosphatases, as well as proteases. In the present study we demonstrate that the gingipain proteases can proteolytically process β-catenin and GSK3β in gingival epithelial cells. Processed β-catenin can enter the nucleus and activate the TCF/LEF promoter element. Further characterization of the role of noncanonical activation of β-catenin may provide novel insights into the pathogenesis of local and systemic diseases associated with P. gingivalis.
Supplementary Material
ACKNOWLEDGMENTS
The support of the National Institutes of Health through grants DE01111 and DE017921 (R.J.L.), DE022597 and DE023207 (J.P.), DE023633 (H.W.), and DE017680 (D.A.S.) is gratefully acknowledged.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00302-15.
REFERENCES
- 1.Darveau RP, Hajishengallis G, Curtis MA. 2012. Porphyromonas gingivalis as a potential community activist for disease. J Dent Res 91:816–820. doi: 10.1177/0022034512453589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hajishengallis G, Lamont RJ. 2012. Beyond the red complex and into more complexity: the polymicrobial synergy and dysbiosis (PSD) model of periodontal disease etiology. Mol Oral Microbiol 27:409–419. doi: 10.1111/j.2041-1014.2012.00663.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lamont RJ, Jenkinson HF. 1998. Life below the gum line: pathogenic mechanisms of Porphyromonas gingivalis. Microbiol Mol Biol Rev 62:1244–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tribble GD, Lamont RJ. 2010. Bacterial invasion of epithelial cells and spreading in periodontal tissue. Periodontol 2000 52:68–83. doi: 10.1111/j.1600-0757.2009.00323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yilmaz O, Verbeke P, Lamont RJ, Ojcius DM. 2006. Intercellular spreading of Porphyromonas gingivalis infection in primary gingival epithelial cells. Infect Immun 74:703–710. doi: 10.1128/IAI.74.1.703-710.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jauregui CE, Wang Q, Wright CJ, Takeuchi H, Uriarte SM, Lamont RJ. 2013. Suppression of T-cell chemokines by Porphyromonas gingivalis. Infect Immun 81:2288–2295. doi: 10.1128/IAI.00264-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lamont RJ, Hajishengallis G. 2015. Polymicrobial synergy and dysbiosis in inflammatory disease. Trends Mol Med 21:172–183. doi: 10.1016/j.molmed.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moffatt CE, Lamont RJ. 2011. Porphyromonas gingivalis induction of microRNA-203 expression controls suppressor of cytokine signaling 3 in gingival epithelial cells. Infect Immun 79:2632–2637. doi: 10.1128/IAI.00082-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takeuchi H, Hirano T, Whitmore SE, Morisaki I, Amano A, Lamont RJ. 2013. The serine phosphatase SerB of Porphyromonas gingivalis suppresses IL-8 production by dephosphorylation of NF-κB RelA/p65. PLoS Pathog 9:e1003326. doi: 10.1371/journal.ppat.1003326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hajishengallis G. 2014. The inflammophilic character of the periodontitis-associated microbiota. Mol Oral Microbiol 29:248–257. doi: 10.1111/omi.12065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Curtis MA, Aduse-Opoku J, Rangarajan M. 2001. Cysteine proteases of Porphyromonas gingivalis. Crit Rev Oral Biol Med 12:192–216. doi: 10.1177/10454411010120030101. [DOI] [PubMed] [Google Scholar]
- 12.Nakayama K. 2015. Porphyromonas gingivalis and related bacteria: from colonial pigmentation to the type IX secretion system and gliding motility. J Periodontal Res 50:1–8. doi: 10.1111/jre.12255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Veith PD, Chen YY, Gorasia DG, Chen D, Glew MD, O'Brien-Simpson NM, Cecil JD, Holden JA, Reynolds EC. 2014. Porphyromonas gingivalis outer membrane vesicles exclusively contain outer membrane and periplasmic proteins and carry a cargo enriched with virulence factors. J Proteome Res 13:2420–2432. doi: 10.1021/pr401227e. [DOI] [PubMed] [Google Scholar]
- 14.Mantri CK, Chen CH, Dong X, Goodwin JS, Pratap S, Paromov V, Xie H. 2015. Fimbriae-mediated outer membrane vesicle production and invasion of Porphyromonas gingivalis. Microbiologyopen 4:53–65. doi: 10.1002/mbo3.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haurat MF, Aduse-Opoku J, Rangarajan M, Dorobantu L, Gray MR, Curtis MA, Feldman MF. 2011. Selective sorting of cargo proteins into bacterial membrane vesicles. J Biol Chem 286:1269–1276. doi: 10.1074/jbc.M110.185744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Glew MD, Veith PD, Peng B, Chen YY, Gorasia DG, Yang Q, Slakeski N, Chen D, Moore C, Crawford S, Reynolds EC. 2012. PG0026 is the C-terminal signal peptidase of a novel secretion system of Porphyromonas gingivalis. J Biol Chem 287:24605–24617. doi: 10.1074/jbc.M112.369223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bostanci N, Belibasakis GN. 2012. Porphyromonas gingivalis: an invasive and evasive opportunistic oral pathogen. FEMS Microbiol Lett 333:1–9. doi: 10.1111/j.1574-6968.2012.02579.x. [DOI] [PubMed] [Google Scholar]
- 18.Whitmore SE, Lamont RJ. 2014. Oral bacteria and cancer. PLoS Pathog 10:e1003933. doi: 10.1371/journal.ppat.1003933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Atanasova KR, Yilmaz O. 2014. Looking in the Porphyromonas gingivalis cabinet of curiosities: the microbium, the host and cancer association. Mol Oral Microbiol 29:55–66. doi: 10.1111/omi.12047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mao S, Park Y, Hasegawa Y, Tribble GD, James CE, Handfield M, Stavropoulos MF, Yilmaz O, Lamont RJ. 2007. Intrinsic apoptotic pathways of gingival epithelial cells modulated by Porphyromonas gingivalis. Cell Microbiol 9:1997–2007. doi: 10.1111/j.1462-5822.2007.00931.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yao L, Jermanus C, Barbetta B, Choi C, Verbeke P, Ojcius DM, Yilmaz O. 2010. Porphyromonas gingivalis infection sequesters proapoptotic Bad through Akt in primary gingival epithelial cells. Mol Oral Microbiol 25:89–101. doi: 10.1111/j.2041-1014.2010.00569.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuboniwa M, Hasegawa Y, Mao S, Shizukuishi S, Amano A, Lamont RJ, Yilmaz O. 2008. Porphyromonas gingivalis accelerates gingival epithelial cell progression through the cell cycle. Microbes Infect 10:122–128. doi: 10.1016/j.micinf.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Handfield M, Mans JJ, Zheng G, Lopez MC, Mao S, Progulske-Fox A, Narasimhan G, Baker HV, Lamont RJ. 2005. Distinct transcriptional profiles characterize oral epithelium-microbiota interactions. Cell Microbiol 7:811–823. doi: 10.1111/j.1462-5822.2005.00513.x. [DOI] [PubMed] [Google Scholar]
- 24.Clevers H, Nusse R. 2012. Wnt/beta-catenin signaling and disease. Cell 149:1192–1205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 25.Kikuchi A, Kishida S, Yamamoto H. 2006. Regulation of Wnt signaling by protein-protein interaction and posttranslational modifications. Exp Mol Med 38:1–10. doi: 10.1038/emm.2006.1. [DOI] [PubMed] [Google Scholar]
- 26.Dierick H, Bejsovec A. 1999. Cellular mechanisms of wingless/Wnt signal transduction. Curr Top Dev Biol 43:153–190. [DOI] [PubMed] [Google Scholar]
- 27.Fang D, Hawke D, Zheng Y, Xia Y, Meisenhelder J, Nika H, Mills GB, Kobayashi R, Hunter T, Lu Z. 2007. Phosphorylation of β-catenin by AKT promotes beta-catenin transcriptional activity. J Biol Chem 282:11221–11229. doi: 10.1074/jbc.M611871200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang H, He X. 2008. Wnt/β-catenin signaling: new (and old) players and new insights. Curr Opin Cell Biol 20:119–125. doi: 10.1016/j.ceb.2008.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brembeck FH, Rosario M, Birchmeier W. 2006. Balancing cell adhesion and Wnt signaling, the key role of β-catenin. Curr Opin Genet Dev 16:51–59. doi: 10.1016/j.gde.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 30.Fracalossi AC, Silva MDS, Oshima CT, Ribeiro DA. 2010. Wnt/beta-catenin signaling pathway following rat tongue carcinogenesis induced by 4-nitroquinoline 1-oxide. Exp Mol Pathol 88:176–183. doi: 10.1016/j.yexmp.2009.11.004. [DOI] [PubMed] [Google Scholar]
- 31.Lo Muzio L. 2001. A possible role for the WNT-1 pathway in oral carcinogenesis. Crit Rev Oral Biol Med 12:152–165. doi: 10.1177/10454411010120020501. [DOI] [PubMed] [Google Scholar]
- 32.Rubinstein MR, Wang X, Liu W, Hao Y, Cai G, Han YW. 2013. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/beta-catenin signaling via its FadA adhesin. Cell Host Microbe 14:195–206. doi: 10.1016/j.chom.2013.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yilmaz O, Watanabe K, Lamont RJ. 2002. Involvement of integrins in fimbria-mediated binding and invasion by Porphyromonas gingivalis. Cell Microbiol 4:305–314. doi: 10.1046/j.1462-5822.2002.00192.x. [DOI] [PubMed] [Google Scholar]
- 34.Nakayama K, Kadowaki T, Okamoto K, Yamamoto K. 1995. Construction and characterization of arginine-specific cysteine proteinase (Arg-gingipain)-deficient mutants of Porphyromonas gingivalis: evidence for significant contribution of Arg-gingipain to virulence. J Biol Chem 270:23619–23626. [DOI] [PubMed] [Google Scholar]
- 35.Shi Y, Ratnayake DB, Okamoto K, Abe N, Yamamoto K, Nakayama K. 1999. Genetic analyses of proteolysis, hemoglobin binding, and hemagglutination of Porphyromonas gingivalis: construction of mutants with a combination of rgpA, rgpB, kgp, and hagA. J Biol Chem 274:17955–17960. [DOI] [PubMed] [Google Scholar]
- 36.Moffatt-Jauregui CE, Robinson B, de Moya AV, Brockman RD, Roman AV, Cash MN, Culp DJ, Lamont RJ. 2013. Establishment and characterization of a telomerase immortalized human gingival epithelial cell line. J Periodontal Res 48:713–721. 10.1111/jre.12059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Veillard F, Sztukowska M, Mizgalska D, Ksiazek M, Houston J, Potempa B, Enghild JJ, Thogersen IB, Gomis-Ruth FX, Nguyen KA, Potempa J. 2013. Inhibition of gingipains by their profragments as the mechanism protecting Porphyromonas gingivalis against premature activation of secreted proteases. Biochim Biophys Acta 1830:4218–4228. doi: 10.1016/j.bbagen.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen JS, Li HC, Lin SI, Yang CH, Chien WY, Syu CL, Lo SY. 2015. Cleavage of dicer protein by I7 protease during vaccinia virus infection. PLoS One 10:e0120390. doi: 10.1371/journal.pone.0120390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hirano T, Beck DA, Wright CJ, Demuth DR, Hackett M, Lamont RJ. 2013. Regulon controlled by the GppX hybrid two-component system in Porphyromonas gingivalis. Mol Oral Microbiol 28:70–81. doi: 10.1111/omi.12007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moffatt CE, Inaba H, Hirano T, Lamont RJ. 2012. Porphyromonas gingivalis SerB-mediated dephosphorylation of host cell cofilin modulates invasion efficiency. Cell Microbiol 14:577–588. doi: 10.1111/j.1462-5822.2011.01743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang H, Kumar A, Lamont RJ, Scott DA. 2014. GSK3β and the control of infectious bacterial diseases. Trends Microbiol 22:208–217. doi: 10.1016/j.tim.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Duan Y, Liao AP, Kuppireddi S, Ye Z, Ciancio MJ, Sun J. 2007. beta-Catenin activity negatively regulates bacterium-induced inflammation. Lab Invest 87:613–624. [DOI] [PubMed] [Google Scholar]
- 43.Lu R, Wu S, Zhang YG, Xia Y, Liu X, Zheng Y, Chen H, Schaefer KL, Zhou Z, Bissonnette M, Li L, Sun J. 2014. Enteric bacterial protein AvrA promotes colonic tumorigenesis and activates colonic beta-catenin signaling pathway. Oncogenesis 3:e105. doi: 10.1038/oncsis.2014.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Silva-Garcia O, Valdez-Alarcon JJ, Baizabal-Aguirre VM. 2014. The Wnt/beta-catenin signaling pathway controls the inflammatory response in infections caused by pathogenic bacteria. Mediators Inflamm 2014:310183. doi: 10.1155/2014/310183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Larabee JL, DeGiusti K, Regens JL, Ballard JD. 2008. Bacillus anthracis edema toxin activates nuclear glycogen synthase kinase 3β. Infect Immun 76:4895–4904. doi: 10.1128/IAI.00889-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bezerra Lima B, Faria Fonseca B, da Graca Amado N, Moreira Lima D, Albuquerque Ribeiro R, Garcia Abreu J, de Castro Brito GA. 2014. Clostridium difficile toxin A attenuates Wnt/beta-catenin signaling in intestinal epithelial cells. Infect Immun 82:2680–2687. doi: 10.1128/IAI.00567-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rims CR, McGuire JK. 2014. Matrilysin (MMP-7) catalytic activity regulates beta-catenin localization and signaling activation in lung epithelial cells. Exp Lung Res 40:126–136. doi: 10.3109/01902148.2014.890681. [DOI] [PubMed] [Google Scholar]
- 48.Scragg MA, Alsam A, Rangarajan M, Slaney JM, Shepherd P, Williams DM, Curtis MA. 2002. Nuclear targeting of Porphyromonas gingivalis W50 protease in epithelial cells. Infect Immun 70:5740–5750. doi: 10.1128/IAI.70.10.5740-5750.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Furuta N, Takeuchi H, Amano A. 2009. Entry of Porphyromonas gingivalis outer membrane vesicles into epithelial cells causes cellular functional impairment. Infect Immun 77:4761–4770. doi: 10.1128/IAI.00841-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Inaba H, Amano A, Lamont RJ, Murakami Y. Involvement of protease-activated receptor 4 in overexpression of matrix metalloproteinase 9 induced by Porphyromonas gingivalis. Med Microbiol Immunol, in press. [DOI] [PubMed] [Google Scholar]
- 51.Inaba H, Sugita H, Kuboniwa M, Iwai S, Hamada M, Noda T, Morisaki I, Lamont RJ, Amano A. 2014. Porphyromonas gingivalis promotes invasion of oral squamous cell carcinoma through induction of proMMP9 and its activation. Cell Microbiol 16:131–145. doi: 10.1111/cmi.12211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tokuda M, Karunakaran T, Duncan M, Hamada N, Kuramitsu H. 1998. Role of Arg-gingipain A in virulence of Porphyromonas gingivalis. Infect Immun 66:1159–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tancharoen S, Matsuyama T, Kawahara KI, Tanaka K, Lee LJ, Machigashira M, Noguchi K, Ito T, Imamura T, Potempa J, Kikuchi K, Maruyama I. 2015. Cleavage of host cytokeratin-6 by lysine-specific gingipain induces gingival inflammation in periodontitis patients. PLoS One 10:e0117775. doi: 10.1371/journal.pone.0117775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nakayama M, Inoue T, Naito M, Nakayama K, Ohara N. 2015. Attenuation of the PI3 kinase/Akt signaling pathway by Porphyromonas gingivalis gingipains RgpA, RgpB, and Kgp. J Biol Chem 290:5190–5202. doi: 10.1074/jbc.M114.591610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stafford P, Higham J, Pinnock A, Murdoch C, Douglas CW, Stafford GP, Lambert DW. 2013. Gingipain-dependent degradation of mammalian target of rapamycin pathway proteins by the periodontal pathogen Porphyromonas gingivalis during invasion. Mol Oral Microbiol 28:366–378. doi: 10.1111/omi.12030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kinane JA, Benakanakere MR, Zhao J, Hosur KB, Kinane DF. 2012. Porphyromonas gingivalis influences actin degradation within epithelial cells during invasion and apoptosis. Cell Microbiol 14:1085–1096. doi: 10.1111/j.1462-5822.2012.01780.x. [DOI] [PubMed] [Google Scholar]
- 57.Inaba H, Kuboniwa M, Sugita H, Lamont RJ, Amano A. 2012. Identification of signaling pathways mediating cell cycle arrest and apoptosis induced by Porphyromonas gingivalis in human trophoblasts. Infect Immun 80:2847–2857. doi: 10.1128/IAI.00258-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Palm E, Khalaf H, Bengtsson T. 2015. Suppression of inflammatory responses of human gingival fibroblasts by gingipains from Porphyromonas gingivalis. Mol Oral Microbiol 30:74–85. doi: 10.1111/omi.12073. [DOI] [PubMed] [Google Scholar]
- 59.Nishikawa K, Duncan MJ. 2010. Histidine kinase-mediated production and autoassembly of Porphyromonas gingivalis fimbriae. J Bacteriol 192:1975–1987. doi: 10.1128/JB.01474-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Aduse-Opoku J, Slaney JM, Hashim A, Gallagher A, Gallagher RP, Rangarajan M, Boutaga K, Laine ML, Van Winkelhoff AJ, Curtis MA. 2006. Identification and characterization of the capsular polysaccharide (K-antigen) locus of Porphyromonas gingivalis. Infect Immun 74:449–460. doi: 10.1128/IAI.74.1.449-460.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tribble GD, Kerr JE, Wang BY. 2013. Genetic diversity in the oral pathogen Porphyromonas gingivalis: molecular mechanisms and biological consequences. Future Microbiol 8:607–620. doi: 10.2217/fmb.13.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang J, Dai J, Liu B, Gu S, Cheng L, Liang J. 2013. Porphyromonas gingivalis lipopolysaccharide activates canonical Wnt/β-catenin and p38 MAPK signaling in stem cells from the apical papilla. Inflammation 36:1393–1402. doi: 10.1007/s10753-013-9679-y. [DOI] [PubMed] [Google Scholar]
- 63.Tang Y, Zhou X, Gao B, Xu X, Sun J, Cheng L, Zheng L. 2014. Modulation of Wnt/beta-catenin signaling attenuates periapical bone lesions. J Dent Res 93:175–182. doi: 10.1177/0022034513512507. [DOI] [PubMed] [Google Scholar]
- 64.Xing Q, Ye Q, Fan M, Zhou Y, Xu Q, Sandham A. 2010. Porphyromonas gingivalis lipopolysaccharide inhibits the osteoblastic differentiation of preosteoblasts by activating Notch1 signaling. J Cell Physiol 225:106–114. doi: 10.1002/jcp.22201. [DOI] [PubMed] [Google Scholar]
- 65.Kandasamy AD, Chow AK, Ali MA, Schulz R. 2010. Matrix metalloproteinase-2 and myocardial oxidative stress injury: beyond the matrix. Cardiovasc Res 85:413–423. doi: 10.1093/cvr/cvp268. [DOI] [PubMed] [Google Scholar]
- 66.Herbst A, Jurinovic V, Krebs S, Thieme SE, Blum H, Goke B, Kolligs FT. 2014. Comprehensive analysis of beta-catenin target genes in colorectal carcinoma cell lines with deregulated Wnt/beta-catenin signaling. BMC Genomics 15:74. doi: 10.1186/1471-2164-15-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Debelec-Butuner B, Alapinar C, Ertunc N, Gonen-Korkmaz C, Yorukoglu K, Korkmaz KS. 2014. TNFα-mediated loss of beta-catenin/E-cadherin association and subsequent increase in cell migration is partially restored by NKX3.1 expression in prostate cells. PLoS One 9:e109868. doi: 10.1371/journal.pone.0109868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Keerthivasan S, Aghajani K, Dose M, Molinero L, Khan MW, Venkateswaran V, Weber C, Emmanuel AO, Sun T, Bentrem DJ, Mulcahy M, Keshavarzian A, Ramos EM, Blatner N, Khazaie K, Gounari F. 2014. β-Catenin promotes colitis and colon cancer through imprinting of proinflammatory properties in T cells. Sci Transl Med 6:225ra28. doi: 10.1126/scitranslmed.3007607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Uraguchi M, Morikawa M, Shirakawa M, Sanada K, Imai K. 2004. Activation of WNT family expression and signaling in squamous cell carcinomas of the oral cavity. J Dent Res 83:327–332. doi: 10.1177/154405910408300411. [DOI] [PubMed] [Google Scholar]
- 70.Chaw SY, Majeed AA, Dalley AJ, Chan A, Stein S, Farah CS. 2012. Epithelial to mesenchymal transition (EMT) biomarkers—E-cadherin, beta-catenin, APC and Vimentin—in oral squamous cell carcinogenesis and transformation. Oral Oncol 48:997–1006. doi: 10.1016/j.oraloncology.2012.05.011. [DOI] [PubMed] [Google Scholar]
- 71.Tanaka N, Odajima T, Ogi K, Ikeda T, Satoh M. 2003. Expression of E-cadherin, alpha-catenin, and beta-catenin in the process of lymph node metastasis in oral squamous cell carcinoma. Br J Cancer 89:557–563. doi: 10.1038/sj.bjc.6601124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nagy KN, Sonkodi I, Szoke I, Nagy E, Newman HN. 1998. The microflora associated with human oral carcinomas. Oral Oncol 34:304–308. doi: 10.1016/S1368-8375(98)00008-6. [DOI] [PubMed] [Google Scholar]
- 73.Katz J, Onate MD, Pauley KM, Bhattacharyya I, Cha S. 2011. Presence of Porphyromonas gingivalis in gingival squamous cell carcinoma. Int J Oral Sci 3:209–215. doi: 10.4248/IJOS11075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lang M, Borgmann M, Oberhuber G, Evstatiev R, Jimenez K, Dammann KW, Jambrich M, Khare V, Campregher C, Ristl R, Gasche C. 2013. Thymoquinone attenuates tumor growth in ApcMin mice by interference with Wnt-signaling. Mol Cancer 12:41. doi: 10.1186/1476-4598-12-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Peng Y, Zhao S, Song L, Wang M, Jiao K. 2013. Sertad1 encodes a novel transcriptional coactivator of SMAD1 in mouse embryonic hearts. Biochem Biophys Res Commun 441:751–756. doi: 10.1016/j.bbrc.2013.10.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.