

Abstract
Rhodococcus equi is a facultative intracellular pathogen of macrophages, relying on the presence of a conjugative virulence plasmid harboring a 21-kb pathogenicity island (PAI) for growth in host macrophages. The PAI encodes a family of 6 virulence-associated proteins (Vaps) in addition to 20 other proteins. The contribution of these to virulence has remained unclear. We show that the presence of only 3 virulence plasmid genes (of 73 in total) is required and sufficient for intracellular growth. These include a single vap family member, vapA, and two PAI-located transcriptional regulators, virR and virS. Both transcriptional regulators are essential for wild-type-level expression of vapA, yet vapA expression alone is not sufficient to allow intracellular growth. A whole-genome microarray analysis revealed that VirR and VirS substantially integrate themselves into the chromosomal regulatory network, significantly altering the transcription of 18% of all chromosomal genes. This pathoadaptation involved significant enrichment of select gene ontologies, in particular, enrichment of genes involved in transport processes, energy production, and cellular metabolism, suggesting a major change in cell physiology allowing the bacterium to grow in the hostile environment of the host cell. The results suggest that following the acquisition of the virulence plasmid by an avirulent ancestor of R. equi, coevolution between the plasmid and the chromosome took place, allowing VirR and VirS to regulate the transcription of chromosomal genes in a process that ultimately promoted intracellular growth. Our findings suggest a mechanism for cooption of existing chromosomal traits during the evolution of a pathogenic bacterium from an avirulent saprophyte.
INTRODUCTION
The genus Rhodococcus comprises a large number of metabolically diverse species that have attracted considerable biotechnological interest because of their ability to metabolize a wide variety of substrates, which finds applications in bioremediation and in the synthesis of precursors of pharmaceutical compounds (1). The genus contains only two pathogenic species: the plant pathogen Rhodococcus fascians and the animal pathogen Rhodococcus equi (2). Although the latter species was initially isolated from young foals, it has subsequently been isolated from a wide range of animals and humans (3). Disease in foals and immunosuppressed humans usually presents as pyogranulomatous pneumonia, although other manifestations, including osteomyelitis, may also occur. In pigs and cattle, R. equi is usually associated with submandibular lymphadenitis (3). In addition to having a pathogenic lifestyle, R. equi grows readily as a saprophyte in soils, as well as in the equine intestinal tract (3).
R. equi is a parasite of macrophages, which prevents killing by the host cell through inhibition of phagosomal maturation, resulting in the formation of R. equi-containing vacuoles devoid of lysosomal markers, including cathepsin D and the proton-pumping vacuolar ATPase (vATPase) complex (4–6). The growth of R. equi is cytotoxic, resulting in the necrotic death of the phagocytic cell (7, 8). The inhibition of phagosomal maturation as a strategy for survival in macrophages is shared with the closely related organism Mycobacterium tuberculosis (9). However, despite the superficial similarities in virulence strategy, the underlying mechanisms are different. The subversion of the normal functioning of macrophages by R. equi is dependent on the presence of a conjugative plasmid harboring a pathogenicity island encoding a family of six virulence-associated proteins (Vap) that do not occur in M. tuberculosis (10–12). One of these, the cell envelope-associated protein VapA, is required, but not sufficient, for the inhibition of phagosomal maturation and for intracellular growth (13–16). It is not clear whether the remaining five vap genes (vapC, vapD, vapE, vapG, and vapH) play any role in macrophage parasitism.
The vap genes are organized into five transcriptional units (17–19). One of these, the virR operon, contains four genes in addition to vapH: virR, icgA, orf7, and virS (18). The icgA gene encodes a transport protein belonging to the major facilitator family. Expression of this protein reduces the intracellular growth rate of R. equi, resulting in increased macrophage viability, allowing R. equi to reside longer in macrophages (20). VirR and VirS are transcriptional regulators belonging to the LysR-type transcriptional regulators and response regulators of two-component systems, respectively (12). Both VirR and VirS are required for wild-type-level expression of VapA, which has been offered as an explanation for the attenuation of mutants lacking either virR or virS (21–23).
In addition to the plasmid-encoded virulence factors, a number of virulence-associated factors, including cytoadhesive pili (24), the hydroxamate siderophore rhequichelin (25), and isocitrate lyase (26), are encoded by the chromosome. Comparative genome analysis demonstrated that many of these have orthologues in nonpathogenic actinobacteria. This suggests that in addition to the acquisition of a conjugative plasmid containing a pathogenicity island, virulence evolved by cooption of existing chromosomally encoded traits via an as yet unknown mechanism (24).
In this study, we demonstrate that the presence of just three pathogenicity island genes, vapA, virR, and virS, is required and sufficient for intracellular growth in macrophages. While this study confirms the central role of VapA in virulence, it also reveals that the two virulence plasmid-encoded transcriptional regulators, VirR and VirS, modify the chromosomal transcriptome, thus altering R. equi physiology to allow macrophage parasitism. This study thus provides evidence for the coevolution of the conjugative virulence plasmid and the ancestor of R. equi. In addition, it suggests a mechanism for the cooption of existing chromosomally encoded traits in the emergence of R. equi virulence.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
The bacterial strains and plasmids used in this study are listed in Table S1 in the supplemental material. Bacteria were grown on either brain heart infusion (BHI) or lysogeny broth (LB) medium. Antibiotics, when necessary, were used at the following concentrations: apramycin, 80 μg/ml (R. equi) or 30 μg/ml (Escherichia coli); hygromycin, 180 μg/ml.
Mutant construction.
R. equi was made electrocompetent and was transformed by electroporation as described previously (20). Unmarked deletion mutants were constructed using the positive selection vector pSelAct (27). The construction of the R. equi multiple-deletion mutant (MDM) is described in the supporting Materials and Methods in the supplemental material.
Expression of virR, virS, and vapA.
Plasmids pVirRSvapA, pVirRvapA, and pVirSvapA were constructed in the integrative vector pSET152 and contain the native promoters of the virR and vapA operons. The expression of vapA from its native promoter is dependent on both virR and virS. To drive the expression of vapA in the absence of either virR or virS, vapA was fused to the mycobacterial hsp60 promoter. The construction of these expression plasmids is described in the supporting Materials and Methods in the supplemental material.
Intracellular growth of R. equi in macrophages.
Murine J774.1 macrophages were maintained in Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal calf serum (FCS) and 2 mM glutamine and were incubated at 37°C under 5% CO2. Murine bone marrow-derived macrophages were isolated and cultured as described previously (14). For intracellular growth assays, macrophages were seeded in 24-well tissue culture plates at 2 × 105 per well and were incubated at 37°C under 5% CO2 overnight. Macrophages were infected at a multiplicity of infection (MOI) of 10 as described previously (20). At various times postinfection, macrophage monolayers were lysed with water (14), dilutions of the lysate were spread onto BHI agar, and plates were incubated (at 37°C for 48 h) to determine associated CFU.
Microarray expression analysis.
Custom 12×135K gene expression microarrays (Roche NimbleGen) were designed with coverage of 4,525 (100%) chromosomal genes, 73 (100%) virulence plasmid-borne genes, and 20 pseudogenes. For each of these, seven individual probes (n = 32,319) of 60 nucleotides (nt) were designed and were spotted in quadruplicate. REQ01550 and REQ02340 are 100% identical and were covered by the same probes.
Total RNA was isolated from logarithmically grown R. equi bacteria using the guanidinium isothiocyanate method as described previously (20). Double-stranded cDNA was synthesized using the cDNA Synthesis System using random hexameric primers (Roche) and was cleaned using the High Pure PCR product purification kit (Roche). cDNA labeling, microarray hybridization, and signal detection were performed at Roche NimbleGen Systems (Iceland) (see the supplemental material for details). Network analysis was performed using BioLayout Express3D using log2 robust multiarray average (RMA) expression values as described elsewhere (28). Four biological replicates were analyzed per strain. Gene Ontology (GO) enrichment analysis was performed with the customized microarray analysis tool of the Gene Ontology Enrichment Analysis Software Toolkit (GOEAST). P values were obtained using a Fisher exact test, adjusted for the false discovery rate (FDR) by the Benjamini-Yekutieli method, and corrected for overrepresentation of neighboring GO terms by the method of Alexa et al. (29, 30). R. equi Gene Ontology annotation was obtained from UniProt. Only 3,040 of 4,511 chromosomal genes have annotated GO terms.
RT-qPCR analysis.
RNA isolation and reverse transcription-PCR (RT-PCR) using gene-specific primers (see Table S2 in the supplemental material) were carried out as described previously (19, 31). Quantitative PCR (qPCR) was carried out as described previously (20). The geNorm method was employed to determine the optimal number of reference targets from an initial set of 9 candidates. Normalization factor based on the REQ04990 and REQ23320 genes yielded a geNorm V value of <0.15 and a geNorm M value of ≤0.2, satisfying the requirements for use as reference targets for normalization under the experimental conditions used (32). qbasePlus software was used to evaluate quantitative variation in gene expression among strains (33). To rule out the presence of contaminating genomic DNA, controls without reverse transcriptase were routinely performed. In all experiments, each strain was represented by at least three independent biological replicates, and each sample was analyzed in duplicate.
Statistical analysis.
Statistical analyses for bacterial growth assays were performed using the SigmaPlot (version 11.2.0.5) statistical package (Systat Software, San Jose, CA). The mean intracellular numbers of different bacterial strains were compared using one-way analysis of variance (ANOVA). When appropriate, multiple pairwise comparisons were carried out using Tukey's honestly significant difference (HSD) test. Significance was set at a P value of <0.05.
RESULTS
VapA is the sole virulence-associated protein family member essential for intramacrophage growth.
The pathogenicity island encodes six Vaps, one of which, VapA, is essential for intracellular growth (14). To determine whether the remaining Vap are also required, a multiple-deletion mutant (MDM) was constructed in which 14 of the 26 genes of the pathogenicity island, including all vap genes except vapA, were removed (Fig. 1; see also Fig. S1 in the supplemental material). The intracellular growth of the MDM was compared to that of the wild-type strain R. equi 103S and the avirulent plasmid-cured strain R. equi 103SP− in murine bone marrow-derived macrophages (Fig. 1). The R. equi MDM displayed a significantly (P = 0.004) better intracellular growth phenotype than wild-type R. equi. We showed previously that deletion of icgA, which is also absent in the R. equi MDM, resulted in enhanced intracellular growth (20). As expected, complementation of the R. equi MDM with icgA returned intracellular growth to wild-type levels (see Fig. S2 in the supplemental material). The data thus demonstrate that growth in macrophages does not require additional vap genes and that expression of vapA in the presence of the remaining pathogenicity island genes is sufficient for intracellular growth.
FIG 1.

VapA is the only Vap family member required for growth in macrophages. (A) Schematic representation of the pathogenicity island in wild-type R. equi and the MDM, indicating the positions of genes deleted from the R. equi MDM. Open rectangles represent vap genes and the transcriptional regulators virR and virS. vapI and vapF are pseudogenes. Shaded rectangles represent all remaining genes of the pathogenicity island. (B and C) The intracellular growth of R. equi strains was assessed in murine bone marrow-derived macrophages infected with R. equi 103S, 103SP−, or the MDM at an MOI of 10. Following incubation for 1 h to allow phagocytosis, monolayers were washed and were treated with amikacin to kill remaining extracellular bacteria (t = 0 h). Monolayers were lysed in triplicate 24 h and 48 h postinfection. (B) Intracellular growth of R. equi strains following infection of macrophages. (C) Fold changes in the CFU count of intracellular bacteria at 24 h and 48 h postinfection relative to that at 1 h postinfection. Error bars represent the standard deviations of the means. The statistical significances of the differences in the fold change in CFU per monolayer between different R. equi strains are given. Data are representative of the results of three independent experiments.
VapA and the transcriptional regulators VirR and VirS are the only virulence plasmid-encoded proteins required for intracellular growth.
We subsequently postulated that vapA and the genes encoding the transcriptional activators VirR and VirS are the only genes of the pathogenicity island required for intracellular growth. To test this hypothesis, the avirulent, virulence plasmid-cured strain R. equi 103SP− was transformed with pVirRSvapA, containing vapA, virR, and virS under the control of their native promoters. The transcription of virR, virS, and vapA was confirmed by RT-PCR (see Fig. S3 in the supplemental material). Macrophages were infected with R. equi 103SP−/pVirRSvapA, and its growth relative to that of the wild-type R. equi strain 103S and the virulence plasmid-free R. equi strain 103SP− was assessed. As expected, the avirulent R. equi strain 103SP− did not grow in macrophages (Fig. 2). Remarkably, however, expression of virR, virS, and vapA in the 103SP− background restored intracellular growth, leading to levels of intracellular bacteria similar to those of the wild-type strain 103S at 48 h postinfection. These data demonstrate that the virulence plasmid-encoded proteins VapA, VirR, and VirS are sufficient to allow intramacrophage growth.
FIG 2.
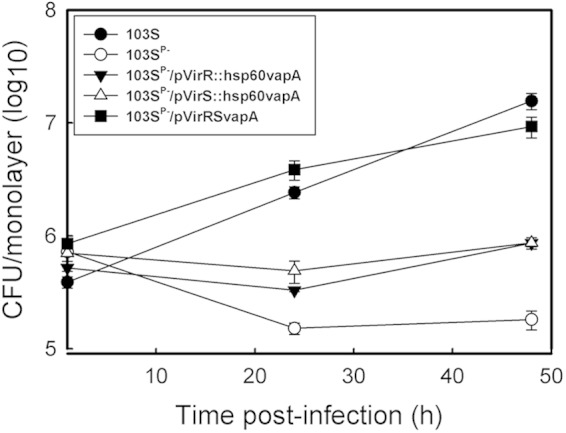
The transcriptional regulators VirR and VirS and the virulence-associated protein VapA are the only plasmid-encoded proteins required for growth in macrophages. Murine J774.1 macrophage monolayers were infected with R. equi 103S or 103SP−, or with R. equi 103SP− expressing vapA along with an individual regulator (103SP−/pVirR::hsp60vapA or 103SP−/pVirS::hsp60vapA) or with both regulators (103SP−/pVirRSvapA). Intracellular bacterial growth was followed for 48 h postinfection. Values are means ± standard deviations for monolayers, each assessed in triplicate.
To determine if the presence of both transcriptional regulators was required for growth in macrophages, the plasmid-cured R. equi strain 103SP− was engineered to express either virR or virS in the presence of constitutively expressed vapA by transformation of R. equi 103SP−/pVirR or 103SP−/pVirS with pMV261vapA, an episomal plasmid expressing vapA from the constitutive mycobacterial hsp60 promoter (34). Expression of VapA in the resultant strains, 103SP−/pVirR::hsp60vapA and 103SP−/pVirS::hsp60vapA, respectively, was confirmed by Western blotting (see Fig. S4 in the supplemental material). In contrast to R. equi 103SP−/pVirRSvapA, strains expressing either VirR or VirS in conjunction with vapA did not grow in macrophages, suggesting that both regulators are required for intramacrophage replication (Fig. 2). We therefore conclude that vapA, virR, and virS represent the minimum number of virulence plasmid genes that are required and sufficient for the intracellular growth of R. equi.
The virulence plasmid-encoded transcriptional regulators VirR and VirS remodel the R. equi transcriptome.
Since vapA expression in the absence of VirR and VirS is not sufficient to allow intracellular growth, we hypothesized that in addition to regulating vapA expression, these two transcriptional regulators control the transcription of chromosomal genes. This was tested by comparing the transcriptional profiles of R. equi 103S and 103SP−/pVirRSvapA to that of the virulence plasmid-cured strain R. equi 103SP− grown under conditions that induce the transcription of vapA (pH 5.5, 37°C) (35). The presence of just these two virulence plasmid-encoded transcriptional regulators, VirR and VirS, had a significant effect (at least a 2-fold change [P < 0.05]) on the transcript levels of 18% of the transcriptome (Table 1). Introduction of the entire virulence plasmid had a similar effect, resulting in significantly altered transcript levels of 20% of the transcriptome (Table 1). The microarray data were validated by determining the changes in the transcript levels of 18 genes using reverse transcriptase qPCR (see Fig. S5 in the supplemental material).
TABLE 1.
The virulence plasmid and the transcriptional regulators VirR and VirS have similar effects on the R. equi transcriptome
| Strain | No. of genes with a >2-fold change in expression (P < 0.05) from that in R. equi 103SP− |
||
|---|---|---|---|
| Upregulated | Downregulated | Total (%)a | |
| R. equi 103S | 347 | 572 | 919 (20) |
| R. equi 103SP−/pVirRSvapA | 386 | 406 | 792 (18) |
Percentage of the total number of chromosomal genes in the R. equi genome (n = 4,525).
Comparison of these transcriptomes with matrix scatter plots and a Euclidian distance-based dendrogram showed that expression of virR and virS in R. equi 103SP−/pVirRSvapA resulted in a transcription profile closely resembling that of the virulence plasmid-containing R. equi strain 103S (Fig. 3). This was further underpinned by a 3-dimensional (3D) network analysis, which showed that introduction of the virulence plasmid resulted in the formation of two clusters of transcripts with highly correlated expression patterns (Fig. 4). The nodes at the core of each of these clusters represent genes with significantly increased (red) or decreased (blue) transcript levels. A network with very similar topology was observed when virR and virS were expressed in R. equi 103SP− (Fig. 4). Inspection of individual genes showed that the presence of either the virulence plasmid or a plasmid driving the combined expression of virR and virS altered the expression levels (at least a 2-fold change; P < 0.05) of 614 genes in a similar manner (see Table S3 in the supplemental material). These data thus show that the modulation of the transcriptome by the virulence plasmid is due predominantly to the transcriptional regulators VirR and VirS.
FIG 3.

Transformation of R. equi 103SP− with virR, virS, and vapA mimics the effect of the virulence plasmid on R. equi gene expression. Bacteria were grown to an optical density at 600 nm of 1 under vapA-inducing growth conditions (LB at 37°C and pH 5.5). RNA isolation, cDNA synthesis, and labeling and microarray hybridization were performed for four biological replicates as described in Materials and Methods. (A) Matrix of scatter plots comparing the log2 expression values between strains and between biological replicates. (B) Euclidian distance-based dendrogram obtained with the whole transcriptome, showing that the expression profile of R. equi 103SP−/pVirRSvapA is more similar to that of R. equi 103S than to that of the parental strain lacking the virulence plasmid.
FIG 4.

Network analysis to show the impact of the pathogenicity island regulators VirR and VirS on the expression of chromosomal genes. Shown are pairwise comparisons of the transcriptomes of R. equi 103S (plasmid containing, virulent), R. equi 103SP− (plasmid cured, avirulent), and R. equi 103SP− expressing the virulence plasmid pathogenicity island genes virR, virS, and vapA (R. equi 103SP−/pVirRSvapA). 3D networks were made in BioLayout Express3D, version 2.2, using log2 RMA expression values at a correlation threshold of 0.85, with an MCL inflation of 2.2, and an edge/node filter setting of 10. Each network shows a pairwise comparison of the transcriptomes between different strains or between biological replicates of strain 103SP−. Genes are represented by nodes, which are connected by edges according to gene expression interrelationships above the threshold. Red and blue nodes represent genes that are up- and downregulated, respectively, in R. equi 103S relative to their expression in the plasmid-free avirulent strain R. equi 103SP− (>2-fold change [P < 0.05]). The colored nodes in each of the four networks refer to the same set of genes. Genes with nonsignificant differences in transcript levels are shown in gray. Node size: gray, 1; colored, 3. Edge thickness, 0.1. The analysis was performed with log2 RMA expression values obtained by analysis of oligonucleotide microarrays for four biological replicates.
VirR and VirS coopt chromosomally encoded regulatory networks.
Among the chromosomal genes that were differentially expressed in the presence of VirR and VirS are 31 regulatory genes, encoding 28 transcriptional regulators, 2 sigma factors, and 1 antitermination regulator (Table 2). The transcriptional activity of these chromosomal regulators in R. equi 103SP− transformed with virR, virS, and vapA closely mimics that of the plasmid-containing wild-type strain 103S (Fig. 5). While some of these regulators may control only a small regulon, others, in particular sigma factors, may control the transcription of a large number of genes. Of particular interest is the finding that VirR and VirS affected the transcript levels of 3 histidine sensor kinase- and 3 response regulator-encoding genes. These are part of two-component regulatory systems, which may respond to a wide range of environmental stimuli and often play a role in virulence, as highlighted by the response regulator VirS. The changes in the transcript levels of these regulatory genes resulting from the presence of VirR and VirS were tremendous, ranging from −805- to 24-fold. This undoubtedly has a profound impact on the regulatory network of the cell, offering an explanation for the major effect of VirR and VirS on the transcriptome of R. equi.
TABLE 2.
Regulatory genes displaying at least a 2-fold (P < 0.05) change in transcript levels in both R. equi 103S and R. equi 103SP−/pVirRSvapA from those in the virulence plasmid-cured strain R. equi 103SP−
| Gene name | Annotation | Expression in: |
|||
|---|---|---|---|---|---|
| 103S |
103SP−/pVirRSvapA |
||||
| Fold changea | P | Fold changea | P | ||
| REQ00140 | TetR family transcriptional regulator | 3.66 | 0.0001 | 2.11 | 0.0012 |
| REQ00980 | TetR family transcriptional regulator | 2.97 | 0.0019 | 4.93 | 0.0007 |
| REQ02910 | MarR family transcriptional regulator | −2.15 | 0.0044 | −2.60 | 0.0004 |
| REQ03830 | IclR family transcriptional regulator | −2.47 | <0.0001 | −2.52 | <0.0001 |
| REQ08990 | IclR family transcriptional regulator | −3.06 | <0.0001 | −2.87 | 0.0003 |
| REQ10540 | TCSb response regulator | 2.58 | 0.0006 | 2.15 | 0.002 |
| REQ13000 | TetR family transcriptional regulator | 2.68 | 0.0042 | 2.21 | 0.0072 |
| REQ14460 | Sigma 70 factor | 24.20 | <0.0001 | 15.61 | 0.0003 |
| REQ15290 | Transcriptional regulator | 2.56 | <0.0001 | 2.76 | <0.0001 |
| REQ15500 | IclR family transcriptional regulator | −2.65 | 0.0006 | −2.91 | 0.0002 |
| REQ15980 | LuxR family transcriptional regulator | 4.96 | 0.0001 | 4.78 | 0.0002 |
| REQ21040 | TetR family transcriptional regulator | −2.42 | 0.0023 | −2.60 | 0.0012 |
| REQ23970 | AraC family transcriptional regulator | 2.96 | 0.0001 | 2.47 | 0.0008 |
| REQ26080 | RNA binding sensor regulator | 2.05 | 0.0005 | 2.01 | 0.0001 |
| REQ26260 | MarR family transcriptional regulator | 2.23 | 0.0016 | 3.57 | 0.0004 |
| REQ31030 | TCS response regulator KdpE | 4.35 | 0.0002 | 4.29 | 0.0008 |
| REQ31040 | TCS sensor kinase KdpD | 4.21 | <0.0001 | 5.42 | 0.0002 |
| REQ31260 | AsnC family transcriptional regulator | 2.26 | 0.0004 | 2.26 | 0.0009 |
| REQ31580 | Two-component system sensor kinase | −2.45 | 0.0007 | −2.13 | 0.0013 |
| REQ33120 | Two-component system sensor kinase | 2.85 | <0.0001 | 2.95 | 0.0001 |
| REQ34960 | Sigma 70 factor | 5.00 | <0.0001 | 4.95 | <0.0001 |
| REQ38490 | NmrA family transcriptional regulator | −7.68 | <0.0001 | −3.02 | 0.0019 |
| REQ40160 | PadR family transcriptional regulator | −2.92 | 0.001 | −2.27 | 0.005 |
| REQ41890 | GntR family transcriptional regulator | −29.26 | <0.0001 | −22.74 | <0.0001 |
| REQ41960 | IclR family transcriptional regulator | −805.27 | <0.0001 | −926.06 | <0.0001 |
| REQ41970 | ArsR family transcriptional regulator | −176.15 | <0.0001 | −172.14 | <0.0001 |
| REQ42130 | MarR family transcriptional regulator | −2.60 | 0.0027 | −2.05 | 0.0031 |
| REQ44180 | TetR family transcriptional regulator | −3.16 | <0.0001 | −3.05 | <0.0001 |
| REQ45770 | Purine catabolism regulatory protein | −4.25 | <0.0001 | −3.96 | <0.0001 |
| REQ46220 | Transcriptional regulator | 5.72 | 0.0001 | 3.84 | 0.0043 |
| REQ46610 | PadR family transcriptional regulator | −2.33 | 0.002 | −2.42 | 0.0004 |
From expression in the virulence plasmid-cured strain R. equi 103SP−.
TCS, two-component system.
FIG 5.
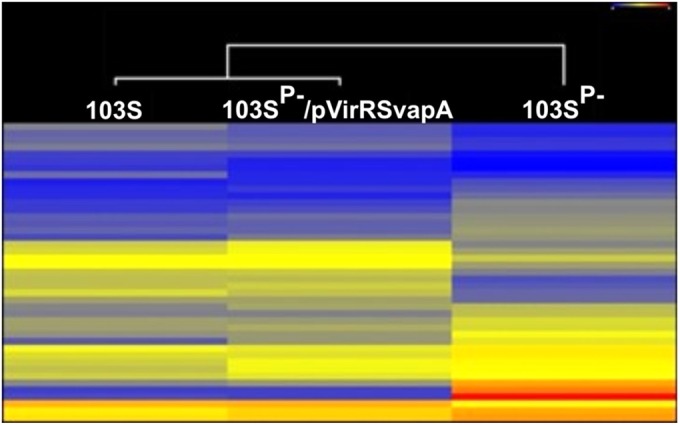
The regulatory transcriptome of R. equi 103SP− transformed with virR, virS, and vapA closely resembles the wild-type R. equi regulatory transcriptome. Bacteria were grown to an optical density at 600 nm of 1 under vapA-inducing growth conditions (LB at 37°C and pH 5.5). RNA isolation, cDNA synthesis, and labeling and microarray hybridization were performed for four biological replicates as described in Materials and Methods. Shown is a heat map of differentially expressed genes annotated as regulators. Above the heat map is a Euclidian distance-based dendrogram of strains based on changed regulators. Microarray data were analyzed with ArrayStar (DNAStar).
Enrichment in Gene Ontology terms of differentially expressed genes.
GOEAST analysis of the 614 genes that were similarly differentially expressed in R. equi 103SP−/pVirRSvapA and R. equi 103S compared to R. equi strain 103SP− showed an enrichment in Gene Ontology terms (see Table S4 in the supplemental material). Within the “cellular component” domain, genes annotated with the terms GO:0005886 (“plasma membrane”) and GO:0016021 (“integral to membrane”) were enriched. Many of the genes within these ontologies encode transport proteins (44% and 63% of genes in GO:0005886 and GO:0016021, respectively), suggesting a major reconfiguration of transport activities when VirR and VirS are expressed. Interestingly, the majority of the genes encoding major facilitator superfamily (MFS) (88%) and ABC (64%) transporters were downregulated. Differentially expressed genes annotated with Gene Ontology terms that were enriched in the “biological process” and “molecular function” domains were predominantly related to energy and nitrogen metabolism. The enrichment of a limited number of Gene Ontology terms suggests that the effects of VirR and VirS on the transcriptome are not random but rather target a discrete number of processes that play a critical role in the adaptation of R. equi to an intracellular lifestyle.
DISCUSSION
Although it is well established that the pathogenicity island of the virulence plasmid is essential for the host cell parasitism of R. equi (19), it remained unclear what role individual pathogenicity island genes play in this process, and which pathogenicity island genes are required and sufficient for intracellular growth. In addition to deepening our understanding of the virulence mechanisms of this pathogen, answering these questions also provides insight into the possible sequence of events in the evolution of this pathogenic bacterium from a saprophytic ancestor.
The pathogenicity island encodes six homologous Vaps, which share a high degree of sequence similarity, in particular in their carboxy-terminal domains (12, 36). The recent structural determination of the C-terminal domains of VapD and VapB showed a novel 8-stranded β-barrel topology that is conserved among Vaps (37, 38). In addition to their structural relatedness, transcription of the vap genes is also regulated in a similar manner in response to changes in the pH and temperature of the growth medium (15, 35, 39). Deletion of vapA abolishes the capacity for intracellular growth and results in an attenuated phenotype in the mouse R. equi infection model (14). To date, it has remained unclear whether the VapA homologues encoded by the pathogenicity island also play a role in intracellular growth. Deletion of individual vap genes other than vapA (vapG, vapH) did not show a phenotype (19, 20). The similarities these proteins share in amino acid sequence, extracellular location, and expression patterns initially suggested that functional redundancy may exist. It was therefore surprising that deletion of all vap genes except vapA did not negatively affect the growth characteristics of R. equi in macrophages. Our data thus unequivocally demonstrate that VapA is the only Vap that is required and sufficient for intracellular growth in in vitro-cultured murine macrophages.
The transcriptional regulators VirR and VirS are required for wild-type-level vapA expression (21, 22). However, as was observed previously, VirR- and VirS-independent expression of vapA from the Hsp60 promoter in a virulence plasmid-cured strain did not result in intracellular growth (10). Surprisingly, introduction of a plasmid (pVirRSvapA) expressing all three genes did restore normal levels of intracellular growth. This observation has major implications for our understanding of the mechanisms by which R. equi blocks endosomal maturation. Analysis of R. equi-containing vacuoles demonstrated that early endosomal markers are acquired and lost normally; however, these phagosomal vacuoles do not acquire certain late endosomal markers, including cathepsin D and the proton-pumping vATPase complex (5). Deletion of vapA from the virulence plasmid allows phagosomes to develop into mature acidified phagolysosomes, resulting in the killing of R. equi (16). Although this clearly demonstrates that VapA is required for the prevention of phagosomal maturation, it does not exclude the possibility that other pathogenicity island-encoded proteins, including Vap homologues, may also be involved. Nonetheless, the data presented here demonstrate that VapA is the only structural pathogenicity island-encoded protein that is required and sufficient for normal intracellular growth. This suggests that VapA is critical for the inhibition of endosomal maturation without the need for other virulence plasmid-encoded structural proteins.
The results from this study show that VirR and VirS do not just regulate the expression of vapA and other pathogenicity island genes but have a major impact on global transcription. The VirR- and VirS-induced change in cellular physiology is a prerequisite for host cell parasitism, because expression of VapA in the absence of these two regulators does not support intracellular growth. Although a previous study demonstrated that the presence of the virulence plasmid affects the R. equi transcriptome (24), it remained unclear whether this was due to specific or pleiotropic effects, or whether these changes in gene expression are required for intracellular growth. We demonstrate here that the impact of the virulence plasmid on the transcriptome is specific and is due predominantly to the expression of just 2 (virR and virS) of the 63 genes of the virulence plasmid, affecting the transcript levels of 18% of chromosomal genes.
The global effect of VirR and VirS on transcription may be explained in part by the fact that their expression significantly altered the transcript levels of 31 transcriptional regulators, including those of 2 sigma factors. It therefore appears likely that in many cases, VirR and VirS have an indirect effect on gene expression by altering the transcription levels of other transcriptional regulators. Nevertheless, an analysis of gene ontology showed that the impact of VirR and VirS on global transcription was not random but resulted in enrichment of Gene Ontology term among differentially expressed genes. Most notably, these include genes playing roles in transport processes, e.g., ABC and MFS transporters, and in nitrogen and energy metabolism.
Acquisition of the pathogenicity island on a conjugative plasmid by a nonpathogenic ancestor of R. equi was undoubtedly the initial event in the evolution of this pathogen. However, the genome of this ancestral R. equi organism would not have had VirS and VirR DNA binding sites to allow these regulators to affect chromosomal gene expression in a nonrandom manner. In addition, VirS is an orphan response regulator, which may interact with a chromosomally encoded sensor kinase (12).
It is generally accepted that bacterial strategies to prevent or survive grazing by predatory protozoa were a major driving force in the evolution of pathogenesis (40–42). We therefore hypothesize that in the evolution of R. equi, acquisition of the pathogenicity island on a conjugative plasmid may have given an R. equi ancestor a selective advantage in surviving predation by protozoa, which are abundant both in the equine intestinal tract and in soil (43). Continued selective pressure exerted by these predatory protozoa may have subsequently selected for chromosomal mutations facilitating the binding of VirR and VirS to promoters that control the expression of genes influencing intracellular survival by altering bacterial physiology to enable adaptation to the host. In addition, predation may have selected for mutations in two-component systems allowing VirS to interact with one or more sensor kinase proteins, thus tapping into the sensor network of the ancestral R. equi organism. This scenario is supported by comparative analysis of the R. equi genome, which showed that the majority of genes implicated in virulence are conserved in nonpathogenic actinobacteria (24). Although the acquisition of the pathogenicity island may have been the initial event, the virulence of R. equi must therefore have evolved subsequently via adaptation of existing traits to the requirements of intracellular growth through nonrandom, VirR- and VirS-induced changes in the physiology of R. equi.
In conclusion, the pathogenicity island of the virulence plasmid fulfills at least three functions in relation to intracellular growth, by encoding (i) VapA, which alters the maturation of the early endosome, thus creating a niche conducive to the growth of R. equi, (ii) IcgA, an MFS transport protein, which tempers the intracellular growth rate of R. equi, resulting in increased macrophage viability, allowing R. equi to stay longer in this intracellular niche, and (iii) VirR and VirS, which selectively adjust chromosomal gene expression, thereby modifying the physiology of R. equi to enable survival and growth in an intracellular niche.
Supplementary Material
ACKNOWLEDGMENTS
We thank Kimberly Goldbach and Monica LaGatta for technical assistance. We are grateful to Vibhay Tripathi for intellectual input and discussions of this work.
This study was funded by the National Institutes of Health (grant R01 AI060469, to M.K.H.) and Science Foundation Ireland (grant 02/IN.1/B203, to W.G.M.).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00230-15.
REFERENCES
- 1.van der Geize R, Dijkhuizen L. 2004. Harnessing the catabolic diversity of rhodococci for environmental and biotechnological applications. Curr Opin Microbiol 7:255–261. doi: 10.1016/j.mib.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 2.Bell KS, Philp JC, Aw DWJ, Christofi N. 1998. The genus Rhodococcus. J Appl Microbiol 85:195–210. doi: 10.1046/j.1365-2672.1998.00525.x. [DOI] [PubMed] [Google Scholar]
- 3.Muscatello G, Leadon DP, Klay M, Ocampo-Sosa A, Lewis DA, Fogarty U, Buckley T, Gilkerson JR, Meijer WG, Vázquez-Boland JA. 2007. Rhodococcus equi infection in foals: the science of ‘rattles.’ Equine Vet J 39:470–478. [DOI] [PubMed] [Google Scholar]
- 4.Hietala SK, Ardans AA. 1987. Interaction of Rhodococcus equi with phagocytic cells from R. equi-exposed and non-exposed foals. Vet Microbiol 14:307–320. doi: 10.1016/0378-1135(87)90118-0. [DOI] [PubMed] [Google Scholar]
- 5.Fernandez-Mora E, Polidori M, Lührmann A, Schaible UE, Haas A. 2005. Maturation of Rhodococcus equi-containing vacuoles is arrested after completion of the early endosome stage. Traffic 6:635–653. doi: 10.1111/j.1600-0854.2005.00304.x. [DOI] [PubMed] [Google Scholar]
- 6.Zink MC, Yager JA, Prescott JF, Fernando MA. 1987. Electron microscopic investigation of intracellular events after ingestion of Rhodococcus equi by foal alveolar macrophages. Vet Microbiol 14:295–305. doi: 10.1016/0378-1135(87)90117-9. [DOI] [PubMed] [Google Scholar]
- 7.Nordmann P, Zinzendorf N, Keller M, Lair I, Ronco E, Guenounou M. 1994. Interaction of virulent and non-virulent Rhodococcus equi human isolates with phagocytes, fibroblast- and epithelial-derived cells. FEMS Immunol Med Microbiol 9:199–205. doi: 10.1111/j.1574-695X.1994.tb00494.x. [DOI] [PubMed] [Google Scholar]
- 8.Lührmann A, Mauder N, Sydor T, Fernandez-Mora E, Schulze-Luehrmann J, Takai S, Haas A. 2004. Necrotic death of Rhodococcus equi-infected macrophages is regulated by virulence-associated plasmids. Infect Immun 72:853–862. doi: 10.1128/IAI.72.2.853-862.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Soldati T, Neyrolles O. 2012. Mycobacteria and the intraphagosomal environment: take it with a pinch of salt(s)! Traffic 13:1042–1052. doi: 10.1111/j.1600-0854.2012.01358.x. [DOI] [PubMed] [Google Scholar]
- 10.Giguère S, Hondalus MK, Yager JA, Darrah P, Mosser DM, Prescott JF. 1999. Role of the 85-kilobase plasmid and plasmid-encoded virulence-associated protein A in intracellular survival and virulence of Rhodococcus equi. Infect Immun 67:3548–3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tripathi VN, Harding WC, Willingham-Lane JM, Hondalus MK. 2012. Conjugal transfer of a virulence plasmid in the opportunistic intracellular actinomycete Rhodococcus equi. J Bacteriol 194:6790–6801. doi: 10.1128/JB.01210-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takai S, Hines SA, Sekizaki T, Nicholson VM, Alperin DA, Osaki M, Osaki D, Nakamura M, Suzuki K, Ogino N, Kakuka T, Dan H, Prescott JF. 2000. DNA sequence and comparison of virulence plasmids from Rhodococcus equi ATCC 33701 and 103. Infect Immun 68:6840–6847. doi: 10.1128/IAI.68.12.6840-6847.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tan C, Prescott JF, Patterson MC, Nicholson VM. 1995. Molecular characterization of a lipid-modified virulence-associated protein of Rhodococcus equi and its potential in protective immunity. Can J Vet Res 59:51–59. [PMC free article] [PubMed] [Google Scholar]
- 14.Jain S, Bloom BR, Hondalus MK. 2003. Deletion of vapA encoding virulence associated protein A attenuates the intracellular actinomycete Rhodococcus equi. Mol Microbiol 50:115–128. doi: 10.1046/j.1365-2958.2003.03689.x. [DOI] [PubMed] [Google Scholar]
- 15.Takai S, Iie M, Watanabe Y, Tsubaki S, Sekizaki T. 1992. Virulence-associated 15- to 17-kilodalton antigens in Rhodococcus equi: temperature-dependent expression and location of the antigens. Infect Immun 60:2995–2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.von Bargen K, Polidori M, Becken U, Huth G, Prescott JF, Haas A. 2009. Rhodococcus equi virulence-associated protein A is required for diversion of phagosome biogenesis but not for cytotoxicity. Infect Immun 77:5676–5681. doi: 10.1128/IAI.00856-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Byrne GA, Boland CA, O'Connell EP, Meijer WG. 2008. Differential mRNA stability of the vapAICD operon of the facultative intracellular pathogen Rhodococcus equi. FEMS Microbiol Lett 280:89–94. doi: 10.1111/j.1574-6968.2007.01055.x. [DOI] [PubMed] [Google Scholar]
- 18.Byrne GA, Russell DA, Chen X, Meijer WG. 2007. Transcriptional regulation of the virR operon of the intracellular pathogen Rhodococcus equi. J Bacteriol 189:5082–5089. doi: 10.1128/JB.00431-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coulson GB, Agarwal S, Hondalus MK. 2010. Characterization of the role of the pathogenicity island and vapG in the virulence of the intracellular actinomycete pathogen Rhodococcus equi. Infect Immun 78:3323–3334. doi: 10.1128/IAI.00081-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang X, Coulson GB, Miranda-Casoluengo AA, Miranda-Casoluengo R, Hondalus MK, Meijer WG. 2014. IcgA is a virulence factor of Rhodococcus equi that modulates intracellular growth. Infect Immun 82:1793–1800. doi: 10.1128/IAI.01670-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kakuda T, Hirota T, Takeuchi T, Hagiuda H, Miyazaki S, Takai S. 2014. VirS, an OmpR/PhoB subfamily response regulator, is required for activation of vapA gene expression in Rhodococcus equi. BMC Microbiol 14:243. doi: 10.1186/s12866-014-0243-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Russell DA, Byrne GA, O'Connell EP, Boland CA, Meijer WG. 2004. The LysR-type transcriptional regulator VirR is required for expression of the virulence gene vapA of Rhodococcus equi ATCC 33701. J Bacteriol 186:5576–5584. doi: 10.1128/JB.186.17.5576-5584.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ren J, Prescott JF. 2004. The effect of mutation on Rhodococcus equi virulence plasmid gene expression and mouse virulence. Vet Microbiol 103:219–230. doi: 10.1016/j.vetmic.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 24.Letek M, Gonzalez P, Macarthur I, Rodriguez H, Freeman TC, Valero-Rello A, Blanco M, Buckley T, Cherevach I, Fahey R, Hapeshi A, Holdstock J, Leadon D, Navas J, Ocampo A, Quail MA, Sanders M, Scortti MM, Prescott JF, Fogarty U, Meijer WG, Parkhill J, Bentley SD, Vazquez-Boland JA. 2010. The genome of a pathogenic rhodococcus: cooptive virulence underpinned by key gene acquisitions. PLoS Genet 6:e1001145. doi: 10.1371/journal.pgen.1001145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miranda-Casoluengo R, Coulson GB, Miranda-Casoluengo A, Vazquez-Boland JA, Hondalus MK, Meijer WG. 2012. The hydroxamate siderophore rhequichelin is required for virulence of the pathogenic actinomycete Rhodococcus equi. Infect Immun 80:4106–4114. doi: 10.1128/IAI.00678-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wall DM, Duffy PS, Dupont C, Prescott JF, Meijer WG. 2005. Isocitrate lyase activity is required for virulence of the intracellular pathogen Rhodococcus equi. Infect Immun 73:6736–6741. doi: 10.1128/IAI.73.10.6736-6741.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van der Geize R, de Jong W, Hessels GI, Grommen AW, Jacobs AA, Dijkhuizen L. 2008. A novel method to generate unmarked gene deletions in the intracellular pathogen Rhodococcus equi using 5-fluorocytosine conditional lethality. Nucleic Acids Res 36:e151. doi: 10.1093/nar/gkn811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Theocharidis A, van Dongen S, Enright AJ, Freeman TC. 2009. Network visualization and analysis of gene expression data using BioLayout Express3D. Nat Protoc 4:1535–1550. doi: 10.1038/nprot.2009.177. [DOI] [PubMed] [Google Scholar]
- 29.Alexa A, Rahnenfuhrer J, Lengauer T. 2006. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 22:1600–1607. doi: 10.1093/bioinformatics/btl140. [DOI] [PubMed] [Google Scholar]
- 30.Zheng Q, Wang XJ. 2008. GOEAST: a web-based software toolkit for Gene Ontology enrichment analysis. Nucleic Acids Res 36:W358–W363. doi: 10.1093/nar/gkn276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miranda-Casoluengo R, Miranda-Casoluengo AA, O'Connell EP, Fahey RJ, Boland CA, Vazquez-Boland JA, Meijer WG. 2011. The vapA co-expressed virulence plasmid gene vcgB (orf10) of the intracellular actinomycete Rhodococcus equi. Microbiology 157:2357–2368. doi: 10.1099/mic.0.049759-0. [DOI] [PubMed] [Google Scholar]
- 32.Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. 2002. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 3:RESEARCH0034. doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hellemans J, Mortier G, De Paepe A, Speleman F, Vandesompele J. 2007. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol 8:R19. doi: 10.1186/gb-2007-8-2-r19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hong Y, Hondalus MK. 2008. Site-specific integration of Streptomyces FC31 integrase-based vectors in the chromosome of Rhodococcus equi. FEMS Microbiol Lett 287:63–68. doi: 10.1111/j.1574-6968.2008.01298.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takai S, Fukunaga N, Kamisawa K, Imai Y, Sasaki Y, Tsubaki S. 1996. Expression of virulence-associated antigens of Rhodococcus equi is regulated by temperature and pH. Microbiol Immunol 40:591–594. doi: 10.1111/j.1348-0421.1996.tb01113.x. [DOI] [PubMed] [Google Scholar]
- 36.Letek M, Ocampo-Sosa AA, Sanders M, Fogarty U, Buckley T, Leadon DP, Gonzalez P, Scortti M, Meijer WG, Parkhill J, Bentley S, Vázquez-Boland JA. 2008. Evolution of the Rhodococcus equi vap pathogenicity island seen through comparison of host-associated vapA and vapB virulence plasmids. J Bacteriol 190:5797–5805. doi: 10.1128/JB.00468-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Whittingham JL, Blagova EV, Finn CE, Luo H, Miranda-Casoluengo R, Turkenburg JP, Leech AP, Walton PH, Murzin AG, Meijer WG, Wilkinson AJ. 2014. Structure of the virulence-associated protein VapD from the intracellular pathogen Rhodococcus equi. Acta Crystallogr D Biol Crystallogr 70:2139–2151. doi: 10.1107/S1399004714012632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Geerds C, Wohlmann J, Haas A, Niemann HH. 2014. Structure of Rhodococcus equi virulence-associated protein B (VapB) reveals an eight-stranded antiparallel beta-barrel consisting of two Greek-key motifs. Acta Crystallogr F Struct Biol Commun 70:866–871. doi: 10.1107/S2053230X14009911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Byrne BA, Prescott JF, Palmer GH, Takai S, Nicholson VM, Alperin DC, Hines SA. 2001. Virulence plasmid of Rhodococcus equi contains inducible gene family encoding secreted proteins. Infect Immun 69:650–656. doi: 10.1128/IAI.69.2.650-656.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brown MRW, Barker J. 1999. Unexplored reservoirs of pathogenic bacteria: protozoa and biofilms. Trends Microbiol 7:46–50. doi: 10.1016/S0966-842X(98)01425-5. [DOI] [PubMed] [Google Scholar]
- 41.Brüssow H. 2007. Bacteria between protists and phages: from antipredation strategies to the evolution of pathogenicity. Mol Microbiol 65:583–589. doi: 10.1111/j.1365-2958.2007.05826.x. [DOI] [PubMed] [Google Scholar]
- 42.Matz C, Kjelleberg S. 2005. Off the hook—how bacteria survive protozoan grazing. Trends Microbiol 13:302–307. doi: 10.1016/j.tim.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 43.Dougal K, Harris PA, Edwards A, Pachebat JA, Blackmore TM, Worgan HJ, Newbold CJ. 2012. A comparison of the microbiome and the metabolome of different regions of the equine hindgut. FEMS Microbiol Ecol 82:642–652. doi: 10.1111/j.1574-6941.2012.01441.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.