

Abstract
Borrelia burgdorferi, the causative agent of Lyme disease in the United States, is able to persist in the joint, heart, skin, and central nervous system for the lifetime of its mammalian host. Borrelia species achieve dissemination to distal sites in part by entry into and travel within the bloodstream. Much work has been performed in vitro describing the roles of many B. burgdorferi outer surface proteins in adhesion to host cell surface proteins and extracellular matrix components, although the biological relevance of these interactions is only beginning to be explored in vivo. A need exists in the field for an in vivo model to define the biological roles of B. burgdorferi adhesins in tissue-specific vascular interactions. We have developed an in vivo model of vascular interaction of B. burgdorferi in which the bacteria are injected intravenously and allowed to circulate for 1 h. This model has shown that the fibronectin binding protein BB0347 has a tropism for joint tissue. We also have shown an importance of the integrin binding protein, P66, in binding to vasculature of the ear and heart. This model also revealed unexpected roles for Borrelia adhesins BBK32 and OspC in bacterial burdens in the bloodstream. The intravenous inoculation model of short-term infection provides new insights into critical B. burgdorferi interactions with the host required for initial survival and tissue colonization.
INTRODUCTION
Borrelia burgdorferi, the causative agent of Lyme disease in the United States, is a vector-borne disease transmitted via the bite of an Ixodes scapularis tick (1, 2). Human infection with this bacterium often results in debilitating chronic arthritis, carditis, and/or neurologic symptoms if left untreated (3, 4). Upon entry into the host, Lyme disease Borrelia spirochetes are able to disseminate into the tissues and persist in the joints, heart, skin, and central nervous system (1). It is likely that Borrelia spp. are able to achieve rapid dissemination within the host by entry into and travel within the bloodstream. Colonization of specific host tissues is thought to occur through interactions of the bacteria with host cells of the microvasculature. A large variety of vascular beds are available in the host, determined by the size and type of vessel, as well as the organ with which they are associated (5, 6). One possible mechanism of B. burgdorferi attachment to various vascular beds and bacterial extravasation at particular tissue sites is through preferential interactions of various adhesive outer surface proteins on the bacterial surface with different types of endothelial cell surface proteins, carbohydrates, and/or extracellular matrix components. It has been shown that adhesion of B. burgdorferi to the vasculature occurs in a series of interactions (7). We hypothesize that the action of vascular binding and tissue colonization is not a random event but rather is determined by adhesion of B. burgdorferi surface proteins to tissue bed-specific endothelial cell (EC) surface receptors, such as VCAM1 on liver ECs, CD36 on lung and heart ECs, L-selectin (CD62L/SELL) on spleen ECs, and CD133 on ECs of the skin, brain, eye, and testis (5).
Infectious B. burgdorferi is known to express at least 19 adhesive outer surface proteins, many of which are known to bind to host cells and extracellular matrix components (reviewed in references 8–10). At least three outer surface proteins of B. burgdorferi have been shown to have the ability to bind fibronectin in vitro. One such protein is the 47-kDa protein, BBK32, which has recently been shown to bind both fibronectin and glycosaminoglycans (GAGs) using two distinct domains (11, 12). These binding domains of BBK32 also have been shown to be involved in binding to the host vasculature by intravital microscopy (7, 13), and the GAG binding domain specifically has been shown to confer a tropism of the protein for joint tissue (14). Other outer surface proteins of B. burgdorferi, such as BB0347 (15) and RevA (16), also have been found to bind with lower affinity to fibronectin in vitro (11). RevA and BB0347 were found previously to have a minimal effect on vascular binding in mouse flank skin in vivo, although the effect on binding to vasculature associated with other tissue types has not yet been addressed (11). Recent work on the decorin and GAG binding adhesins, decorin binding proteins A and B (Dbp), has revealed a role for these proteins in tissue-specific colonization of the host after subcutaneous inoculation with B. burgdorferi (14, 17). B. burgdorferi outer surface proteins also have been identified that bind to other host ligands, including P66 and BBB07, which bind to integrins (18, 19), and OspC, which was recently found to bind plasminogen (20) in vitro.
B. burgdorferi regulates the production of its surface proteins depending on the environment in which the bacterium resides. One example of this regulation is the tight control of production of the virulence determinant, OspC. Expression of OspC is induced inside the tick midgut upon tick feeding but is turned off prior to 28 days postinfection (21). This early induction of OspC expression is required to establish but not to maintain a mammalian infection (22, 23). In fact, B. burgdorferi that cannot decrease OspC production is cleared by the host adaptive immune system prior to 6 weeks postinoculation (22). Interestingly, artificial production of OspA, OspE, VlsE, and DbpA lipoproteins can in part substitute for OspC in allowing the OspC mutant to survive in host tissues 72 h postinoculation (24). Recent work has identified E61 of OspC as being an amino acid important for mammalian infection by B. burgdorferi through an unknown mechanism (25). Like OspC, BBK32 and P66 also are induced in the tick midgut during feeding, but unlike OspC, the production of these outer surface proteins is maintained throughout the duration of mammalian infection (26, 27). The Vls antigenic variation system present in B. burgdorferi enables evasion of adaptive immune system recognition through continual and segmental recombination of silent vls gene cassettes encoding different VlsE variants into the expression site (28–31).
Aside from evading the adaptive immune system of the host by surface protein variation, B. burgdorferi also is able to resist killing by the innate immune response by expressing several surface proteins that subvert the alternative complement cascade activity indirectly. The differentially regulated B. burgdorferi surface protein CspA (32–35) and some of the OspE-related protein family members, including ErpA (36, 37), ErpP (37), and ErpC (38, 39), all act similarly by recruitment of complement regulator factor H (FH) and factor H-like protein-1 (FHL-1) or complement factor H-related proteins (CFHR), presumably to the surface of the bacterium, although the majority of studies have been performed using recombinant protein. FH, FHL-1, and the CFHRs inhibit the alternative complement cascade by cleaving C3b. Although not expressed in the mammalian host, cspA expression is highly upregulated in the tick (40). This is likely important in enabling B. burgdorferi to survive in the midgut of a feeding tick prior to transmission into host tissue (40) and for maintenance of the tick-mammal life cycle of Borrelia.
Although much work has been done to identify the roles of B. burgdorferi outer surface proteins in bacterial adhesion to host cells and the surrounding matrix in vitro, the biological relevance of these adhesive proteins is only beginning to be explored in vivo. In order to more closely determine the roles of select outer surface proteins of B. burgdorferi in vivo, we have developed a short-course infection model. With this model we are able to observe the tissue-specific binding capacity of individual B. burgdorferi adhesins down to the level of the protein domain during early time points after inoculation, when the bacteria are interacting with the vasculature but have not transmigrated out into the surrounding tissues (14) (D. Kumar, L. C. Ristow, M. Shi, J. A. Ritchie, W. Y. Lee, P. Kubes, J. Coburn, and G. Chaconas, unpublished data). Our model also has enabled us to observe bloodstream survival of various B. burgdorferi strains in the mouse, revealing the multifaceted roles of specific outer surface proteins in both tissue-specific vascular adhesion and bloodstream survival in vivo.
MATERIALS AND METHODS
Animals.
Female BALB/c and C3H/HeN mice were acquired from Charles River Laboratories (Charles River Laboratories International, Inc., Wilmington, MA) and used at 8 weeks of age. All work with animals was approved by the IACUC at the Medical College of Wisconsin.
Bacterial culture and strains.
All strains used in this study are described in Table 1. B. burgdorferi strains were grown in Barbour-Stoenner-Kelly (BSKII) medium (43) at 33°C to a density of 1 × 108 spirochetes/ml. When antibiotic selection was required, kanamycin was used at 200 μg/ml and gentamicin was used at 40 μg/ml. The presence of genomic plasmids was determined in each culture by PCR prior to inoculation into mice (41, 44).
TABLE 1.
B. burgdorferi strains
| Strain | Description | Plasmid contenta | Reference |
|---|---|---|---|
| B31-A3 | Infectious low-passage clone of tick isolate B31 | Missing cp9 | 41 |
| B31A (GCB706) | High-passage noninfectious B. burgdorferi expressing GFP | Missing cp9, cp32-6, cp32-7, lp25, lp28-1, lp28-4, lp36, lp21 | 11 |
| B314 | Noninfectious high-passage clone of B31 | Missing lp54, lp17, lp25, lp28-1, lp28-2, lp28-3, lp28-4, lp38, lp36, lp56, lp5, lp21, cp9, cp32-6, cp32-9, cp32-7 | 42 |
| B31ApBB0347 (GCB1574) | B31A expressing BB0347 and GFP | 11 | |
| B31ApRevA (GCB 1570) | B31A expressing RevA and GFP | 11 | |
| B31ApBBK32 (GCB1585) | B31A expressing full-length BBK32 and GFP | 11 | |
| B31ApBBK32ΔGAG (GCB1580) | B31A expressing BBK32 deficient in GAG binding and GFP | 11 | |
| B31ApBBK32ΔFn (GCB1583) | B31A expressing BBK32 deficient in Fn binding and GFP | 11 | |
| B31-A3Δp66 | B31-A3 deficient in P66 expression | 18 | |
| B31-A3ΔospC | B31-A3 deficient in OspC expression | 22 | |
| B31-A3ΔospC pOspC | B31-A3ΔospC with restored OspC expression | 22 |
cp, circular plasmid; lp, linear plasmid.
OspC expression was induced by growth in BSK-H medium (45) (Sigma-Aldrich, St. Louis, MO) at 33°C to a density of 1 × 108/ml to 2 × 108/ml. Cultures were diluted 1:1 in fresh BSK-H and incubated at 37°C to a density of 1 × 108/ml to 2 × 108/ml (46, 47).
Gain-of-function derivatives made in B. burgdorferi noninfectious strain B31A to exogenously express adhesive outer surface proteins BB0347 (GCB1574), BBK32 (GCB1585), BBK32ΔGAG (GCB1580), BBK32ΔFn (GCB1583), and RevA (GCB1570) of B. burgdorferi from the flagellar (flaB) promoter were previously described (11). The naming scheme for the gain-of-function strains has been carried over from the original publication, but for the sake of clarity the strains have been designated by genotype in this report; both names are listed in Table 1 for clarification. Strains bearing deletions in p66 and ospC were made in the infectious B. burgdorferi strain B31-A3 and were described previously (18, 22).
Preparation of bacteria for inoculation into mice.
B. burgdorferi cells in each culture were enumerated using a Petroff-Hausser counting chamber. The cultures were pelleted (11,900 × g for 10 min at ambient temperature). Supernatants were decanted, and bacterial pellets were resuspended in 30 ml phosphate-buffered saline (PBS) plus 0.2% heat-inactivated mouse serum from the mouse strain to be used in the experiment. The wash solution containing mouse strain-specific serum was used to avoid nonspecific host responses to the proteins present in B. burgdorferi culture medium and rabbit serum, which is frequently used for preparation of B. burgdorferi stocks for animal inoculations. Resuspended bacteria were pelleted (11,900 × g for 10 min at ambient temperature) and washed by resuspension in 1 ml PBS plus 0.2% mouse serum and pelleted (16,100 × g for 8 min at ambient temperature). Supernatants were pipetted off bacterial pellets, and cells were diluted in PBS plus 0.2% mouse serum to a final density of 1 × 109 bacteria/ml. When heat-killed B. burgdorferi was used for inoculation into mice, the bacteria were incubated at 56°C for 30 min followed by the washing steps described above. The success of heat killing of cultures was confirmed by plating 1 × 108 heat-killed bacteria in 20 ml of semisolid plating medium. After 8 weeks of incubation at 33°C, no bacterial colonies formed on the plate, while the expected numbers of colonies of control B. burgdorferi were present after 2 weeks of incubation.
Retro-orbital inoculation of mice.
C3H/HeN or BALB/c mice were anesthetized with 100 mg ketamine/kg of body weight and 10 mg/kg xylazine, delivered by intraperitoneal injection. Anesthesia was confirmed by toe pinch and maintained throughout the duration of the experiment with one additional dose when necessary. Anesthetized mice were retro-orbitally inoculated in the vasculature behind the right eye with 100 μl of bacterial suspension using a 29-gauge needle. Each mouse was inoculated with 1 × 108 total bacteria, a dose which was empirically determined to be optimal for bacterial load quantification (data not shown) (7, 11, 13). A high dose of bacteria is required to allow observation and quantification of interactions with the vasculature that fall within the linear range of the assay. Due to the technical challenges of the experiments described here, experiments were not necessarily performed on the same days. To allow comparisons to be made, we carefully controlled the density at which the bacterial cultures were harvested, the bacterial inoculum, the culture conditions, and the bacterial growth medium.
Cardiac perfusion.
One hour after inoculation, anesthetized mice underwent a thoracotomy, and blood was collected by cardiac puncture through the right ventricle into a 10% final volume of anticoagulant (60 mM sodium citrate plus 40 mM citric acid). The time point of one hour was experimentally determined to be optimal for quantification of interactions of B. burgdorferi with the vasculature in this early infection model and was previously reported to be within the time range optimal for imaging of B. burgdorferi interactions with the vasculature in living mice (7, 13). After blood collection, a small cut was made in the right atrium to act as a drain, and mice were perfused with 0.9% sterile sodium chloride solution at a drip rate of 4.4 ml/min for 6 min to wash away unbound bacteria from the vasculature. Successful perfusion was evident by blanching of the tissues. After perfusion was complete, the right lung, median lobe of the liver, heart, spleen, bladder, right tibiotarsal joint, and left ear skin representing a disseminated skin site (ear opposite the inoculated eye) were collected, rinsed with PBS, and immediately stored on dry ice. Tissues were kept at −80°C until DNA was isolated. The efficiency of perfusion was confirmed by quantifying the number of infectious wild-type (WT) bacteria (B31-A3) present in each tissue of perfused and unperfused mice (data not shown). With these experiments, we found perfusion to significantly decrease the number of bacteria in the lung, spleen, and joint. Perfusion of the ear, heart, and bladder also decreased bacterial burdens in these tissues, although the difference was not statistically significant by Mann-Whitney unpaired t test. This may be due to stable binding interactions of WT bacteria with the heart, bladder, and ear that are not affected by perfusion. Therefore, tropism to liver, heart, ear, or bladder by infectious strain B31-A3 suggested by this model should be considered with caution, although data from mutant strains deficient in heart and ear binding indicate that differences in tropism in these tissues can be detected using this model.
DNA preparation and genome enumeration.
Total genomic DNA was isolated from each blood and tissue sample using the Qiagen DNA minikit (Qiagen, Valencia, CA). One hundred microliters of blood, 100 μl of inoculum, or 20 mg of tissue was processed in each column of the kit. The concentration of eluted DNA containing both bacterial and mouse genomes was determined using a spectrophotometer (at an optical density of 230 nm [OD230]), and the samples were diluted to 20 ng/μl. Samples in which total DNA was less than 20 ng/μl were used without dilution. All quantitative PCRs (qPCRs) were performed in triplicate using 100 ng of DNA per reaction for all tissue except ear, which was performed on 2 ng of isolated DNA due to the presence of unknown PCR inhibitors in those samples. B. burgdorferi genomes were quantified by qPCR using Qiagen QuantiFast SYBR green master mix at 95.0°C for 5 min (95.0°C for 10 s at 63.3°C [recA, B31-A3] or 58.3°C [recA, B31A] for 30 s, repeated 39 times), 95°C for 1 min, and 50°C for 1 min (see Table S1 in the supplemental material). Mouse genomes in each DNA sample also were quantified using the same qPCR protocol to ensure successful DNA isolation but with primers designed to amplify a region of the mouse β-actin gene (see Table S1). Genomes were quantified using a standard curve generated from purified B. burgdorferi B31A or B31-A3 genomic DNA (gDNA) or mouse gDNA. Mouse gDNA isolated from liver tissue from the mouse strain used in the particular experiment was added to each bacterial DNA standard at a concentration of 20 ng/μl to mimic the mouse gDNA present in each tissue DNA sample. B. burgdorferi genomes quantified for each sample were normalized to the target inoculum each mouse received in the experiment, as determined by qPCR of the inoculum. B. burgdorferi genome values were normalized to those for inoculum for each sample and then were divided by total nanograms of DNA contained in each PCR (100 ng). A value of 1 was added to each normalized value to allow values of zero to be represented on log-scale graphs. Medians and ranges were plotted for each data set and analyzed for statistical significance by Mann-Whitney unpaired t test or one-way analysis of variance (ANOVA) using GraphPad Prism 5.01. Bacteria were enumerated in blood samples from B31-A3-inoculated C3H/HeN mice over a 1-h time course, and CFU levels were found to be about 10-fold lower than those of the genomes, as quantified by qPCR at each time point.
We have good evidence that the bacterial genomes detected in the bloodstream of mice by qPCR in our model are representative of the number of living bacteria, as CFU values are consistently about 10-fold lower than qPCR values over a 1-h time course of infection, although it should be noted that quantification by qPCR cannot differentiate between living and dead bacteria. Variable plating efficiencies have been reported for B. burgdorferi, which could explain this 10-fold difference in bacterial enumeration between CFU and qPCR (48, 49).
Serum susceptibility assays.
Serum harvested from C3H/HeN or BALB/c mice was inactivated by 30 min of incubation at 56°C or was left unheated. B. burgdorferi cultures were enumerated and diluted to a density of 5 × 106/ml. Diluted cultures were incubated in a 40% final volume of active or heat-inactivated serum in BSKII or with BSKII medium alone at 33°C. At 1, 2, 4, and 16 h of incubation, cultures were enumerated and observed for motility. At each time point, cultures were diluted 1/200 in BSKII medium, and 5 μl of diluted culture was plated in 10 ml BSKII plus 1.7% agarose on top of 10 ml of BSKII plus 1.7% agarose, which was plated and allowed to solidify immediately before. CFU were enumerated after 2 weeks of incubation at 33°C. All experiments were performed with three technical replicates.
SDS-PAGE, silver staining, and immunoblotting.
Bacterial cultures were centrifuged at 11,200 × g for 8 min at ambient temperature, and supernatants were removed. Cell pellets were resuspended in 1 ml of PBS and centrifuged at 11,200 × g for 8 min at ambient temperature. Cell pellets were resuspended in Laemmli buffer plus 0.1% β-mercaptoethanol and boiled at 100°C for 10 min. Total bacteria (1 × 107) were loaded into each well of two replicate 15% SDS-PAGE gels. One gel was silver stained for visualization of total protein (50), and the other was used to perform immunoblotting. For the immunoblot, proteins were transferred to a polyvinylidene difluoride (PVDF) membrane, and nonspecific binding sites were blocked with 5% milk in TBS for 1 h at ambient temperature. Membranes were incubated overnight at 4°C in rabbit anti-OspC (200-401-C11; Rockland Immunochemicals, Inc., Limerick, PA) and mouse anti-flagellin antibodies, washed 3 times, and incubated in alkaline phosphatase-conjugated anti-rabbit and alkaline phosphatase-conjugated anti-mouse antibodies (both from Promega Corp., Madison, WI) for 1 h at ambient temperature. Washed membranes were incubated with chromogenic substrates nitroblue tetrazolium (NBT) and 5-bromo-4-chloro-3-indolylphosphate (BCIP; Sigma-Aldrich, St. Louis, MO) to visualize the bands.
RESULTS
Comparison of infectious and noninfectious B. burgdorferi B31-derived strains in binding to the vasculature in BALB/c mice in vivo.
High-passage noninfectious laboratory strains of B. burgdorferi have lost many genomic plasmids (Table 1); therefore, they encode fewer adhesive proteins. Presumably due to the reduced adhesive proteins on the surface of the bacterium, noninfectious strains have a lesser ability to interact with the vasculature of the host, as determined by intravital microscopy (7). In fact, noninfectious B. burgdorferi strains are commonly used as a background to generate gain-of-function derivatives due to their lower adhesive capabilities (7, 11–13, 51). We evaluated infectious strain B31-A3 and noninfectious strains B31A and B314 of B. burgdorferi in our short-term quantitative infection model. Bacterial burdens in the spleen (P = 0.0193), bladder (P = 0.0007), joint (P = 0.0007), and ear (P = 0.0027) were significantly decreased in BALB/c mice inoculated with B31A compared to levels for B31-A3 (Fig. 1). Unexpectedly, noninfectious strain B314 showed higher burdens in the blood than infectious strain B31-A3 or noninfectious strain B31A (Fig. 1). No significant differences in vascular adhesion to the other tissues analyzed were seen between noninfectious strains B31A and B314 in BALB/c mice (Fig. 1).
FIG 1.

Tissue burdens of infectious and noninfectious B. burgdorferi 1 h after i.v. inoculation. BALB/c mice were inoculated retro-orbitally with 1 × 108 B31-A3 (n = 10), B31A (n = 5), or B314 (n = 5). Bacterial burdens in the spleen, bladder, joint, and ear are significantly lower for B31A than for B31-A3. Blood burdens of B314 are significantly higher than those of B31A and B31-A3. Bacterial burdens are similar between noninfectious strains B314 and B31A as well as infectious strain B31-A3 in all other tissues analyzed. An asterisk indicates statistical significance determined by Mann-Whitney unpaired t test (P < 0.05).
To ensure that effective perfusion of the tissues was occurring and that the bacterial interactions with the vasculature in each tissue were specific, mice were inoculated with 1 × 108 heat-killed B31-A3. Bacterial burdens in all tissues except the ear were significantly lower in mice inoculated with heat-killed bacteria than those inoculated with live bacteria (see Fig. S1 in the supplemental material).
Mouse strain affects vascular adhesion of B. burgdorferi to joint tissue in vivo.
To determine whether differences in vascular interactions are affected by the mouse strain, C3H/HeN and BALB/c mice, both commonly used in the Lyme disease field, were compared for use in our short-term infection model. Mice were retro-orbitally inoculated with 1 × 108 total B. burgdorferi infectious strain B31-A3. After 1 h of circulation, the mice underwent whole-body perfusion, as described in Materials and Methods, and tissues were harvested. Quantification of bacterial burdens revealed significantly more infectious B. burgdorferi in the joints of arthritis-susceptible C3H/HeN mice than in BALB/c mice (P = 0.0280) (Fig. 2). Burdens of infectious B. burgdorferi in the heart and ear were not significantly different between mouse strains (Fig. 2). This effect is not due to differences in the ability of the bacterial strains to survive in the blood, as bacterial burdens in the blood were not significantly different between the mouse strains (see Fig. S2 in the supplemental material).
FIG 2.

Mouse strain affects tissue burdens of B. burgdorferi. BALB/c and C3H/HeN mice were retro-orbitally inoculated with infectious B31-A3 or noninfectious B31A. One hour after inoculation, C3H/HeN mice have significantly higher burdens of B31-A3 and B31A in the joint tissue than BALB/c mice. An asterisk indicates statistical significance determined by Mann-Whitney unpaired t test (P < 0.05; B31-A3 in BALB/c, n = 10; all others, n = 5). BALB/c data for B31-A3- and B31A-inoculated mice are duplicated from Fig. 1.
These experiments were repeated using noninfectious B. burgdorferi strain B31A. After 1 h of circulation, followed by perfusion, significantly higher numbers of bacteria were detected in the joint of C3H/HeN than in those of BALB/c mice (P = 0.0119) (Fig. 2). Bacterial burdens in the heart and ear were not significantly different between mouse strains (Fig. 2).
BB0347 expression confers tropism to joint tissue in vivo.
BB0347 is a fibronectin binding protein expressed on the surface of B. burgdorferi (15). To determine whether BB0347 has a role in the tissue tropism of B. burgdorferi, a gain-of-function derivative was made in the noninfectious strain B31A (11). B31A expressing BB0347 (pBB0347) or the parental B31A were retro-orbitally inoculated into BALB/c mice at an inoculum of 1 × 108 total bacteria. BALB/c mice were chosen for these experiments, because they had lower bacterial burdens in the tibiotarsal joint than C3H/HeN mice (Fig. 2), providing the opportunity to examine possible gain of function upon adhesin expression. After 1 h, animals were perfused and tissues were collected, and B. burgdorferi genomes present in each tissue sample were determined by qPCR. We observed significantly higher bacterial burdens of B31ApBB0347 in the joint than with the parental strain B31A (P = 0.0119) (Fig. 3). This tropism appears to be specific to the joint, as we did not observe any significant differences in bacterial burden in any other tissues collected, including heart and ear (Fig. 3 and data not shown). This joint-specific binding is similar to what we have previously observed with another fibronectin binding protein, BBK32 (14).
FIG 3.

BB0347 is tropic for joint tissue. One hour postinoculation of BALB/c mice with noninfectious B. burgdorferi strain B31A or B31A exogenously expressing BB0347 (pBB0347), significantly higher burdens of B31ApBB0347 are present in the joint tissue. This tropism is specific to the joint, as B31A and B31ApBB0347 burdens are not significantly different in heart or ear. An asterisk indicates statistical significance determined by Mann-Whitney unpaired t test (n = 5, P = 0.0119). B31A data are duplicated from Fig. 1.
RevA expression decreases B. burgdorferi adhesion to heart and ear tissue in BALB/c mice.
RevA is a 17-kDa outer surface protein of B. burgdorferi that has also been shown to bind host fibronectin (16). To explore the role of RevA in vascular binding of B. burgdorferi in vivo, a gain-of-function derivative was made to exogenously express the RevA protein (pRevA) (11). Noninfectious parental strain B31A and the RevA-expressing derivative were inoculated into BALB/c mice. Bacterial burdens of perfused tissues revealed significantly lower numbers of B31ApRevA in the heart (P = 0.0079) and ear tissue (P = 0.0449) than those of B31A (Fig. 4), suggesting that RevA is not involved in adhesion to these target tissues in vivo early in infection. Burdens of B31ApRevA were similar to those of B31A in the joint tissue of inoculated animals (Fig. 4).
FIG 4.

RevA inhibits adhesion of B. burgdorferi to the vasculature in the heart and ear. One hour postinoculation of BALB/c mice with noninfectious B. burgdorferi strain B31A or B31A exogenously expressing RevA (pRevA), significantly lower burdens of B31ApRevA are present in the heart and ear compared to those of B31A. RevA expression does not affect adhesion of B31A to joint tissue, as shown by similar bacterial burdens of B31A and B31ApRevA in this tissue. An asterisk indicates statistical significance determined by Mann-Whitney unpaired t test (P < 0.05, n = 5). B31A data are duplicated from Fig. 1.
Elimination of an adhesin, P66, from infectious B. burgdorferi can be used to reveal tissue tropisms.
We have shown the ability of our model system to decipher tissue specific binding properties of three B. burgdorferi adhesins using gain-of-function derivatives of noninfectious B. burgdorferi strains (11 and this work). We also were interested in testing the ability of our system to detect loss of vascular binding function due to mutations in an infectious strain background. To achieve this, we chose to look at the integrin-binding adhesin, P66, a B. burgdorferi protein which we have previously shown to be required for infectivity in mice (18). Infectious strain B31-A3 deficient in expression of P66 was used to inoculate BALB/c mice for our short-term infection model. Mice were inoculated with 1 × 108 total B31-A3 or B31-A3Δp66 mutant. After 1 h, tissues were perfused and collected. qPCR quantitation of B. burgdorferi genomes revealed significantly lower numbers of the B31-A3Δp66 mutant in the heart (P = 0.0029) and ear tissue (P = 0.002) than those for B31-A3 (Fig. 5). P66 mutant bacteria were present at levels similar to those of the WT in the joint tissue of inoculated animals (Fig. 5).
FIG 5.

P66 promotes adhesion to the vasculature in heart and ear tissue 1 h after i.v. inoculation. BALB/c mice were retro-orbitally inoculated with infectious B. burgdorferi strain B31-A3 or the B31-A3Δp66 mutant, which is not infectious in mice. Bacterial burdens of B31-A3Δp66 were significantly lower than those of the WT in the heart and ear as determined by qPCR. Bacterial burdens of the mutant were similar to those of the WT in the joint tissue. An asterisk indicates statistical significance determined by Mann-Whitney unpaired t test (P < 0.05, n = 10). B31-A3 data are duplicated from Fig. 1.
B. burgdorferi adhesins influence bloodstream burdens in vivo.
It has been shown previously that B. burgdorferi expresses surface proteins that recruit host complement regulatory factors to the bacterial surface (52–55). In this way, B. burgdorferi achieves resistance to the alternative complement cascade active in the blood of the host. We found that freshly isolated serum from both BALB/c and C3H/HeN mice was unable to kill infectious B. burgdorferi strain B31-A3 as well as E. coli M1061 in vitro (56 and data not shown). Using our short-term infection model, we were able to assess differences in bloodstream burdens of infectious and noninfectious Borrelia strains in vivo. Blood was collected 1 h after inoculation of BALB/c mice with strain B31A or gain-of-function derivatives expressing RevA (pRevA), BB0347 (pBB0347), or BBK32 (pBBK32). We also assessed BBK32 lacking the GAG binding domain (pBBK32ΔGAG) or the fibronectin binding domain (pBBK32ΔFn) (11). One hour postinoculation, there were significantly fewer B31ApRevA than parental B31A bacteria present in the blood (P = 0.0079) (Fig. 6). The expression of full-length BBK32 by B31A allowed for a significant increase in bacterial presence in the blood compared to that of parental B31A (P = 0.04) (Fig. 6). This increased blood burden is reduced to parental B31A levels upon the elimination of the GAG binding domain of BBK32 (Fig. 6). The gain-of-function derivative expressing BB0347 also appeared to show increased blood burdens compared to those of parental B31A, although this difference was not statistically significant (Fig. 6).
FIG 6.
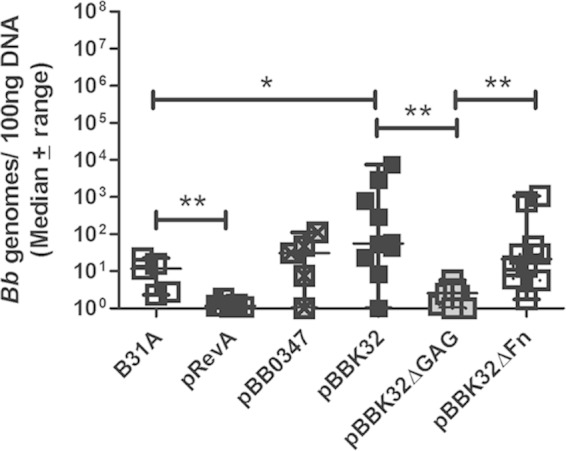
Adhesins influence bloodstream burdens of B. burgdorferi in vivo. BALB/c mice were inoculated with 1 × 108 total noninfectious B. burgdorferi strain B31A or B31A exogenously expressing adhesive protein RevA (pRevA), BB0347 (pBB0347), BBK32 (pBBK32), or BBK32 with a deletion in the GAG (pBBK32ΔGAG) or fibronectin (pBBK32ΔFn) binding domains. The expression of BBK32 significantly increased the number of bacteria present in the blood compared to that of the WT. The GAG binding domain of BBK32 is responsible for this phenotype. Statistical significance was determined by Mann-Whitney unpaired t test for B31A, pRevA, and pBB0347 (n = 5) and for pBBK32, pBBK32ΔGAG, and pBBK32ΔFn (n = 10). **, P < 0.01; *, P < 0.05. B31A data are duplicated from Fig. 1.
We had assessed the bloodstream burdens of another noninfectious strain of B. burgdorferi, B314, early in the course of developing this short-term infection model (Fig. 1). Unexpectedly, we observed an increase in bloodstream burdens of noninfectious strain B314 compared to those of B31-A3 and B31A 1 h after intravenous inoculation into both BALB/c and C3H/HeN mice (Fig. 1; also see Fig. S2 in the supplemental material), although the burdens of B314 in other tissues did not differ significantly from those of B31-A3 or B31A. To identify candidate proteins responsible for this phenotype, we examined total cell lysates of B31-A3, B31A, and B314 on a reducing SDS-PAGE gel and performed a silver stain (see Fig. S3A). Analysis of the silver-stained gel revealed an abundant protein running at 24 kDa that is present in the B314 lysate. Immunoblotting was performed to determine whether this abundant protein was the outer surface protein OspC. We found that OspC is produced at higher levels in B314 than B31-A3 and B31A (see Fig. S3B). In fact, OspC was undetectable in the B31A lysate by immunoblotting (see Fig. S3B). To determine the impact of OspC on bloodstream burdens of infectious WT B. burgdorferi, we inoculated BALB/c mice with infectious strain B31-A3, which is deficient in OspC expression (22). Previous work using this strain has shown that OspC is required for the early stages of mammalian infection but is not required for survival of B. burgdorferi in the tick (21, 22). One hour postinoculation, we saw significantly higher burdens of B31-A3ΔospC mutant than infectious WT bacteria in the blood of BALB/c mice (P = 0.0004) (Fig. 7). As we have shown previously, the mouse strain used for our short-term infection model can have an impact on vascular adhesion of B. burgdorferi (Fig. 2). With this in mind, we inoculated C3H/HeN mice with WT infectious strain B31-A3, B31-A3ΔospC mutant, OspC mutant with OspC expression restored (B31-A3ΔospCpOspC) (22), or B31-A3Δp66 mutant (18) bacteria. In this short-term infection model, a median of 30.75 B31-A3 genomes per 100 ng total DNA was present in the blood of C3H/HeN mice after 1 h of circulation. In contrast, we were unable to detect any B31-A3ΔospC mutant bacteria in the blood of C3H/HeN mice (P = 0.0013) (Fig. 7), similar to the results seen when C3H/HeN mice were inoculated with heat-killed bacteria (see Fig. S1). Bacterial burdens in the bloodstream were increased upon restoration of OspC expression (Fig. 7). Just as was seen in BALB/c mice, P66 mutant bacteria survived as well as the infectious WT did in the bloodstream of C3H/HeN mice at 1 h postinoculation (Fig. 7).
FIG 7.

Elimination of OspC expression decreases bloodstream burdens of infectious B. burgdorferi in C3H/HeN but not BALB/c mice. C3H/HeN and BALB/c mice were inoculated with 1 × 108 total infectious B. burgdorferi strain B31-A3, B31-A3 deficient in OspC expression (ΔospC), or B31-A3ΔospC with OspC expression restored (pOspC). After 1 h, blood was collected and bacteria were enumerated in each sample by qPCR. B31-A3ΔospC mutant bacteria were undetectable in the bloodstream of C3H/HeN mice 1 h postinoculation. In BALB/c mice, significantly higher numbers of the OspC mutant than of WT bacteria were present. Restoration of OspC expression partially restores burdens of the mutant in the blood of C3H/HeN mice. No significant differences in blood burden were seen between the P66 mutant and infectious WT-inoculated BALB/c or C3H/HeN mice. An asterisk indicates statistical significance determined by nonparametric one-way ANOVA with Dunn's posttest (B31-A3 and the ΔospC mutant, n = 10; pOspC, n = 5; Δp66 mutant, n = 5 [C3H/HeN]; n = 10 [BALB/c]). B31-A3 data from BALB/c mice are duplicated from Fig. 1.
DISCUSSION
B. burgdorferi is known to cause persistent infections in the joint, heart, skin, and central nervous system of the human host. It is thought that at least a portion of the transmitted spirochetes disseminate from the site of tick bite by entering the bloodstream in the vicinity of the tick feeding site and then exit the vasculature by binding to the walls of small vessels and transmigrating out into the surrounding tissue. It has been shown that B. burgdorferi binds to the host microvasculature as well as to cells and extracellular matrix components using adhesive proteins that are expressed on the bacterial cell surface (7, 11). To discern the biological roles of different outer surface proteins of B. burgdorferi in tissue-specific vascular adhesion of the pathogen, a new type of in vivo model was required. We have developed an animal model to allow us to study bacterial binding to the vasculature, the precursor step to tissue invasion and colonization by the bacteria. The optimal time point for observation of bacterial adhesion in our model was 1 h postinoculation, similar to that used for intravital microscopic studies of infectious B. burgdorferi (7, 13).
In our model, the bacterial burdens in the tissue likely are attributable to vascular adhesion by the spirochetes, as transmigration out of the vasculature and subsequent tissue colonization has not yet occurred (Kumar et al., unpublished data). Low bacterial burdens in the blood and tissues could be due to an inability of the bacteria to bind to the vasculature, resulting in continued circulation in the bloodstream, bacterial lysis by complement components in the bloodstream, or uptake by circulating phagocytes. However, the ability of the bacteria to resist phagocytosis or lysis due to actions of components of the innate immune system also may affect B. burgdorferi burdens in all tissues, including the blood. Using our model, we have confirmed previous findings that noninfectious strains of B. burgdorferi adhere less to the host vasculature than infectious strains (7). Additionally, adhesion to the vascular wall seems to be an active process initiated and/or maintained by living bacteria, as we detect significantly less heat-killed B31-A3 than live B31-A3 in all collected mouse tissues except ear (Fig. S1). At this time, the kinetics of bacterial DNA breakdown are not known, but we suspect it occurs in less than an hour, as this would explain the reduced burdens of the heat-killed bacteria in all tissues (see Fig. S1 in the supplemental material).
Vascular adhesion by B. burgdorferi appears to differ based on the strain of mouse used for the model. BALB/c mice show significantly lower burdens of both infectious and noninfectious B. burgdorferi in the joint than C3H/HeN mice (Fig. 2). The difference in joint adhesion that we see between BALB/c and C3H/HeN mice early after inoculation with B. burgdorferi may have effects on the immune response of the host late in infection and may in part contribute to the previous observations that BALB/c mice are less susceptible to B. burgdorferi-mediated arthritis than C3H/HeN mice (57, 58).
We used our short-term infection model to differentiate the in vivo roles of several adhesive outer surface proteins of B. burgdorferi that have been shown to bind fibronectin in vitro. Using gain-of-function strains generated in the otherwise noninfectious strain B31A, we were able to observe increased binding to the vasculature in joint tissue attributable to expression of BB0347 but not of RevA. It remains to be determined whether the adhesion of BB0347 to the vasculature of joint tissue is attributable to its fibronectin binding capacity. A similar phenotype of joint-specific binding was seen with B31A expressing BBK32 (14), an outer surface protein of B. burgdorferi that has been shown to bind both glycosaminoglycans (GAGs) and fibronectin via distinct domains (11). The resolution of our short-term infection model allowed us to determine that the GAG binding domain of BBK32 is required for the joint-specific vascular binding observed for bacteria expressing full-length BBK32 (14). This phenotype was confirmed by a lack of joint colonization after 21 days in mice needle inoculated with BBK32 mutant bacteria in an otherwise infectious B. burgdorferi strain (14). The flexibility of the model also has allowed us to determine the contribution of B. burgdorferi outer surface proteins to tissue-tropic binding of infectious B. burgdorferi through use of a deletion mutant in an infectious strain background. A role for P66 in binding to the vasculature in heart and ear tissue was found through use of a B31-A3Δp66 mutant generated previously (18, 56).
When considering all of the data together, it becomes evident that vascular binding of B. burgdorferi is more complex than just that of a simple protein-receptor model. We observed that the expression of RevA on the surface of B31A decreases bacterial burdens of B. burgdorferi in the heart and skin compared to levels for parental B31A. This may be due to decreased adhesion to the vasculature at those tissue sites or increased bacterial clearance by the innate immune system. However, we saw a similar phenotype in the heart and skin of mice inoculated with the B31-A3Δp66 mutant. It is possible that the decrease observed in the heart and skin burdens of mice inoculated with B31ApRevA actually is due to a shielding of the surface-exposed regions of P66 on these bacteria, resulting in a phenotype mirroring that of the B31-A3Δp66 mutant.
Survival of B. burgdorferi in the blood of the mammal likely is important in the life cycle of this pathogen. During a tick blood meal, B. burgdorferi within the tick come into contact with blood of the host. Without survival in the feeding tick, the bacteria would not be transmitted to cause successful infection in new vertebrate hosts. Efficient dissemination from the site of tick bite to distal tissue sites in the vertebrate host involves a bacteremic phase, i.e., survival in the bloodstream. This bacteremic phase has been documented in the mouse model (57, 59) as well as in human patients (1, 2, 60). To better survive travel in the bloodstream of the host, B. burgdorferi has developed resistance to killing by serum complement proteins, a feature shared by B. afzelii (61). Serum resistance is not, however, absolutely required for mammalian infection, as serum-sensitive B. garinii still is able to survive in the natural enzootic cycle and to cause Lyme borreliosis in humans (61). The study of B. burgdorferi proteins and their roles in serum resistance has not been addressable in mouse serum in vitro due to the inactivity of mouse serum proteins (presumably complement proteins) ex vivo. Our mouse model has proven useful in circumventing this complication. Using gain-of-function and gene knockout mutant strains, we were able to address the roles of specific outer surface proteins in affecting bloodstream burdens of B. burgdorferi, as the bacteria were directly inoculated into the bloodstream in our animal model. The expression of BBK32 on the surface of noninfectious strain B31A increased the bacterial burden in blood compared to that of parental strain B31A, whereas the expression of RevA decreased the bacterial burdens in the blood compared to those of the parental strain (Fig. 6). The differences that we see in bloodstream survival of the bacteria in the presence or absence of BBK32 may be due to decreased bacterial clearance or to decreased binding to the vasculature of the host. We believe the latter case is unlikely, as strain B31A showed lower bacterial adhesion to the vasculature in several tissues but no increase in bloodstream burden of bacteria. Finally, we see increased binding of B31ApBBK32 specifically to the vasculature of the joint (14) and increased bloodstream survival with the same strain. Using targeted gene knockouts, we were able to find a novel role for a well-studied outer surface protein, OspC, in bloodstream burdens of B. burgdorferi. With deletion of the ospC gene in the otherwise infectious strain B31-A3, we saw complete clearance of the bacteria within 1 h of inoculation directly into the vasculature of C3H/HeN mice. This phenotype was due directly to the presence of OspC on the bacterial surface, as the restoration of OspC expression partially restored bloodstream burdens back to WT levels. It has been shown that OspC-deficient B. burgdorferi is able to persist at the inoculation site for 24 h after infection (23), but survival of this mutant in the mammalian bloodstream has never been addressed. The increased blood burden of noninfectious strain B314, which we showed to express high levels of OspC, supports our conclusion that the OspC protein is important for bloodstream survival of B. burgdorferi. This increase in burdens of B314 in the bloodstream likely is not due to a decrease in vascular binding, as we did not observe a significant difference in B314 burdens compared to those of the infectious WT in any other tissue collected. Interestingly, we saw increased bacterial burdens in the blood of BALB/c mice inoculated with B31-A3ΔospC mutant bacteria. Although blood burdens of both B314 and the B31-A3ΔospC mutant both were significantly higher than that of B31-A3 in BALB/c mice, burdens of B314 were lower than those seen in the blood of ospC mutant-inoculated BALB/c mice. However, although B31A does not express detectable levels of OspC, bloodstream burdens of this strain in BALB/c mice are much lower than what is seen with B31-A3ΔospC mutant-inoculated mice. These contradictory results could be explained by differences in the other surface proteins encoded in the genomes of noninfectious B314 and B31A compared to infectious strain B31-A3, as well as differential regulation of expression of surface proteins in the three B. burgdorferi strains. Our data suggest that there is a protein or phagocytic cell type present in the bloodstream of C3H/HeN mice but absent from BALB/c mice that is involved in controlling B. burgdorferi burdens in the bloodstream through direct or indirect interactions with OspC on the surface of the pathogen. More work will need to be done to define the mechanism of OspC and its effects on bloodstream survival.
This short-term model has proven to be very powerful in identifying not only unanticipated differences between mouse strains in B. burgdorferi infection but also the binding specificities of individual outer surface proteins of B. burgdorferi in an in vivo setting. This infection model also has revealed unanticipated roles in serum resistance, as inferred by increased bloodstream burdens, for two B. burgdorferi proteins, OspC and BBK32. The strength and versatility of this model will be useful in understanding the complex mechanisms of bloodstream survival and tissue colonization by Borrelia species.
Supplementary Material
ACKNOWLEDGMENTS
We thank George Chaconas (University of Calgary, Calgary, Alberta, Canada), Tara Moriarty (University of Toronto, Toronto, Ontario, Canada), Patricia Rosa (Rocky Mountain Laboratories, Hamilton, MT), and John Leong and Yi-Pin Lin (Tufts University, Boston, MA) for their generous donations of B. burgdorferi strains, Tom Schwan (Rocky Mountain Laboratories, Hamilton, MT) for providing antibodies necessary for our work, and Hiromi Sato (Medical College of Wisconsin, Milwaukee, WI) for review of the manuscript.
Research reported in this publication was supported by the National Institute Of Allergy and Infectious Diseases of the National Institutes of Health under award numbers R01AI093104 and R01AI084873.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00349-15.
REFERENCES
- 1.Steere AC. 2001. Lyme disease. N Engl J Med 345:115–125. doi: 10.1056/NEJM200107123450207. [DOI] [PubMed] [Google Scholar]
- 2.Steere AC, Grodzicki RL, Kornblatt AN, Craft JE, Barbour AG, Burgdorfer W, Schmid GP, Johnson E, Malawista SE. 1983. The spirochetal etiology of Lyme disease. N Engl J Med 308:733–740. [DOI] [PubMed] [Google Scholar]
- 3.Steere AC. 1997. Diagnosis and treatment of Lyme arthritis. Med Clin North Am 81:179–194. doi: 10.1016/S0025-7125(05)70510-1. [DOI] [PubMed] [Google Scholar]
- 4.Steere AC. 1995. Musculoskeletal manifestations of Lyme disease. Am J Med 98:44S–51S. doi: 10.1016/S0002-9343(99)80043-6. [DOI] [PubMed] [Google Scholar]
- 5.Nolan DJ, Ginsberg M, Israely E, Palikuqi B, Poulos MG, James D, Ding B-S, Schachterle W, Liu Y, Rosenwaks Z, Butler JM, Xiang J, Rafii A, Shido K, Rabbany SY, Elemento O, Rafii S. 2013. Molecular signatures of tissue-specific microvascular endothelial cell heterogeneity in organ maintenance and regeneration. Dev Cell 26:204–219. doi: 10.1016/j.devcel.2013.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yano K, Gale D, Massberg S, Cheruvu PK, Monahan-Earley R, Morgan ES, Haig D, von Andrian UH, Dvorak AM, Aird WC. 2007. Phenotypic heterogeneity is an evolutionarily conserved feature of the endothelium. Blood 109:613–615. doi: 10.1182/blood-2006-05-026401. [DOI] [PubMed] [Google Scholar]
- 7.Moriarty TJ, Norman MU, Colarusso P, Bankhead T, Kubes P, Chaconas G. 2008. Real-time high resolution 3D imaging of the Lyme disease spirochete adhering to and escaping from the vasculature of a living host. PLoS Pathog 4:e1000090. doi: 10.1371/journal.ppat.1000090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pulzova L, Bhide M. 2014. Outer surface proteins of Borrelia: peerless immune evasion tools. Curr Protein Pept Sci 15:75–88. doi: 10.2174/1389203715666140221124213#sthash.TIQ5HbGd.dpuf. [DOI] [PubMed] [Google Scholar]
- 9.Coburn J, Leong J, Chaconas G. 2013. Illuminating the roles of the Borrelia burgdorferi adhesins. Trends Microbiol 21:372–379. doi: 10.1016/j.tim.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brissette CA, Gaultney RA. 2014. That's my story, and I'm sticking to it–an update on B. burgdorferi adhesins. Front Cell Infect Microbiol 4:41. doi: 10.3389/fcimb.2014.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moriarty TJ, Shi M, Lin Y-P, Ebady R, Zhou H, Odisho T, Hardy P-O, Salman-Dilgimen A, Wu J, Weening EH, Skare JT, Kubes P, Leong J, Chaconas G. 2012. Vascular binding of a pathogen under shear force through mechanistically distinct sequential interactions with host macromolecules. Mol Microbiol 86:1116–1131. doi: 10.1111/mmi.12045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fischer JR, LeBlanc KT, Leong JM. 2006. Fibronectin binding protein BBK32 of the Lyme disease spirochete promotes bacterial attachment to glycosaminoglycans. Infect Immun 74:435–441. doi: 10.1128/IAI.74.1.435-441.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Norman MU, Moriarty TJ, Dresser AR, Millen B, Kubes P, Chaconas G. 2008. Molecular mechanisms involved in vascular interactions of the Lyme disease pathogen in a living host. PLoS Pathog 4:e1000169. doi: 10.1371/journal.ppat.1000169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin Y-P, Chen Q, Ritchie JA, Dufour NP, Fischer JR, Coburn J, Leong JM. 2015. Glycosaminoglycan binding by Borrelia burgdorferi adhesin BBK32 specifically and uniquely promotes joint colonization. Cell Microbiol 17:860–875. doi: 10.1111/cmi.12407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gaultney RA, Gonzalez T, Floden AM, Brissette CA. 2013. BB0347, from the Lyme disease spirochete Borrelia burgdorferi, is surface exposed and interacts with the CS1 heparin-binding domain of human fibronectin. PLoS One 8:e75643. doi: 10.1371/journal.pone.0075643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brissette CA, Bykowski T, Cooley AE, Bowman A, Stevenson B. 2009. Borrelia burgdorferi RevA antigen binds host fibronectin. Infect Immun 77:2802–2812. doi: 10.1128/IAI.00227-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fortune DE, Lin Y-P, Deka RK, Groshong AM, Moore BP, Hagman KE, Leong JM, Tomchick DR, Blevins JS. 2014. Identification of lysine residues in the Borrelia burgdorferi DbpA adhesin required for murine infection. Infect Immun 82:3186–3198. doi: 10.1128/IAI.02036-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ristow LC, Miller HE, Padmore LJ, Chettri R, Salzman N, Caimano MJ, Rosa PA, Coburn J. 2012. The β3-integrin ligand of Borrelia burgdorferi is critical for infection of mice but not ticks. Mol Microbiol 85:1105–1118. doi: 10.1111/j.1365-2958.2012.08160.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Behera AK, Durand E, Cugini C, Antonara S, Bourassa L, Hildebrand E, Hu LT, Coburn J. 2008. Borrelia burgdorferi BBB07 interaction with integrin α3β1 stimulates production of pro-inflammatory mediators in primary human chondrocytes. Cell Microbiol 10:320–331. doi: 10.1111/j.1462-5822.2007.01043.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Önder Ö, Humphrey PT, McOmber B, Korobova F, Francella N, Greenbaum DC, Brisson D. 2012. OspC is potent plasminogen receptor on surface of Borrelia burgdorferi. J Biol Chem 287:16860–16868. doi: 10.1074/jbc.M111.290775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grimm D, Tilly K, Byram R, Stewart PE, Krum JG, Bueschel DM, Schwan TG, Policastro PF, Elias AF, Rosa PA. 2004. Outer-surface protein C of the Lyme disease spirochete: a protein induced in ticks for infection of mammals. Proc Natl Acad Sci U S A 101:3142–3147. doi: 10.1073/pnas.0306845101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tilly K, Krum JG, Bestor A, Jewett MW, Grimm D, Bueschel D, Byram R, Dorward D, VanRaden MJ, Stewart P, Rosa P. 2006. Borrelia burgdorferi OspC protein required exclusively in a crucial early stage of mammalian infection. Infect Immun 74:3554–3564. doi: 10.1128/IAI.01950-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tilly K, Bestor A, Jewett MW, Rosa P. 2007. Rapid clearance of Lyme disease spirochetes lacking OspC from skin. Infect Immun 75:1517–1519. doi: 10.1128/IAI.01725-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu Q, McShan K, Liang FT. 2008. Essential protective role attributed to the surface lipoproteins of Borrelia burgdorferi against innate defences. Mol Microbiol 69:15–29. doi: 10.1111/j.1365-2958.2008.06264.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Earnhart CG, LeBlanc DV, Alix KE, Desrosiers DC, Radolf JD, Marconi RT. 2010. Identification of residues within ligand-binding domain 1 (LBD1) of the Borrelia burgdorferi OspC protein required for function in the mammalian environment. Mol Microbiol 76:393–408. doi: 10.1111/j.1365-2958.2010.07103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fikrig E, Feng W, Barthold SW, Telford SR, Flavell RA. 2000. Arthropod- and host-specific Borrelia burgdorferi BBK32 expression and the inhibition of spirochete transmission. J Immunol 164:5344–5351. doi: 10.4049/jimmunol.164.10.5344. [DOI] [PubMed] [Google Scholar]
- 27.Cugini C, Medrano M, Schwan TG, Coburn J. 2003. Regulation of expression of the Borrelia burgdorferi β3-chain integrin ligand, P66, in ticks and in culture. Infect Immun 71:1001–1007. doi: 10.1128/IAI.71.2.1001-1007.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang J-R, Hardham JM, Barbour AG, Norris SJ. 1997. Antigenic variation in Lyme disease Borreliae by promiscuous recombination of VMP-like sequence cassettes. Cell 89:275–285. doi: 10.1016/S0092-8674(00)80206-8. [DOI] [PubMed] [Google Scholar]
- 29.Lawrenz MB, Hardham JM, Owens RT, Nowakowski J, Steere AC, Wormser GP, Norris SJ. 1999. Human antibody responses to VlsE antigenic variation protein of Borrelia burgdorferi. J Clin Microbiol 37:3997–4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou W, Brisson D. 2014. Potentially conflicting selective forces that shape the vls antigenic variation system in Borrelia burgdorferi. Infect Genet Evol 27:559–565. doi: 10.1016/j.meegid.2014.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liang FT, Yan J, Mbow ML, Sviat SL, Gilmore RD, Mamula M, Fikrig E. 2004. Borrelia burgdorferi changes its surface antigenic expression in response to host immune responses. Infect Immun 72:5759–5767. doi: 10.1128/IAI.72.10.5759-5767.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kraiczy P, Stevenson B. 2013. Complement regulator-acquiring surface proteins of Borrelia burgdorferi: structure, function and regulation of gene expression. Ticks Tick Borne Dis 4:26–34. doi: 10.1016/j.ttbdis.2012.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kenedy MR, Vuppala SR, Siegel C, Kraiczy P, Akins DR. 2009. CspA-mediated binding of human factor H inhibits complement deposition and confers serum resistance in Borrelia burgdorferi. Infect Immun 77:2773–2782. doi: 10.1128/IAI.00318-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hammerschmidt C, Koenigs A, Siegel C, Hallström T, Skerka C, Wallich R, Zipfel PF, Kraiczy P. 2014. Versatile roles of CspA orthologs in complement inactivation of serum-resistant Lyme disease spirochetes. Infect Immun 82:380–392. doi: 10.1128/IAI.01094-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hallström T, Siegel C, Mörgelin M, Kraiczy P, Skerka C, Zipfel PF. 2013. CspA from Borrelia burgdorferi inhibits the terminal complement pathway. mBio 4:e00481-13. doi: 10.1128/mBio.00481-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alitalo A, Meri T, Lankinen H, Seppälä I, Lahdenne P, Hefty PS, Akins D, Meri S. 2002. Complement inhibitor factor H binding to Lyme disease spirochetes is mediated by inducible expression of multiple plasmid-encoded outer surface protein E paralogs. J Immunol 169:3847–3853. doi: 10.4049/jimmunol.169.7.3847. [DOI] [PubMed] [Google Scholar]
- 37.Kraiczy P, Hartmann K, Hellwage J, Skerka C, Kirschfink M, Brade V, Zipfel PF, Wallich R, Stevenson B. 2004. Immunological characterization of the complement regulator factor H-binding CRASP and Erp proteins of Borrelia burgdorferi. Int J Med Microbiol 293:152–157. [DOI] [PubMed] [Google Scholar]
- 38.Caesar JJE, Johnson S, Kraiczy P, Lea SM. 2013. ErpC, a member of the complement regulator-acquiring family of surface proteins from Borrelia burgdorferi, possesses an architecture previously unseen in this protein family. Acta Crystallogr F Struct Biol 69:624–628. doi: 10.1107/S1744309113013249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hammerschmidt C, Hallström T, Skerka C, Wallich R, Stevenson B, Zipfel PF, Kraiczy P. 2012. Contribution of the infection-associated complement regulator-acquiring surface protein 4 (ErpC) to complement resistance of Borrelia burgdorferi. Clin Dev Immunol 2012:349657. doi: 10.1155/2012/349657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bykowski T, Woodman ME, Cooley AE, Brissette CA, Brade V, Wallich R, Kraiczy P, Stevenson B. 2007. Coordinated expression of Borrelia burgdorferi complement regulator-acquiring surface proteins during the Lyme disease spirochete's mammal-tick infection cycle. Infect Immun 75:4227–4236. doi: 10.1128/IAI.00604-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Elias AF, Stewart PE, Grimm D, Caimano MJ, Eggers CH, Tilly K, Bono JL, Akins DR, Radolf JD, Schwan TG, Rosa P. 2002. Clonal polymorphism of Borrelia burgdorferi strain B31 MI: implications for mutagenesis in an infectious strain background. Infect Immun 70:2139–2150. doi: 10.1128/IAI.70.4.2139-2150.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sadziene A, Wilske B, Ferdows MS, Barbour AG. 1993. The cryptic ospC gene of Borrelia burgdorferi B31 is located on a circular plasmid. Infect Immun 61:2192–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barbour AG. 1984. Isolation and cultivation of Lyme disease spirochetes. Yale J Biol Med 57:521–525. [PMC free article] [PubMed] [Google Scholar]
- 44.Bunikis I, Kutschan-Bunikis S, Bonde M, Bergström S. 2011. Multiplex PCR as a tool for validating plasmid content of Borrelia burgdorferi. J Microbiol Methods 86:243–247. doi: 10.1016/j.mimet.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 45.Tokarz R, Anderton JM, Katona LI, Benach JL. 2004. Combined effects of blood and temperature shift on Borrelia burgdorferi gene expression as determined by whole genome DNA array. Infect Immun 72:5419–5432. doi: 10.1128/IAI.72.9.5419-5432.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schwan TG, Piesman J, Golde WT, Dolan MC, Rosa PA. 1995. Induction of an outer surface protein on Borrelia burgdorferi during tick feeding. Proc Natl Acad Sci U S A 92:2909–2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stevenson B, Schwan TG, Rosa PA. 1995. Temperature-related differential expression of antigens in the Lyme disease spirochete, Borrelia burgdorferi. Infect Immun 63:4535–4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Frank KL, Bundle SF, Kresge ME, Eggers CH, Samuels DS. 2003. aadA confers streptomycin resistance in Borrelia burgdorferi. J Bacteriol 185:6723–6727. doi: 10.1128/JB.185.22.6723-6727.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sadziene A, Thomas DD, Barbour AG. 1995. Borrelia burgdorferi mutant lacking Osp: biological and immunological characterization. Infect Immun 63:1573–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shevchenko A, Wilm M, Vorm O, Mann M. 1996. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem 68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 51.Fischer JR, Parveen N, Magoun L, Leong JM. 2003. Decorin-binding proteins A and B confer distinct mammalian cell type-specific attachment by Borrelia burgdorferi, the Lyme disease spirochete. Proc Natl Acad Sci U S A 100:7307–7312. doi: 10.1073/pnas.1231043100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kraiczy P, Skerka C, Kirschfink M, Zipfel PF, Brade V. 2001. Mechanism of complement resistance of pathogenic Borrelia burgdorferi isolates. Int Immunopharmacol 1:393–401. doi: 10.1016/S1567-5769(00)00041-2. [DOI] [PubMed] [Google Scholar]
- 53.Kraiczy P, Skerka C, Brade V, Zipfel PF. 2001. Further characterization of complement regulator-acquiring surface proteins of Borrelia burgdorferi. Infect Immun 69:7800–7809. doi: 10.1128/IAI.69.12.7800-7809.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kraiczy P, Rossmann E, Brade V, Simon MM, Skerka C, Zipfel PF, Wallich R. 2006. Binding of human complement regulators FHL-1 and factor H to CRASP-1 orthologs of Borrelia burgdorferi. Wien Klin Wochenschr 118:669–676. doi: 10.1007/s00508-006-0691-1. [DOI] [PubMed] [Google Scholar]
- 55.Kraiczy P, Skerka C, Kirschfink M, Brade V, Zipfel PF. 2001. Immune evasion of Borrelia burgdorferi by acquisition of human complement regulators FHL-1/Reconectin and factor H. Eur J Immunol 31:1674–1684. doi:. [DOI] [PubMed] [Google Scholar]
- 56.Ristow LC, Bonde M, Lin Y-P, Sato H, Curtis M, Geissler E, Hahn BL, Fang J, Wilcox DA, Leong JM, Bergström S, Coburn J. 16 February 2015. Integrin binding by Borrelia burgdorferi p66 facilitates dissemination but is not required for infectivity. Cell Microbiol doi: 10.1111/cmi.12418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Barthold SW, Beck DS, Hansen GM, Terwilliger GA, Moody KD. 1990. Lyme borreliosis in selected strains and ages of laboratory mice. J Infect Dis 162:133–138. doi: 10.1093/infdis/162.1.133. [DOI] [PubMed] [Google Scholar]
- 58.Wooten RM, Weis JJ. 2001. Host–pathogen interactions promoting inflammatory Lyme arthritis: use of mouse models for dissection of disease processes. Curr Opin Microbiol 4:274–279. doi: 10.1016/S1369-5274(00)00202-2. [DOI] [PubMed] [Google Scholar]
- 59.Barthold S. 1991. Kinetics of Borrelia burgdorferi dissemination and evolution of disease after intradermal inoculation of mice. Am J Pathol 139:263–273. [PMC free article] [PubMed] [Google Scholar]
- 60.Schmid GP. 1989. Epidemiology and clinical similarities of human spirochetal diseases. Rev Infect Dis 11:S1460–S1469. [DOI] [PubMed] [Google Scholar]
- 61.van Dam AP, Oei A, Jaspars R, Fijen C, Wilske B, Spanjaard L, Dankert J. 1997. Complement-mediated serum sensitivity among spirochetes that cause Lyme disease. Infect Immun 65:1228–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.