

Abstract
Listeria monocytogenes is a highly adaptive bacterium that replicates as a free-living saprophyte in the environment as well as a facultative intracellular pathogen that causes invasive foodborne infections. The intracellular life cycle of L. monocytogenes is considered to be its primary virulence determinant during mammalian infection; however, the proportion of L. monocytogenes that is intracellular in vivo has not been studied extensively. In this report, we demonstrate that the majority of wild-type (strain EGDe) and mouse-adapted (InlAm-expressing) L. monocytogenes recovered from the mesenteric lymph nodes (MLN) was extracellular within the first few days after foodborne infection. In addition, significantly lower burdens of L. monocytogenes were recovered from the colon, spleen, and liver of gentamicin-treated mice than of control mice. This led us to investigate whether intracellular replication of L. monocytogenes was essential during the intestinal phase of infection. We found that lipoate protein ligase-deficient L. monocytogenes (ΔlplA1) mutants, which display impaired intracellular growth, were able to colonize the colon but did not persist efficiently and had a significant defect in spreading to the MLN, spleen, and liver. Together, these data indicate that the majority of the L. monocytogenes burden in the gastrointestinal tract is extracellular, but the small proportion of intracellular L. monocytogenes is essential for dissemination to the MLN and systemic organs.
INTRODUCTION
Listeria monocytogenes is a highly adaptable bacterial pathogen that can grow in diverse environments, including the cytosol of mammalian cells (1, 2). Much research effort has focused on defining the factors that allow cell-to-cell spread of L. monocytogenes without encountering the extracellular environment, since this is thought to be the primary virulence strategy of L. monocytogenes in vivo. However, as a facultative intracellular pathogen, L. monocytogenes can readily survive and multiply in extracellular spaces, and there may be multiple environments that harbor extracellular bacteria during infection. For example, we previously showed that extracellular L. monocytogenes was present in the lamina propria of both the ileum and the colon after foodborne infection (3). The liver, spleen, and placenta were each shown to contain gentamicin-sensitive L. monocytogenes during systemic infection of mice or guinea pigs (4, 5). There is also evidence that L. monocytogenes replicates extracellularly in the lumen of the murine gallbladder, and it was suggested that the presence of these organisms may prolong intestinal infection if infected bile is released into the small intestine (6). These previous studies indicate that extracellular L. monocytogenes can be present in a variety of tissues during mammalian infection; however, the relative proportion of extracellular L. monocytogenes and a role in virulence have not been clearly established.
Studies using signature-tagged bacteria have been fundamental in modeling the systemic spread of enteric pathogens by highlighting two routes of spread from the gut (7–9). One route presumably involves direct invasion of the bloodstream and dissemination via the portal vein since it results in colonization primarily in the liver. The mechanisms used by bacteria to promote this invasion are unclear, and it is possible that the use of excessively large inocula or physically traumatic transmission methods can facilitate rapid spread by this route. Bacteria can also reach the spleen and liver after colonizing the draining mesenteric lymph nodes (MLN) (7–9). Melton-Witt et al. showed that this indirect route of spread led to continual seeding of the spleen, presumably due to the flow of efferent lymphatic fluid into the bloodstream via the thoracic duct (9). They found that MLN contained the highest percentage of bacterial clones of all other organs tested and proposed that the MLN represented a bottleneck for a secondary wave of L. monocytogenes dissemination to the spleen and liver (9). To spread via this indirect route, L. monocytogenes could be transported inside a migratory phagocyte, or it is possible that extracellular L. monocytogenes could traffic within afferent lymphatic vessels to the MLN. Lymph nodes also represent an important bottleneck for systemic spread during bacterial infections that occur via other routes of transmission. For example, Gonzalez et al. recently showed that extracellular Yersinia pestis disseminated from the dermis of the skin to draining lymph nodes and that only a subset of the clones that reached the lymph nodes could spread to the spleen (10).
Based on retrospective analysis of foodborne listeriosis outbreaks, the infectious dose for humans is thought to be approximately 1 × 106 CFU (11). Mice appear to be more resistant to oral infection than humans, and this has led investigators to use much higher inocula (109 to 1011 CFU) to establish an intestinal infection with L. monocytogenes. The relative resistance of mice has been attributed mainly to the species specificity of the interaction between E-cadherin expressed on intestinal epithelial cells and the bacterial surface protein internalin A (InlA) (12, 13). Two approaches have been developed to circumvent this species barrier. The Lecuit group generated “humanized” mice with a single amino acid substitution (E16P) in murine E-cadherin (14). Wollert et al. constructed a mouse-adapted L. monocytogenes strain expressing a modified InlA protein (InlAm) that binds murine E-cadherin with a similar affinity as native InlA binds human E-cadherin (15). Using this mouse-adapted strain, intestinal infection can be established with doses as low as 106 to 107 CFU in susceptible animals (3, 15). Tsai et al. recently reported that the mouse-adapted L. monocytogenes strain has an altered cell tropism for intestinal invasion compared to infection in the E16P humanized mice, but it is not entirely clear how that may affect dissemination to the MLN during foodborne infection (16).
In this study, we used a foodborne model of listeriosis to test how both the mouse-adapted and wild-type L. monocytogenes strains spread to the MLN. Surprisingly, we found that there was very little intracellular L. monocytogenes within the MLN during the first few days after infection. This led us to investigate whether intracellular growth was essential for the dissemination of L. monocytogenes to the MLN or other peripheral tissues. The results presented here using lipoate protein ligase A1 (lplA1)-deficient bacteria, which are unable to replicate in cells, demonstrate that the minimal fraction of intracellular L. monocytogenes present during the intestinal phase of the infection is crucial for efficient spread to the MLN, spleen, and liver after foodborne infection.
MATERIALS AND METHODS
Bacteria.
L. monocytogenes EGDe and an isogenic inlA deletion (ΔinlA) mutant were provided by Cormac Gahan (University College Cork, Ireland). All L. monocytogenes isolates used in this study were derivatives of this strain. All strains and plasmids used are listed in Table 1. L. monocytogenes was routinely grown in brain heart infusion (BHI) broth or agar (Difco). Intestinal homogenates were plated on BHI agar supplemented with 15 g/liter LiCl and 10 g/liter glycine (BHI/L+G) to inhibit the growth of intestinal microbiota. Suspect colonies were confirmed to be L. monocytogenes by plating on CHROMagar Listeria plates. For selection of L. monocytogenes, antibiotics were used at the following concentrations: chloramphenicol, 5 μg/ml (pKSV7) or 7.5 μg/ml (pPL2 or pIMC3); erythromycin, 5 μg/ml; kanamycin, 50 μg/ml; and tetracycline, 10 μg/ml. For selection of Escherichia coli, the following antibiotic concentrations were used: carbenicillin, 100 μg/ml; chloramphenicol, 10 μg/ml (pGJ-cGFP) or 100 μg/ml (pTML1); erythromycin, 250 μg/ml; kanamycin, 50 μg/ml; and tetracycline, 10 μg/ml. IPTG (isopropyl-β-d-thiogalactopyranoside; final concentration, 1 mM) was added to induce the expression of antibiotic resistance genes carried on pIMC3 derivatives. Recombinant plasmids were transformed into E. coli DH5α or E. coli SURE. All DNA purifications were done using Qiagen kits. For each strain, aliquots were prepared and stored at −80°C and thawed prior to use in either in vivo or in vitro infections (17).
TABLE 1.
Plasmids and strains used in this study
| Plasmid or strain | Description | Antibiotic resistancea | Source or reference |
|---|---|---|---|
| Plasmids | |||
| pKSV7 | Temp-sensitive shuttle vector | Cb, Cm | 18 |
| pPL2 | Site-specific integration vector | Cm | 43 |
| pIMC3ery, pIMC3tet, pIMC3kan | Site-specific integration vectors with IPTG-induced expression of Ery, Tet, Kan | Cm, Ery, Tet, Kan | 44 |
| pAD1-cGFP | Phyper-driven expression of GFP (constitutive) in pPL2 derivative | Cm | 45 |
| pTM2 | InlAm subcloned into PstI/BamHI-digested pKSV7 | Cb, Cm | This study |
| pGJ-cGFP | Phelp from pIMC3 subcloned into SacI/EagI-digested pAD1-cGFP | Cm | This study |
| pAF1a | 0.98-kb fragment upstream of lplA1 (−988 to −1) in pKSV7 | Cb, Cm | This study |
| pAF1-1 | 1.042-kb fragment from bp 954 of lplA1 to bp +1001 downstream adjacent to 0.98-kb fragment upstream of lplA1 in pAF1a | Cb, Cm | This study |
| pTML1 | lplA1 plus 988 bp of upstream DNA from L. monocytogenes InlAm in SalI/PstI-digested pIMC3ery | Cm, Ery | This study |
| Bacterial strains | |||
| E. coli | |||
| DH5α | F− endA1 hsdR17(rK− mK−) supE44 thi-1 λ− recA1 gyrA96 relA1 ϕ80dlacZΔM15 | None | M. N. Starnbach |
| SURE | mcrA mcrCB mcrF mrr hsdR endA recB recJ F′ lacIqZΔM15 | Kan, Tet, Cm40 | Agilent Technologies |
| L. monocytogenes | |||
| EGDe | Wild-type L. monocytogenes | None | C. G. Gahan |
| ΔinlA | inlA deletion mutant derived from strain EGDe | None | C. G. Gahan |
| InlAm | Mouse-adapted L. monocytogenes; InlA S192N, Y369S | None | W.-D. Schubert |
| SD1902 | InlAm::pIMC3ery | Cm, Ery | This study |
| SD2000 | ΔinlA::pTM2 (InlAm) | None | This study |
| SD2001 | SD2000::pIMC3kan | Cm, Kan | This study |
| SD2002 | SD2000::pIMC3ery | Cm, Ery | This study |
| SD2300 | SD2000 ΔlplA1 | None | This study |
| SD2301 | SD2300::pIMC3kan | Cm, Kan | This study |
| SD2302 | SD2300::pTML1 (+lplA1) | Cm, Ery | This study |
| SD2710 | SD2000::pGJ-cGFP | Cm | This study |
| SD2800 | ΔinlA::pIMC3tet | Cm, Tet | This study |
| SD2900 | EGDe::pIMC3ery | Cm, Ery | This study |
Cb, carbenicillin; Cm, chloramphenicol; Ery, erythromycin; Kan, kanamycin; Tet, tetracycline.
Construction of recombinant L. monocytogenes strains.
PCR primers were purchased from Integrated DNA Technologies (Coralville, IA) or Sigma-Aldrich (St. Louis, MO). The temperature-sensitive shuttle vector pKSV7 was used to generate integrations and deletions on the L. monocytogenes chromosome as described previously (18). Electrocompetent L. monocytogenes strains were generated as described by Monk et al. (19) using either filter-sterilized BHI or vegetable peptone broth (VGP; Oxoid) supplemented with 500 mM sucrose to improve bacterial growth rate and electroporation efficiency. After electroporation, bacteria were immediately recovered in 1 ml of room temperature BHI or VGP supplemented with 500 mM sucrose (filter sterilized) and incubated statically for 1.5 h at 37°C (or 30°C for pKSV7) prior to plating.
(i) Isogenic InlAm and ΔinlA strains.
A 4.4-kb DNA fragment comprising InlAm with flanking regions (∼1 kb upstream and downstream) was amplified from mouse-adapted L. monocytogenes strain EGDe (15) using Platinum Taq DNA Polymerase High Fidelity (Invitrogen) and primers that had PstI and BamHI sites added. The PCR product was ligated into PstI- and BamHI-digested pKSV7 and transformed into E. coli SURE, resulting in pTM2. InlAm was integrated into the chromosome of L. monocytogenes ΔinlA to create L. monocytogenes SD2000, and the integration was confirmed by determining the DNA sequence of the region spanning 500 bp upstream and 924 bp downstream of InlAm.
(ii) GFP-expressing L. monocytogenes.
Phyper was excised from pAD1-cGFP by digestion with SacI and EagI. The Phelp promoter was amplified from the pIMC backbone (bp 4379 to 4581; GenBank accession number AM940001.1) using primers containing SacI and NotI sites to create compatible ends and ligated to SacI- and EagI-digested pAD1-cGFP. The resulting plasmid, pGJ-cGFP, was electroporated into SD2000 to create L. monocytogenes SD2710. Green fluorescent protein (GFP) expression was verified using flow cytometry.
(iii) lplA1-deficient L. monocytogenes.
A DNA fragment spanning from −988 bp to −1 bp upstream of lplA1 was amplified from L. monocytogenes InlAm and ligated into HindIII- and XbaI-digested pKSV7, resulting in pAF1a. Next, a 1.042-kb DNA fragment spanning from 954 bp of the lplA1 coding sequence to 1,001 bp downstream of lplA1 was amplified from L. monocytogenes InlAm using primers that had EcoRI and XbaI sites added. The PCR product was ligated into EcoRI- and XbaI-digested pAF1a, resulting in pAF1-1. L. monocytogenes SD2000 was electroporated with pAF1-1 to create SD2300. After the recovery of Cms mutants, the chromosomal deletion of lplA1 was confirmed by determining the DNA sequence of the region spanning 988 bp upstream and 192 bp downstream of lplA1. To complement the lplA1 mutation, lplA1 plus 988 bp of upstream DNA was amplified from L. monocytogenes InlAm and ligated into PstI- and SalI-digested pIMC3ery, resulting in pTML1.
Mice.
Female BALBc/By/J (BALB) and C57BL/6/J (B6) mice were purchased from The Jackson Laboratory (Bar Harbor, ME) at 4 weeks of age. Mice were housed in a specific-pathogen free facility with a 10-h dark cycle (8 a.m. to 6 p.m.) and a 14-h light cycle (6 p.m. to 8 a.m.) for at least 2 weeks before being used in experiments when they were 6 to 10 weeks old. All procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Kentucky.
Foodborne infection.
Mice were infected using a natural feeding model as previously described (17, 20). Briefly, mice were transferred to cages with raised wire flooring to prevent coprophagy and fasted for 16 to 24 h. Aliquots of late-exponential-phase L. monocytogenes were recovered in either BHI or improved minimal medium (IMM) for 1.5 h at 30°C. Bacteria suspended in a mixture of phosphate-buffered saline (PBS) and salted butter (2:3 ratio) were used to saturate a 2- to 3-mm bread piece (Kroger). Mice were fed L. monocytogenes-contaminated bread pieces near the onset of their dark cycle. For coinfections, two L. monocytogenes strains with different antibiotic resistances were mixed in a 1:1 ratio and added to a single bread piece.
Bacterial loads in tissue homogenates.
Colons and ileums (approximated by cutting the terminal third of the small intestine) were harvested aseptically, flushed with 8 ml PBS, and squeezed with sterile forceps to remove intestinal contents (17). Flushed intestines were cut longitudinally with a sterile scalpel blade and then cut laterally into several small pieces. Intestinal fragments were homogenized for 1 min in 2 ml sterile water using a PowerGen 1000 homogenizer at 60% power. Serial dilutions were prepared in sterile water and plated on BHI/L+G agar. Spleen and liver were harvested aseptically and homogenized in 2.5 ml sterile water for 30 s. MLN were mashed through sterile steel screens (mesh no. 80) into 1.5 ml sterile water. Gallbladders were placed in 1 ml sterile water in a microcentrifuge tube, ruptured with sterile scissors, and vortexed for 30 s. Serial dilutions of spleen, liver, MLN, and gallbladder samples were plated on BHI agar. For coinfections, competitive index (CI) ratios were determined by dividing the number of CFU recovered for the mutant strain by the number of CFU recovered for the reference strain. If no CFU were recovered, the limit of detection was used for the calculation.
Dissociation of mesenteric lymph nodes.
MLN were cut into 3 or 4 pieces with a sterile scalpel and digested with collagenase type IV (300 U/ml; Worthington) in 4 ml of RPMI 1640 (Invitrogen no. 21870) supplemented with 20 μM HEPES and 5% fetal bovine serum (FBS) for 20 min at 37°C in 7% CO2. The partially digested MLN were then mashed through sterile steel screen (mesh no. 80) or cell strainers (BD Falcon; 40-μm pore size) using the end of a 3-ml syringe plunger. In some experiments, mechanical dissociation was avoided by digesting with collagenase and DNase I (120 U/ml; Worthington) for 30 min at 37°C with orbital shaking (250 rpm) in a 50-ml conical tube containing a sterile stir bar (2 cm). Trypan blue staining indicated that digestion with collagenase and DNase I resulted in 95% viability of dissociated MLN cells.
Antibodies and flow cytometry.
Cells were incubated with antibodies specific for the following proteins purchased from eBioscience: CD16/CD32, F4/80 (BM8), CD11c (N418), and CD11b (M1/70). For phagocyte enrichment, MLN cells were preincubated with anti-CD16/32 (Fc block) and then stained with either F4/80-PE or CD11c-APC. CD11c+ or F4/80+ cells were positively selected by incubating with either phosphatidylethanolamine (PE) or allophycocyanin (APC)-specific magnetic particles-DM (BD Bioscience). Magnetic selection was performed using three consecutive 6-min incubations, and the cells were recovered in RPMI 1640 (Invitrogen no. 21870), l-glutamine, HEPES, 2-mercaptoethanol (2-ME), and 10% FBS (RP-10 medium) supplemented with 25 μg/ml gentamicin. For GFP+ L. monocytogenes experiments, flow cytometry data were acquired using an iCyt Synergy sorter and analyzed using FlowJo software (Tree Star). Debris and cell aggregates were excluded by using forward scatter (FSC) versus side scatter (SSC) and FSC-A versus FSC-H parameters, respectively, resulting in “singlets.” The percentage of GFP+ cells in each population was determined by using mice infected with L. monocytogenes SD2001 as a negative gating control.
Determination of intracellular and extracellular CFU.
For phagocyte enrichment experiments, the number of extracellular bacteria (“supernatant”) was determined after MLN cells were centrifuged for 10 min at 300 × g. The supernatant was collected, and serial dilutions were prepared in sterile water and plated on BHI. The number of intracellular bacteria associated with enriched phagocyte populations was determined after gentamicin-treated cells were lysed in sterile water.
Following enzymatic processing of MLN, intracellular L. monocytogenes was identified by centrifuging a sample of MLN cells (10% of total volume) for 6 min at 300 × g. The cell pellet was suspended in 1 ml of RP-10 medium supplemented with gentamicin. Cells were incubated statically for 20 min at 37°C in 7% CO2 and then washed once with RP-10 medium. The cells were centrifuged for 8 min (20,000 × g) and suspended in sterile water before being plated on BHI. To quantify L. monocytogenes, a sample of untreated MLN cells (10% of total volume) was centrifuged for 8 min at 20,000 × g. Bacteria and cells were suspended in sterile water, and serial dilutions were plated on BHI. The number of extracellular L. monocytogenes was calculated by subtracting the number of intracellular CFU from the total number of CFU recovered from the MLN of each mouse.
For determination of minimal bactericidal concentration, exponential-phase L. monocytogenes SD2000 was resuspended in PBS at 4 × 107/ml and seeded in triplicate (25 μl/well) in a 96-well plate. RP-10 medium supplemented with various concentrations of gentamicin was added, and the plate was incubated for 20 min at 37°C in 7% CO2. Serial dilutions were prepared in sterile water and plated on BHI. For in vivo gentamicin experiments, mice were given a single intraperitoneal (i.p.) injection of 2 mg gentamicin in PBS and control mice received an injection of 500 μl PBS. To determine if residual gentamicin present in tissue homogenates could kill L. monocytogenes during in vitro processing, uninfected mice were treated with gentamicin or PBS for 12 h and then tissue homogenates were prepared. L. monocytogenes SD2000 (1.5 × 103 CFU) was added to each homogenate and then incubated for 1 h on ice (to mimic normal harvest conditions) before plating on BHI.
Lipoate starvation.
For lipoate starvation, L. monocytogenes was grown in IMM, which was prepared fresh from concentrated stocks and used within 2 weeks (21). Fresh isolated colonies grown on BHI agar were used to inoculate 3 ml of BHI broth in 16- by 150-mm glass tubes and incubated at 37°C in a rotating rack for 8 h. Growth was normalized by optical density at 600 nm (OD600), and bacteria were washed with PBS before inoculating 20 ml IMM without lipoic acid and incubated for ∼16 h at 37°C shaking. For growth curves, lipoate-starved bacteria were back-diluted to an OD600 of 0.05 and growth was monitored over time. Aliquots of lipoate-starved L. monocytogenes were prepared as previously described for animal infections (17) with the exceptions that L. monocytogenes was suspended in IMM without lipoic acid before storage at −80°C and was recovered in IMM before use in infections.
Intracellular growth assays.
J774 cells were maintained in RP-10 medium supplemented with penicillin and streptomycin. The day before infection, cells were washed once with warm RPMI and suspended in RP-10 without antibiotics. Cells (5 × 105/ml) were seeded onto round glass coverslips (12-mm diameter) in wells of 24-well plates. Lipoate-starved L. monocytogenes was added at various multiplicities of infection (MOI), and the plates were centrifuged for 5 min at 300 × g to synchronize infection. After 30 min, cells were washed three times with prewarmed PBS. Cells were suspended in RP-10 medium plus 10 μg/ml gentamicin and incubated at 37°C in 7% CO2. At each time point indicated, coverslips were removed, placed in 5 ml sterile water, and vortexed for 30 s, and serial dilutions were plated on BHI agar.
Statistics.
Statistical analysis was performed using Prism software for Macintosh (version 6; Graph Pad), and the specific tests used are indicated in each figure legend. P values of <0.05 were considered significant and are indicated as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
RESULTS
Wild-type L. monocytogenes EGDe spreads beyond the intestine similarly to a murinized strain after foodborne infection in mice.
The mouse-adapted strain of L. monocytogenes is ideal for studying oral transmission of listeriosis in mice because it can be used at doses that are 10- to 100-fold lower than the 109 CFU typically required to establish intestinal infection with wild-type L. monocytogenes EGDe (3, 15). However, Tsai et al. recently showed that the InlAm protein expressed by the mouse-adapted strain altered the tropism for L. monocytogenes in the intestinal epithelium by promoting binding to N-cadherin as well as E-cadherin (16). It is unclear how this may affect subsequent dissemination to peripheral tissues and the remainder of the infection in mice. To find out if wild-type L. monocytogenes EGDe had a similar course of infection to that previously published using the murinized strain (3, 15), we fed both susceptible BALB/cBy/J (BALB) and resistant C57BL/6J (B6) mice 3 × 109 CFU and determined the bacterial loads in various tissues over the course of 8 days. As expected, a small percentage of the initial inoculum was recovered from the ileum and colon 24 h postinfection (Fig. 1A). Three days postinfection, the colon harbored more L. monocytogenes than the ileum in both mouse strains. These findings were similar to what was previously observed in the gut after mice were fed a 10-fold-lower dose (108 CFU) of the mouse-adapted L. monocytogenes strain (3). However, L. monocytogenes EGDe did not continue to multiply exponentially in the intestines of susceptible BALB mice (Fig. 1A).
FIG 1.

L. monocytogenes EGDe invaded the intestine and spread to systemic organs after foodborne infection of mice. (A) BALB/c/By/J (BALB) or C57BL/6J (B6) mice were fed 3 × 109 CFU of L. monocytogenes SD2900 (Eryr EGDe). Tissue-associated CFU (in ileum and colon) or total CFU was determined at the indicated time points. Data from two separate experiments were pooled (n = 9 or 10 per group); mean values ± standard deviations (SD) are shown. (B) Total CFU recovered from the spleen or liver of individual mice fed 3 × 109 CFU of L. monocytogenes SD2900 (data shown in panel A) or 3 × 109 L. monocytogenes InlAm. Horizontal lines indicate mean values for each group. Dashed horizontal lines indicate the limit of detection for each tissue.
Once L. monocytogenes EGDe disseminated beyond the intestines, the growth curves in the spleen and liver (Fig. 1A) closely mimicked previously published time course experiments that were performed using L. monocytogenes InlAm (3). Exponential growth of L. monocytogenes was observed in the gallbladders of susceptible BALB, but not resistant B6 mice (Fig. 1A). The peak bacterial burden occurred 5 days postinfection in all three of these tissues. Small numbers of L. monocytogenes EGDe were detected in the brain starting at 5 days postinfection in both BALB and B6 mice (Fig. 1A). Together, these results suggested that oral infection with wild-type L. monocytogenes resulted in a pattern of dissemination from the gut similar to that of the mouse-adapted strain shown in a previous study, but wild-type L. monocytogenes required a 10-fold-higher inoculum to achieve bacterial loads comparable to that of the InlAm strain.
Although the course of infection in mice fed 109 CFU L. monocytogenes EGDe was similar to previous results using L. monocytogenes InlAm, there was one noticeable difference. During foodborne infection with 108 CFU of the mouse-adapted strain, there was a reproducible delay of at least 36 to 48 h until L. monocytogenes reached the spleen or liver (3). In contrast, when mice were fed 3 × 109 L. monocytogenes EGDe CFU, spread to the liver occurred within 18 h in about half of the mice (Fig. 1B). To determine if this was related to the bacterial strain difference or the higher inoculum size, BALB and B6 mice were fed 3 × 109 CFU of the mouse-adapted strain, and total CFU in the spleen and liver was determined 1 day later. As shown in Fig. 1B, L. monocytogenes InlAm spread to the liver in 3 of 8 mice tested. Thus, an inoculum of 109 CFU promoted rapid spread to the liver, regardless of which bacterial strain was used, suggesting that inocula greater than or equal to 109 CFU may overwhelm innate immune defenses and promote direct spread from the gut to the liver via the portal vein. Therefore, a key advantage of using the L. monocytogenes InlAm strain is that it can be fed to mice at low enough doses to study the bottleneck that occurs in the gut as bacteria spread via the MLN to the spleen.
The majority of L. monocytogenes in the MLN was extracellular.
We previously showed that when BALB mice were coinfected with wild-type EGDe and the mouse-adapted strain, the wild-type L. monocytogenes had a bimodal distribution in the MLN, with some mice having low or undetectable numbers while others had high bacterial loads comparable to mice infected with L. monocytogenes InlAm (3). The bimodal distribution suggested that there was a bottleneck in the intestinal lamina propria and that expression of InlAm enhanced dissemination of L. monocytogenes to the MLN. One mechanism to explain this could be that InlAm promoted invasion of a migratory cell type that could transport intracellular L. monocytogenes from the intestinal lamina propria to the MLN.
To test this, we assessed the amount of L. monocytogenes associated with either CD11c+ cells, which represent primarily migratory subsets of dendritic cells, or F4/80+ cells, which represent mainly tissue-resident macrophages. BALB mice were coinfected with an equal ratio of L. monocytogenes InlAm and an InlA deletion (ΔinlA) mutant strain that had been tagged with two different antibiotic resistance genes. Three days postinfection, CD11c+ or F4/80+ cells were enriched from the MLN by positive selection, and the total number of either InlAm or ΔinlA mutant CFU associated with these cells was determined. As shown in Fig. 2A, CD11c-enriched MLN cells harbored approximately 10-fold more InlAm than ΔinlA mutant CFU. In contrast, similar numbers of InlAm and ΔinlA mutant CFU were recovered from F4/80-enriched MLN cells (Fig. 2B). These results suggested that expression of InlAm may enhance invasion of dendritic cells but not macrophages. However, the combined number of L. monocytogenes CFU associated with either CD11c+ or F480+ cells was surprisingly small, ranging from ∼400 to 2,300 CFU per mouse. Typically, the total number of L. monocytogenes cells found in the MLN 3 days postinfection for either the wild type (Fig. 2C) or the mouse-adapted strain (3) is approximately 105 CFU. To find out if the remaining CFU were associated with other cell types or were simply present in the extracellular environment, supernatant fractions from the processing of the CD11c+ and F480+ enriched cells were collected and plated. As shown in Fig. 2A and B, the majority of the total CFU was found in the supernatant. Since the number of CFU recovered from the intracellular and supernatant fractions was approximately equal to the total MLN burdens shown in Fig. 2C, this suggests that adherent CFU were likely to be only a minor proportion of L. monocytogenes in the MLN.
FIG 2.
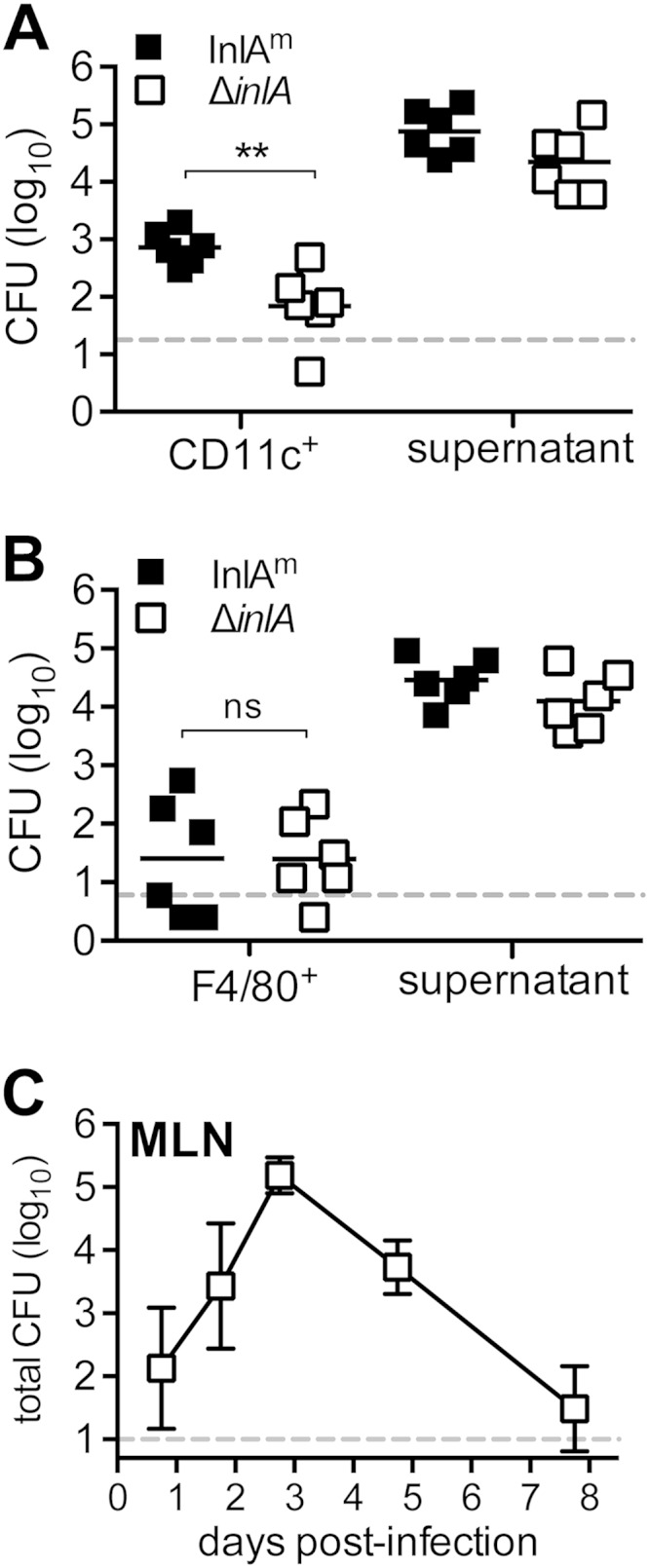
Intracellular L. monocytogenes was a minor proportion of total CFU in the MLN. BALB mice were coinfected with equal proportions of L. monocytogenes SD1902 (InlAm) and L. monocytogenes SD2800 (ΔinlA) for a total inoculum of 5 × 108 CFU, and MLN were harvested 3 days postinfection. The number of extracellular CFU (supernatant) and the total number of intracellular (gent25r) CFU associated with either CD11c+ (A) or F4/80+ (B) MLN cells are shown. CFU for individual mice are shown; horizontal lines indicate mean values. Statistical significance was determined by two-tailed Mann-Whitney analysis. (C) BALB mice were infected with 2 × 109 to 3 × 109 CFU of L. monocytogenes SD2900 (Eryr EGDe), and the total number of CFU in the MLN was determined. Mean values (±SD) for pooled data from 9 mice (1 and 8 days postinfection [dpi]), 4 mice (2 dpi), or 10 mice (3 and 5 dpi) are shown. Dashed lines indicate limits of detection.
It was possible that the large proportion of extracellular L. monocytogenes in the MLN was an artifact of the ex vivo processing techniques used to enrich for either CD11c+ or F4/80+ cells. Mechanical dissociation, in particular, could have lysed heavily infected, fragile cells. To avoid mechanical forces, MLN from infected mice were digested into single-cell suspensions using only collagenase. A portion of the cell suspension was lysed with water and plated to determine the number of total L. monocytogenes CFU in the MLN. Another portion of MLN cells was treated with gentamicin (25 μg/ml) for 20 min and then lysed and plated to determine the number of intracellular L. monocytogenes CFU. The number of extracellular L. monocytogenes CFU was calculated by subtracting the number of intracellular L. monocytogenes CFU from the total CFU. Using this approach, intracellular L. monocytogenes InlAm comprised only 1% of the total bacterial load in the MLN at both 2 and 3 days postinfection (Fig. 3A).
FIG 3.

The majority of L. monocytogenes in the MLN was predominantly gentamicin sensitive. (A) BALB mice were fed 3 × 108 to 8 × 108 CFU L. monocytogenes InlAm. Mean percentages (±SD) of intracellular (gent25r) and extracellular CFU in collagenase-treated MLN of mice harvested 2 (n = 6) and 3 (n = 11) days postinfection are shown. (B) Mean number (±SD) of L. monocytogenes SD2000 CFU surviving after 20 min of incubation at 37°C with CO2 at the indicated concentration of gentamicin. (C) BALB mice were fed 2 × 108 to 7 × 108 CFU L. monocytogenes InlAm, and MLN were dissociated using only enzymatic digestion (n = 7 to 11 per group). Mean percentages (±SD) of intracellular (gent10r) and extracellular CFU in the MLN are shown. (D) BALB mice were fed 3 × 108 or 3 × 109 L. monocytogenes EGDe (n = 4 per group), and MLN were dissociated using enzymatic digestion 3 days postinfection. Mean percentages (±SD) of intracellular (gent10r) and extracellular CFU in the MLN are shown. (E, F) BALB mice were fed 2 × 108 to 5 × 108 CFU of L. monocytogenes SD2710 (GFP+) or L. monocytogenes SD2001. MLN were collected 2 days postinfection and analyzed by flow cytometry. Representative dot plots showing the percentage of GFP+ cells in either total MLN (singlets) or the CD11chiCD11b−/+ and CD11b+CD11c−/+ populations are shown in panel E; the gating strategy is shown in panel F. (G) Mean percentages (±SD) of GFP+ cells in each myeloid-derived MLN population (n = 5 mice) are shown. Statistical significance was determined by two-tailed Mann-Whitney analysis. (H) Symbols indicate the total number of GFP+ cells identified in the MLN of each mouse; the shaded bar represents the mean value for the group.
Gentamicin treatment is commonly used to selectively kill extracellular bacteria; however, gentamicin can penetrate mammalian cells when used at high enough concentrations. To determine the minimal bactericidal concentration for our experimental conditions, we exposed 1 × 106 CFU of L. monocytogenes InlAm to increasing concentrations of gentamicin. The lowest concentration that killed 100% of the inoculum was 10 μg/ml (Fig. 3B). We repeated the analysis of intracellular and extracellular L. monocytogenes in the MLN 2 and 3 days postinfection using 10 μg/ml gentamicin and recovered approximately 10-fold-higher percentages of intracellular L. monocytogenes (Fig. 3C versus 3A). Thus, 10% of the bacterial burden in the MLN was intracellular, and approximately 90% was extracellular (gentamicin resistant). To ensure that these results applied to both the wild-type and murinized strains, we fed groups of mice two different doses of L. monocytogenes EGDe. Slightly higher proportions of intracellular L. monocytogenes EGDe than L. monocytogenes InlAm were found, but the majority of the bacteria in the MLN were still extracellular (Fig. 3D).
To confirm these findings, flow cytometry was used to quantify cell-associated L. monocytogenes in the MLN. Mice were fed 108 CFU of either an L. monocytogenes InlAm derivative that constitutively expressed GFP (L. monocytogenes SD2710) or a vector control strain (L. monocytogenes SD2001). As expected, only a small fraction of MLN cells were infected. It was difficult to detect the small number of GFP+ cells when bulk populations were analyzed, due to some autofluorescence of MLN cells using a 525/50-nm filter (Fig. 3E). Therefore, myeloid-derived cell subsets were analyzed by gating on either CD11chi cells (dendritic cells) or CD11b+CD11c−/+ cells, which included monocytes, macrophages, and neutrophils (Fig. 3F). As shown in representative dot plots (Fig. 3E) and in collected data from groups of mice (Fig. 3G), clear shifts in GFP expression were visible when analyzing these myeloid-derived subsets. However, even when these two subsets were combined, an average of only 4.3 × 104 GFP+ MLN cells were detected in each mouse (Fig. 3H), even though the total number of L. monocytogenes recovered from the MLN in these experiments ranged from 1.5 × 105 to 7 × 105 CFU. This suggested either that each infected cell contained at least 4 and up to 16 bacteria or that some portion of the total CFU burden was extracellular.
In vivo treatment with gentamicin significantly reduced L. monocytogenes burdens following foodborne challenge.
Since all in vitro approaches involve some degree of processing and handling, we next used an in vivo approach to assess the degree to which extracellular L. monocytogenes was present in the gut and the draining lymph nodes. To do this, mice were fed L. monocytogenes InlAm, and 3 days later, half of the animals received an intraperitoneal injection of 2 mg of gentamicin and the other half were injected with PBS. The number of viable bacteria present in each tissue was determined 12 h later. As shown in Fig. 4A, significantly lower numbers of L. monocytogenes were recovered from mice treated with gentamicin than from control mice. The average number of L. monocytogenes CFU recovered from the colons of gentamicin-treated mice represented only 14% of the bacterial load in the colons of PBS-treated mice. Likewise, a significantly reduced number of CFU were observed in the MLN, spleens, and livers of mice treated with gentamicin (Fig. 4A). In each tissue, gentamicin-sensitive L. monocytogenes represented approximately 60 to 80% of the bacterial load recovered. It was possible that the lower number of CFU recovered from gentamicin-treated mice occurred because of residual gentamicin present in the tissue homogenates, which may have killed intracellular L. monocytogenes that was released during cell lysis and plating. To test this, uninfected mice were treated with gentamicin or PBS. Tissue homogenates were prepared 12 h later and inoculated in vitro with L. monocytogenes. As shown in Fig. 4B, there was no inhibition of L. monocytogenes growth in the gentamicin-treated homogenates compared to the PBS-treated homogenates. Therefore, the results of both the in vitro and in vivo approaches strongly suggested that intracellular L. monocytogenes represented only a minimal fraction of the total L. monocytogenes burden during the first few days following oral challenge.
FIG 4.

Gentamicin treatment reduced bacterial burdens in mice fed L. monocytogenes. (A) BALB mice were orally infected with 2 × 108 to 5 × 108 CFU of L. monocytogenes SD2000 (InlAm). Three days postinfection, mice were injected (i.p.) with either 2 mg of gentamicin (Gent) or PBS, and the colons, MLN, spleens, and livers were harvested 12 h later. Symbols indicate CFU values for individual mice; gray bars indicate the median for each group. The mean percentage of CFU recovered from gentamicin-treated mice relative to the CFU recovered from PBS-treated mice is shown in parentheses. Data from two separate experiments were pooled, and statistical significance was determined by one-tailed Mann-Whitney analysis. (B) Uninfected BALB mice were treated with 2 mg gentamicin or PBS, and tissues were harvested 12 h later. Homogenates were inoculated with 1.5 × 103 L. monocytogenes SD2000 CFU and incubated on ice for 1 h before plating on BHI agar. The horizontal dashed line indicates the inoculum, and the bars indicate the mean CFU (±SD) recovered from each homogenate (n = 3 mice per group).
Intracellular replication was not required for L. monocytogenes to establish intestinal infection.
The relatively small proportion of intracellular L. monocytogenes in the intestines and MLN led us to question if intracellular replication was necessary during the intestinal phase of foodborne infection. Therefore, we constructed a mutant strain of mouse-adapted L. monocytogenes that had a defect only in intracellular replication, with normal growth in extracellular environments. O'Riordan et al. previously showed that lipoate protein ligase A1 (LplA1)-deficient L. monocytogenes strains were unable to scavenge lipoate from host cell-derived lipoyl peptides and, thus, had a significant defect in intracellular growth in J774 macrophages (22). Therefore, we generated an InlAm-expressing ΔlplA1 mutant to study when intracellular replication was important during foodborne infection.
L. monocytogenes stores large quantities of lipoic acid, and the intracellular growth phenotype of the ΔlplA1 mutant can be observed only when these reserves have been depleted (23). To do this, L. monocytogenes was grown overnight in improved minimal media (IMM) in the absence of lipoic acid. L. monocytogenes ΔlplA1 grew as well as either the parental strain InlAm (L. monocytogenes SD2000) or the complemented strain (+lplA1) when lipoate-starved bacteria were transferred to rich media (BHI) (Fig. 5A). During further growth in minimal medium with limiting quantities of nutrients, all strains had an extended lag phase of 12 to 15 h (Fig. 5B and C). Lipoate-starved L. monocytogenes InlAm required supplementation with at least 0.25 nM lipoic acid to reach late exponential phase (Fig. 5B). As expected, none of the lipoate-starved bacteria grew in minimal medium lacking lipoate, and all three strains reached similar optical densities after the addition of 25 nM lipoic acid (Fig. 5C). To verify that the lipoate-starved ΔlplA1 mutant did not replicate inside mammalian cells, we conducted intracellular growth assays with J774 macrophages. Lipoate-starved L. monocytogenes ΔlplA1 strains were able to survive but not grow in these cells (Fig. 5D). In contrast, the complemented mutant grew exponentially in J774 cells after an extended lag phase.
FIG 5.

Lipoate-starved L. monocytogenes grew slowly in minimal medium but was able to establish intestinal infection in mice. Freshly streaked colonies of L. monocytogenes were incubated in IMM(-) overnight to deplete lipoate reserves and then diluted into fresh medium with or without lipoic acid (A to C) or frozen at −80°C prior to infection of cells (D) or mice (E). (A) The rates of growth in a rich medium (BHI) with shaking at 37°C were similar for L. monocytogenes SD2301 (ΔlplA1 mutant), the complemented mutant L. monocytogenes SD2302 (+ lplA1), and the parental strain (L. monocytogenes SD2000). (B) Lipoate-starved L. monocytogenes SD2000 had a long lag phase in IMM but achieved exponential growth in at least 0.25 nM lipoic acid. (C) The lplA1 deletion strain (ΔlplA1) and the complemented mutant (+lplA1) did not grow in the absence of exogenous lipoate but reached growth densities similar to those of the parental strain in IMM supplemented with 25 nM lipoic acid. (D) J774 cells were infected in triplicate, and the mean number (±SD) of gentamicin-resistant (10 μg/ml) CFU per well was determined over time. Statistical significance was determined by Mann-Whitney analysis. For panels A to D, data from one of at least two separate experiments are shown. (E) Mice were coinfected with a 1:1 ratio of L. monocytogenes SD2001 (Kanr InlAm prepared in IMM) and L. monocytogenes SD2002 (Eryr InlAm prepared in BHI) for a total inoculum of 2 × 108 CFU. The population of tissue-associated L. monocytogenes in the ileum or colon was determined 24 h later and is shown both as a competitive index (CI) and as the absolute number of cell-associated CFU recovered from each mouse. (F) BALB mice were coinfected with a 1:1 mixture of L. monocytogenes SD2301 (ΔlplA1) and the complemented mutant L. monocytogenes SD2302 (+lplA1) for an average total inoculum of 8 × 108 CFU. The tissue-associated L. monocytogenes population was determined and is shown as a CI; the fold difference from the hypothetical value of 1.0 is shown in the parentheses above. Pooled data from at least two separate experiments are shown.
Since lipoate-starved L. monocytogenes had an extended lag phase both in liquid medium and in J774 cells, we were concerned that this might reduce the ability of the bacteria to establish infection in mice. To test this, BALB mice were fed an equal ratio of antibiotic-tagged parental strain L. monocytogenes InlAm that was grown either in rich medium (BHI) or under lipoate starvation conditions (IMM without lipoic acid). These bacteria were fully capable of scavenging lipoate from host cells but were transmitted to the gastrointestinal tract in a lipoate-starved state that might require a considerable lag time before bacterial replication could occur. The ileum and colon from each mouse were harvested 20 h later and flushed extensively, and the total number of each bacterial strain present in the flushed tissue was determined by plating on BHI supplemented with either erythromycin or kanamycin. As shown in Fig. 5E, the lipoate-starved L. monocytogenes InlAm did not have a defect in establishing intestinal invasion and, in fact, had a slight advantage compared to L. monocytogenes InlAm grown in BHI. Therefore, although lipoate starvation did cause L. monocytogenes to have an extended lag phase during growth in lipoate-limiting conditions, it did not reduce bacterial fitness to colonize the intestinal tract in mice.
Next, to establish the importance of intracellular growth during the gastrointestinal phase of foodborne listeriosis, BALB mice were fed lipoate-starved L. monocytogenes ΔlplA1 mutant and the complemented strain mixed in a 1:1 ratio. The total number of each strain present in either the ileum or the colon was determined and is presented as a competitive index (CI) ratio in Fig. 5F. The ΔlplA1 strain had very little defect in establishing infection in the colon, but on average, 14-fold fewer lplA1 mutant bacteria were recovered from the ileum 1 day postinfection. By 3 days after infection, however, the complemented strain outcompeted the mutant by an average of ∼500-fold in the ileum and ∼30-fold in the colon (Fig. 5F). Together, these results suggested that intracellular replication was not necessary for L. monocytogenes to establish intestinal infection in mice but that the ability to grow inside a host cell strongly promoted persistence, particularly in the small intestine.
Intracellular replication of L. monocytogenes was vital for spread beyond the intestine.
To find out if intracellular replication was essential for L. monocytogenes to disseminate beyond the intestine, CFU counts in the MLN, spleen, and liver were determined 3 days after coinfection with a 1:1 mixture of lipoate-starved L. monocytogenes ΔlplA1 and the complemented strain. As shown in Fig. 6A, L. monocytogenes ΔlplA1 had a dramatic defect (2,500-fold) in reaching the MLN compared to the complemented (+lplA1) strain. The MLN is thought to be a bottleneck for further dissemination to the spleen and liver via the bloodstream, and accordingly, no L. monocytogenes ΔlplA1 was recovered from the liver 3 days postinfection, while an average of 104 CFU of the +lplA1 complemented strain was detected in the spleen (Fig. 6B). Likewise, only a few mice had any L. monocytogenes ΔlplA1 in the spleen, whereas, on average, 104 to 105 CFU of the complemented strain was recovered 3 days postinfection (Fig. 6B).
FIG 6.

lplA1-deficient L. monocytogenes had a severe dissemination defect in mice. BALB mice were coinfected with a 1:1 mixture of L. monocytogenes SD2301 (ΔlplA1) and the complemented mutant L. monocytogenes SD2302 (+lplA1) for a total inoculum of 3 × 108 to 7 × 108 CFU. Pooled data from at least two separate experiments are shown. (A) The percentage of each strain recovered from the MLN 3 days postinfection is expressed as a competitive index (CI); the solid horizontal line indicates the geometric mean. (B and C) Absolute number of CFU of each strain recovered 3 (B) or 4 (C) days postinfection. Solid horizontal lines indicate mean values, and dashed lines indicate limits of detection.43
Although dissemination to the MLN was greatly reduced for L. monocytogenes unable to replicate intracellularly, approximately 60 CFU of L. monocytogenes ΔlplA1 was recovered from the MLN 3 days postinfection (data not shown). Therefore, it was possible that dissemination of these bacteria was simply delayed, rather than inhibited. To test this, we also evaluated bacterial burdens both in the gut and in peripheral tissues 4 days after coinfection. As shown in Fig. 6C, very few of the ΔlplA mutants were detected in either the ileum, colon, MLN, spleen, or liver 4 days postinfection. In contrast, the complemented strain had increased numbers in all tissues relative to the day 3 counts. Thus, dissemination beyond the intestinal lamina propria was severely limited for L. monocytogenes ΔlplA. Together, these results indicate that the minor proportion of L. monocytogenes isolates that invade cells in the gut and replicate is crucial for L. monocytogenes to disseminate via the MLN during foodborne infection.
DISCUSSION
Invasion of mammalian cells is considered to be the main virulence strategy of L. monocytogenes, but a significant portion of the bacterial burden in L. monocytogenes-infected animals appears to be extracellular. Although the focus of this study was primarily the MLN, the predominance of extracellular L. monocytogenes was noted in all tissues examined and is consistent with previous reports using other small animal models of listeriosis (4, 5). We conclude from these studies that intracellular L. monocytogenes is actually a minimal component of the bacterial load during the early stages of infection. Silva and Pestana recently suggested that the extracellular phase of many facultative intracellular pathogens could be important for virulence and that the presence of extracellular bacteria was greatly underappreciated in most infection models (24). The data presented here suggest that virulence strategies used by extracellular L. monocytogenes may be particularly important for initial colonization and survival in the gastrointestinal tract. In that regard, Travier et al. recently reported that ActA, the sole L. monocytogenes protein required to mediate actin-based motility in the host cell cytosol (25), was also required for extracellular aggregation and biofilm formation in vitro (26). The authors further showed that ActA-dependent aggregation promoted both colonization and persistence of extracellular L. monocytogenes in the intestinal lumen of mice infected by the intragastric route.
In this study, intracellular L. monocytogenes was not crucial for bacterial survival in the gastrointestinal tract until relatively late in the infection (3 days after foodborne challenge). During this time frame, L. monocytogenes will have invaded the intestinal epithelium, penetrated the underlying lamina propria, and potentially replicated exponentially either in the interstitial fluid or within a phagocyte. Most available data suggest that L. monocytogenes does not replicate extensively within intestinal epithelial cells. For example, Nikitas et al. demonstrated that InlA-mediated uptake could occur rapidly with L. monocytogenes transcytosing across goblet cells in the murine small intestine and being deposited into the lamina propria without ever leaving the endocytic vacuole (27). Accordingly, we previously showed that the number of intracellular L. monocytogenes CFU in the colonic epithelium peaked 3 days postinfection but L. monocytogenes in the colonic lamina propria continued to increase exponentially until 5 days postinfection (3). The fate of L. monocytogenes once the bacteria cross the mucosal barrier is not well understood and may depend on the route used for invasion. For example, uptake via M cells in the small intestine would result in deposition within a lymphoid follicle or Peyer's patch, where rapid phagocytosis is likely to occur by a unique subset of dendritic cells localized in the subepithelial dome (28, 29). In contrast, InlA-mediated transcytosis across the goblet cells prominent in the colon may increase the chance that extracellular L. monocytogenes could avoid phagocytosis in the underlying lamina propria and traffic in the lymphatic fluid to the draining lymph node. In support of this idea, we showed here that intracellular replication of L. monocytogenes was more important for colonization and persistence in the ileum than in the colon.
The primary strategy used here for determining the proportion of intracellular L. monocytogenes was treatment with gentamicin, an aminoglycoside that does not penetrate mammalian cells at low concentrations. Although this is a widely used technique in the field of bacterial pathogenesis, one must be cautious in interpreting data from gentamicin protection assays, since excessively high concentrations of gentamicin can kill or stress intracellular bacteria, possibly due to the pinocytosis of extracellular fluid containing antibiotics (30–32). As we showed here, even treatment with 25 μg/ml gentamicin in vitro slightly underestimated the proportion of intracellular organisms in the MLN compared to using only 10 μg/ml in tissue culture medium. Glomski et al. reported serum gentamicin levels of 5.6 μg/ml 12 h after the subcutaneous injection of 1 mg gentamicin (4). Thus, we treated mice with 2 mg gentamicin to approximate the minimal bactericidal concentration determined in vitro (10 μg/ml) (Fig. 3B). Using this approach, it is difficult to confirm that the antibiotic penetrated all tissues efficiently; however, we did recover significantly lower CFU from the colon, MLN, spleen, and liver of gentamicin-treated mice than from control mice. Notably, we found that the liver harbored a larger fraction of extracellular L. monocytogenes than the spleen, a result that is consistent with previous studies that utilized different animal models of listeriosis (4, 5, 33). All of the approaches that we used to quantify extracellular L. monocytogenes in the MLN have the caveat that some processing of the tissue was required, and we cannot rule out the possibility that some of the extracellular bacteria that we identified were present in fragile, heavily infected cells that lysed in vitro. However, the combined results from both the in vitro and in vivo gentamicin treatments strongly suggest that a large proportion of the L. monocytogenes burden in the gut is extracellular during the first few days following foodborne challenge.
To address the role of intracellular L. monocytogenes during the early stages of infection in the gut, we used lipoate-starved L. monocytogenes ΔlplA1 mutants. These bacteria were able to invade cells, escape from the vacuole, and mediate actin-based motility to avoid autophagy (34–36) but could not replicate efficiently due to an inability to scavenge lipoate from the host cell cytosol. A more common approach to study L. monocytogenes that cannot survive intracellularly has been to use listeriolysin O (LLO) mutants that are killed following invasion of murine cells because they cannot mediate escape from the vacuole (37, 38). But our primary objective was to determine if intracellular localization of L. monocytogenes was needed for dissemination to the MLN, and use of an LLO mutant could abort infection prior to colonization of the lamina propria (39, 40), making it difficult to distinguish invasion defects from dissemination defects. When grown in minimal defined medium, lipoate-starved L. monocytogenes ΔlplA1 required a minimum of 0.25 nM lipoate to replicate (Fig. 5B). Although the concentration of lipoic acid present in the tissues of mice is unknown, it was previously reported that concentrations of lipoic acid in plasma in healthy humans ranged from 3.1 to 50 ng/ml (∼15 to 242 nM) and the concentration in normal human liver tissue was 198 ng/mg protein (41, 42). Therefore, it is likely that during in vivo growth in mice, extracellular L. monocytogenes ΔlplA1 can readily obtain free lipoate from the host and that the persistence and dissemination defects that we observed were due to a lack of intracellular replication.
It is possible that invasion of a particular cell type in the intestinal lamina propria is critical for L. monocytogenes to be transported to MLN. For example, InlA may enhance invasion into a subset of dendritic cells that express E-cadherin. E-cadherin-positive dendritic cells were shown to be recruited to the intestine and MLN using a T cell-mediated model of colitis in mice (46), but their role in inflammation induced by infection has not yet been explored. However, the ΔlplA1 mutant data reported here suggest that it is replication, and not just intracellular localization of L. monocytogenes, that is important for persistence and spread beyond the gut. Exponential replication in the cytosol of a more permissive cell type in the gut, such as a tissue-resident macrophage, may serve as an amplification step to increase L. monocytogenes burdens above a particular threshold that is needed for efficient dissemination. These results further highlight the importance of studying dissemination in the context of natural foodborne transmission, using relatively low doses, because innate immune defenses that limit bacterial spread can be overwhelmed by excessively large intragastric inocula. In support of this idea, Gonzalez et al. showed that the dermis represented a significant bottleneck for the spread of Y. pestis to the skin-draining lymph nodes, but this bottleneck was partially ablated when higher doses of bacteria were used (10).
Extracellular L. monocytogenes predominated in the gut whether we infected mice with wild-type L. monocytogenes EGDe or with a mouse-adapted derivative of this strain. Tsai et al. recently raised the concern that InlAm-expressing strains of L. monocytogenes may cause more inflammation than wild-type L. monocytogenes during intestinal infection and that this could lead to prolonged colonization in the gut (16). In this regard, we showed here that foodborne transmission of wild-type L. monocytogenes resulted in a more transient infection of the intestines than was previously observed with L. monocytogenes InlAm-expressing strains. Likewise, we did find that approximately twice as much L. monocytogenes InlAm as wild-type L. monocytogenes EGDe was extracellular, and it is possible that this was due to increased cellular damage in the inflamed gut. However, during infection with the wild-type strain, we still found that the vast majority of L. monocytogenes in the MLN (70 to 80%) was extracellular, and the kinetics of systemic spread and clearance in peripheral tissues was similar to that previously published for the mouse-adapted strain (3). We propose that extracellular localization of L. monocytogenes during the early stages of intestinal infection is a feature that is likely to be shared by all L. monocytogenes strains and that the large proportion of extracellular bacteria in the gut may be involved in promoting dissemination.
ACKNOWLEDGMENTS
We thank Hilary Denney, Michelle Pitts, Jennifer Strange, and Jessie Zhu for technical assistance and Michelle Pitts for critical reading of the manuscript.
This work was supported by Public Health Service grant AI101373 to S.E.F.D.
REFERENCES
- 1.Xayarath B, Freitag NE. 2012. Optimizing the balance between host and environmental survival skills: lessons learned from Listeria monocytogenes. Future Microbiol 7:839–852. doi: 10.2217/fmb.12.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chaturongakul S, Raengpradub S, Wiedmann M, Boor KJ. 2008. Modulation of stress and virulence in Listeria monocytogenes. Trends Microbiol 16:388–396. doi: 10.1016/j.tim.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bou Ghanem EN, Jones GS, Myers-Morlaes T, Patil PN, Hidayatullah AN, D'Orazio SEF. 2012. InlA promotes dissemination of Listeria monocytogenes to the mesenteric lymph nodes during food borne infection of mice. PLoS Pathog 8:e1003015. doi: 10.1371/journal.ppat.1003015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Glomski IJ, Decatur AL, Portnoy DA. 2003. Listeria monocytogenes mutants that fail to compartmentalize listerolysin O activity are cytotoxic, avirulent, and unable to evade host extracellular defenses. Infect Immun 71:6754–6765. doi: 10.1128/IAI.71.12.6754-6765.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bakardjiev AI, Theriot JA, Portnoy DA. 2006. Listeria monocytogenes traffics from maternal organs to the placenta and back. PLoS Pathog 2:e66. doi: 10.1371/journal.ppat.0020066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hardy J, Francis KP, DeBoer M, Chu P, Gibbs K, Contag CH. 2004. Extracellular replication of Listeria monocytogenes in the murine gall bladder. Science 303:851–853. doi: 10.1126/science.1092712. [DOI] [PubMed] [Google Scholar]
- 7.Barnes PD, Bergman MA, Mecsas J, Isberg RR. 2006. Yersinia pseudotuberculosis disseminates directly from a replicating bacterial pool in the intestine. J Exp Med 203:1591–1601. doi: 10.1084/jem.20060905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lim CH, Voedisch S, Wahl B, Rouf SF, Geffers R, Rhen M, Pabst O. 2014. Independent bottlenecks characterize colonization of systemic compartments and gut lymphoid tissue by salmonella. PLoS Pathog 10:e1004270. doi: 10.1371/journal.ppat.1004270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Melton-Witt JA, Rafelski SM, Portnoy DA, Bakardjiev AI. 2012. Oral infection with signature-tagged Listeria monocytogenes reveals organ-specific growth and dissemination routes in guinea pigs. Infect Immun 80:720–732. doi: 10.1128/IAI.05958-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gonzalez RJ, Lane MC, Wagner NJ, Weening EH, Miller VL. 2015. Dissemination of a highly virulent pathogen: tracking the early events that define infection. PLoS Pathog 11:e1004587. doi: 10.1371/journal.ppat.1004587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.FAO/WHO. 2004. Risk assessment of Listeria monocytogenes in ready-to-eat foods. http://www.fao.org/3/a-y5394e.pdf Accessed 1 April 2015. [Google Scholar]
- 12.Gaillard JL, Berche P, Frehel C, Gouin E, Cossart P. 1991. Entry of L. monocytogenes into cells is mediated by internalin, a repeat protein reminiscent of surface antigens from gram-positive cocci. Cell 65:1127–1141. [DOI] [PubMed] [Google Scholar]
- 13.Lecuit M, Dramsi S, Gottardi C, Fedor-Chaiken M, Gumbiner B, Cossart P. 1999. A single amino acid in E-cadherin responsible for host specificity towards the human pathogen Listeria monocytogenes. EMBO J 18:3956–3963. doi: 10.1093/emboj/18.14.3956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Disson O, Grayo S, Huillet E, Nikitas G, Langa-Vives F, Dussurget O, Ragon M, Le Monnier A, Babinet C, Cossart P, Lecuit M. 2008. Conjugated action of two species-specific invasion proteins for fetoplacental listeriosis. Nature 455:1114–1118. doi: 10.1038/nature07303. [DOI] [PubMed] [Google Scholar]
- 15.Wollert T, Pasche B, Rochon M, Deppenmeier S, van den Heuvel J, Gruber AD, Heinz DW, Lengeling A, Schubert WD. 2007. Extending the host range of Listeria monocytogenes by rational protein design. Cell 129:891–902. doi: 10.1016/j.cell.2007.03.049. [DOI] [PubMed] [Google Scholar]
- 16.Tsai YH, Disson O, Bierne H, Lecuit M. 2013. Murinization of internalin extends its receptor repertoire, altering Listeria monocytogenes cell tropism and host responses. PLoS Pathog 9:e1003381. doi: 10.1371/journal.ppat.1003381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bou Ghanem EN, Myers-Morales T, D'Orazio SE. 2013. A mouse model of foodborne Listeria monocytogenes infection. Curr Protoc Microbiol 31:9B.3.1–9B.3.16. doi: 10.1002/9780471729259.mc09b03s31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith K, Youngman P. 1992. Use of a new integrational vector to investigate compartment-specific expression of Bacillus subtilis spoIIM gene. Biochimie 74:705–711. doi: 10.1016/0300-9084(92)90143-3. [DOI] [PubMed] [Google Scholar]
- 19.Monk IR, Gahan CG, Hill C. 2008. Tools for functional postgenomic analysis of listeria monocytogenes. Appl Environ Microbiol 74:3921–3934. doi: 10.1128/AEM.00314-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bou Ghanem EN, Myers-Morales T, Jones GS, D'Orazio SEF. 2013. Oral transmission of Listeria monocytogenes in mice via ingestion of contaminated food. J Vis Exp 75:e50381. doi: 10.3791/50381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Phan-Thanh L, Gormon T. 1997. A chemically defined minimal medium for the optimal culture of Listeria. Int J Food Microbiol 35:91–95. doi: 10.1016/S0168-1605(96)01205-6. [DOI] [PubMed] [Google Scholar]
- 22.O'Riordan M, Moors MA, Portnoy DA. 2003. Listeria intracellular growth and virulence require host-derived lipoic acid. Science 302:462–464. doi: 10.1126/science.1088170. [DOI] [PubMed] [Google Scholar]
- 23.Keeney KM, Stuckey JA, O'Riordan MX. 2007. LplA1-dependent utilization of host lipoyl peptides enables Listeria cytosolic growth and virulence. Mol Microbiol 66:758–770. doi: 10.1111/j.1365-2958.2007.05956.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Silva MT, Pestana NT. 2013. The in vivo extracellular life of facultative intracellular bacterial parasites: role in pathogenesis. Immunobiology 218:325–337. doi: 10.1016/j.imbio.2012.05.011. [DOI] [PubMed] [Google Scholar]
- 25.Smith GA, Portnoy DA, Theriot JA. 1995. Asymmetric distribution of the Listeria monocytogenes ActA protein is required and sufficient to direct actin-based motility. Mol Microbiol 17:945–951. doi: 10.1111/j.1365-2958.1995.mmi_17050945.x. [DOI] [PubMed] [Google Scholar]
- 26.Travier L, Guadagnini S, Gouin E, Dufour A, Chenal-Francisque V, Cossart P, Olivo-Marin JC, Ghigo JM, Disson O, Lecuit M. 2013. ActA promotes Listeria monocytogenes aggregation, intestinal colonization and carriage. PLoS Pathog 9:e1003131. doi: 10.1371/journal.ppat.1003131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nikitas G, Deschamps C, Disson O, Niault T, Cossart P, Lecuit M. 2011. Transcytosis of Listeria monocytogenes across the intestinal barrier upon specific targeting of goblet cell accessible E-cadherin. J Exp Med 208:2263–2277. doi: 10.1084/jem.20110560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lelouard H, Henri S, De Bovis B, Mugnier B, Chollat-Namy A, Malissen B, Meresse S, Gorvel JP. 2010. Pathogenic bacteria and dead cells are internalized by a unique subset of Peyer's patch dendritic cells that express lysozyme. Gastroenterology 138:173–184.e1-3. doi: 10.1053/j.gastro.2009.09.051. [DOI] [PubMed] [Google Scholar]
- 29.Lelouard H, Fallet M, de Bovis B, Meresse S, Gorvel JP. 2012. Peyer's patch dendritic cells sample antigens by extending dendrites through M cell-specific transcellular pores. Gastroenterology 142:592–601 e593. doi: 10.1053/j.gastro.2011.11.039. [DOI] [PubMed] [Google Scholar]
- 30.Drevets DA, Canono BP, Leenen PJ, Campbell PA. 1994. Gentamicin kills intracellular Listeria monocytogenes. Infect Immun 62:2222–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qazi SN, Harrison SE, Self T, Williams P, Hill PJ. 2004. Real-time monitoring of intracellular Staphylococcus aureus replication. J Bacteriol 186:1065–1077. doi: 10.1128/JB.186.4.1065-1077.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Menashe O, Kaganskaya E, Baasov T, Yaron S. 2008. Aminoglycosides affect intracellular Salmonella enterica serovars Typhimurium and Virchow. Antimicrob Agents Chemother 52:920–926. doi: 10.1128/AAC.00382-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Drevets DA, Jelinek TA, Freitag NE. 2001. Listeria monocytogenes-infected phagocytes can initiate central nervous system infection in mice. Infect Immun 69:1344–1350. doi: 10.1128/IAI.69.3.1344-1350.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mitchell G, Ge L, Huang Q, Chen C, Kianian S, Roberts MF, Schekman R, Portnoy DA. 2015. Avoidance of autophagy mediated by PlcA or ActA is required for Listeria monocytogenes growth in macrophages. Infect Immun 83:2175–2184. doi: 10.1128/IAI.00110-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tattoli I, Sorbara MT, Yang C, Tooze SA, Philpott DJ, Girardin SE. 2013. Listeria phospholipases subvert host autophagic defenses by stalling pre-autophagosomal structures. EMBO J 32:3066–3078. doi: 10.1038/emboj.2013.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yoshikawa Y, Ogawa M, Hain T, Yoshida M, Fukumatsu M, Kim M, Mimuro H, Nakagawa I, Yanagawa T, Ishii T, Kakizuka A, Sztul E, Chakraborty T, Sasakawa C. 2009. Listeria monocytogenes ActA-mediated escape from autophagic recognition. Nat Cell Biol 11:1233–1240. doi: 10.1038/ncb1967. [DOI] [PubMed] [Google Scholar]
- 37.Gaillard JL, Berche P, Sansonetti P. 1986. Transposon mutagenesis as a tool to study the role of hemolysin in the virulence of Listeria monocytogenes. Infect Immun 52:50–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cossart P, Vicente MF, Mengaud J, Baquero F, Perez-Diaz JC, Berche P. 1989. Listeriolysin O is essential for virulence of Listeria monocytogenes: direct evidence obtained by gene complementation. Infect Immun 57:3629–3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krawczyk-Balska A, Bielecki J. 2005. Listeria monocytogenes listeriolysin O and phosphatidylinositol-specific phospholipase C affect adherence to epithelial cells. Can J Microbiol 51:745–751. doi: 10.1139/w05-058. [DOI] [PubMed] [Google Scholar]
- 40.Vadia S, Arnett E, Haghighat AC, Wilson-Kubalek EM, Tweten RK, Seveau S. 2011. The pore-forming toxin listeriolysin O mediates a novel entry pathway of L. monocytogenes into human hepatocytes. PLoS Pathog 7:e1002356. doi: 10.1371/journal.ppat.1002356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baker H, Deangelis B, Baker ER, Hutner SH. 1998. A practical assay of lipoate in biologic fluids and liver in health and disease. Free Radic Biol Med 25:473–479. doi: 10.1016/S0891-5849(98)00087-2. [DOI] [PubMed] [Google Scholar]
- 42.Carlson DA, Young KL, Fischer SJ, Ulrich H. 2008. An evaluation of the stability and plasma pharmacokinetics of R-lipoic acid (RLA) and R-dihydrolipoic acid (R-DHLA) dosage forms in human plasma from healthy volunteers, p 235–270. In Patel MS, Packer L (ed), Lipoic acid: energy production, antioxidant activity & health effects. Taylor and Francis Publishers, London, England. [Google Scholar]
- 43.Lauer P, Chow MY, Loessner MJ, Portnoy DA, Calendar R. 2002. Construction, characterization, and use of two Listeria monocytogenes site-specific phage integration vectors. J Bacteriol 184:4177–4186. doi: 10.1128/JB.184.15.4177-4186.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Monk IR, Casey PG, Cronin M, Gahan CG, Hill C. 2008. Development of multiple strain competitive index assays for Listeria monocytogenes using pIMC; a new site-specific integrative vector. BMC Microbiol 8:96. doi: 10.1186/1471-2180-8-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Balestrino D, Hamon MA, Dortet L, Nahori MA, Pizarro-Cerda J, Alignani D, Dussurget O, Cossart P, Toledo-Arana A. 2010. Single-cell techniques using chromosomally tagged fluorescent bacteria to study Listeria monocytogenes infection processes. Appl Environ Microbiol 76:3625–3636. doi: 10.1128/AEM.02612-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Siddiqui KR, Laffont S, Powrie F. 2010. E-cadherin marks a subset of inflammatory dendritic cells that promote T cell-mediated colitis. Immunity 32:557–567. doi: 10.1016/j.immuni.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]