

Abstract
Schizophrenia is thought to be caused, at least in part, by dysfunction in striatal dopamine neurotransmission. Both clinical studies and animal research have implicated the dopamine neuromodulator neurotensin (NT) in the pathophysiology of schizophrenia. Utilizing male mice lacking the NT gene (NT−/−), these studies examined the consequences of NT deficiency on dopaminergic tone and function, investigating (1) dopamine concentrations and dopamine receptor and transporter expression and binding in dopaminergic terminal regions, and (2) the behavioral effects of selective dopamine receptor agonists on locomotion and sensorimotor gating in adult NT−/− mice compared to wildtype (NT+/+) mice. NT−/− mice did not differ from NT+/+ mice in concentrations of dopamine or its metabolite DOPAC in any brain region examined. However, NT−/− mice showed significantly increased D1 receptor, D2 receptor, and dopamine transporter (DAT) mRNA in the caudate putamen compared to NT+/+ controls. NT−/− mice also showed elevated D2 receptor binding densities in both the caudate putamen and nucleus accumbens shell compared to NT+/+ mice. In addition, some of the behavioral effects of the D1-type receptor agonist SKF-82958 and the D2-type receptor agonist quinpirole on locomotion, startle amplitude, and prepulse inhibition were dose-dependently altered in NT−/− mice, showing altered D1-type and D2-type receptor sensitivity to stimulation by agonists in the absence of NT. The results indicate that NT deficiency alters striatal dopamine receptor expression, binding, and function. This suggests a critical role for the NT system in the maintenance of striatal DA system homeostasis and implicates NT deficiency in the etiology of dopamine-associated disorders such as schizophrenia.
Keywords: Neurotensin, Dopamine, Dopamine receptor, Striatum, Schizophrenia
1. Introduction
Disrupted dopamine (DA) neurotransmission has long been hypothesized to play a role in the pathophysiology of schizophrenia. The ‘dopamine hypothesis’ of schizophrenia posits dysfunctional hyperactivity in the striatal DA system to be responsible for psychotic symptoms of schizophrenia [1–3]. This hypothesis is supported by early studies noting that administration of amphetamine, an indirect DA agonist that increases synaptic concentrations of DA in striatal regions, exacerbated psychotic symptoms in schizophrenic patients [3]. Also in support of this theory, all clinically effective antipsychotic drugs act as antagonists at the dopamine D2 receptor, producing decreased striatal DAergic activity [1,4]. Finally, many imaging and post mortem studies have shown increases in striatal D2 receptor binding densities in the brains of schizophrenic patients, confirming a disruption in the striatal DA system in schizophrenia [5–7]. These studies support the dopamine hypothesis of schizophrenia and implicate the D2 receptor in particular in schizophrenia neuropathology. However it remains unclear whether striatal DA disruption is a primary dysfunction in schizophrenia or whether it is a compensatory alteration due to some other defect [8].
In addition to DA system dysfunction, many other neurotransmitters have been implicated in the neuropathology of schizophrenia, including the tridecapeptide neurotensin (NT). Several clinical studies implicate NT in the neurobiology of schizophrenia. Decreased concentrations of NT are found in the cerebrospinal fluid (CSF) of a subset of schizophrenic subjects [9–12], and NT levels normalize in these subjects following effective treatment with antipsychotic drugs [13,14]. These studies led to the hypothesis that NT may act as an endogenous antipsychotic [15]. Preclinical studies utilizing animal models of psychosis also supported the use of NT receptor agonists as potential novel antipsychotic drugs [16].
Laboratory animal studies have shown endogenous NT to be a neuromodulator of nigrostriatal and mesocorticolimbic DA neurotransmission (for review see [17]). Increasing NT neurotransmission by administration of NT receptor agonists such as NT69L alter the behavioral effects of DA receptor agonists such as amphetamine, an indirect DA receptor agonist, and haloperidol, a D2 receptor antagonist [16]. For example, NT69L administration prevents haloperidol-induced catalepsy in rats [18]. In addition, studies utilizing inhibition of NT neurotransmission also suggest the effects of pharmacological manipulation of the DA systems are in fact dependent on intact NT neurotransmission [19–21]. Specifically, NT gene knockout in mice results in blunted effects of pharmacological activation of striatal DA neurons by amphetamine and by the antipsychotic drug haloperidol [22,23]. In addition, the behavioral effects of amphetamine and some antipsychotic drugs on prepulse inhibition (PPI), a measure of sensorimotor gating which is regulated by the nigrostriatal and mesocorticolimbic DA systems, are altered in NT−/− mice [21]. These findings are paralleled by studies utilizing pharmacological antagonism of NT neurotransmission that demonstrate that NT receptor antagonism also alters the physiological and behavioral effects of DA receptor agonists and antagonists [19,20,23–25]. These studies suggest functional alterations in both the mesocorticolimbic and nigrostriatal DA systems in the absence of NT neurotransmission.
Although disrupting NT neurotransmission by NT gene knockout is known to result in altered physiological and behavioral responses to activation of the DA systems, it is unknown whether a deficit in NT produces any developmental changes in these circuits. The experiments detailed in this paper sought to evaluate DAergic tone in DA terminal regions in NT−/− mice compared to wildtype (NT+/+) mice. Specifically, these studies measured DA receptor and dopamine transporter (DAT) gene expression and binding and the concentrations of DA and its metabolite 3,4-dihydroxyphenylacetic acid (DOPAC) in the nucleus accumbens (NAcc), caudate putamen (CP), and frontal cortex (FCTX). In light of the previously observed altered behavioral response to amphetamine on PPI in NT−/− mice [21], these studies also sought to investigate possible changes in DA receptor function or sensitivity in NT−/− mice by measuring behavioral response to selective DA receptor agonists compared to NT+/+ mice.
2. Materials and methods
2.1. Animals
NT−/− mice were generated as previously described [22]. Only male mice were used in these experiments. Mice (60 days of age and older) from the lab’s NT−/− breeding colony backcrossed against the C57BL/6J strain were used for these studies. Mice heterozygous for the NT gene were bred to generate wildtype (NT+/+) and NT−/− mice. Animals were housed in an environmentally controlled animal facility with a 12 h light–dark cycle (lights off: 10:00, lights on: 22:00). Food and water were available ad libitum. Mice were weaned on postnatal day 21 and housed in same sex groups of two to six per cage. All behavioral testing and euthanasia procedures were completed in the dark phase. All animal protocols were approved by the Emory University Institutional Animal Care and Use Committee (IACUC) in compliance with the National Institutes of Health and AAALAC guidelines for use of laboratory animals.
2.2. Genotyping
At weaning, ear punches were obtained from all mice and DNA was extracted from the tissue. The presence or absence of the NT gene was identified using custom PCR primers (Invitrogen) to amplify the wildtype NT gene or the disrupted NT gene construct. Primer sequences to detect the wildtype NT gene allele were 5′-CATCCCTCACAGTTCACTCACTTTG-3′ (25 mer, Tm = 74 °C) and 5′-CCTGGATTCATTTACCTGAGTAGCA-3′ (25 mer Tm = 72 °C). Primer sequences to detect the NT−/− gene allele were 5′-CATCCCTCACAGTTCACTCACTTTG-3′ (25 mer, Tm = 74 °C) and 5′-CCCAGTCACGACGTTGTAAAACGAC-3′ (25 mer, Tm = 76 °C). The PCR products for the wildtype NT gene and for the NT−/− gene allele were 270 bp and 188 bp, respectively. PCR products were run on gel electrophoresis to identify the genotype for each animal.
2.3. HPLC
Concentrations of DA and its metabolite DOPAC were assayed in NT+/+ (n = 6–8) and NT−/− (n = 8–12) mice by high-pressure liquid chromatography (HPLC). All mice used for these studies first underwent behavioral testing for startle amplitude and PPI as described below. One week after testing, mice were euthanized by decapitation and brains were collected and quickly frozen on dry ice. Brains were later dissected according to a mouse brain atlas [26]. NAcc, CP, and FCTX regions were collected. Samples of mouse brains were prepared by adding 200 μl of ice-cold 0.1 N perchloric acid containing 0.01% sodium metabisulfite and 25 ng/ml internal standard 3,4-dihydroxybenzylamine hydrobromide (DHBA) to the tissue. Samples were then homogenized and centrifuged at 15,000 × g for 10 min at 4 °C. The supernatant was injected at a constant flow rate of 1 mL/min onto an Ultrasphere ODS 250 mm × 4.6 mm column, 5 μm (Beckman Coulter, Fullerton, CA) with mobile phase (0.1 mM EDTA; 0.35 mM sodium octyl sulfate; 50 mM phosphoric acid; 5% acetonitrile, adjusted to pH 2.7 with NaOH). A coulometric detector (ESA Inc., Chelmsford, MA; guard cell set at 600 mV and analytical cell at 300 mV) was used to detect the DA, DOPAC, and DHBA chromatographic peaks. The retention time, height, and area of DA and DOPAC peaks were compared with reference standard solutions (Sigma-Aldrich, St. Louis, MO) and quantified by ChemStation chromatography software (Agilent Technologies, Santa Clara, CA). For each sample, DA and DOPAC amounts were normalized to total protein as determined by the method of Lowry [27] using bovine serum albumin as standard.
2.4. Real time RT-PCR
Messenger RNA expression levels of the DAT, D1 receptor, and D2 receptor in NT+/+ (n = 8 pairs) and NT−/− (n = 8 pairs) mice were determined by real time reverse transcription polymerase chain reaction (RT-PCR). All mice used in these studies first underwent behavioral testing for startle amplitude and PPI as described below. One week following testing, mice were euthanized by decapitation and brains were collected and quickly frozen on dry ice. Brains were later dissected according to a mouse brain atlas [26]. NAcc, CP, and FCTX regions were collected and pooled together in pairs from the same genotype matched on overall % PPI values to generate enough tissue for RNA extraction. RNA was extracted by the TRIzol method (Invitrogen, Carlsbad, CA) and reverse transcribed with the High Capacity RNA-to-cDNA kit (Applied Biosystems, Foster City, CA). Before running samples, a mouse endogenous control plate (Applied Biosystems, Foster City, CA) was utilized to determine the ideal endogenous control. The gene showing the least variation in expression between the genotypes was Polr2a (gene encoding polymerase (RNA) II (DNA directed) polypeptide A), and it was selected as the endogenous control gene for this experiment. cDNA was quantified with a NanoDrop spectrophotometer (Thermo Fisher Scientific Inc, Pittsburgh, PA). Primers for Slc6a3 (gene encoding DAT), Drd1a (gene encoding D1), and Drd2 (gene encoding D2) targets were purchased from Applied Biosystems Assays on Demand (Applied Biosystems, Foster City, CA). RT-PCR was performed on the Applied Biosystems 7900HT system (Applied Biosystems, Foster City, CA). dCT values were calculated by subtracting the CT of the target gene from the CT of the endogenous control gene for each sample. ddCT values were calculated by subtracting the mean dCT of NT+/+ from the mean dCT of NT−/−. Gene expression changes were then assessed with the following formula: Fold change in gene expression = 2−ddCT.
2.5. Receptor and transporter binding autoradiography
DAT, D1 receptor, and D2 receptor binding were assayed in NT+/+ and NT−/− mice (n = 8/genotype). All mice used in these studies first underwent behavioral testing for startle amplitude and PPI as described below. One week following testing, mice were euthanized by decapitation and brains were collected and quickly frozen on dry ice. Brains were sectioned on a cryostat at 25 μm thickness and mounted on Superfrost slides (Fisher Scientific, Pittsburgh, PA). Slides were stored at −80 °C until autoradiography, and alternate sections from the same brains were used for separate binding assays. For D1 binding, sections were incubated for 90 min at room temperature in the presence of 4 nM [3H]-SCH23390 (Perkin Elmer, Waltham, MA) and 1 μM mianserin (MP Biomedicals Inc), in order to avoid the binding of [3H]-SCH23390 to 5-HT2 and 5-HT1c receptors. Non-specific binding was determined in the presence of 10 μM unlabeled cis-flupenthixol (Santa Cruz Biotechnology). To label D2 receptors, sections were incubated for 60 min in the presence of 4 nM [3H]-raclopride (Perkin Elmer, Waltham, MA) and 10 nM 7-OH-DPAT (Sigma-Aldrich, St. Louis, MO) in order to prevent the binding of [3H]-raclopride to D3 receptors. Non-specific binding was determined in the presence of 10 μM sulpiride (Sigma-Aldrich, St. Louis, MO). Slides were exposed to BAS-5000 phosphorimaging plates (FujiFilm) for 48 hours (D1 binding) or 12 days (D2 binding) along with tritium standards (American Radiolabeled Chemicals, Inc., St. Louis, MO). For DAT binding, sections were incubated for 60 min in 20 pM [125I]-RT1-121 (Perkin Elmer, Waltham, MA). Non-specific binding was determined in the presence of 200 μM unlabeled nomifensine maleate (Sigma–Aldrich, St. Louis, MO). The sections and 14C plastic standards (Amersham Biosciences) were exposed to BioMax MR film (Kodak) for 48 h.
2.6. Image and film quantification
For D1 and D2 receptor autoradiography, the BAS-5000 plates (FujiFilm) were developed in a BAS-5000 phosphorimager (Fuji-Film) and images were analyzed using MultiGauge software (FujiFilm). Photostimulated luminescence per mm2 (PSL/mm2) was measured for regions of interest bilaterally. PSL/mm2 were converted to nanocuries/mg protein with tritium standards (American Radiolabeled Chemicals, Inc., St. Louis, MO). For DAT autoradiography, BioMax MR films (Kodak) were analyzed by quantitative densitometry using AIS computerized software (AIS, St. Catherines, Ontario, Canada). Optical densities were measured for regions of interest bilaterally, and were converted to nanocuries/mg protein with the 14C standards (Amersham Biosciences). To determine regions of interest, slide-mounted sections were stained with Neutral Red and compared to a mouse atlas [26]. For all sections, two densitometry measurements were made in the NAcc bilaterally, one at bregma +1.34 mm and one between bregma +1.18 mm and +1.1 mm. Densitometries were sampled in the NAcc core in a rectangular area and in the NAcc shell using the free hand tracing tools in MultiGauge and AIS (Fig. 3A). The CP was measured bilaterally between bregma +1.18 mm and +0.98 mm. Densitometries were sampled in CP subregions (dorsomedial, dorsolateral, ventromedial, and ventrolateral regions) in rectangular areas and in the total CP using the free hand tracing tools in MultiGauge and AIS (Fig. 3A). Background was subtracted from regions of interest from adjacent areas lacking specific binding on the same section.
Fig. 3.

Striatal DAT, D1-like, and D2-like binding in NT+/+ and NT−/− mice. (A) Brain regions were sampled bilaterally for quantitative densitometry as shown in the (a) NAcc shell and (b) NAcc core (Bregma +1.18 mm), and (c) Dorsomedial (dm), (d) dorsolateral (dl), (e) ventromedial (vm), and (f) ventrolateral (vl) CP (Bregma +0.98 mm). There were no significant differences in (B) DAT or (C) D1-like binding between NT+/+ and NT−/− mice. (D) NT−/− mice showed significantly increased D2-like binding in the NAcc shell, dm CP, and total CP. (E) NT−/− mice also showed significantly increased D2/D1 in several striatal regions. Data are expressed as mean binding density (nCi/mg or nCi/g protein) ± S.E.M. Abbreviations: dm: dorsomedial, dl: dorsolateral, vm: ventromedial, vl: ventrolateral. *p < 0.05, **p < 0.01, NT−/− compared to NT+/+ mice within brain region.
2.7. Behavioral testing
Startle amplitude and PPI were measured in San Diego Instruments startle chambers (San Diego, CA). All mice in the behavioral study described below and in the tissue studies described above underwent startle testing. Startle amplitude was measured from vibrations of a Plexiglas cylinder (resting on a platform) caused by whole-body response. Vibrations were converted into analog signals using a piezoelectric unit attached to the platform. These signals were digitized and stored in a personal computer. The testing session began with a 5 min acclimatization to the startle chamber in the presence of 65 dB background white noise. Testing sessions consisted of eleven 120 dB pulses alone, eleven no stimulus trials, and 18 pulses preceded (100 ms) by a prepulse of 4, 8, or 12 dB above background. Pulses were presented in a pseudorandom order with an average of 15 s between pulses. Percent PPI for each mouse at each prepulse intensity was calculated using the following formula: %PPI = 100 − (startle amplitude with prepulse × 100/startle amplitude with pulse alone).
Mice in the behavioral study described below underwent locomotor testing. Locomotor activity measurements were evaluated by placing mice in an open field consisting of a white plastic bucket (24.5 cm in diameter, 26.5 cm in height) and videotaped under red light conditions. Activity was recorded for 90 min, and videos were post-processed to quantify time-dependent spontaneous behavior. Distance moved by each animal in the arena was automatically determined using TopScan (Clever Sys Inc., Reston, VA).
2.8. DA receptor agonist study
The behavioral effects of the D1-type receptor agonist SKF-82958 and the D2-type receptor agonist quinpirole on locomotor activity and PPI were tested in NT+/+ (n = 8) and NT−/− mice (n = 13). Drugs were dissolved in 0.9% saline and injected i.p. at a volume of 1.0 ml/kg body weight. Mice were weighed before each testing session to determine the appropriate dose for each animal. SKF-82958 hydrobromide was obtained through the NIMH Chemical Synthesis and Drug Supply Program (RTI International, Research Triangle Park, NC), and quinpirole was purchased from Sigma-Aldrich. For all tests, drugs or saline were injected i.p. 10 min before PPI testing, which was immediately followed by locomotor testing. Before drug testing, NT+/+ and NT−/− mice first underwent baseline testing twice (receiving injections of 0.9% saline i.p. before testing) to obtain baseline PPI and locomotor values for each animal. One week after baseline testing, all animals underwent weekly PPI and locomotor testing, receiving each treatment (0.1 mg/kg quinpirole, 1 mg/kg quinpirole, 0.3 mg/kg SKF-82958, 1 mg/kg SKF-82958, or saline) in a counterbalanced, within-subjects design.
2.9. Statistical analysis
For the HPLC, RT-PCR, and autoradiography experiments, differences between genotypes for each measure were analyzed by a Student’s t test. For the gene expression studies, Pearson correlations were calculated between −dCT values across brain regions.
For the DA receptor agonist study, the behavioral effects of quinpirole and SKF-82958 on locomotor behavior, startle response, and PPI were analyzed utilizing ANOVAs. Following ANOVAs, planned comparisons were tested using Tukey’s HSD post-test. The effect of each drug dose on each behavior was analyzed separately. For locomotor behavior and PPI for each drug dose, the effects of genotype and drug were analyzed using three-way repeated measures mixed ANOVAs (SAS PROC GLIMMIX specifying a gamma distribution and a log link for the dependent variable and robust standard errors) with the factors: drug, genotype, and timepoint for locomotor behavior and drug, genotype, and prepulse for PPI. For startle amplitude, the effects of genotype and drug dose were analyzed using two-way ANOVAs (factors: genotype and drug). When there was a significant genotype × drug interaction, Tukey’s post-tests were used for pairwise comparisons of saline controls to each drug dose within each genotype to assess drug response within genotype. When there was a significant effect of drug but no significant drug × genotype interaction, saline and drug groups were compared collapsed across genotypes for each timepoint or prepulse using Tukey’s post-tests. Baseline saline control values for startle amplitude and PPI values were not statistically different and, thus, were averaged to generate control values for startle amplitude and PPI comparisons. Significance was set at p < 0.05 for all analyses. Statistical analyses were performed with GraphPad Prism 3.0 (GraphPad Software, San Diego, CA), SysStat SigmaPlot 12.3 (San Jose, CA), and SAS (Cary, NC) statistical software.
3. Results
3.1. DA and DOPAC concentrations in NT+/+ and NT−/− mice
To compare DAergic tone in NT+/+ and NT−/− mice, DA and DOPAC concentrations in the NAcc, CP, and FCTX in NT+/+ and NT−/− mice were examined by HPLC. There were no significant differences in DA (Fig. 1A; CP: t(18) = 0.30), p > 0.05; NAcc: t(14) = 1.27, p > 0.05; FCTX: t(16) = 0.01, p > 0.05) or DOPAC concentrations (Fig. 1B; CP: t(18) = 0.05, p > 0.05; NAcc: t(14) = 0.06, p > 0.05; FCTX: t(15) = 0.22, p > 0.05) in any of the brain regions examined. To evaluate DA turnover in NT+/+ and NT−/− mice, the DOPAC/DA ratio was calculated and compared between genotypes. There was no difference in the DOPAC/DA ratio (Fig. 1C; CP: t(18) = 0.69, p > 0.05; NAcc: t(14) = 1.37, p > 0.05; FCTX: t(15) = 0.81, p > 0.05) between NT+/+ and NT−/− mice in any of the brain regions examined. The NAcc and FCTX are anatomically and functionally connected directly via corticostriatal projections and indirectly through striatopallidothalamic projections [28], and experimental disruption of frontocortical DA neurotransmission in rodent models has been shown to produce hyperactivity of DAergic function in the striatum, especially the ventral striatum [29]. For this reason, DA and DOPAC concentrations were compared between NAcc and FCTX regions by ratio analyses in NT+/+ and NT−/− mice. There was no difference in DA (t(13) = 0.58, p > 0.05) and DOPAC (t(12) = 1.13, p > 0.05) concentrations in the NAcc compared to the FCTX (NAcc/FCTX ratio), consistent with the idea that there is no major disruption of frontocortical DA transmission in NT−/− mice.
Fig. 1.

Regional DA and DOPAC concentrations in NT+/+ and NT−/− mice. NT+/+ and NT−/− mice did not differ significantly in (A) DA, (B) DOPAC, or (C) DOPAC/DA ratio (p > 0.05). Data are expressed as mean concentration (ng/mg protein, A and B) ±S.E.M or mean DOPAC/DA ratio (C) ±S.E.M.
3.2. DA receptor and DAT gene expression in NT+/+ and NT−/−mice
Differences in mRNA levels of the dopamine D1 receptor (Fig. 2A), dopamine D2 receptor (Fig. 2B), and the DAT (Fig. 2C) between NT+/+ and NT−/− mice were examined by real time RT-PCR. NT−/− mice had significantly increased D1 (t(13) = 5.04, p < 0.001), D2 (t(13) = 2.39, p < 0.05), and DAT mRNA levels (t(13) = 2.52, p < 0.05) compared to NT+/+ mice in the CP. NT−/− mice showed a trend for increased DAT mRNA in the FCTX compared to NT+/+ mice, but this difference did not reach significance (t(13) = 1.98, p = 0.07). There were no significant differences in mRNA levels of any of the targets in the NAcc between genotypes (p > 0.05).
Fig. 2.

Gene expression in NT+/+ and NT−/− mice. NT−/− showed increased (A) D1, (B) D2, and (C) DAT mRNA compared to NT+/+ mice in the CP. There were no significant differences in gene expression in the NAcc or FCTX. Data are expressed as mean fold change ± S.E.M. *p < 0.05, ***p < 0.001, NT−/− compared to NT+/+ mice within brain region.
Again, as DA neurotransmission in the FCTX and NAcc are functionally related, D1 and D2 mRNA levels (−dCT values) were analyzed for regional correlation between the FCTX and NAcc. Receptor expression in the FCTX was not correlated with its expression in the NAcc in either genotype (p > 0.05).
3.3. DA receptor and transporter binding in NT+/+ and NT−/− mice
Differences in striatal DAT, D1-like, and D2-like binding densities in NT+/+ and NT−/− mice were examined (Fig. 3). There were no significant differences in DAT binding (Fig. 3B) or D1-like (Fig. 3C) binding densities in the NAcc or CP between the genotypes (p > 0.05). However, NT−/− mice had significantly increased D2-like binding in the CP (at Bregma 1.18–0.98 mm) and NAcc (at Bregma 1.18–1.1 mm but not at Bregma 1.34 mm). Specifically, NT−/− mice showed increased D2-like densities in the NAcc shell (t(13) = 3.11, p < 0.01), dorsomedial CP (t(13) = 2.17, p < 0.05), and total CP (t(13) = 2.17, p < 0.05) regions compared to NT+/+ mice (Fig. 3D). NT−/− mice showed trends for increased D2-like binding in the NAcc core (t(13) = 1.95, p = 0.07), dorsolateral CP (t(13) = 2.11, p = 0.06), and ventromedial CP (t(13) = 1.98, p = 0.07) that approached significance. NT−/− mice also showed a significantly increased D2/D1 ratio in the NAcc shell (t(13) = 2.28, p < 0.05) and in the dorsomedial (t(13) = 2.39, p < 0.05), dorsolateral (t(13) = 2.34, p < 0.05), ventrolateral (t(13) = 2.40, p < 0.05), and total CP regions (t(13) = 2.42, p < 0.05) (Fig. 3E). NT−/− mice showed a trend for increased D2/D1 ratio in the ventromedial CP (t(13) = 2.13, p = 0.05).
3.4. Baseline behavior in NT+/+ and NT−/− mice
Pulse alone startle amplitude and PPI were measured in all animals prior to tissue collection in the experiments described above. For startle amplitude, t-tests showed no differences between NT+/+ and NT−/− mice (HPLC mice: t(16) = 1.05, p > 0.05; RT-PCR mice: t(14) = 0.32, p > 0.05; autoradiography mice: t(14) = 0.04, p > 0.05). Likewise, in the DA receptor agonist study (described below), baseline startle amplitude did not differ between genotypes (p > 0.05). In all experimental cohorts, there were no significant differences in PPI between NT+/+ and NT−/− at any of the prepulse intensities (p > 0.05). Likewise, there was no difference in overall % PPI (all prepulses combined) between NT+/+ and NT−/− mice (HPLC mice: t(16) = 0.42, p > 0.05; RT-PCR mice: t(14) = 1.21, p > 0.05; autoradiography mice: t(14) = 0.36, p > 0.05; DA receptor agonist study mice: t(19) = 0.64, p > 0.05). In addition, in the DA receptor agonist study, baseline locomotor activity as measured by distance moved did not differ between genotypes (described below).
3.5. Effects of selective DA receptor agonists on locomotor activity in NT+/+ and NT−/− mice
Baseline locomotor testing showed no differences in distance moved between NT+/+ and NT−/− mice (t(16) = 1.02, p > 0.05). The effects of SKF-82958 (0.3 mg/kg and 1 mg/kg) and quinpirole (0.1 mg/kg and 1 mg/kg) on distance moved were analyzed separately by three-way repeated measures ANOVAs (genotype × drug × timepoint) across three 30 min time windows (0–30 min, 30–60 min and 60–90 min post injection, Fig. 4).
Fig. 4.

Effects of SKF-82958 and quinpirole on distance moved (m) in NT+/+ and NT−/− mice. Data are expressed as mean distance moved (m)/30 min time period ± S.E.M. 0.3 mg/kg SKF-82958 significantly increased locomotion at the 0–30 min timepoint (A), and 1 mg/kg SKF-82958 significantly increased locomotion at the 0–30 min and 30–60 min timepoints (B), but NT knockout did not significantly affect SKF-82958-induced hyperlocomotion at either dose. (C) 0.1 mg/kg quinpirole significantly decreased locomotion similarly in both NT+/+ and NT−/− mice at the 0–30 min timepoint. (D) 1 mg/kg quinpirole significantly decreased locomotion at the 30–60 min timepoint and 60–90 min timepoint in NT+/+ mice (**p < 0.01, drug compared to saline control within genotype), but the hypolocomotor effect of 1 mg/kg was absent in NT−/− mice (n.s., p > 0.05).
SKF-82958 had significant effects on locomotor activity at both doses (Fig. 4A and B). At the lower dose (0.3 mg/kg), there was a significant drug × time interaction (F(2,80) = 6.08, p < 0.01), and Tukey’s post-tests showed locomotion (as measured by distance moved) was significantly increased at the 0–30 min time interval (p < 0.05) but not at the other timepoints (p > 0.05). At the higher dose (1 mg/kg) there was a significant effect of drug (F(1,75) = 11.42, p < 0.01) and a drug × time interaction (F(2,75) = 31.91,p < 0.0001), and Tukey’s post-tests showed locomotion was significantly increased at the 0–30 min (p < 0.01) and 30–60 min timepoints (p < 0.01). However, at both doses of SKF-82958, there were no significant effects of genotype and no genotype × drug interactions (p > 0.05), indicating there was no effect of NT knockout on the hyperlocomotor effects of SKF-82958, and that SKF-82958 increased locomotion in NT+/+ and NT−/− similarly.
The effects of quinpirole on locomotor activity were dose-dependent; both doses (0.1 mg/kg and 1 mg/kg) had a significant hypolocomotor effect on locomotor activity at distinct timepoints. At the lower dose (0.1 mg/kg, Fig. 4C), the ANOVA showed a significant effect of drug (F(1,80) = 20.01, p < 0.0001), a drug × time interaction (F(2,80) = 27.62, p < 0.0001), and a drug × genotype × time interaction (F(2,80) = 4.99, p < 0.01). Tukey’s post-tests showed quinpirole significantly reduced locomotor activity compared to saline in the first time block (0–30 min) in both NT+/+ and NT−/− mice (p < 0.01), but did not significantly reduce locomotor activity in the NT+/+ or NT−/−mice (p > 0.05) in the 30–60 min time block although there was a non-significant trend in the NT+/+ mice at this timepoint. These results indicate the hypolocomotor effects of 0.1 mg/kg quinpirole are similar in NT+/+ and NT−/− mice. At the higher dose (1 mg/kg, Fig. 4D), the ANOVA showed a significant effect of drug (F(1,80) = 10.93, p < 0.01) and a significant drug × genotype interaction (F(1,80) = 5.58, p < 0.05). Post-tests showed 1 mg/kg quinpirole significantly decreased locomotor activity compared to saline in the 30–60 min time block and the 60–90 min time block in the NT+/+ mice (p < 0.01), but not in the NT−/− mice (p > 0.05). These results indicate a diminished hypolocomotor response to 1 mg/kg quinpirole in the NT−/− mice.
3.6. Effects of selective DA receptor agonists on startle amplitude in NT+/+ and NT−/− mice
There were no differences in baseline pulse alone startle amplitude between NT+/+ and NT−/− mice (t(19) = 1.71, p > 0.05). The effects of SKF-82958 (0.3 mg/kg, 1 mg/kg) and quinpirole (0.1 mg/kg, 1 mg/kg) on startle amplitude were analyzed by two-way repeated measures ANOVAs (genotype × drug) (Fig. 5).
Fig. 5.
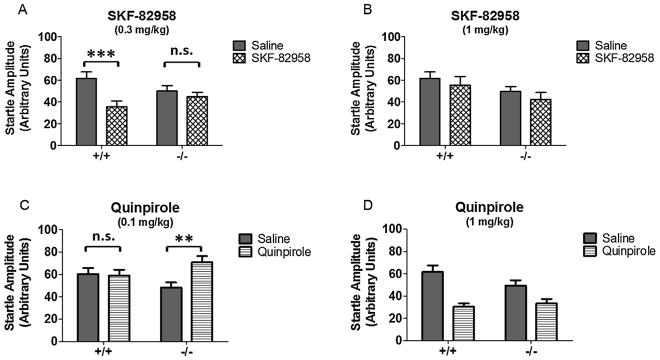
Effects of SKF-82958 and quinpirole on startle amplitude in NT+/+ and NT−/− mice. Data are expressed as mean startle amplitude ± S.E.M. (A) 0.3 mg/kg SKF-82958 suppressed startle amplitude in NT+/+ mice (***p < 0.001) but not NT−/− mice (n.s., p > 0.05). (B) 1 mg/kg SKF-82958 did not significantly affect startle amplitude. (C) 0.1 mg/kg quinpirole increased startle amplitude in NT−/− mice (**p < 0.01) but not in the NT+/+ mice (n.s., p > 0.05). (D) 1 mg/kg decreased startle amplitude and had similar effects regardless of genotype.
For 0.3 mg/kg SKF-82958 (Fig. 5A), there was a significant effect of drug (F(1,16) = 15.95, p = 0.001) and a genotype × drug interaction (F(1,16) = 7.11, p < 0.05). There was no effect of genotype (p > 0.05). Post-tests indicated that these effects were due to SKF-82958 suppression of startle amplitude in NT+/+ mice (p < 0.001) but not NT−/− mice (p > 0.05). At the higher dose of SKF-82958 (1 mg/kg) (Fig. 5B), there were no significant effects of drug or genotype and no significant genotype × drug interaction (p > 0.05). These results indicate NT knockout blocks the suppressive effect of 0.3 mg/kg SKF-82958 on startle amplitude, and that the suppressive effect of SKF-82958 at this dose requires NT signaling.
Treatment with 0.1 mg/kg quinpirole (Fig. 5C) produced a significant drug effect on startle amplitude (F(1,19) = 5.02, p < 0.05), and there was also a significant genotype × drug interaction at this dose (F(1,19) = 6.41, p < 0.05). Post-tests indicated that these effects were due to a significant quinpirole-induced increase in startle amplitude in the NT−/− mice (p < 0.01) but not in the NT+/+ mice (p > 0.05). In contrast, the higher dose of quinpirole (1 mg/kg) decreased startle amplitude and had similar effects regardless of genotype (Fig. 5D). Treatment with 1 mg/kg quinpirole produced a significant effect of drug (F(1,17) = 24.36, p < 0.001), but no significant effects of genotype and no genotype × drug interaction (p > 0.05). Post-tests showed 1 mg/kg quinpirole significantly decreased startle amplitude regardless of genotype (p < 0.001). Collectively, these results indicate that NT−/− mice are more sensitive to quinpirole, which selectively enhances startle amplitude in these mice at the lower dose, but that at the higher dose of quinpirole suppresses startle amplitude similarly in both genotypes.
3.7. Effects of selective DA receptor agonists on PPI in NT+/+ and NT−/− mice
There were no differences in baseline PPI between NT+/+ and NT−/− mice (% overall PPI: t(19) = 0.64, p > 0.05). The effects of 0.3 mg/kg SKF-82958, 1 mg/kg SKF-82958, 0.1 mg/kg quinpirole, and 1 mg/kg quinpirole on PPI were analyzed by three-way repeated measures ANOVAs (genotype × drug × prepulse). Results are presented in Fig. 6.
Fig. 6.

Effects of SKF-82958 and quinpirole on % PPI in NT+/+ and NT−/− mice. Data are expressed as mean % PPI ± S.E.M. (A) 0.3 mg/kg SKF-82958 significantly disrupted PPI at the 4 dB and 12 dB prepulses in NT+/+ mice (**p < 0.01) but did not significantly disrupt PPI in the NT−/− mice (n.s., p > 0.05). (B) 1 mg/kg SKF-82958 decreased PPI at the 8 dB and 12 dB prepulses, but NT knockout did not affect PPI disruption by SKF-82958 at this dose. (C) 0.1 mg/kg quinpirole only had a significant effect on PPI at the 12 dB prepulse (p < 0.05), and these effects were not altered by NT knockout. (D) 1 mg/kg quinpirole did not significantly affect PPI at any prepulse.
Treatment with SKF-82958 (Fig. 6A,B) produced a significant effect on PPI in the ANOVA at both doses (0.3 mg/kg, F(1,80)= 18.89, p < 0.0001; 1 mg/kg, F(1,85) = 19.44, p < 0.0001). At the lower dose there was a significant drug × genotype interaction (F(1,80) = 4.46, p < 0.05). Post-tests indicated that SKF-82958 significantly decreased PPI at the 4 dB and 12 dB prepulse in the NT+/+ mice (p < 0.01) but not the NT−/− mice at the lower dose (0.3 mg/kg) (Fig. 6A). At the higher dose (1 mg/kg) (Fig. 6B), there was no significant drug × genotype interaction (p > 0.05), and post-tests showed SKF-82958 decreased PPI at the 8 dB and 12 dB prepulse (p < 0.01), but that NT knockout did not affect PPI disruption by SKF-82958 at this dose. These results suggest that NT−/− mice are less sensitive to the disruptive effects of SKF-82958 on PPI.
Treatment with quinpirole (Fig. 6C,D) produced a significant effect in the ANOVA at both doses (0.1 mg/kg, F(1,95) = 4.52, p < 0.05; 1 mg/kg, F(1,85) = 5.09, p < 0.05), but post-tests indicated 0.1 mg/kg quinpirole only had a significant effect on PPI at the 12 dB prepulse, and that 1 mg/kg quinpirole did not significantly affect PPI at any prepulse. Also, there was no significant effect of genotype and no genotype × drug interaction at either dose (p > 0.05). These results indicate that quinpirole only marginally affected PPI and that these effects were not altered by NT knockout.
4. Discussion
The mesolimbic and nigrostriatal DA systems are known to display a great deal of plasticity in response to pharmacological [30–32], genetic [33,34], and developmental manipulations [35]. The experiments described in this paper examined whether a deficit in NT produced alterations in DA system tone and function by investigating striatal and cortical DA and DA metabolite concentrations, DA receptor and transporter gene expression and binding, and behavioral sensitivity to selective DA receptor agonists in NT−/− mice. Indeed, our results show that disruption of NT signaling by NT gene knockout is associated with increased D1 receptor, D2 receptor, and DAT gene expression in the CP (Fig. 2) and increased region-specific D2 binding in the NAcc and CP (Fig. 3). In addition, a lack of NT results in some altered behavioral responses to D1-type and D2-type agonists (Figs. 4–6), demonstrating altered DA receptor function as a consequence of NT deficiency.
4.1. Changes in DA receptor expression and binding in NT−/− mice
D1 receptor, D2 receptor, and DAT mRNA were significantly increased in the CP of NT−/− compared to NT+/+ mice (Fig. 2). No significant changes in gene expression of these targets were noted in the FCTX or NAcc regions in NT−/− mice. In light of these changes in gene expression, D1 receptor, D2 receptor, and DAT binding in the dorsal and ventral striatum were investigated in NT−/− mice (Fig. 3). D2-like, but not D1-like or DAT, binding was significantly elevated in the dorsal CP and NAcc shell in the absence of NT. Increases in D2-like binding most likely reflect increases in D2 receptor binding and not D3 or D4 receptor binding, as the autoradiography experiment was conducted in the presence of unlabeled 7-OH-DPAT to preclude D3 binding, and raclopride (the radioligand used) has a low affinity for D4 receptors [36]. These results partially coincide with results from the gene expression study, as both D2 mRNA and D2 receptor ligand binding were elevated to a similar extent in the dorsal CP of NT−/− mice. However, in the binding experiment, NT−/−mice showed increased D2-like binding in the NAcc shell, while in the gene expression study, D2 mRNA in the NAcc was unaltered. This discrepancy might be due to differences in the methods used; in the gene expression study, D2 mRNA was measured by RT-PCR from the dissected NAcc region while D2 binding was measured by autoradiography. Increased D2 binding in the NAcc of NT−/−mice may reflect increased D2 trafficking to the cell surface without increased D2 receptor expression and protein synthesis. In addition, NT−/− mice showed increased striatal D1 and DAT expression but did not show increases in striatal D1 and DAT ligand binding. Increases in gene expression need not correlate with increases in protein, and it is possible that NT−/− mice possess increased D1 and DAT mRNA but not increased D1 and DAT protein. It is also possible that NT−/− mice may have increased intracellular D1 and DAT protein levels but that these proteins are not trafficked to the cell surface and are thus not detected by autoradiography. Nevertheless, the results from the autoradiography experiment demonstrate that in the absence of NT, striatal D2 receptor levels are increased.
Several studies have shown NT functionally opposes the effects of the D2 receptor, and activation of NT receptors by NT is known to desensitize and internalize D2 receptors [17,37]. In the absence of NT, it is thus not surprising that D2 binding is increased in the striatum. Striatal D2 receptors exist both pre-synaptically (D2S isoform) as autoreceptors as well as post-synaptically (D2L isoform). NT is able to desensitize both isoforms of the receptor in vitro and antagonize the function of both pre-synaptic and post-synaptic D2 receptors [37,38]. From our study, it is not possible to specifically determine whether D2 receptors were increased pre-synaptically or post-synaptically in NT−/− mice. Because the post-synaptic D2L receptor isoform comprises the vast majority of D2 receptor mRNA and protein in the striatum [30,39], we might theorize that an overall increase in D2 receptor binding density in the striatum may be due primarily to an increase in post-synaptic D2L receptors. However, future studies are needed to determine this.
Interestingly, a recent study by Liang and colleagues [40] showed mice lacking the neurotensin receptor 1 (NTS1) and neurotensin receptor 2 (NTS2) also showed changes in striatal DA receptor expression and DAergic tone, although these alterations were different from those found in NT−/− mice (decreased D1 mRNA expression, no changes in D2 mRNA expression, increased basal extracellular DA levels). Studies suggest NT exerts many of its modulatory effects on the DA system via NT receptors, particularly, the NTS1 receptor [37,41]. However, other uncharacterized NT receptors or mechanisms likely exist through which NT modulates DA signaling [42,43], and thus, the consequences of NT knockout on the striatal DA system might be distinct from those of NT receptor knockout. It is also possible that NT and NT receptors may play different roles during neural development of the striatum, and that constitutive knockout of either NT or NT receptor subtypes may perturb the striatal DA system in unique ways.
4.2. Lack of changes in striatal and cortical DA concentrations and metabolites in NT−/− mice
DA denervation by MPTP treatment produces compensatory increases in striatal D1 receptor and D2 receptor transcripts [44], while constitutive hyper-dopaminergic tone in a mutant mouse model (DAT knockout) produces compensatory decreases in D1 and D2 receptor expression [45]. Because NT−/− mice have increased striatal D1 and D2 receptor expression, it might be hypothesized that mice lacking NT may show hypo-dopaminergic tone in the striatum. However, our studies showed no changes in DA concentrations, DOPAC concentrations, or DOPAC/DA ratio in any of the terminal regions in NT−/− mice compared to NT+/+ mice (Fig. 1), indicating NT−/− mice do not show decreases in mesolimbic or nigrostriatal tonic DA or DA metabolism. Nonetheless, our study measured regional tissue concentrations to quantify DA and DOPAC levels. It is possible that while tissue concentrations of DA and DOPAC are unchanged in NT−/− mice, synaptic concentrations of these monoamines or DA release might be altered.
4.3. Baseline behavior in NT+/+ and NT−/− mice
NT+/+ and NT−/− mice did not differ in baseline locomotor activity. In addition, in these studies NT+/+ and NT−/− mice did not significantly differ in PPI or startle amplitude as was previously observed [21]. This lack of replication may be due to the smaller sample sizes utilized in this study (n = 8–13/genotype) compared to the sample sizes used previously (n = 48–52/genotype). PPI is known to vary widely between individual animals, and it is possible that the PPI deficit observed in NT−/− mice in previous studies may only be apparent with large sample sizes. However, some of the behavioral effects of DA receptor agonists on locomotor activity, startle amplitude, and PPI were altered in NT−/− mice compared to NT+/+ mice as discussed below.
4.4. Differences in behavioral response to D1-type and D2-type agonists in NT−/− mice
As discussed above, in NT−/− mice, striatal D2 receptors and the D2/D1 receptor ratio are increased. Thus, it might be predicted that a functional consequence of NT system disruption by NT gene knockout would be altered sensitivity to D1-type and D2 type receptor agonists. This prediction was tested by investigating the effects of low and high doses of selective D1-type and D2-type agonists on behaviors known to be regulated by the nigrostriatal and mesolimbic DA systems (locomotion, startle response, PPI). Our hypothesis was supported by our results in some of the behaviors, which suggest blunted D1-type sensitivity to agonists and altered D2-type function in NT−/− mice. In addition, these results suggest at least some of the behavioral effects of D1-type and D2-type agonists are mediated by the NT system.
SKF-82958, a D1-family agonist, increased locomotion, decreased startle amplitude (at 0.3 mg/kg), and disrupted PPI in NT+/+ mice as was expected from studies in the literature [46]. The effects of SKF-82958 on startle amplitude (Fig. 5) and PPI (Fig. 6) were dose-dependently diminished in NT−/− mice compared to NT+/+ mice. Significant behavioral effects of the low dose of SKF-82958 (0.3 mg/kg) on startle response and PPI were absent in NT−/− compared to NT+/+, although the effects of SKF-82958 on locomotor behavior were similar in NT−/− and NT+/+ mice. Our results indicate D1-type receptor function, as it pertains to startle response and PPI disruption, is blunted in the absence of NT. These results concur with one previous study utilizing an NT receptor antagonist that showed blocking NT neurotransmission decreased the behavioral effects of another selective D1 receptor agonist [47]. These results also parallel the results from a previous study showing a blunted behavioral response to amphetamine in NT−/− mice [21]. It is possible that the decreased response to amphetamine in NT−/− mice might also be due to diminished D1-type function. In contrast, the behavioral effects of the higher dose of SKF-82958 (1 mg/kg) on PPI in NT−/− mice were similar to those in NT+/+ mice. The lack of prominent effect of NT gene knockout on behavioral response to 1 mg/kg SKF-82958 may be due to the overwhelming effect of the agonist at this high dose. Thus, lack of NT diminishes sensitivity to the PPI disrupting effects of the D1 agonist, but this can be compensated for with higher doses. Nonetheless, these results indicate that D1-type function, as it pertains to startle response and PPI disruption, is blunted in the absence of NT. These results also suggest the some of the behavioral effects of D1 agonists are mediated by the NT system.
In addition, some of the behavioral effects of quinpirole (a D2-like agonist) were altered in NT−/− mice. Quinpirole decreased locomotion at both doses in NT+/+ mice (Fig. 4) in agreement with previous studies showing both high and low doses of quinpirole decrease locomotion in mice [48]. Hypolocomotor response to 0.1 mg/kg quinpirole was similar in NT−/− and NT+/+ mice. However, at the 1 mg/kg dose, NT−/− mice did not show a significant hypolocomotor response to quinpirole (Fig. 4D), suggesting altered D2-like receptor function in NT−/− mice.
In NT+/+ mice, the lower dose of quinpirole (0.1 mg/kg) did not significantly affect pulse alone startle amplitude (Fig. 5C). In contrast, the low dose of quinpirole increased pulse alone startle amplitude (Fig. 5C) in NT−/− mice. The effects of quinpirole in NT−/−mice were expected to be exacerbated given the known inhibitory actions of NT on D2 receptor function [49,50]. In the literature, quinpirole has no effect on startle response at low doses in C57BL/6J mice, but decreases startle amplitude in mice at higher doses, which is in line with our results [51]. As quinpirole (0.1 mg/kg) had no effect on startle amplitude in wildtype NT+/+ mice, it might be predicted that quinpirole would decrease startle amplitude in mice lacking NT. Surprisingly, quinpirole (0.1 mg/kg) had the opposite effect of increasing startle amplitude in NT−/− mice. This might be explained by the observation in our autoradiography study that NT−/− mice show increased striatal D2 receptor expression and binding that may alter the behavioral effects of systemic D2 receptor agonism. The altered behavioral effects of quinpirole in NT−/−mice were dose-dependent, as the effects of 1 mg/kg quinpirole on startle amplitude were similar in NT+/+ and NT−/− mice.
In sum, NT−/− mice showed blunted behavioral response to the D1-type agonist SKF-82958 (at the 0.3 mg/kg dose), and altered behavioral response to the D2-type agonist quinpirole that was dose- and behavior-specific. Taken together, these results suggest that NT−/− mice not only show changes in striatal D2 receptor levels, but that these changes have functional consequences for some behaviors.
4.5. Relevance to schizophrenia
Imaging studies have demonstrated increased binding of D2-like receptors in the striata of schizophrenic patients [6,7] and in their non-schizophrenic monozygotic twins, suggesting increased D2 receptor binding might be a biomarker for genetic susceptibility to schizophrenia [52]. However, other discordant studies show no difference in D2-like binding between patients with schizophrenia and healthy controls [53,54]. Furthermore, these studies are sometimes confounded by use of antipsychotic drugs (D2 antagonists), which also upregulate D2-like binding [55]. Nevertheless, many animal models of schizophrenia – including those that are altered genetically, pharmacologically, and developmentally – also show an increase in striatal D2 receptors, suggesting striatal D2 receptor upregulation reflects a neurophysiological alteration in the striatal DA system that is associated with psychosis (for review see See-man et al. [56]). The results from the present study show for the first time that constitutive NT deficiency is sufficient to increase D2 binding in the striata in mice, a schizophrenia-like phenotype. As noted above, decreased CSF NT concentrations have been reported in schizophrenic patients [9,10,12]. These results suggest a decrease in the synaptic availability of NT in some patients with schizophrenia, which may contribute to the observed increases in striatal D2 expression and in the development of psychosis.
Some schizophrenic patients show supersensitivity to indirect DA agonists, and when these drugs are administered in low doses, psychotic symptoms are exacerbated in some patients (for a meta-analysis see [3]). However, some schizophrenics show no response to DA agonists, and there is evidence that DA agonists might actually improve negative symptoms in some schizophrenic patients [57,58]. Most animal models relevant to schizophrenia also show supersensitivity to direct and indirect DA agonists, as measured by the effects of these drugs on locomotor activity and sensorimotor gating, while a few show subsensitivity to DA agonists [59]. NT−/− mice may fall in the latter category, as they show behavioral and physiological subsensitivity to the effects of the indirect DA agonist amphetamine [21,23] and to the selective D1-like agonist SKF-82958, although they also showed altered responsivity to the D2-like agonist quinpirole in our study. These findings suggest NT−/− mice may have potential as a unique animal model of negative schizophrenia-like symptoms. In line with this reasoning, some clinical studies have shown the decreased CSF NT levels observed in some schizophrenics are associated with negative symptoms [12,14]. Historically, most animal studies have emphasized modeling positive symptoms of schizophrenia while neglecting negative symptoms, and negative symptoms are notoriously difficult to treat in schizophrenic patients [60,61]. For this reason, future studies might investigate NT−/− mice for physiology and behaviors relevant to the negative symptoms of schizophrenia.
4.6. Conclusions
The results of these studies indicate that NT deficiency due to NT gene knockout results in DA system plasticity, particularly, changes in DA receptor expression, binding, and function. These results demonstrate a regulatory role for NT in the maintenance of striatal DA system homeostasis. Finally, these studies implicate a deficit in NT neurotransmission in the neurobiology of disorders associated with DA system abnormalities, specifically in schizophrenia.
HIGHLIGHTS.
We investigated dopaminergic tone and function in mice lacking neurotensin (NT−/−).
NT−/− mice show increased striatal D1, D2 and DAT mRNA compared to wildtype mice.
NT−/− mice show increased striatal D2 receptor binding densities.
Some of the behavioral effects of D1- and D2-type receptor agonists are altered in NT−/− mice.
Results implicate NT deficiency in dopamine-related disorders such as schizophrenia.
Acknowledgments
This work was supported by a grant from the National Institute of Mental Health (NIMH grant MH39415). The HPLC work was supported by grants from the National Eye Institute (NEI grants R01EY004864 and P30EY006360). We thank Aaron DeLaRosa from Rutgers University, Department of Animal Sciences for help in creating the brain illustrations in Fig. 3. We also thank Dr. Anthony Pawlak from Rutgers University for consultation on statistical analyses.
Abbreviations
- CP
caudate putamen
- CSF
cerebrospinal fluid
- DA
dopamine
- DAT
dopamine transporter
- DHBA
3,4-dihydroxybenzylamine hydrobromide
- DOPAC
3,4-dihydroxyphenylacetic acid
- FCTX
frontal cortex
- HPLC
high-pressure liquid chromatography
- NAcc
nucleus accumbens
- NT
neurotensin
- NT−/−
neurotensin knockout
- NT+/+
wildtype
- NTS1
neurotensin receptor 1
- NTS2
neurotensin receptor 2
- PPI
prepulse inhibition
- RT-PCR
reverse transcription polymerase chain reaction
Footnotes
Conflicts of interest
Chastain, Qu, Iuvone, Dobner, Kinkead: None; Bourke: Currently works for the FDA; Nemeroff: Consulting: Xhale, Takeda, SK Pharma, Shire, Roche, Lilly, Allergan, Mitsubishi Tanabe Pharma Development America, Taisho Pharmaceutical Inc., Lundbeck, Prismic Pharmaceuticals; Stockholder: CeNeRx, BioPharma, PharmaNeuro-Boost, Revaax Pharma, Xhale, Celgene, Seattle Genetics, Abbvie; Scientific Advisory Boards: American Foundation for Suicide Prevention (AFSP), CeNeRx BioPharma (2012), National Alliance for Research on Schizophrenia and Depression (NARSAD), Xhale, PharmaNeuroBoost (2012), Anxiety Disorders Association of America (ADAA), Skyland Trail; Board of Directors: AFSP, NovaDel (2011), Skyland Trail, Gratitude America, ADAA; Income sources of equity of $10,000 or more: PharmaNeuroBoost, CeNeRx BioPharma, NovaDel Pharma, Reevax Pharma, American Psychiatric Publishing, Xhale; Patents: Method and devices for transdermal delivery of lithium (US 6,375,990B1), Method of assessing antidepressant drug therapy via transport inhibition of monoamine neurotransmitters by ex vivo assay (US 7,148,027B2); Honoraria: Various; Royalties: Various; Expert Witness: Various.
References
- 1.van Rossum JM. The significance of dopamine-receptor blockade for the mechanism of action of neuroleptic drugs. Arch Int Pharmacodyn Ther. 1966;160:492–4. [PubMed] [Google Scholar]
- 2.Meltzer HY, Stahl SM. The dopamine hypothesis of schizophrenia: a review. Schizophr Bull. 1976;2:19–76. doi: 10.1093/schbul/2.1.19. [DOI] [PubMed] [Google Scholar]
- 3.Lieberman JA, Kane JM, Alvir J. Provocative tests with psychostimulant drugs in schizophrenia. Psychopharmacology (Berl) 1987;91:415–33. doi: 10.1007/BF00216006. [DOI] [PubMed] [Google Scholar]
- 4.Grace AA, Bunney BS, Moore H, Todd CL. Dopamine-cell depolarization block as a model for the therapeutic actions of antipsychotic drugs. Trends Neurosci. 1997;20:31–7. doi: 10.1016/S0166-2236(96)10064-3. [DOI] [PubMed] [Google Scholar]
- 5.Seeman P. Dopamine receptors and the dopamine hypothesis of schizophrenia. Synapse. 1987;1:133–52. doi: 10.1002/syn.890010203. [DOI] [PubMed] [Google Scholar]
- 6.Seeman P, Kapur S. Schizophrenia: more dopamine, more D2 receptors. Proc Natl Acad Sci USA. 2000;97:7673–5. doi: 10.1073/pnas.97.14.7673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wong DF, Pearlson GD, Tune LE, Young LT, Meltzer CC, Dannals RF, et al. Quantification of neuroreceptors in the living human brain: IV. Effect of aging and elevations of D2-like receptors in schizophrenia and bipolar illness. J Cereb Blood Flow Metab. 1997;17:331–42. doi: 10.1097/00004647-199703000-00010. [DOI] [PubMed] [Google Scholar]
- 8.Carlsson A, Waters N, Holm-Waters S, Tedroff J, Nilsson M, Carlsson ML. Interactions between monoamines, glutamate, and GABA in schizophrenia: new evidence. Ann Rev Pharmacol Toxicol. 2001;41:237–60. doi: 10.1146/annurev.pharmtox.41.1.237. [DOI] [PubMed] [Google Scholar]
- 9.Breslin NA, Suddath RL, Bissette G, Nemeroff CB, Lowrimore P, Weinberger DR. CSF concentrations of neurotensin in schizophrenia: an investigation of clinical and biochemical correlates. Schizophr Res. 1994;12:35–41. doi: 10.1016/0920-9964(94)90082-5. [DOI] [PubMed] [Google Scholar]
- 10.Lindström LH, Widerlöv E, Bissette G, Nemeroff CB. Reduced CSF neurotensin concentration in drug-free schizophrenic patients. Schizophr Res. 1988;1:55–9. doi: 10.1016/0920-9964(88)90040-0. [DOI] [PubMed] [Google Scholar]
- 11.Nemeroff CB, Bissette G, Widerlov E, Beckmann H, Gerner R, Manberg PJ, et al. Neurotensin-like immunoreactivity in cerebrospinal fluid of patients with schizophrenia, depression, anorexia nervosa-bulimia, and premenstrual syndrome. J Neuropsychiatry Clin Neurosci. 1989;1:16–20. doi: 10.1176/jnp.1.1.16. [DOI] [PubMed] [Google Scholar]
- 12.Sharma RP, Janicak PG, Bissette G, Nemeroff CB. CSF neurotensin concentrations and antipsychotic treatment in schizophrenia and schizoaffective disorders. Am J Psychiatry. 1997;154:1019–21. doi: 10.1176/ajp.154.7.1019. [DOI] [PubMed] [Google Scholar]
- 13.Widerlöv E, Lindström LH, Besev G, Manberg PJ, Nemeroff CB, Breese GR, et al. Subnormal CSF levels of neurotensin in a subgroup of schizophrenic patients: normalization after neuroleptic treatment. Am J Psychiatry. 1982;139:1122–6. doi: 10.1176/ajp.139.9.1122. [DOI] [PubMed] [Google Scholar]
- 14.Garver DL, Bissette G, Yao JK, Nemeroff CB. Relation of CSF neurotensin concentrations to symptoms and drug response of psychotic patients. Am J Psychiatry. 1991;148:484–8. doi: 10.1176/ajp.148.4.484. [DOI] [PubMed] [Google Scholar]
- 15.Nemeroff CB. Neurotensin: perchance an endogenous neuroleptic. Biol Psychiatry. 1980;15:283–302. [PubMed] [Google Scholar]
- 16.Boules M, Li Z, Smith K, Fredrickson P, Richelson E. Diverse roles of neurotensin agonists in the central nervous system. Front Endocrinol (Lausanne) 2013;4:36. doi: 10.3389/fendo.2013.00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Binder EB, Kinkead B, Owens MJ, Nemeroff CB. Neurotensin and dopamine interactions. Pharmacol Rev. 2001;53:453–86. [PubMed] [Google Scholar]
- 18.Cusack B, Boules M, Tyler BM, Fauq A, McCormick DJ, Richelson E. Effects of a novel neurotensin peptide analog given extracranially on CNS behaviors mediated by apomorphine and haloperidol. Brain Res. 2000;856:48–54. doi: 10.1016/s0006-8993(99)02363-x. [DOI] [PubMed] [Google Scholar]
- 19.Costa FG, Frussa-Filho R, Felicio LF. The neurotensin receptor antagonist, SR48692, attenuates the expression of amphetamine-induced behavioural sensitisation in mice. Eur J Pharmacol. 2001;428:97–103. doi: 10.1016/s0014-2999(01)01271-7. [DOI] [PubMed] [Google Scholar]
- 20.Cáceda R, Binder EB, Kinkead B, Nemeroff CB. The role of endogenous neurotensin in psychostimulant-induced disruption of prepulse inhibition and locomotion. Schizophr Res. 2012;136:88–95. doi: 10.1016/j.schres.2011.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kinkead B, Dobner PR, Egnatashvili V, Murray T, Deitemeyer N, Nemeroff CB. Neurotensin-deficient mice have deficits in prepulse inhibition: restoration by clozapine but not haloperidol, olanzapine or quetiapine. J Pharmacol Exp Ther. 2005;315:256–64. doi: 10.1124/jpet.105.087437. [DOI] [PubMed] [Google Scholar]
- 22.Dobner PR, Fadel J, Deitmeyer N, Carraway RE, Deutch AY. Neurotensin-deficient mice show altered responses to antipsychotic drugs. Proc Natl Acad Sci U S A. 2001;98:8048–53. doi: 10.1073/pnas.141042198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fadel J, Dobner PR, Deutch AY. Amphetamine-elicited striatal Fos expression is attenuated in neurotensin null mutant mice. Neurosci Lett. 2006;402:97–101. doi: 10.1016/j.neulet.2006.03.043. [DOI] [PubMed] [Google Scholar]
- 24.Binder EB, Kinkead B, Owens MJ, Nemeroff CB. Neurotensin receptor antagonist SR 142948A alters Fos expression and extrapyramidal side effect profile of typical and atypical antipsychotic drugs. Neuropsychopharmacology. 2004;29:2200–7. doi: 10.1038/sj.npp.1300546. [DOI] [PubMed] [Google Scholar]
- 25.Fadel J, Dobner PR, Deutch AY. The neurotensin antagonist SR 48692 attenuates haloperidol-induced striatal Fos expression in the rat. Neurosci Lett. 2001;303:17–20. doi: 10.1016/s0304-3940(01)01708-6. [DOI] [PubMed] [Google Scholar]
- 26.Franklin KBJ, Paxinos G. The Mouse Brain in Stereotaxic Coordinates. San Diego: Academic Press; 1997. [Google Scholar]
- 27.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–75. [PubMed] [Google Scholar]
- 28.Alexander GE, DeLong MR, Strick PL. Parallel organization of functionally segregated circuits linking basal ganglia and cortex. Ann Rev Neurosci. 1986;9:357–81. doi: 10.1146/annurev.ne.09.030186.002041. [DOI] [PubMed] [Google Scholar]
- 29.Deutch AY. Prefrontal cortical dopamine systems and the elaboration of functional corticostriatal circuits: implications for schizophrenia and Parkinson’s disease. J Neural Trans. 1993;91:197–221. doi: 10.1007/BF01245232. [DOI] [PubMed] [Google Scholar]
- 30.Neve KA, Neve RL, Fidel S, Janowsky A, Higgins GA. Increased abundance of alternatively spliced forms of D2 dopamine receptor mRNA after denervation. Proc Natl Acad Sci USA. 1991;88:2802–6. doi: 10.1073/pnas.88.7.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shilling PD, Kelsoe JR, Segal DS. Dopamine transporter mRNA is up-regulated in the substantia nigra and the ventral tegmental area of amphetamine-sensitized rats. Neurosci Lett. 1997;236:131–4. doi: 10.1016/s0304-3940(97)00768-4. [DOI] [PubMed] [Google Scholar]
- 32.Belin D, Deroche-Gamonet V, Jaber M. Cocaine-induced sensitization is associated with altered dynamics of transcriptional responses of the dopamine transporter, tyrosine hydroxylase, and dopamine D2 receptors in C57Bl/6J mice. Psychopharmacology (Berl) 2007;193:567–78. doi: 10.1007/s00213-007-0790-3. [DOI] [PubMed] [Google Scholar]
- 33.Jones SR, Gainetdinov RR, Jaber M, Giros B, Wightman RM, Caron MG. Profound neuronal plasticity in response to inactivation of the dopamine transporter. Proc Natl Acad Sci USA. 1998;95:4029–34. doi: 10.1073/pnas.95.7.4029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fauchey V, Jaber M, Bloch B, Le Moine C. Dopamine control of striatal gene expression during development: relevance to knockout mice for the dopamine transporter. Eur J Neurosci. 2000;12:3415–25. doi: 10.1046/j.1460-9568.2000.00220.x. [DOI] [PubMed] [Google Scholar]
- 35.Gatzke-Kopp LM. The canary in the coalmine: the sensitivity of mesolimbic dopamine to environmental adversity during development. Neurosci Biobehav Rev. 2011;35:794–803. doi: 10.1016/j.neubiorev.2010.09.013. [DOI] [PubMed] [Google Scholar]
- 36.Lahti RA, Evans DL, Stratman NC, Figur LM. Dopamine D4 versus D2 receptor selectivity of dopamine receptor antagonists: possible therapeutic implications. Eur J Pharmacol. 1993;236:483–6. doi: 10.1016/0014-2999(93)90488-4. [DOI] [PubMed] [Google Scholar]
- 37.Thibault D, Albert PR, Pineyro G, Trudeau LE. Neurotensin triggers dopamine D2 receptor desensitization through a protein kinase C and beta-arrestin1-dependent mechanism. J Biol Chem. 2011;286:9174–84. doi: 10.1074/jbc.M110.166454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fuxe K, O’Connor WT, Antonelli T, Osborne PG, Tanganelli S, Agnati LF, et al. Evidence for a substrate of neuronal plasticity based on pre- and postsynaptic neurotensin-dopamine receptor interactions in the neostriatum. Proc Natl Acad Sci U S A. 1992;89:5591–5. doi: 10.1073/pnas.89.12.5591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mack KJ, Todd RD, O’Malley KL. The mouse dopamine D2A receptor gene: sequence homology with the rat and human genes and expression of alternative transcripts. J Neurochem. 1991;57:795–801. doi: 10.1111/j.1471-4159.1991.tb08221.x. [DOI] [PubMed] [Google Scholar]
- 40.Liang Y, Boules M, Li Z, Williams K, Miura T, Oliveros A, et al. Hyperactivity of the dopaminergic system in NTS1 and NTS2 null mice. Neuropharmacology. 2010;58:1199–205. doi: 10.1016/j.neuropharm.2010.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jomphe C, Lemelin PL, Okano H, Kobayashi K, Trudeau LE. Bidirectional regulation of dopamine D2 and neurotensin NTS1 receptors in dopamine neurons. Eur J Neurosci. 2006;24:2789–800. doi: 10.1111/j.1460-9568.2006.05151.x. [DOI] [PubMed] [Google Scholar]
- 42.Trudeau LE. Neurotensin regulates intracellular calcium in ventral tegmental area astrocytes: evidence for the involvement of multiple receptors. Neuroscience. 2000;97:293–302. doi: 10.1016/s0306-4522(99)00597-7. [DOI] [PubMed] [Google Scholar]
- 43.Mandell AJ, Owens MJ, Selz KA, Morgan WN, Shlesinger MF, Nemeroff CB. Mode matches in hydrophobic free energy eigenfunctions predict peptide–protein interactions. Biopolymers. 1998;46:89–101. doi: 10.1002/(SICI)1097-0282(199808)46:2<89::AID-BIP4>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 44.Smith TS, Trimmer PA, Khan SM, Tinklepaugh DL, Bennett JP., Jr Mitochondrial toxins in models of neurodegenerative diseases. II: Elevated zif268 transcription and independent temporal regulation of striatal D1 and D2 receptor mRNAs and D1 and D2 receptor-binding sites in C57BL/6 mice during MPTP treatment. Brain Res. 1997;765:189–97. doi: 10.1016/s0006-8993(97)00430-7. [DOI] [PubMed] [Google Scholar]
- 45.Fauchey V, Jaber M, Caron MG, Bloch B, Le Moine C. Differential regulation of the dopamine D1, D2 and D3 receptor gene expression and changes in the phenotype of the striatal neurons in mice lacking the dopamine transporter. Eur J Neurosci. 2000;12:19–26. doi: 10.1046/j.1460-9568.2000.00876.x. [DOI] [PubMed] [Google Scholar]
- 46.Frau R, Pillolla G, Bini V, Tambaro S, Devoto P, Bortolato M. Inhibition of 5alpha-reductase attenuates behavioral effects of D(1)-, but not D(2)-like receptor agonists in C57BL/6 mice. Psychoneuroendocrinology. 2012;38:542–51. doi: 10.1016/j.psyneuen.2012.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Poncelet M, Souilhac J, Gueudet C, Terranova JP, Gully D, Le Fur G, et al. Effects of SR 48692, a selective non-peptide neurotensin receptor antagonist, on two dopamine-dependent behavioural responses in mice and rats. Psychopharmacology (Berl) 1994;116:237–41. doi: 10.1007/BF02245067. [DOI] [PubMed] [Google Scholar]
- 48.Halberda JP, Middaugh LD, Gard BE, Jackson BP. DAD1- and DAD2-like agonist effects on motor activity of C57 mice: differences compared to rats. Synapse. 1997;26:81–92. doi: 10.1002/(SICI)1098-2396(199705)26:1<81::AID-SYN9>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 49.Shi WS, Bunney BS. Neurotensin attenuates dopamine D2 agonist quinpirole-induced inhibition of midbrain dopamine neurons. Neuropharmacology. 1990;29:1095–7. doi: 10.1016/0028-3908(90)90119-c. [DOI] [PubMed] [Google Scholar]
- 50.Shi WX, Bunney BS. Neurotensin modulates autoreceptor mediated dopamine effects on midbrain dopamine cell activity. Brain Res. 1991;543:315–21. doi: 10.1016/0006-8993(91)90043-u. [DOI] [PubMed] [Google Scholar]
- 51.Ralph-Williams RJ, Lehmann-Masten V, Geyer MA. Dopamine D1 rather than D2 receptor agonists disrupt prepulse inhibition of startle in mice. Neuropsychopharmacology. 2003;28:108–18. doi: 10.1038/sj.npp.1300017. [DOI] [PubMed] [Google Scholar]
- 52.Hirvonen J, van Erp TG, Huttunen J, Aalto S, Nagren K, Huttunen M, et al. Increased caudate dopamine D2 receptor availability as a genetic marker for schizophrenia. Arch Gen Psychiatry. 2005;62:371–8. doi: 10.1001/archpsyc.62.4.371. [DOI] [PubMed] [Google Scholar]
- 53.Nordstrom AL, Farde L, Eriksson L, Halldin C. No elevated D2 dopamine receptors in neuroleptic-naive schizophrenic patients revealed by positron emission tomography and [11C]N-methylspiperone. Psychiatry Res. 1995;61:67–83. doi: 10.1016/0925-4927(95)02732-d. [DOI] [PubMed] [Google Scholar]
- 54.Farde L, Wiesel FA, Stone-Elander S, Halldin C, Nordstrom AL, Hall H, et al. D2 dopamine receptors in neuroleptic-naive schizophrenic patients. A positron emission tomography study with [11C]raclopride. Arch Gen Psychiatry. 1990;47:213–9. doi: 10.1001/archpsyc.1990.01810150013003. [DOI] [PubMed] [Google Scholar]
- 55.Burt DR, Creese I, Snyder SH. Antischizophrenic drugs: chronic treatment elevates dopamine receptor binding in brain. Science. 1977;196:326–8. doi: 10.1126/science.847477. [DOI] [PubMed] [Google Scholar]
- 56.Seeman P, Schwarz J, Chen JF, Szechtman H, Perreault M, McKnight GS, et al. Psychosis pathways converge via D2high dopamine receptors. Synapse. 2006;60:319–46. doi: 10.1002/syn.20303. [DOI] [PubMed] [Google Scholar]
- 57.Lindenmayer JP, Nasrallah H, Pucci M, James S, Citrome L. A systematic review of psychostimulant treatment of negative symptoms of schizophrenia: challenges and therapeutic opportunities. Schizophr Res. 2013;147:241–52. doi: 10.1016/j.schres.2013.03.019. [DOI] [PubMed] [Google Scholar]
- 58.Sanfilipo M, Wolkin A, Angrist B, van Kammen DP, Duncan E, Wieland S, et al. Amphetamine and negative symptoms of schizophrenia. Psychopharmacology (Berl) 1996;123:211–4. doi: 10.1007/BF02246180. [DOI] [PubMed] [Google Scholar]
- 59.Seeman P. All roads to schizophrenia lead to dopamine supersensitivity and elevated dopamine D2(high) receptors. CNS Neurosci Ther. 2011;17:118–32. doi: 10.1111/j.1755-5949.2010.00162.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Foussias G, Remington G. Negative symptoms in schizophrenia: avolition and Occam’s razor. Schizophr Bull. 2010;36:359–69. doi: 10.1093/schbul/sbn094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hanson E, Healey K, Wolf D, Kohler C. Assessment of pharmacotherapy for negative symptoms of schizophrenia. Curr Psychiatry Rep. 2010;12:563–71. doi: 10.1007/s11920-010-0148-0. [DOI] [PubMed] [Google Scholar]