

Abstract
Background
Identification of the causative genes of retinitis pigmentosa (RP) is important for the clinical care of patients with RP. However, a comprehensive genetic study has not been performed in Korean RP patients. Moreover, the genetic heterogeneity found in sensorineural genetic disorders makes identification of pathogenic mutations challenging. Therefore, high throughput genetic testing using massively parallel sequencing is needed.
Results
Sixty-two Korean patients with nonsyndromic RP (46 patients from 18 families and 16 simplex cases) who consented to molecular genetic testing were recruited in this study and targeted exome sequencing was applied on 53 RP-related genes. Causal variants were characterised by selecting exonic and splicing variants, selecting variants with low allele frequency (below 1 %), and discarding the remaining variants with quality below 20. The variants were additionally confirmed by an inheritance pattern and cosegregation test of the families, and the rest of the variants were prioritised using in-silico prediction tools. Finally, causal variants were detected from 10 of 18 familial cases (55.5 %) and 7 of 16 simplex cases (43.7 %) in total. Novel variants were detected in 13 of 20 (65 %) candidate variants. Compound heterozygous variants were found in four of 7 simplex cases.
Conclusion
Panel-based targeted re-sequencing can be used as an effective molecular diagnostic tool for RP.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-1723-x) contains supplementary material, which is available to authorized users.
Keywords: Retinitis pigmentosa, Targeted re-sequencing, Genetic diagnosis, Familial case, Sporadic case
Background
Retinitis pigmentosa (RP) is a group of hereditary retinal diseases characterised by progressive loss of rod and cone photoreceptor cells. It is the most common hereditary retinal disease with a frequency of approximately 1 in 3000–4000 people, with an estimated total of 1.5 million people affected worldwide [1, 2]. Patients with RP usually present with a loss of night vision and complain of progressive constriction of the visual field. Eventually, central vision is affected and worsens. Clinical features such as age of onset and rate of progression vary greatly between individuals.
The identification of causative genetic variants is expected to be the starting point of RP treatment. Genetic counselling and gene therapy or other patient-specific treatment options can be suggested based on genetic background. In addition to treating the disease, molecular diagnosis is helpful for informing the differential diagnosis. Hereditary retinal dystrophies have a slowly progressive nature and complete clinical features may not be present at the time of examination. Therefore, many other hereditary retinal dystrophies may be confused with RP at certain phases of disease progression.
However, the genetic heterogeneity of RP is a hurdle for the easy application of molecular diagnosis. At the time of September 2011, 53 genes and 7 mapped loci are known to be associated with nonsyndromic RP [3] (RetNet). These genes show considerable phenotypic variability and multiple inherited retinal disease shares certain causative genes with RP [4]. Because of this phenotypic heterogeneity and confusion between genotypic and phenotypic correlations, the clinical features of individual patients provide few clues to narrow down the list of candidate genes [2, 5]. Therefore, these genes and loci should be screened simultaneously.
Massively parallel sequencing, also called next-generation sequencing (NGS) is a recent technical breakthrough in genetics [6, 7]. NGS-based high-throughput sequencing even makes whole-genome sequencing available in a single facility, which would have required tremendous cost and time in the past. RP can be a good target for massively parallel sequencing as an appropriate number of candidate genes are known for RP. Therefore, this study attempts to detect candidate variants in RP by applying a targeted enrichment and re-sequencing strategy.
Results
Quality validation of sequencing
A total of 217.5 kbp of targeting and flanking regions from 53 RP-related genes and 748 RP-related exons were captured; 97.1 % of the 2.89 M reads obtained on average per individual were properly paired and mapped (Additional file 1: Table S1). The average read depth was 146.2 ± 26.2, and 98.6 % and 96.5 % of the captured target exons showed more than 1X and 10X coverage, respectively (Additional file 1: Table S1).
Detection of variants in familial RP
Strong candidate variants of RP were identified in 10 families (Table 1). Among 18 families, 10 had an autosomal dominant, five had an autosomal recessive and three had an X-linked inheritance pattern. Briefly, five known pathogenic variants (c.421-1G > A in PRPF31, p.P347L in RHO, p.A153V in KLHL7, p.Y178C in RHO, p.R354X in PRPF31 were detected [8–13]. The known variant, pC3416G in USH2A, was initially detected, but the other variant, p.G4647R in the XF4 family under the inheritance pattern of autosomal recessive (compound heterozygote), was predicted as non-pathogenic. Therefore, the XF4 family was excluded. The other five candidate variants in RP1, RP2 and TOPORS have not been reported in dbSNP and were co-segregated in the family members.
Table 1.
Strong candidate variants in familiar cases
| Family | Inheritance | Gene | Genotype | Chr | Exon | nucleotide | amino acid | Mutation Type | Reference | EVS | In-house | Class |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| F03 | AD | RP1 | Hetero | 8 | 4 | c.1455 T > G | p.Y485X | nonsense | novel | - | - | II |
| F04 | X-L | RP2 | Hemi | X | 2 | c.340 T > C | p.C114R | nonsynonymous | novel | - | - | II |
| F06 | AD | RP1 | Hetero | 8 | 4 | c.2296C > T | p.Q766X | nonsense | novel | - | - | II |
| F07 | AD | PRPF31 | Hetero | 19 | 6 | c.421-1G > A | splicing | Xia et al. [11] | - | - | I | |
| F09 | AD | RHO | Hetero | 3 | 5 | c.1040C > T | p.P347L | nonsynonymous | rs29001566 | - | - | I |
| F10 | AD | KLHL7 | Hetero | 7 | 5 | c.458C > T | p.A153V | nonsynonymous | rs137853113 | - | - | I |
| F12 | X-L | RP2 | Hemi | X | 2 | c.560_561delGC | p.Ser187fs | Frameshift deletion | novel | - | - | II |
| F13 | AD | RHO | Hetero | 3 | 3 | c.533A > G | p.Y178C | nonsynonymous | rs104893776 | - | - | I |
| XF1 | AD | TOPORS | Hetero | 9 | 2 | c.2344C > T | p.R782X | nonsense | novel | - | 0.0052083 | II |
| XF3 | AD | PRPF31 | Hetero | 19 | 10 | c.1060C > T | p.R354X | nonsense | Sullivan et al. [12] | - | - | I |
Chr: chromosome; Homo: homozygous; Hetero: heterozygous; Hemi: hemizygous; AD: Autosomal Dominant; X-L X-linked
In-house: Korean normal reference consisting of 192 exomes
The 10 variants were comprised of four nonsynonymous variants, four nonsense mutations, one frameshift deletion, and one splicing variant. All the nonsense mutations (p.Y485X in RP1, p.Q766X in RP1, p.R782X in TOPORS, and p.R354X in PRPF31) were located at the upstream exons (exon 4 of 4, exon 3 of 4, exon 2 of 3 and exon 10 of 16 respectively), which might lead to nonsense-mediated decay of mRNA. A frameshift variant (p. Ser187fs in RP2) was also located at the 5′ upstream exon, and all the nonsense and frameshift variants were classified as possible pathogenic variants (category II).
The clinical features were relatively severe in patients having X-linked variants, F04 (p.C114R in RP2) and F12 (p.Ser187fs in RP2) (Additional file 1: Table S2). The affected family members experienced early onset of symptoms and presented with rapid progression. In the F04 family, the proband (VI-1) was a 6-year-old male whose visual acuity was 20/200 (OD) and 20/1000 (OS). The visual field was mildly constricted using a Goldmann perimetry V4 target, and the rod responses were extinguished and photopic responses were severely decreased on the electroretinogram. The photoreceptor inner segment and outer segment junction was not detectable using OCT (optical coherence tomography). The patient’s grandfather (II-1) had no light perception in both eyes. Fundus examination and OCT scan showed a severely degenerative retina. For family F12, a possible pathogenic missense variant was found in the X-linked RP2 gene. Hemizygous male patients (II-5, II-16 and III-2) showed poor visual acuity at less than counting fingers. The retina was totally degenerated such that the retina and choroid were not distinguishable on OCT. Scoptopic and photopic electroretinogram (ERG) were all extinguished. Female patients of the F12 family (II-8, II-13, III-11 and III-20) showed variable penetrance. Three of them (II-13, III-11 and III-20) were confirmed to have heterozygous variants. Two female patients (III-11 and III-20) had mild peripheral pigmentary deposits and decreased rod and cone amplitude on electroretinography. Two other female patients (II-8 and II-13) showed complete expression of RP. They had visual acuity limited to light perception in both eyes, typical bony spicule pigment deposits, attenuated retinal vessels. Scoptopic and photopic ERG amplitude were also abolished.
F06 (p.Q766X in RP1) showed a milder phenotype. In family F06, all patients experienced their first symptoms in their 30s. Two patients older than age 50 (II-3, 75 years old; III-2, 54 years old) had visual loss, while the other patients (III-6, III-10 and III-13) had normal vision. Patient III-6 and III-10 showed a normal macula on OCT and a mildly constricted visual field. The clinical features of the patients and pedigrees are summarised in Additional file 1: Table S2 and Figure S1. Fundus photograph and OCT are presented in Additional file 1: Figure S2.
Detection of variants in simplex RP
The possible pathogenic variants were also identified in 7 of 16 patients with simplex RP (43.7 %) (Table 2). These patients denied a familial history of RP or related hereditary disorders. The inheritance pattern of candidate genes was based on the sequencing results (Table 2). Initially four known pathogenic variants, p.H278Y in PDE6B, p.Y2935X and p.G2186E in EYS, and p.C3416G in USH2A, were detected. [13–16]. However a possible second variant in EYS (patient 435) and USH2A (patient 450) was not found, p.Y2935X (EYS) and p.C3416G (USH2A) were not included in final results. Eight causal candidate variants were newly identified. Overall, ten possible pathogenic variants were detected. They were comprised of six nonsynonymous, three nonsense, and one frameshift insertion variants. Compound heterozygous variants (in trans) were detected in five patients (PDE6B in 436 and 445, EYS in 439 and 440, USH2A in 438). The same frameshift variant of EYS (p.S1653fs) was identified in two unrelated patients with simplex RP (439 and 440).
Table 2.
Strong candidate variants in sporadic cases
| No | Inheritance* | Gene | Genotype | Chr | Exon | nucleotide | amino acid | Mutation Type | Reference | EVS | In-house | Class |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 430 | AD | PRPF31 | Hetero | 19 | 1 | c.310G > A | p.E104K | nonsynonymous | Novel | - | - | II |
| 432 | AD | PRPH2 | Hetero | 6 | 1 | c.380A > G | p.E127G | nonsynonymous | Novel | - | - | II |
| 436 | AR | PDE6B | Compound hetero | 4 | 8 | c.832C > T | p.H278Y | nonsynonymous | rs121918581 | - | - | Ι |
| 1 | c.32G > A | p.W11X | nonsense | Novel | - | - | II | |||||
| 438 | AR | USH2A | Compound hetero | 1 | 42 | c.8885 T > G | p.L2962R | nonsynonymous | Novel | - | - | II |
| 18 | c.4460G > A | p.W1487X | nonsense | Novel | - | - | II | |||||
| 439 | AR | EYS | Compound hetero | 6 | 8 | c.1750G > T | p.E584X | nonsense | Novel | - | - | II |
| 26 | c.4958_4959insA | p.S1653fs | frameshift insertion | Novel | - | - | II | |||||
| 440 | AR | EYS | Compound hetero | 6 | 29 | c.6557G > A | p.G2186E | nonsynonymous | 10 Littink | - | - | Ι |
| 26 | c.4958_4959insA | p.S1653fs | frameshift insertion | Novel | - | - | II | |||||
| 445 | AR | PDE6B | Compound hetero | 4 | 8 | c.832C > T | p.H278Y | nonsynonymous | rs121918581 | - | - | I |
| 8 | c.767 T > A | p.I256N | nonsynonymous | Novel | - | - | II |
*Inheritance is not inferred from pedigrees of the patients. These patients stated that there is no affected individual in their family tree other than indexed patients. Inheritance pattern described the results suggested by sequencing data
In-house: Korean normal reference consisting of 192 exomes
Chr chromosome; AD Autosomal dominant; AR Autosomal recessive; X-L X-linked
Regarding the clinical feature, patient 430 with the PRPF31 mutation (p.E104K) showed a mild clinical phenotype. Although he was 42 years old, his visual acuity was 20/20 in both eyes, but a paracentral scotoma was detected in the visual field test. The electroretinogram showed a small decrease only in rod response. Most of the photoreceptor inner segment and outer segment junction (5 mm in the horizontal scan) was observed intact using an OCT macula scan (Additional file 1: Table S3). Fundus photograph and OCT are presented in Additional file 1: Figure S2.
Discussion
In this study, targeted exome capture and massively parallel sequencing (MPS) was implemented for genetic diagnosis of RP. Strong candidate variants were identified in 10 (55.5 %) of 18 families and 7 (43.7 %) of 16 sporadic patients. The overall detection rate was 50 %. This method seems to be highly efficient and cost effective in comparison with PCR amplicon sequencing and other conventional methods such as microarray analysis. It is certainly much higher than the 7–16 % detection rate of microarray genotyping [17–21]. A previous genetic analysis of Korean RP patients using microarrays revealed a causative mutation in 26 out of 336 patients (2 %) [19]. The subjects of this microarray study were different from the current study. Patients from the F09 and F13 families had also been screened for detection of the RHO mutation by using direct sequencing in a previous study [22]. The current study analysed 53 target genes and showed exactly the same results as the direct sequencing of the RHO gene for these particular individuals, thereby supporting the accuracy and efficiency of the current strategy. The current results are also comparable with other RP genetic analysis using targeted resequencing, in which the detection rate ranged from 36 to 82 % [23–28]. Unbiased sequencing, including whole-genome or exome sequencing, can provide comprehensive genetic data that reveal novel causal genes of RP such as NEK2, HK1 and MVK. However the sequencing and data processing burden is still the bottleneck for widespread use of unbiased sequencing. Moreover, detection rates using whole exome sequencing have not proven to be significantly better than targeted resequencing as previously expected [29]. This also supports the usefulness of targeted exome sequencing in RP genetic screening.
Twenty possible causal variants were detected from 10 genes. Of these, 13 variants (65 %) were novel and this proportion is comparable with recent works reporting novel allele rates of 62–68 % [25–27, 29]. The considerable number of novel variants further emphasises the use of MPS as a reasonable tool for RP molecular diagnosis. The PRPF31 mutation (17.6 %) is most frequently found in current study, followed by mutations in EYS, PDE6B, RHO, RP1, and RP2 (11.8 % respectively). PRPF31 (30 %), RHO (20 %), and RP1 (20 %) were frequently affected genes in autosomal dominant RP (adRP), whereas EYS (40 %) and PDE6B (40 %) were frequently affected genes for autosomal recessive RP (arRP). Despite the limitation of extending this result to the general distribution of RP mutations in Koreans, frequently affected genes are similar to those reported in recent works as well as previous reports by Hartong et al. [2, 23, 27, 29]. Possible causal genes responsible for simplex cases had primarily autosomal recessive inheritance (5 out of 7 cases, 71 %) as suggested previously [1, 2]. It could not be confirmed whether causal alleles were de novo because segregation analysis was not able to be performed in simplex cases.
In X-linked RP, the RPGR gene is thought to be the most common causal gene, accounting for over 70 % of cases [30]. Although the RPGR gene was included in the capture library of the present study, the candidate causal variant could not be found. ORF15 is a mutation hotspot of RPGR, where roughly two thirds of disease-causing mutations are found [30]. This repetitive purine-rich ORF15 region is rarely covered by next-generation sequencing. Therefore, the RPGR mutation could have been missed and, instead, only the RP2 variants detected. A missense variant (p.C114R) in family F04 and a truncating variant (p.S187Tfs*31) in family F12 was identified. The missense variant (p.C114R) was located in the conserved tubulin binding cofactor C (TBCC) domain and was evolutionarily conserved. Interestingly, family F12 showed an autosomal dominant pattern without male to male transmission. Unlike the expected pattern of an X-linked disease affecting only males, recent studies have reported that RP2 and RPGR mutation cause variable RP phenotypes in heterozygous female patients.[30–32] Furthermore, it is reported that mutations in RP2 and RPGR account for 8.5 % of patients with RP in provisional autosomal dominant families [33].
PRPF31 variants were identified in two families and one simplex RP patient. One splicing site variation (c.421-1G > A in family F07) and truncating variant (p.R354X in family XF3) were known pathogenic variants [11, 12]. Haploinsufficiency from a null allele rather than gain of function is assumed to be the major mechanism of PRPF31 mutation-derived RP [34]. However, missense variants also have been suggested as possibly pathogenic in several reports [34–37]. A missense variant (p.E104K) was also detected in a simplex RP patient (430) in the current study. This substitution is located at the highly conserved NOSIK domain and this sequence is evolutionarily conserved.
An EYS variant was identified in two simplex patients. The same frameshift insertion variant (p.S1653Kfs*) was identified in two unrelated patients. As a second candidate allele, 439 had a premature truncating variant (p.E584X) and 440 had a missense variant (p.G2186E). Nonsense mediated decay from a heterozygous compound truncating mutation is assumed to be a possible pathogenic mechanism in 439. The missense variant (p.G2186E) in patient 440 was located in the highly conserved Laminin G domain. Interestingly, this mutation was previously reported in a patient with Korean ancestry [16]. A frameshift variant (p.S1653Kfs*) and missense variant (p.G2186E) were recurrent in a relatively small cohort and it is assumed that these could be founder variants in Koreans. Iwanami et al. suggested that patients having a homozygous or compound heterozygous truncating mutation of EYS show a more severe decline of visual function than patients having only one allele of the truncating variant [15]. However, both cases (439 and 440) did not show any difference in progression in the current study independent from number of truncating variants.
A known pathogenic missense variant (p.H278Y) of PDE6B was detected in two unrelated patients (436 and 445). A truncating variant (p.W11X) was found in patient 436 as a second variant and a novel missense variant (p.I256N) was detected in patient 445. This missense variant is located in the highly conserved GAF domain. Loss of function of cGMP phosphoodiesterase activity is a possible mechanism in these two patients.
Just one possible pathogenic allele was found in the XF4 family and simplex patients 433, 435 and 450. (Data not shown in Tables 1 and 2) The identified genes in these patients have an autosomal recessive inheritance pattern. Known pathogenic variants (p.C3416G of USH2A in XF4 and 450, p.Y2935X of EYS in 435) were found in just one allele. As a second pathogenic candadate, missense variants p.T2465S of EYS and p.V2228E of USH2A were detected and predicted to be pathogenic in patient 435 and 450, respectively. (Additional file 1: Table S4) These were relatively common variants (more than 1 %) in the genomic data of 1000 Asians. Considering that the counter allele, p.Y2935X and p.C3416G, is rare and pathogenic, these candidate variants may also be pathogenic alleles. Regarding other cases in the current study (XF4 and 433), a second pathogenic allele could not be identified. Meaningful second variants may not have been identified because they were large indels or located in an untranslated region. Alternatively, detected pathogenic variants might be accidental carriers and causal variants are located in other genes. Accidental carrier mutations are increasingly being reported as the use of multigenic screening with MPS increases [23, 27]. Although the current study revealed a possible causative variant in half of cases, obviously a candidate causal allele could not be found in the other half of patients. Since the target library was designed, three more causative genes have been added to the RP gene list. Unknown causal genes for RP can still exist and including these targets would increase the detection yield. However, whole exome sequencing or the use of an extended target library of over 100 genes has not revealed a higher detection rate [25, 29, 38]. Unbiased sequencing not only poses an enormous data processing burden, but also impedes deep sequencing. Covering the non-coding exons of target genes and complete coverage of the target may be better way to increase diagnostic yield rather than extending the target library. Eisenberg et al. reported high detection rates using high coverage, copy number variance (CNV) analysis and sequencing the 5′ UTR [27]. Xu et al. reported that complete coverage using deep sequencing and PCR amplicon for less covered regions yielded an 82 % detection rate [23]. Although a ≥ 10X sequencing depth was reached in 96 % of the sequenced region, 4 % had a less than 10X depth and 1.4 % was not covered (Additional file 1: Table S1). GC-rich regions or highly repetitive regions are usually under-captured by MPS. The mutational hotspot, ORF15 of RPGR, is one of those regions. Complete coverage assisted by direct sequencing and deep sequencing is needed to raise the causative variant detection yield.
Identifying a causal mutation is the starting point of RP treatment, followed by proper genetic counselling and prognostic data. For instance, patients can be informed that they have a certain mutation, which puts them at risk for rapid progression of RP, and aggressive therapy can be initiated on the basis of confirmative genetic data, although there are limited treatment options available to help RP patients as of yet. Gene therapy, one of the possible treatment options for RP, is largely dependent on genetic data, indicating that identifying the causal mutation will become an increasingly important step in RP treatment.
Conclusion
Using the targeted re-sequencing of known genes for RP, pathogenic and possibly pathogenic variants were identified in more than the half of patients and families. This strategy will become an increasingly efficient and cost-effective molecular diagnostic test for retinitis pigmentosa.
Methods
Patients
Sixty-two Korean patients with RP were selected for the RP cohort. This cohort consisted of 46 patients from 18 families and 16 patients with simplex RP. All the patients had nonsyndromic RP. Comprehensive ocular examinations, including visual acuity assessment, slit-lamp examination, fundus examination, electroretinograms, Goldmann Perimetry (Haag-Streit, Bern, Switzerland) and optical coherence tomography (Carl Zeiss Meditec Inc., Dublin, CA, USA) were used for clinical diagnosis. RP was diagnosed when the patients had the typical retinal appearance of RP and had attenuated or abolished rod and cone signal on electroretinography. Bony spicule-like pigment deposits, retinal vessel attenuation and optic disc pallor were included in the characterisation of typical retinal features of RP. It was assumed that the mode of inheritance was autosomal dominant when a patient appeared in every generation of the pedigree, and when affected fathers and mothers transmitted the phenotype to both sons and daughters. Autosomal recessive patterns were suspected when a phenotype appeared in the male and female progeny of unaffected parents. X-linked recessive inheritance was suspected when affected males passed the condition on to all of their daughters, but to none of their sons, and female patients married to unaffected males passed the condition on to half of their sons and daughters. Written informed consent was obtained from all patients before they were enrolled in the study. This study followed the tenets of the declaration of Helsinki (Edinburgh, 2000) and was approved by the Institutional Review Board of Seoul National University Hospital.
Targeted exome sequencing of RP-related genes
Customised baits were designed to capture all exons of the 53 genes known to be associated with RP at the time of panel design. (Table 3, Roche NimbleGen Inc, Madison, WI). Genomic DNA from 62 patients in total was extracted from peripheral blood as described previously [39] and was sequenced using the Genome Analyzer II. Sequencing reads were aligned to the UCSC hg19 reference genome using BWA-0.6.1 with default settings. Duplications were removed via Picard v1.93, and local realignment was done by GATK v2.4-7. Variants were identified by the Unified Genotyper from GATK for the SNVs and indels. ANNOVAR was used to annotate the variants. Coverage of TES data was calculated by the ‘Depth of Coverage’ module from GATK. Sanger sequencing for candidate variants was performed using specific primers for each exon as demonstrated.
Table 3.
53 RP-related genes selected for targeted resequencing
| Gene | Inheritance | RefSeq | Cytogenetic Loci | Exon Count | Description |
|---|---|---|---|---|---|
| ABCA4 | AR/AD | NM_000350 | 1p22.1 | 50 | ATP-binding cassette, sub-family A (ABC1), member 4 (ABCA4) |
| ARL6 | AR | NM_177976 | 3q11.2 | 9 | ADP-ribosylation factor-like 6 (ARL6), transcript variant 2 |
| BEST1 | AD | NM_001139443 | 11q12.3 | 9 | bestrophin 1 (BEST1), transcript variant 2 |
| C2ORF71 | AR | NM_001029883 | 2p23.2 | 2 | chromosome 2 open reading frame 71 (C2orf71) |
| CA4 | AD | NM_000717 | 17q23.1 | 8 | carbonic anhydrase IV (CA4) |
| CERKL | AR | NM_201548 | 2q31.3 | 13 | ceramide kinase-like (CERKL), transcript variant 1 |
| CLRN1 | AR | NM_001195794 | 3q25.1 | 4 | clarin 1 (CLRN1), transcript variant 5 |
| CNGA1 | AR | NM_001142564 | 4p12 | 10 | cyclic nucleotide-gated channel alpha 1 (CNGA1), transcript variant 1 |
| CNGB1 | AR | NM_001297 | 16q21 | 33 | Homo sapiens cyclic nucleotide-gated channel beta 1 (CNGB1), transcript variant 1 |
| CRB1 | AR | NM_201253 | 1q31.3 | 12 | crumbs homolog 1 (Drosophila) (CRB1), transcript variant 1 |
| CRX | AD | NM_000554 | 19q13.33 | 4 | cone-rod homeobox (CRX) |
| DHDDS | AR | NM_024887 | 1p36.11 | 9 | Homo sapiens dehydrodolichyl diphosphate synthase (DHDDS), transcript variant 2 |
| EYS | AR | NM_001142800 | 6q12 | 43 | eyes shut homolog (Drosophila) (EYS), transcript variant 1 |
| FAM161A | AR | NM_001201543 | 2p15 | 7 | family with sequence similarity 161, member A (FAM161A), transcript variant 1 |
| FSCN2 | AD | NM_001077182 | 17q25.3 | 5 | fascin homolog 2, actin-bundling protein, retinal (Strongylocentrotus purpuratus) (FSCN2), transcript variant 2 |
| GUCA1B | AD | NM_002098 | 6p21.1 | 4 | guanylate cyclase activator 1B (retina) (GUCA1B) |
| IDH3B | AR | NM_006899 | 20p13 | 12 | isocitrate dehydrogenase 3 (NAD+) beta (IDH3B), nuclear gene encoding mitochondrial protein, transcript variant 1 |
| IMPDH1 | AD | NM_000883 | 7q32.1 | 17 | IMP (inosine 5′-monophosphate) dehydrogenase 1 (IMPDH1), transcript variant 1 |
| IMPG2 | AR | NM_016247 | 3q12.3 | 19 | interphotoreceptor matrix proteoglycan 2 (IMPG2) |
| KLHL7 | AD | NM_001031710 | 7p15.3 | 11 | kelch-like family member 7 (KLHL7), transcript variant 1 |
| LRAT | AR | NM_004744 | 4q32.1 | 3 | lecithin retinol acyltransferase (phosphatidylcholine–retinol O-acyltransferase) (LRAT) |
| MERTK | AR | NM_006343 | 2q13 | 19 | c-mer proto-oncogene tyrosine kinase (MERTK) |
| NR2E3 | AR/AD | NM_016346 | 15q23 | 8 | nuclear receptor subfamily 2, group E, member 3 (NR2E3), transcript variant 1 |
| NRL | AD | NM_006177 | 14q11.2 | 3 | neural retina leucine zipper (NRL) |
| PDE6A | AR | NM_000440 | 5q32 | 22 | phosphodiesterase 6A, cGMP-specific, rod, alpha (PDE6A) |
| PDE6B | AR | NM_000283 | 4p16.3 | 22 | phosphodiesterase 6B, cGMP-specific, rod, beta (PDE6B), transcript variant 1 |
| PDE6G | AR | NM_002602 | 17q25.3 | 4 | phosphodiesterase 6G, cGMP-specific, rod, gamma (PDE6G), transcript variant 1 |
| PRCD | AR | NR_033357 | 17q25.1 | 5 | Homo sapiens progressive rod-cone degeneration (PRCD), transcript variant 2 |
| PROM1 | AR | NM_006017 | 4p15.32 | 27 | prominin 1 (PROM1), transcript variant 1 |
| PRPF3 | AD | NM_004698 | 1q21.2-q21.3 | 16 | PRP3 pre-mRNA processing factor 3 homolog (S. cerevisiae) (PRPF3) |
| PRPF31 | AD | NM_015629 | 19q13.42 | 14 | PRP31 pre-mRNA processing factor 31 homolog (S. cerevisiae) (PRPF31) |
| PRPF8 | AD | NM_006445 | 17p13.3 | 43 | PRP8 pre-mRNA processing factor 8 homolog (S. cerevisiae) (PRPF8) |
| PRPH2 | AD | NM_000322 | 6p21.1 | 3 | peripherin 2 (retinal degeneration, slow) (PRPH2) |
| PRPH2-ROM1 | digenic | NM_000327 | 11q12.3 | 3 | retinal outer segment membrane protein 1 (ROM1) |
| RBP3 | AR | NM_002900 | 10q11.22 | 4 | retinol binding protein 3, interstitial (RBP3) |
| RDH12 | AR | NM_152443 | 14q24.1 | 9 | Homo sapiens retinol dehydrogenase 12 (all-trans/9-cis/11-cis) (RDH12) |
| RGR | AR/AD | NM_002921 | 10q23.1 | 7 | G protein coupled receptor (RGR), transcript variant 1 |
| RHO | AR/AD | NM_000539 | 3q22.1 | 5 | rhodopsin (RHO) |
| RLBP1 | AR | NM_000326 | 15q26.1 | 9 | retinaldehyde-binding protein 1 (RLBP1) |
| RP1 | AR/AD | NM_006269 | 8q12.1 | 4 | retinitis pigmentosa 1 (autosomal dominant) (RP1) |
| RP2 | X-linked | NM_006915 | Xp11.23 | 5 | retinitis pigmentosa 2 (X-linked recessive) (RP2) |
| RP9 | AD | NM_203288 | 7p14.3 | 6 | retinitis pigmentosa 9 (autosomal dominant) (RP9) |
| RPE65 | AR | NM_000329 | 1p31.3-p31.2 | 14 | retinal pigment epithelium-specific protein 65 kDa (RPE65) |
| RPGR | X-linked | NM_001034853 | Xp11.4 | 15 | retinitis pigmentosa GTPase regulator (RPGR), transcript variant C |
| SAG | AR | NM_000541 | 2q37.1 | 16 | S-antigen; retina and pineal gland (arrestin) (SAG) |
| SEMA4A | AR/AD | NM_001193300 | 1q22 | 15 | sema domain, immunoglobulin domain (Ig), transmembrane domain (TM) and short cytoplasmic domain, (semaphorin) 4A (SEMA4A), transcript variant 2 |
| SNRNP200 | AD | NM_014014 | 2q11.2 | 45 | small nuclear ribonucleoprotein 200 kDa (U5) (SNRNP200) |
| SPATA7 | AR | NM_018418 | 14q31.3 | 12 | spermatogenesis-associated 7 (SPATA7), transcript variant 1 |
| TOPORS | AD | NM_005802 | 9p21.1 | 3 | topoisomerase I binding, arginine/serine-rich, E3 ubiquitin protein ligase (TOPORS), transcript variant 1 |
| TTC8 | AR | NM_144596 | 14q31.3 | 15 | tetratricopeptide repeat domain 8 (TTC8), transcript variant 1 |
| TULP1 | AR | NM_003322 | 6p21.31 | 15 | Tubby-like protein 1 (TULP1) |
| USH2 / USH2A | AR | NM_206933 | 1q41 | 72 | Usher syndrome 2A (autosomal recessive, mild) (USH2A), transcript variant 2 |
| ZNF513 | AR | NM_144631 | 2p23.3 | 4 | zinc finger protein 513 (ZNF513), transcript variant 1 |
Prioritization of variants
A flowchart for candidate causal variant detection is shown in Fig. 1. Exonic and splicing variants were first selected if they had an allele frequency below 1 % reported in the NHLBI-ESP 6500 (evs.gs.washington.edu), 1000 Genome Project (www.1000genomes.org), and an in-house database with exomes of 192 Korean individuals. Variants with a quality score below 20 were excluded. Candidate variants were confirmed by Sanger sequencing and co-segregation analysis was performed in cases of familial RP. The potential pathogenicity of variants was categorised into four classes (Table 4). Briefly, Class I included pathogenic variants previously known to cause RP, and Class II variants were expected to cause severe damage to protein structure via frameshift, nonsense, and missense variants, which were likely to cause severe functional change via Polyphen 2 [40], SIFT [41], and MutPred [42]. Class III included variants least likely to be causative and consisted of missense variations that were predicted to be benign or tolerable. All other types of variants were categorised as Class IV. Novel nonsynonymous variants were assumed to be possibly pathogenic if the variant was predicted to be pathogenic by at least two of the three methods. Both probably and possibly damaging mutations were classified as suspected pathogenic variants by Polyphen2. With regard to SIFT, damaging mutations were classified as pathogenic. For Mutpred, a general score higher than 0.5 was categorised as possibly pathogenic.
Fig. 1.
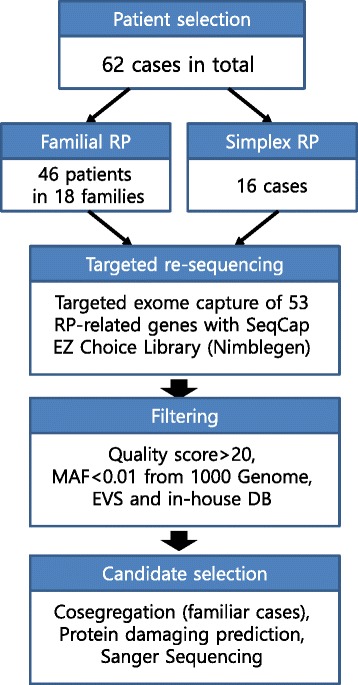
Schematic workflow of the diagnostic application of targeted exome sequencing in familial and simplex retinitis pigmentosa. 62 cases consisting of 46 patients in 18 families and 16 cases in simplex families were recruited, and targeted re-sequencing was performed for 53 RP-related genes. Candidate variants were identified by filtering based on variant quality > 20 and minor allele frequency < 0.01 from 1000 Genome Project (www.1000genomes.org), Exome Variant Server (evs.gs.washington.edu) and our in-house DB consisting of 192 Korean exomes. The variants were finally confirmed by a cosegregation test if familiar cases, in silico tools and Sanger sequencing
Table 4.
Classification of candidate variants in this study
| Category | Explanation |
|---|---|
| Class I | Previously reported pathogenic variants |
| Class II | Single Nucleotide Variants (SNVs) predicted to cause serious protein deformity by using in silico analysis |
| Stopgain, and frameshift mutations | |
| Class III | Mutations causing only protein change |
| Class IV | Other mutations |
Acknowledgments
This study was supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (HT12C001420014) and Samsung Medical Center grant.
Additional files
Quality of sequencing results. Table S2. Clinical features of 10 familial RP cases whose strong variants were detected by targeted resequencing. Table S3. Clinical features of 7 sporadic RP cases whose strong variants were detected by targeted resequencing. Table S4. In silico prediction for nonsysnonymous variants. Figure S1. Pedigrees of 10 familial cases whose strong variants were detected by targeted re-sequencing. Figure S2. Fundus photograph and optical coherence tomography of the interesting cases.
Footnotes
Chang-Ki Yoon and Nayoung K. D. Kim contributed equally to this work.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
CKY participated in sequence alignment and clinical assessment and drafted the manuscript. JYS and JHP participated in clinical analysis and helped to draft the manuscript. NKDK and JGJ participated in bioinformatics analysis of TES data and wrote the manuscript. HHE and HL participated in sequence alignment and Sanger sequencing. WYP and HGY conceived the study and participated in its design and coordination, as well as helping to draft the manuscript. All authors read and approved the final manuscript.
Contributor Information
Woong-Yang Park, Phone: +82-2-3410-6128, Email: woongyang@skku.edu.
Hyeong Gon Yu, Phone: +82 2 2072 2438, Email: hgonyu@snu.ac.kr.
References
- 1.Daiger SP, Bowne SJ, Sullivan LS. Perspective on genes and mutations causing retinitis pigmentosa. Arch Ophthalmol. 2007;125(2):151–8. doi: 10.1001/archopht.125.2.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368(9549):1795–809. doi: 10.1016/S0140-6736(06)69740-7. [DOI] [PubMed] [Google Scholar]
- 3.Retinal Information Network website. Available: https://sph.uth.edu/retnet/.
- 4.Berger W, Kloeckener-Gruissem B, Neidhardt J. The molecular basis of human retinal and vitreoretinal diseases. Prog Retin Eye Res. 2010;29(5):335–75. doi: 10.1016/j.preteyeres.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 5.Daiger SP, Sullivan LS, Bowne SJ. Genes and mutations causing retinitis pigmentosa. Clin Genet. 2013;84(2):132–41. doi: 10.1111/cge.12203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA, Shendure J. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet. 2011;12(11):745–55. doi: 10.1038/nrg3031. [DOI] [PubMed] [Google Scholar]
- 7.Metzker ML. Sequencing technologies - the next generation. Nat Rev Genet. 2010;11(1):31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 8.Friedman JS, Ray JW, Waseem N, Johnson K, Brooks MJ, Hugosson T, Breuer D, Branham KE, Krauth DS, Bowne SJ, et al. Mutations in a BTB-Kelch protein, KLHL7, cause autosomal-dominant retinitis pigmentosa. Am J Hum Genet. 2009;84(6):792–800. doi: 10.1016/j.ajhg.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dryja TP, McGee TL, Reichel E, Hahn LB, Cowley GS, Yandell DW, Sandberg MA, Berson EL. A point mutation of the rhodopsin gene in one form of retinitis pigmentosa. Nature. 1990;343(6256):364–6. doi: 10.1038/343364a0. [DOI] [PubMed] [Google Scholar]
- 10.Sung CH, Davenport CM, Hennessey JC, Maumenee IH, Jacobson SG, Heckenlively JR, Nowakowski R, Fishman G, Gouras P, Nathans J. Rhodopsin mutations in autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci U S A. 1991;88(15):6481–5. doi: 10.1073/pnas.88.15.6481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xia K, Zheng D, Pan Q, Liu Z, Xi X, Hu Z, Deng H, Liu X, Jiang D, Deng H, et al. A novel PRPF31 splice-site mutation in a Chinese family with autosomal dominant retinitis pigmentosa. Mol Vis. 2004;10:361–5. [PubMed] [Google Scholar]
- 12.Sullivan LS, Bowne SJ, Reeves MJ, Blain D, Goetz K, Ndifor V, Vitez S, Wang X, Tumminia SJ, Daiger SP. Prevalence of mutations in eyeGENE probands with a diagnosis of autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2013;54(9):6255–61. doi: 10.1167/iovs.13-12605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang XF, Xiang P, Chen J, Xing DJ, Huang N, Min Q, Gu F, Tong Y, Pang CP, Qu J, et al. Targeted exome sequencing identified novel USH2A mutations in usher syndrome families. PLoS One. 2013;8(5):e63832. doi: 10.1371/journal.pone.0063832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McLaughlin ME, Sandberg MA, Berson EL, Dryja TP. Recessive mutations in the gene encoding the beta-subunit of rod phosphodiesterase in patients with retinitis pigmentosa. Nat Genet. 1993;4(2):130–4. doi: 10.1038/ng0693-130. [DOI] [PubMed] [Google Scholar]
- 15.Iwanami M, Oshikawa M, Nishida T, Nakadomari S, Kato S. High prevalence of mutations in the EYS gene in Japanese patients with autosomal recessive retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2012;53(2):1033–40. doi: 10.1167/iovs.11-9048. [DOI] [PubMed] [Google Scholar]
- 16.Littink KW, van den Born LI, Koenekoop RK, Collin RW, Zonneveld MN, Blokland EA, Khan H, Theelen T, Hoyng CB, Cremers FP, et al. Mutations in the EYS gene account for approximately 5 % of autosomal recessive retinitis pigmentosa and cause a fairly homogeneous phenotype. Ophthalmology. 2010;117(10):2026–33. doi: 10.1016/j.ophtha.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 17.Blanco-Kelly F, Garcia-Hoyos M, Corton M, Avila-Fernandez A, Riveiro-Alvarez R, Gimenez A, Hernan I, Carballo M, Ayuso C. Genotyping microarray: mutation screening in Spanish families with autosomal dominant retinitis pigmentosa. Mol Vis. 2012;18:1478–83. [PMC free article] [PubMed] [Google Scholar]
- 18.Jaijo T, Aller E, Garcia-Garcia G, Aparisi MJ, Bernal S, Avila-Fernandez A, Barragan I, Baiget M, Ayuso C, Antinolo G, et al. Microarray-based mutation analysis of 183 Spanish families with Usher syndrome. Invest Ophthalmol Vis Sci. 2010;51(3):1311–7. doi: 10.1167/iovs.09-4085. [DOI] [PubMed] [Google Scholar]
- 19.Kim C, Chung H, Yu HG. Association of p.P347L in the rhodopsin gene with early-onset cystoid macular edema in patients with retinitis pigmentosa. Ophthalmic Genet. 2012;33(2):96–9. doi: 10.3109/13816810.2011.642453. [DOI] [PubMed] [Google Scholar]
- 20.Avila-Fernandez A, Cantalapiedra D, Aller E, Vallespin E, Aguirre-Lamban J, Blanco-Kelly F, Corton M, Riveiro-Alvarez R, Allikmets R, Trujillo-Tiebas MJ, et al. Mutation analysis of 272 Spanish families affected by autosomal recessive retinitis pigmentosa using a genotyping microarray. Mol Vis. 2010;16:2550–8. [PMC free article] [PubMed] [Google Scholar]
- 21.Gonzalez-del Pozo M, Borrego S, Barragan I, Pieras JI, Santoyo J, Matamala N, Naranjo B, Dopazo J, Antinolo G. Mutation screening of multiple genes in Spanish patients with autosomal recessive retinitis pigmentosa by targeted resequencing. PLoS One. 2011;6(12):e27894. doi: 10.1371/journal.pone.0027894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim KJ, Kim C, Bok J, Kim KS, Lee EJ, Park SP, Chung H, Han BG, Kim HL, Kimm K, et al. Spectrum of rhodopsin mutations in Korean patients with retinitis pigmentosa. Mol Vis. 2011;17:844–53. [PMC free article] [PubMed] [Google Scholar]
- 23.Wang J, Zhang VW, Feng Y, Tian X, Li FY, Truong C, Wang G, Chiang PW, Lewis RA, Wong LJ. Dependable and efficient clinical utility of target capture based deep sequencing in molecular diagnosis of retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2014;55(10):6213–23. doi: 10.1167/iovs.14-14936. [DOI] [PubMed] [Google Scholar]
- 24.Glockle N, Kohl S, Mohr J, Scheurenbrand T, Sprecher A, Weisschuh N, Bernd A, Rudolph G, Schubach M, Poloschek C, et al. Panel-based next generation sequencing as a reliable and efficient technique to detect mutations in unselected patients with retinal dystrophies. Eur J Hum Genet. 2014;22(1):99–104. doi: 10.1038/ejhg.2013.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang F, Wang H, Tuan HF, Nguyen DH, Sun V, Keser V, Bowne SJ, Sullivan LS, Luo H, Zhao L, et al. Next generation sequencing-based molecular diagnosis of retinitis pigmentosa: identification of a novel genotype-phenotype correlation and clinical refinements. Hum Genet. 2014;133(3):331–45. doi: 10.1007/s00439-013-1381-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fu Q, Wang F, Wang H, Xu F, Zaneveld JE, Ren H, Keser V, Lopez I, Tuan HF, Salvo JS, et al. Next-generation sequencing-based molecular diagnosis of a chinese patient cohort with autosomal recessive retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2013;54(6):4158–66. doi: 10.1167/iovs.13-11672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eisenberger T, Neuhaus C, Khan AO, Decker C, Preising MN, Friedburg C, Bieg A, Gliem M, Charbel Issa P, Holz FG, et al. Increasing the yield in targeted next-generation sequencing by implicating CNV analysis, non-coding exons and the overall variant load: the example of retinal dystrophies. PLoS One. 2013;8(11):e78496. doi: 10.1371/journal.pone.0078496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Neveling K, Collin RW, Gilissen C, van Huet RA, Visser L, Kwint MP, Gijsen SJ, Zonneveld MN, Wieskamp N, de Ligt J, et al. Next-generation genetic testing for retinitis pigmentosa. Hum Mutat. 2012;33(6):963–72. doi: 10.1002/humu.22045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu Y, Guan L, Shen T, Zhang J, Xiao X, Jiang H, Li S, Yang J, Jia X, Yin Y, et al. Mutations of 60 known causative genes in 157 families with retinitis pigmentosa based on exome sequencing. Hum Genet. 2014;133(10):1255–71. doi: 10.1007/s00439-014-1460-2. [DOI] [PubMed] [Google Scholar]
- 30.Branham K, Othman M, Brumm M, Karoukis AJ, Atmaca-Sonmez P, Yashar BM, Schwartz SB, Stover NB, Trzupek K, Wheaton D, et al. Mutations in RPGR and RP2 account for 15 % of males with simplex retinal degenerative disease. Invest Ophthalmol Vis Sci. 2012;53(13):8232–7. doi: 10.1167/iovs.12-11025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Souied E, Segues B, Ghazi I, Rozet JM, Chatelin S, Gerber S, Perrault I, Michel-Awad A, Briard ML, Plessis G, et al. Severe manifestations in carrier females in X linked retinitis pigmentosa. J Med Genet. 1997;34(10):793–7. doi: 10.1136/jmg.34.10.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pomares E, Riera M, Castro-Navarro J, Andres-Gutierrez A, Gonzalez-Duarte R, Marfany G. Identification of an intronic single-point mutation in RP2 as the cause of semidominant X-linked retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2009;50(11):5107–14. doi: 10.1167/iovs.08-3208. [DOI] [PubMed] [Google Scholar]
- 33.Churchill JD, Bowne SJ, Sullivan LS, Lewis RA, Wheaton DK, Birch DG, Branham KE, Heckenlively JR, Daiger SP. Mutations in the X-linked retinitis pigmentosa genes RPGR and RP2 found in 8.5 % of families with a provisional diagnosis of autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2013;54(2):1411–6. doi: 10.1167/iovs.12-11541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rio Frio T, Wade NM, Ransijn A, Berson EL, Beckmann JS, Rivolta C. Premature termination codons in PRPF31 cause retinitis pigmentosa via haploinsufficiency due to nonsense-mediated mRNA decay. J Clin Invest. 2008;118(4):1519–31. doi: 10.1172/JCI34211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yuan L, Kawada M, Havlioglu N, Tang H, Wu JY. Mutations in PRPF31 inhibit pre-mRNA splicing of rhodopsin gene and cause apoptosis of retinal cells. J Neurosci. 2005;25(3):748–57. doi: 10.1523/JNEUROSCI.2399-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sullivan LS, Bowne SJ, Birch DG, Hughbanks-Wheaton D, Heckenlively JR, Lewis RA, Garcia CA, Ruiz RS, Blanton SH, Northrup H, et al. Prevalence of disease-causing mutations in families with autosomal dominant retinitis pigmentosa: a screen of known genes in 200 families. Invest Ophthalmol Vis Sci. 2006;47(7):3052–64. doi: 10.1167/iovs.05-1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chakarova CF, Hims MM, Bolz H, Abu-Safieh L, Patel RJ, Papaioannou MG, Inglehearn CF, Keen TJ, Willis C, Moore AT, et al. Mutations in HPRP3, a third member of pre-mRNA splicing factor genes, implicated in autosomal dominant retinitis pigmentosa. Hum Mol Genet. 2002;11(1):87–92. doi: 10.1093/hmg/11.1.87. [DOI] [PubMed] [Google Scholar]
- 38.Wang X, Wang H, Sun V, Tuan HF, Keser V, Wang K, Ren H, Lopez I, Zaneveld JE, Siddiqui S, et al. Comprehensive molecular diagnosis of 179 Leber congenital amaurosis and juvenile retinitis pigmentosa patients by targeted next generation sequencing. J Med Genet. 2013;50(10):674–88. doi: 10.1136/jmedgenet-2013-101558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Min BJ, Kim N, Chung T, Kim OH, Nishimura G, Chung CY, Song HR, Kim HW, Lee HR, Kim J, et al. Whole-exome sequencing identifies mutations of KIF22 in spondyloepimetaphyseal dysplasia with joint laxity, leptodactylic type. Am J Hum Genet. 2011;89(6):760–6. doi: 10.1016/j.ajhg.2011.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–9. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–81. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 42.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome Project Data Processing S The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]