

Abstract
Background
In phylogenetic analysis we face the problem that several subclade topologies are known or easily inferred and well supported by bootstrap analysis, but basal branching patterns cannot be unambiguously estimated by the usual methods (maximum parsimony (MP), neighbor-joining (NJ), or maximum likelihood (ML)), nor are they well supported. We represent each subclade by a sequence profile and estimate evolutionary distances between profiles to obtain a matrix of distances between subclades.
Results
Our estimator of profile distances generalizes the maximum likelihood estimator of sequence distances. The basal branching pattern can be estimated by any distance-based method, such as neighbor-joining. Our method (profile neighbor-joining, PNJ) then inherits the accuracy and robustness of profiles and the time efficiency of neighbor-joining.
Conclusions
Phylogenetic analysis of Chlorophyceae with traditional methods (MP, NJ, ML and MrBayes) reveals seven well supported subclades, but the methods disagree on the basal branching pattern. The tree reconstructed by our method is better supported and can be confirmed by known morphological characters. Moreover the accuracy is significantly improved as shown by parametric bootstrap.
Background
There exist many methods for phylogenetic tree reconstruction, based on different concepts and models. Each method has its strengths and weaknesses. Neighbor-joining [1] or other improved distance methods, e.g., WEIGHBOR [2], BIONJ [3], FASTME [4] and a further approach considering maximum-likelihood estimated triplets of sequences [5], are relatively fast (O(n3) for n taxa), but first reduce the information contained in the characters to a matrix of distances.
Character-based methods, such as maximum parsimony [6] or maximum-likelihood [7], would require an evaluation of super-exponentially many topologies, so one reverts to heuristics. There seems to be no universally accepted best method.
All of the above methods aim to estimate a fully resolved tree from scratch. Occasionally, this is more information than one needs, or than the data support. In several analyses, we are only interested in the basal branching pattern of known or clearly separated and fully supported subclades. In other words, given families of closely related sequences, what is the topology showing the relationships between these families?
This problem arises, for example, when estimating the phylogeny of the Chlorophyceae. In recent studies, e.g. by Buchheim et al. [8] and Wolf et al. [9], a data set of 18S or 18S + 26S rRNA genes was examined using maximum parsimony (MP), neighbor-joining (NJ) and maximum-likelihood (ML). These methods show clear support of seven Chlorophyceae subclades (Oedogoniales, Chaetophorales, Chaetopeltidales, Chlamydomonadales, the core Sphaeropleales, the Sphaeropleaceae and one clade incertae sedis called the Cylindrocapsa-clade), but disagree on the the basal branching pattern, i.e., the relationships between these subclades. This is the same with the improved methods BIONJ and FASTME.
In this paper, we complement the traditional methods with a Bayesian approach (MrBayes) by Huelsenbeck and Ronquist [10]. While we do derive a fully resolved tree with high posterior probabilities on each edge (>90), showing only one intermediate posterior value of 70 (see also Shoup and Lewis [11]), up to now it is not clear how posterior probabilities relate to bootstrap values.
MrBayes was the only tool that was able to reconstruct robustly the phylogenetic tree. Our decision to compare our results to the MrBayes tree was strengthened by a fairly well fit to some well known morphological markers (e.g., basal body configuration and pyrenoid invagination type). We conclude that the presented MrBayes tree is the most robust and most accurate tree out of all calculated trees (Parsimony, distance based, maximum likelihood and MrBayes tree) and therefore we choose this tree as our reference tree in this studied case.
However, we propose the following new method to derive more robust and accurate trees: We replace the set of taxa forming a known subclade by a single supertaxon, which we represent by a sequence profile [12]. To estimate the evolutionary distances between supertaxa, we generalize the maximum-likelihood distance estimator of two sequences to evolutionary distances of profiles. The derived distance matrix can be used to reconstruct a tree by the neighbor-joining method, and we refer to the resulting method as Profile Neighbor-Joining (PNJ). Evaluations indicate that PNJ is both more robust and accurate.
We show that the PNJ tree is resolved with bootstrap values greater than obtained by standard methods, and agrees with the MrBayes tree concerning the Cylindrocapsa-clade, the Chlamydomonadales, the core Sphaeropleales and the Sphaeropleaceae.
An extended abstract of this work [13] appeared at the German Conference on Bioinformatics (GCB'03).
Results
Chlorophycean data set (18S + 26S ribosomal RNA)
A multiple 18S + 26S rRNA sequence alignment of 52 Chlorophyceae species, based on secondary structure was given by Buchheim et al. [8] and deposited at TreeBASE [14,15]. We applied Bayesian methods and Profile Neighbor-Joining to this alignment.
Bayesian analysis
The Bayesian approach to tree reconstruction is reviewed by Holder and Lewis [16] and implemented in the "MrBayes" program by Huelsenbeck and Ronquist [10]. Our analysis is based on a general time reversible (GTR) substitution model with a gamma rate distribution estimated from the data set. Starting from random trees, eight Markov chains are run in parallel to sample trees using the Markov Chain Monte Carlo (MCMC) principle. After the burn-in phase every 100-th sample out of 106 generations is considered, and results are compared among the eight chains in order to confirm that stationarity has been reached. Finally, a 50% majority rule consensus tree is constructed. The resulting tree is shown in Fig. 2(B).
Figure 2.

Phylogenetic trees. (A): Neighbor-joining tree based on subclade profiles. (B): Tree produced by MrBayes. In both trees F. perforate, and C. ellipsoidea (Trebouxiophyceae) were chosen as outgroup. Brackets indicate taxonomical groupings. Edges are annotated with bootstrap values, resp. percent posterior probabilities. Both trees show the same branching pattern concerning sister group relations of the Sphaeropleales, the Chlamydomonadales and the Cylindrocapsa-clade. Note that the Sphaeropleales (core Sphaeropleales plus Sphaeropleaceae) are supported by 59% resp. 70%.
PNJ tree
Using the substitution model estimated by the MrBayes software, we apply the profile distance estimator to the known subclades of the Chlorophyceae and obtain a matrix of evolutionary distances between the subclades (shown in Table 1).
Table 1.
Distance matrix. Estimated profile distance matrix on the seven chlorophycean subclades and the outgroup (Trebouxiophyceae).
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | |
| Cylindrocapsa-clade 1 | |||||||
| Chlamydomonadales 2 | 13.65 | ||||||
| Sphaeropleaceae 3 | 13.34 | 12.97 | |||||
| core Sphaeropleales 4 | 12.63 | 11.97 | 10.89 | ||||
| Oedogoniales 5 | 13.08 | 13.57 | 12.50 | 11.05 | |||
| Chaetopeltidales 6 | 12.05 | 11.69 | 10.80 | 9.55 | 10. 10 | ||
| Chaetophorales 7 | 12.01 | 12.05 | 11.53 | 9.97 | 10. 93 | 8.27 | |
| Trebouxiophyceae 8 | 14.98 | 15.18 | 14.42 | 13.14 | 13. 73 | 12.58 | 12.40 |
From this distance matrix we derive the basal branching pattern of the chlorophycean subclades using neighbor-joining. The Bootstrap analysis indicates that all splits in the profile tree are better supported than by standard methods (no support at the branching pattern), see Fig. 2(A). The branching pattern of this tree coincides with the MrBayes tree regarding the Cylindrocapsa-clade, the Chlamydomonadales, the core Sphaeropleales and the Sphaeropleaceae.
Evaluation of Profile Neighbor-Joining
A high bootstrap support is a necessary, but not sufficient condition for high confidence in an estimated tree. As mentioned by Hillis and Bull [17], bootstrap values are sometimes wrongly interpreted as a measure of accuracy, i.e., the probability that a given result represents a true tree. However, bootstrap values only evaluate the robustness of the tree estimation method, or in other words, yield a measure of repeatability. To estimate the accuracy of profile neighbor-joining (PNJ), we perform the following procedure, which is often referred to as parametric bootstrap. We start with the chlorophycean tree topology and substitution model estimated by MrBayes and sample 100 multiple sequence alignments using Rose [18,19]. In this artificial setup we increased the mutation rate 12-fold, such that both methods are getting difficulties in reconstructing the right topology. From each multiple alignment we calculate a phylogenetic tree by NJ and PNJ. The accuracy is measured by the fraction of splits (bipartitions) that are shared by the estimated tree and the simulated tree. As a distance measure between two tree topologies 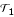 and
and 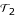 , we use an equivalent form of the Robinson-Foulds distance [20],
, we use an equivalent form of the Robinson-Foulds distance [20],
![]()
where 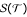 denotes the set of all splits of tree
denotes the set of all splits of tree 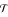 .
.
The PNJ method receives the correct subclades as additional information; the NJ method recovered these in 54 out of 100 sampling runs. In order not to bias the comparison against NJ, we only compare these 54 NJ trees against the 100 PNJ trees. The distribution of the Robinson-Foulds distance for both methods is shown in Fig. 3. The Wilcoxon signed rank test shows that PNJ is significantly more accurate than NJ (p = 6·10-6).
Figure 3.

Robinson-Foulds error distributions. Robinson-Foulds error distributions of NJ and PNJ on the chlorophycean tree topology and substitution model.
Discussion
Contributions
On the methodological side, we recognize a prominent problem in phylogenetic analysis: the reconstruction of the basal topology when several subclades have been well identified. Whereas available tools attempt to estimate a fully resolved tree from scratch, the profile neighbor-joining (PNJ) method we introduce focuses directly on the mentioned problem. Naturally, other approaches are possible: For example, we might simply use one representative taxon from each subclade, or estimate the most likely sequence in the subclade root. The profile-based approach appears preferable because it integrates information from all family members. A similar approach would be to constrain subtrees.
However, consider that, if we generate the profile by implicitly constraining the topology of the considered subtree, we still believe fully in all the estimated distances in the subtree and in their dependencies. However, we have to take into account the possibility that our sequence alignment contains e.g. fast evolving species or strong variation over sites. Then it could happen that one or more sequences in the subtree are connected accidently to the rest by a long branch attraction. This would result in a very strong signal of this sequence in the weighted profile generated from these sequences. In this sense, our approach is more robust.
Moreover in contrast to distances between consensus sequences, profile distances can be viewed more or less as averaged sequence distances. But there is one difference between profile distances and average distances: Average distances are a mean of maximum-likelihood distances of sequences between sequences of both groups, whereas the profile distance is a maximum-likelihood estimation between the "mean" of the sequences in both groups. In this sense the profile distance results in a more computational efficient estimator and leads in contrast to a consensus approach to a more robust formulation.
Summarizing, as we are working with distances, we can directly apply neighbor-joining (or any other improved distance-based method); so we obtain a more robust and time-efficient method. We have also shown that PNJ can be significantly more accurate than simple NJ.
On the biological side, the phylogeny of Chlorophyceae is re-examined using profile-based distances between subclades. The resulting neighbor-joining tree agrees with the MrBayes tree on four of the seven chlorophycean subclades (but note that Bayesian methods are much more time consuming and computationally demanding than neighbor-joining). While Bayesian analysis show high posterior probabilities which are difficult to corelate to bootstrap values, the new proposed method show improved real bootstrap values at the splits in question, Fig. 2(A).
The profile neighbor-joining method supports the first time especially the Sphaeropleales with a bootstrap value higher than 50. This is in contrast to the traditional methods (MP, ML and NJ) used by Buchheim et al. [8] and Wolf et al. [9]. In general, both the PNJ tree and the MrBayes tree can be brought into agreement with the proposed morphological character evolution (absolute orientation of basal bodies and pyrenoid invagination type) as discussed by Buchheim et al. [8] and Wolf et al. [9]. Regarding the topology differences one could argue that the positions of the Oedogoniales, the Chaetophorales, and the Chaetopeltidales can still not be unambiguously resolved. However, the evaluation shows the improved robustness and the high accuracy of PNJ. Therefore we prefer the PNJ tree topology, where (1) the Oedogoniales cluster is at the basis of the Chlorophyceae, (2) the quadriflagellated Chaetopeltidales and Chaetophorales are sister groups, and (3) the biflagellated Sphaeropleales are the most supported as monophyletic compared to the trees derived by other methods (MP, NJ, ML and MrBayes).
Conclusions
The proposed method helps to solve a general problem in phylogeny. It is applicable to all trees with low bootstrap support on the basal branching pattern.
From a computational point of view, there are some promising approaches. For example, we may use the concept of profile distance in a divide-and-conquer algorithm that iteratively applies neighbor-joining on growing well-supported subclades. In this way we may obtain better support on basal branching patterns in supertrees, e.g., in the tree of life or within the crown eukaryotes.
From a statistical point of view, one could investigate more elaborate methods (e.g., sequence weighting) than simple averaging to derive more accurate family profiles. In particular, this could result in even higher bootstrap values. Additionally, the accuracy of the proposed profile neighbor-joining method should be compared with the other traditional tree reconstruction methods as MP, ML and especially MrBayes. Another interesting direction would be the transfer of profile distances into the maximum likelihood tree estimation procedures.
Methods
Profiles
A sequence profile is a stochastic model of a sequence family. It can also be pictured as a fuzzy or "smeared-out" sequence. Formally, a profile is also a sequence, but it is composed of probability distribution vectors instead of characters. Each position k specifies its own nucleotide distribution αk = (αk,A, αk,C, αk,G, αk,T). Nucleotide sequences are special profiles, where each αk is given by one of the unit vectors A = (1, 0, 0, 0), C = (0, 1, 0, 0), and so on.
Several philosophies and methods for estimating profiles from families exist. For example, given a phylogenetic tree of sequence families, we could estimate the most likely profile at the root. In the context of this paper, however, we are more interested in the "center of gravity" of the sequence family distribution. Therefore, we simply take the position-specific relative nucleotide frequencies over all sequence family members. This results in a robust estimate that is independent of estimated subclade topologies.
Distance estimation
Maximum-likelihood methods for tree and distance estimation rely on a model of nucleotide substitution. Substitutions are modeled by an evolutionary Markov process (EMP) acting independently on each site of the sequence [21,22]. The EMP is uniquely described by its starting distribution and its rate matrix Q, often called the substitution model. We assume that Q has a unique stationary distribution π that satisfies π·Q = 0, and that the process starts in its stationary distribution. The model is calibrated in such a way that 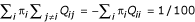 , i.e., that per 100 time units, one substitution is expected to occur.
, i.e., that per 100 time units, one substitution is expected to occur.
Depending on its parameterization, the rate matrix Q is called Jukes-Cantor Model (1 parameter), Kimura Model (2 parameters), or general time reversible (GTR) model (6 parameters). The parameters (such as the transition-transversion ratio in the Kimura model) must be estimated from the data.
We symbolically write "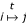 " for the event that nucleotide i has been substituted by nucleotide j after time t. The probability of this substitution is given by (i, j) entry of the time-t transition matrix Pt, which is related to Q via the matrix exponential Pt = exp(tQ).
" for the event that nucleotide i has been substituted by nucleotide j after time t. The probability of this substitution is given by (i, j) entry of the time-t transition matrix Pt, which is related to Q via the matrix exponential Pt = exp(tQ).
To estimate the evolutionary distance between two sequences, we first compute a pairwise alignment and count the number of all nucleotide pair types in the alignment. Let Nij be the number of observed events "i 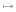 j".
j".
In general, the higher the off-diagonal entries (mismatch counts) are in comparison to the diagonal entries (match counts), the larger is the evolutionary distance. A well-founded framework is given by the maximum likelihood principle. If the distance is t time units, the joint probability of all events is given by 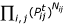 . We seek the value t maximizing this probability, or equivalently, the log-likelihood function,
. We seek the value t maximizing this probability, or equivalently, the log-likelihood function,
![]()
Note that 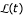 is a sum of log-probabilities log(
is a sum of log-probabilities log(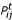 ), weighted by their observed counts Nij. Of course, the counts sum up to the total number n of sites in the alignment. For general models Q, the solution of this one-dimensional maximization problem cannot be given in closed form, but is easily obtained by numerical methods.
), weighted by their observed counts Nij. Of course, the counts sum up to the total number n of sites in the alignment. For general models Q, the solution of this one-dimensional maximization problem cannot be given in closed form, but is easily obtained by numerical methods.
We now generalize this estimator from sequences to profiles. A site need not contribute an integer count of 1 to a single substitution category, but fractional counts summing to 1 can be spread out over all 16 categories. Assume that the profiles at a particular site of the two families are given by α = (αA, αC, αG, αT) and β = (βA, βC, βG, βT), respectively. Intuitively, if the sequence families consist of m1 and m2 sequences and we independently draw a sequence from each family, we observe the nucleotide pair (i, j) on average m1αi·m2βj times, corresponding to a relative frequency of αiβj. The product distribution mirrors the conditionally independent drawing from within the sequence families and does not imply that the families might be unrelated. Thus the total counts Nij for event i j are given by the sum over all n sites: 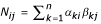 (see Fig. 1). Otherwise, the likelihood function in Eq. (1) and the maximization procedure remain unchanged.
(see Fig. 1). Otherwise, the likelihood function in Eq. (1) and the maximization procedure remain unchanged.
Figure 1.

Counting substitutions. Counting substitutions Nij between sequences a and b (A), and between profiles α and β (B).
Bootstrapping
To assess the robustness of an estimated tree under perturbations of the input alignment, it is customary to perform a bootstrap analysis [23,24], where entire columns of the alignment are resampled with replacement. This immediately carries over to the new setting where we are not given an alignment of sequences, but an alignment of profiles, i.e., we re-sample columns of profiles.
Authors' contributions
TM, SR (from the mathematical point of view) and TD, MW (from the biological point of view) conceived the study and the PNJ algorithm, drafted the manuscript and participated in its design and coordination. All authors read and approved the final manuscript.
Acknowledgments
Acknowledgments
We thank the two anonymous referees for improving the manuscript, as well as Mark A. Buchheim (Tulsa, USA), Hannes Luz (Berlin, Germany), Joachim Friedrich (Aachen, Germany) and Martin Vingron (Berlin, Germany) for fruitful discussions.
Contributor Information
Tobias Müller, Email: tobias.mueller@biozentrum.uni-wuerzburg.de.
Sven Rahmann, Email: sven.rahmann@cebitec.uni-bielefeld.de.
Thomas Dandekar, Email: dandekar@biozentrum.uni-wuerzburg.de.
Matthias Wolf, Email: matthias.wolf@biozentrum.uni-wuerzburg.de.
References
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. J Mol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Bruno WJ, Socci ND, Halpern AL. Weighted Neighbor Joining: A Likelihood-Based Approach to Distance-Based Phylogeny Reconstruction. Mol Biol Evol. 2000;17:189–197. doi: 10.1093/oxfordjournals.molbev.a026231. [DOI] [PubMed] [Google Scholar]
- Gascuel O. BIONJ: an improved version of the NJ algorithm based on a simple model of sequence data. Mol Biol Evol. 1997;14:685–695. doi: 10.1093/oxfordjournals.molbev.a025808. [DOI] [PubMed] [Google Scholar]
- Desper R, Gascuel O. Fast and Accurate Phylogeny Reconstruction Algorithms Based on the Minimum-Evolution Principle. J Comput Biol. 2002;19:687–705. doi: 10.1089/106652702761034136. [DOI] [PubMed] [Google Scholar]
- Ranwez V, Gascuel O. Improvement of Distance-Based Phylogenetic Methods by a Local Maximum Likelihood Approach Using Triplets. Mol Biol Evol. 2002;19:1952–1963. doi: 10.1093/oxfordjournals.molbev.a004019. [DOI] [PubMed] [Google Scholar]
- Camin J, Sokal R. A method for deducing branching sequences in phylogeny. Evolution. 1965;19:311–326. [Google Scholar]
- Felsenstein J. Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol. 1981;17:368–376. doi: 10.1007/BF01734359. [DOI] [PubMed] [Google Scholar]
- Buchheim MA, Michalopulos EA, Buchheim JA. Phylogeny of the Chlorophyceae with special reference to the Sphaeropleales: A study of 18S and 26S rDNA data. J Phycol. 2001;37:819–835. doi: 10.1046/j.1529-8817.2001.00162.x. [DOI] [Google Scholar]
- Wolf M, Buchheim M, Hegewald E, Krienitz L, Hepperle D. Phylogenetic position of the Sphaeropleaceae (Chlorophyta) Plant Syst Evol. 2002;230:161–171. doi: 10.1007/s006060200002. [DOI] [Google Scholar]
- Huelsenbeck J, Ronquist F. MRBAYES: Bayesian inference of phylogeny. Bioinformatics. 2001;17:754–755. doi: 10.1093/bioinformatics/17.8.754. [DOI] [PubMed] [Google Scholar]
- Shoup S, Lewis L. Polyphyletic origin of parallel basal bodies in swimming cells of chlorophycean green algae (Chlorophyta) J Phycol. 2003;39:789–796. doi: 10.1046/j.1529-8817.2003.03009.x. [DOI] [Google Scholar]
- Gribskov M, McLachlan A, Eisenberg D. Profile analysis: detection of distantly related proteins. Proc Natl Acad Sci USA. 1987;84:4355–4358. doi: 10.1073/pnas.84.13.4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller T, Rahmann S, Dandekar T, Wolf M. Robust estimation of the phylogeny of Chlorophyceae (Chlorophyta) based on profile distances. In Proceedings of the German Conference on Bioinformatics (GCB'03) 2003. pp. 97–101.
- Morell V. TreeBASE: the roots of phylogeny. Science. 1996;273:569. [Google Scholar]
- TreeBASE http://www.treebase.org
- Holder M, Lewis P. Phylogeny estimation: traditional and Bayesian approaches. Nature Rev. 2003;4:275–284. doi: 10.1038/nrg1044. [DOI] [PubMed] [Google Scholar]
- Hillis D, Bull J. An empirical test of bootstrapping as a method for assessing confidence in phylogenetic analysis. Syst Biol. 1993;42:182–192. [Google Scholar]
- Stoye J, Evers D, Meyer F. Rose: Generating Sequence Families. Bioinformatics. 1998;14:157–163. doi: 10.1093/bioinformatics/14.2.157. [DOI] [PubMed] [Google Scholar]
- BiBiServ http://bibiserv.techfak.uni-bielefeld.de/rose
- Robinson D, Foulds L. Comparison of phylogenetic trees. Math Biosci. 1981;53:131–147. doi: 10.1016/0025-5564(81)90043-2. [DOI] [Google Scholar]
- Lanave C, Preparata G, Saccone C, Serio G. A new method for calculating evolutionary substitution rates. J Mol Evol. 1984;20:86–93. doi: 10.1007/BF02101990. [DOI] [PubMed] [Google Scholar]
- Müller T, Vingron M. Modeling amino acid replacement. J Comput Biol. 2000;7:761–776. doi: 10.1089/10665270050514918. [DOI] [PubMed] [Google Scholar]
- Efron B, Halloran E, Holmes S. Bootstrap confidence levels for phylogenetic trees. Proc Natl Acad Sci USA. 1996;93:7085–7090. doi: 10.1073/pnas.93.14.7085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39:783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x. [DOI] [PubMed] [Google Scholar]