

Abstract
Background/Aims
The gastrointestinal (GI) tract often becomes involved in patients with systemic amyloidosis. As few GI amyloidosis data have been reported, we describe the clinical features and outcomes of patients with pathologically proven GI amyloidosis.
Methods
We identified 155 patients diagnosed with systemic amyloidosis between April 1995 and April 2013. Twenty-four patients (15.5%) were diagnosed with GI amyloidosis using associated symptoms, and the diagnoses were confirmed by direct biopsy.
Results
Among the 24 patients, 20 (83.3%) had amyloidosis light chain (AL), three (12.5%) had amyloid A, and one (4.2%) had transthyretin-related type amyloidosis. Their median age was 57 years (range, 37 to 72), and 10 patients were female (41.7%). The most common symptoms of GI amyloidosis were diarrhea (11 patients, 45.8%), followed by anorexia (nine patients, 37.5%), weight loss, and nausea and/or vomiting (seven patients, 29.2%). The histologically confirmed GI tract site in AL amyloidosis was the stomach in 11 patients (55.0%), the colon in nine (45.0%), the rectum in seven (35.0%), and the small bowel in one (5.0%). Patients with GI involvement had a greater frequency of organ involvement (p = 0.014). Median overall survival (OS) in patients with GI involvement was shorter (7.95 months; range, 0.3 to 40.54) than in those without GI involvement (15.84 months; range, 0.0 to 114.53; p = 0.069) in a univariate analysis. A multivariate analysis of prognostic factors for AL amyloidosis revealed that GI involvement was not a significant predictor of OS (p = 0.447).
Conclusions
The prognosis of patients with AL amyloidosis and GI involvement was poorer than those without GI involvement, and they presented with more organ involvement and more advanced disease than those without organ involvement.
Keywords: Gastrointestinal amyloidosis, Amyloidosis, Gastrointestinal diseases
INTRODUCTION
Amyloidosis is a group of diseases caused by extracellular deposition of insoluble amyloid fibrils resulting in organ dysfunction. These fibrils are identified by Congo red staining and apple-green birefringence using polarized microscopy [1]. The most common form of systemic amyloidosis is primary (amyloidosis light chain, AL) amyloidosis, which is related to plasma cell dyscrasia and the accumulation of light-chain amyloid fibrils in multiple organs [2]. Secondary (AA) amyloidosis is associated with deposition of serum amyloid A protein, which is an acute-phase reactant protein produced during chronic inflammatory or infectious diseases, such as rheumatoid arthritis, Crohn disease, and osteomyelitis [3,4,5]. The other forms of systemic amyloidosis are classified by the amyloidogenic precursor protein, such as transthyretin, β2-microglobulin, and lysozyme [6].
The kidney and heart are the most commonly involved organs in patients with systemic amyloidosis, followed by the nervous system, soft tissues, lungs, and liver. Although amyloid deposits are frequently observed in blood vessels within the gastrointestinal (GI) tract on biopsy, amyloid deposition in the GI tract and symptoms are infrequent [3]. GI amyloidosis causes various symptoms resulting in a deteriorating quality of life [2,7,8]. The common clinical manifestations of GI amyloidosis are weight loss, bleeding, esophageal reflux, constipation, diarrhea, and abdominal pain. As whether some of these symptoms are caused by autonomic dysfunction is occasionally difficult to determine, verification by direct biopsy is needed for the diagnosis of GI amyloidosis [9]. The most common site of amyloid deposition identified by endoscopic biopsy is the small bowel, followed by the stomach, colorectum, and esophagus [10,11].
As GI involvement is an uncommon manifestation of systemic amyloidosis, few data are available, with the exception of some case reports of patients with GI amyloidosis. The purpose of this study was to describe the characteristics, clinical manifestations, biochemistry, and prognosis of patients with pathologically proven GI amyloidosis.
METHODS
Study population
We included patients aged ≥ 18 years who were diagnosed with systemic amyloidosis between April 1995 and April 2013 at Samsung Medical Center, Seoul, Korea. A total of 171 patients with systemic amyloidosis were identified. To classify the type of amyloid deposition, we performed immunohistochemical staining and evaluated patients for the transthyretin gene mutation if they were suspected of having amyloidosis transthyretin-related (ATTR) amyloidosis. Sixteen patients with unclassifiable types of amyloidosis were excluded. Of 155 patients with systemic amyloidosis, AL amyloidosis was the most common (137 patients, 88.4%), followed by the AA type (11 patients, 7.1%), and the ATTR type (seven patients, 4.5%). Twenty-four patients (15.5%) were confirmed to have GI amyloidosis, which was diagnosed by GI symptoms and verified by direct GI tract biopsy using Congo red staining and apple-green birefringence under a polarized microscope. Specimens from all patients with GI amyloidosis underwent immunohistochemical staining. Although patients were histologically confirmed to have amyloid deposits in the GI tract, and patients without associated symptoms were excluded. We also excluded patients with amyloid angiopathy because the amyloid deposits in these patients occur only in the vascular wall of the GI tract.
Data collection
Of the 24 patients with GI amyloidosis, we collected data from the medical records about the nature of the GI symptoms and organ involvement of systemic amyloidosis including the heart, kidneys, nervous system, and others. We reviewed applicable laboratory results of the patients with AL amyloidosis, including serum and urine immunofixation, serum free light chain (FLC) and bone marrow (BM) biopsy results, the calculated difference between involved and uninvolved serum FLC (dFLC), serum albumin, 24-hour urine protein, serum creatinine, troponin-I (TnI), N-terminal pro-brain natriuretic peptide (NT-proBNP), and serum β2 microglobulin. The Samsung Medical Center Institutional Review Board approved this study, and written informed consent was obtained from all patients with systemic amyloidosis regarding use of their medical records.
Statistical analysis
Continuous variables are reported as medians and ranges. Fisher exact test and the Mann-Whitney U test were used to compare baseline characteristics of the patients with AL amyloidosis with or without GI involvement. Cumulative survival was assessed by the Kaplan-Meier method followed by the log-rank test to analyze differences in survival curves. Significant variables with p < 0.1 in a univariate analysis were included in a multivariate analysis. The Cox proportional hazards regression model was used for the multivariate analysis of overall survival (OS). All tests were two-sided, and p < 0.05 were considered to indicate significance. All statistical analyses were performed using the SPSS version 21 (IBM Co., Armonk, NY, USA).
RESULTS
Patient characteristics
Of the 24 patients, 15.5% of those with systemic amyloidosis were diagnosed with GI amyloidosis (Table 1). The median age of the 24 patients was 57 years (range, 37 to 72), and 10 patients (41.7%) were female. Among the patients with GI amyloidosis, 20 (83.3%) had AL, three (12.5%) had AA, and one (4.2%) had the ATTR type of systemic amyloidosis. The most frequent symptoms were diarrhea in 11 patients (45.8%), anorexia in nine (37.5%), and nausea and/or vomiting and weight loss in seven (29.2%). The heart was the most commonly involved organ of other organs/systems in the patients with GI amyloidosis (15 patients, 62.5%), and the biopsy-confirmed organs in the GI tract were the stomach and colon (13 patients, 54.2%). The median follow-up from the time of diagnosis to the date of data collection was 70.5 months, and median OS was 8.1 months.
Table 1. Patient disease distribution.
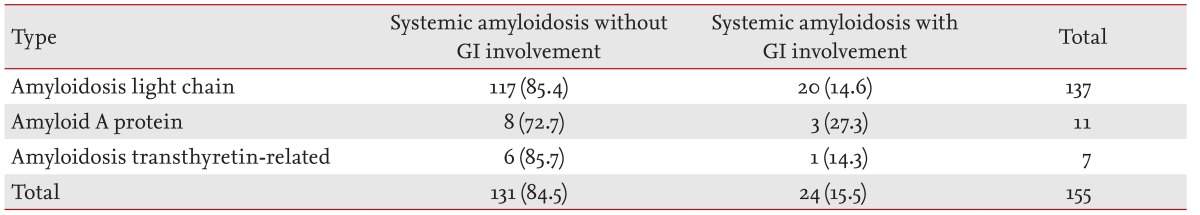
Values are presented as number (%).
GI, gastrointestinal.
Systemic amyloidosis with GI involvement
Twenty patients were diagnosed with AL amyloidosis. Their median age was 56 years (range, 37 to 72), and nine patients (45.0%) were female (Table 2). The histologically confirmed site in the GI tract was the stomach in 11 patients (55.0%), the colon in nine (45.0%), the rectum in seven (35.0%), and the small bowel in one (5.0%). Symptoms were diarrhea and weight loss in seven patients (35.0%), anorexia in six (30.0%), nausea and/or vomiting and GI bleeding in five (25.0%), constipation in four (20.0%), dyspepsia and reflux in three (15.0%), and abdominal pain in two patients (10.0%). Other amyloidosis organ/system involvement included the heart in 13 patients (65.0%), the kidney in six (30.0%), the peripheral nervous system (PNS) in six (30.0%), and the autonomic nervous system (ANS) in two (10.0%). The PNS and ANS were not evaluated in one patient. Half of the patients (10 patients) with AL amyloidosis had three or more organs/systems involved.
Table 2. Clinical presentation of patients with gastrointestinal amyloidosis.
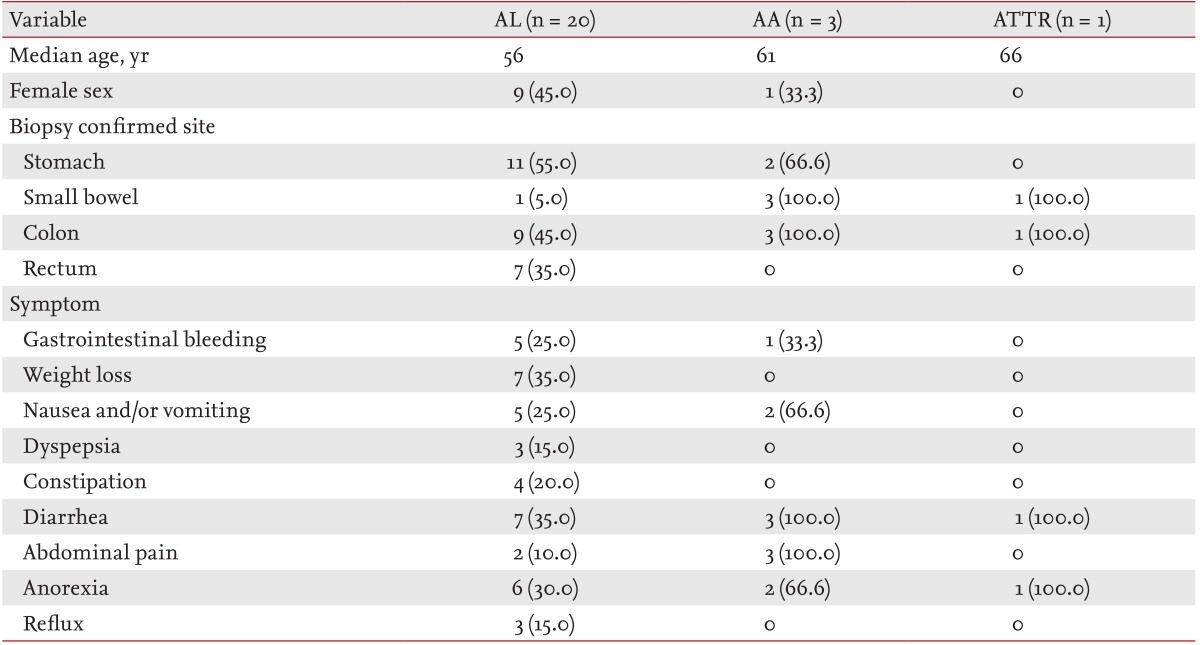
Values are presented as number (%).
AL, amyloidosis light chain; AA, amyloid A protein; ATTR, amyloidosis transthyretin-related.
Three of the 24 patients were diagnosed with AA amyloidosis. Their median age was 61 years (range, 52 to 61), and one patient was female. All three patients had biopsy-verified GI tract involvement in the small bowel and colon. Additional biopsies detected disease in the stomach of two patients. At the time of diagnosis, GI symptoms included diarrhea and abdominal pain in three patients, anorexia and nausea and/or vomiting in two, and GI bleeding in one. Renal involvement was detected in all three patients with AA amyloidosis and GI involvement, and the other organs/systems involved were the heart and PNS in one patient each. The assumed causes of AA amyloidosis were rheumatoid arthritis in two patients and Bechet disease in one. One patient with GI amyloidosis had the ATTR type, which was confirmed by the presence of the TTR gene mutation. The biopsy-confirmed GI tract sites in this patient were the small bowel and colon. This patient also had heart, PNS, and ANS involvement.
Five of the 24 patients with GI amyloidosis are currently alive. Among them, three achieved complete remission, and four showed improved GI symptoms.
Comparison of AL amyloidosis with or without GI involvement
Among the 155 patients with systemic amyloidosis, 137 had AL amyloidosis, including 20 (14.6%) with GI involvement and 117 (85.4%) without GI involvement (Table 3). No differences were detected in age, proportion of females, or Eastern Cooperative Oncology Group (ECOG) performance status between the patients with AL amyloidosis with and without GI involvement. Renal involvement was significantly less frequent in patients with AL amyloidosis and GI involvement than in those without GI involvement (30.0% vs. 57.1%, p = 0.030), and more patients with GI involvement had three or more organs involved compared to those without GI involvement (50.0% vs. 22.2%, p = 0.014). The proportion of patients who underwent stem cell transplantation was not different between the two groups (25.0% vs. 17.1%, p = 0.366). The proportions of heavy chain immunoglobulins (Igs) in the patients with AL amyloidosis and GI involvement were IgG in seven patients (35.0%), IgA in one (5.0%), and light chain only in 11 (55.0%). Patients with the IgA type of heavy chain tended to be less frequent in the AL amyloidosis with GI involvement group than in those without GI involvement group (5.0% vs. 18.8%, p = 0.196), but the difference was not significant.
Table 3. Systemic amyloidosis light chain with or without gastrointestinal involvement.
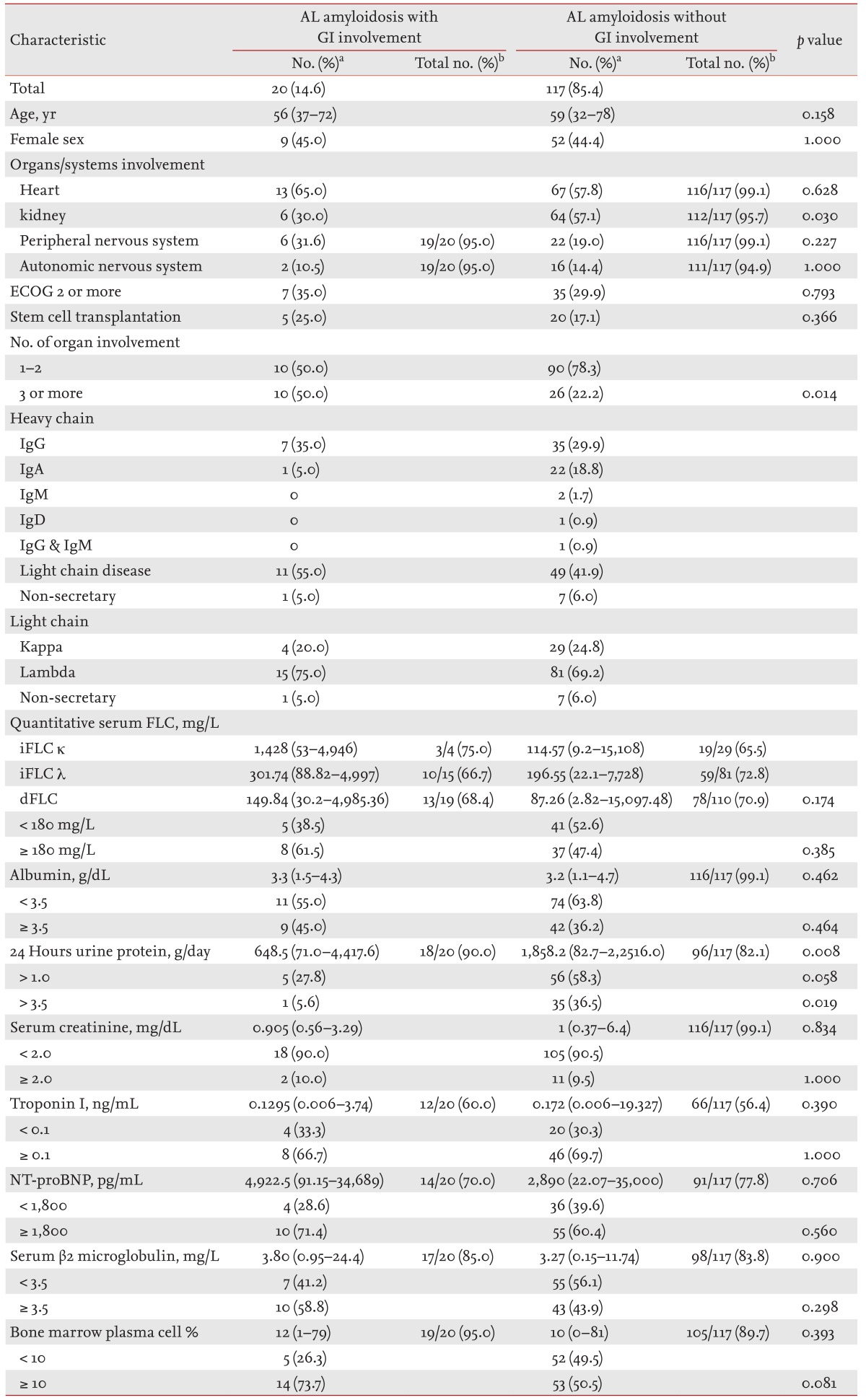
AL, amyloidosis light chain; GI, gastrointestinal; ECOG, Eastern Cooperative Oncology Group; Ig, immunoglobulin; FLC, free light chain; iFLC, involved free light chain; dFLC, difference between involved and uninvolved free light chain; NT-proBNP, N-terminal pro-brain natriuretic peptide.
aValues are presented as number (%) or median (range).
bValues are presented as numbers of patients with available data/total numbers of patients.
Median dFLC level was 149.84 mg/L (range, 30.2 to 4,985.36) in patients with GI involvement and 87.26 mg/L (range, 2.82 to 15,097.48) in those without GI involvement (p = 0.174). No significant differences were observed in the basal laboratory data, including serum albumin, serum creatinine, TnI, and NT-proBNP levels between patients with AL amyloidosis with GI involvement and those without GI involvement. The median 24-hour urine protein level was higher in patients with GI involvement than in those without GI involvement (648.5 g/day [range, 71.0 to 4,417.6] vs. 1,858.2 g/day [range, 82.7 to 22,516.0], p = 0.008). Median serum β2 microglobulin levels were 3.80 mg/L (range, 0.95 to 24.4) in patients with GI involvement and 3.27 mg/L (range, 0.15 to 11.74) in those without GI involvement (p = 0.900). The median percentages of plasma cells in the BM were 12% (range, 1 to 79) in patients with GI involvement and 10% (range, 0 to 81) in patients without GI involvement (p = 0.393).
Patients with AL amyloidosis were categorized according to bone marrow plasma cells (BMPCs) and whether they had hypercalcemia, renal insufficiency, anemia, and bone lesions (CRAB). The study population was categorized into the following three groups: AL-only, AL-CRAB, and AL plasma cell multiple myeloma (AL-PCMM), which had > 10% BMPCs. No differences were observed in the proportion of each type of AL amyloidosis between patients with and without GI involvement (p = 0.484)
Overall survival and prognostic factors in patients with AL amyloidosis
Median OS from diagnosis was 7.95 months (range, 0.3 to 40.54) in patients with AL amyloidosis with GI involvement and 15.84 months (range 0.0 to 114.53) in those without GI involvement (p = 0.069) (Fig. 1). We analyzed the prognostic factors in patients with AL amyloidosis (Table 4). Significant factors with p < 0.1 that predicted OS in the univariate analysis were GI involvement, type of AL amyloidosis, heart or kidney involvement, more than two organs involved, IgA type or not, ECOG performance status > 1, > 3.5 mg/L serum β2 microglobulin, and > 1,800 pg/mL NT-proBNP. The estimated median OS durations according to the type of AL amyloidosis were 48.8 months for AL-only, 9.0 months for AL-CRAB, and 5.4 months for AL-PCMM type (p < 0.001). In the multivariate analysis using Cox proportional hazards regression for the AL-PCMM type of systemic amyloidosis, more than two organs involved and > 1,800 pg/mL NT-proBNP were independent prognostic factors of OS. However, whether the GI tract was involved or not was not significant in the multivariate analysis.
Figure 1. Overall survival of patients with amyloidosis light chain. GI, gastrointestinal.

Table 4. Prognostic factors in patients with amyloidosis light chain.
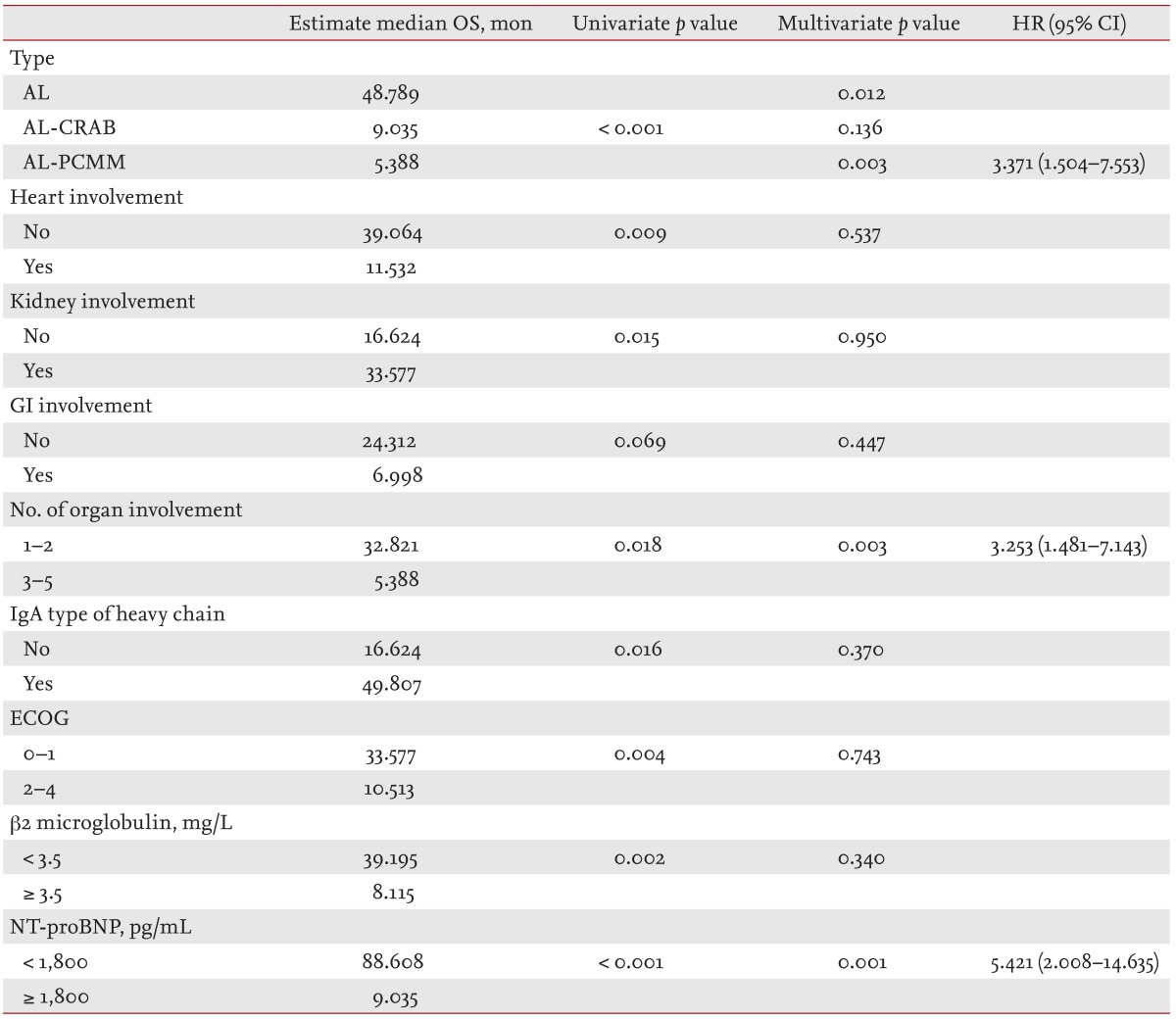
OS, overall survival; HR, hazard ratio; CI, confidence interval; AL, amyloidosis light chain; CRAB, hypercalcemia, renal insufficiency, anemia, and bone lesions; PCMM, plasma cell multiple myeloma; GI, gastrointestinal; ECOG, Eastern Cooperative Oncology Group; NT-proBNP, N-terminal pro-brain natriuretic peptide.
DISCUSSION
Among the 155 patients with systemic amyloidosis, 24 (15.5%) were diagnosed with GI amyloidosis, which was confirmed by GI tract biopsy and associated symptoms. The prominent symptoms in our population were diarrhea, anorexia, and nausea and/or vomiting, which were similar to those reported previously [7,8,12,13]. The most frequently biopsied GI tract sites were the stomach and colon.
GI amyloidosis is relatively rare in western countries (3% to 8% of patients with systemic amyloidosis), whereas our results show a considerably higher incidence of biopsy-proven GI amyloidosis of 15.5% [7,14,15]. An endoscopic examination is an easily available and inexpensive method to identify various GI symptoms for unexplained weight loss or diarrhea in Korea. In addition to the bias of referral to a single center, the ready access to endoscopy may explain the higher incidence of GI involvement in Korean patients with systemic amyloidosis.
Previous studies have shown that small bowel amyloid deposits are most commonly found along the entire GI tract in patients with GI amyloidosis. Cowan et al. [7] showed that almost half of patients with systemic GI amyloidosis have small bowel amyloid deposits [2,7,10,11,16]. However, we found a lower incidence (five cases, 20.8%) of patients with GI amyloidosis diagnosed by small bowel biopsy. The small bowel is rarely inspected during endoscopy due to patient distress and technical difficulties. Thus, patients with small bowel amyloidosis may be detected less frequently.
The median OS of patients with GI amyloidosis was 8.1 months, which was lower than reported previously. The 2-year survival rate of patients with systemic AL amyloidosis of the GI tract was 84% in one study. However, in our study population heart involvement was more frequent, median BMPCs and dFLC levels were increased, and the proportion of patients who underwent stem cell transplantation was lower compared to previous reports [7,12]. Our patients may thus have had more advanced systemic amyloidosis.
Furthermore, OS tended to be shorter in patients with AL amyloidosis with GI involvement than in those without GI involvement. This difference did not reach statistical significance, possibly because only 20 of our cases with systemic AL amyloidosis had GI involvement. A tendency towards more frequent heart involvement was detected in patients with compared to those without GI involvement. In addition, those with GI involvement tended to have higher median levels of NT-proBNP, serum β2 microglobulin, BMPCs, and dFLC, which are known prognostic factors [17,18]. Patients with GI amyloidosis seem to have poor outcomes due to involvement of additional organs and an increased number of poor prognostic factors. Further research is needed, as few OS data are available for patients with GI amyloidosis and comparisons between patients with systemic AL amyloidosis with and without GI involvement have not been reported.
Kourelis et al. [19] showed that patients with AL amyloidosis and > 10% BMPCs and those with AL amyloidosis and CRAB have poorer prognoses than those with only AL amyloidosis based on the consensus that AL amyloidosis and hypercalcemia, renal insufficiency, anemia, and bone lesions can coexist with multiple myeloma [19,20]. However, our patients with AL-PCMM type amyloidosis had a poorer prognosis in the multivariate analysis compared to those with the AL-only type. Although it was not significantly different, the estimated median OS of patients with AL-CRAB tended to be shorter than those with the AL-only type.
Furthermore, the multivariate analysis revealed that the AL-PCMM type, involvement of three or more organs, and > 1,800 pg/mL NT-proBNP are significant prognostic factors, as has been reported previously [17,19]. The number of organs involved has not been shown to be a prognostic factor; however, our patients with three or more amyloidosis-involved organs almost always had heart involvement (86.1%). Because of the high proportion of patients with heart involvement, the number of organs involved could be a significant prognostic factor.
The kidney is affected in 50% to 80% of patients with AL amyloidosis, but our data show that those with GI involvement had less frequent renal involvement [13,21]. There may have been selection bias in our study because it was conducted in a single center. Accordingly, further studies of renal involvement in patients with GI amyloidosis should be conducted.
Several limitations of our study should be mentioned. First, it is difficult to generalize the results because the data were obtained from a relatively small number of patients in a single center. Second, this was a retrospective study, and data for some laboratory parameters were lacking. In particular, TnI was a significant prognostic factor in the univariate analysis, but was excluded from the multivariate analysis due to insufficient data. Third, there was selection bias as our center is a tertiary hospital. Thus, many of the patients had more severe and advanced-stage disease. The shorter OS likely reflects this selection bias.
In summary, we documented the clinical characteristics and outcomes of patients with biopsy-proven GI amyloidosis. Esophagogastroduodenoscopy is the preferred method for confirming GI involvement in patients with amyloidosis. The prognosis of AL amyloidosis with GI involvement was poorer, and those patients presented with more organ involvement and more advanced disease than those without GI involvement. Further work is required to investigate the prognosis of patients with GI amyloidosis because of the lack of studies of OS in this patient group.
KEY MESSAGE
1. The incidence of gastrointestinal amyloidosis is higher in Korea than that in Western countries.
2. The prognosis of amyloidosis light chain with gastrointestinal involvement is poorer, particularly in patients who presented with more organ involvement and more advanced disease.
Acknowledgments
This study was supported by a grant from the Korea Health Technology R&D Project, Ministry of Health and Welfare, Republic of Korea (A120175).
Footnotes
Conflict of interest: No potential conflict of interest relevant to this article was reported.
References
- 1.Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Engl J Med. 2003;349:583–596. doi: 10.1056/NEJMra023144. [DOI] [PubMed] [Google Scholar]
- 2.Ebert EC, Nagar M. Gastrointestinal manifestations of amyloidosis. Am J Gastroenterol. 2008;103:776–787. doi: 10.1111/j.1572-0241.2007.01669.x. [DOI] [PubMed] [Google Scholar]
- 3.Falk RH, Comenzo RL, Skinner M. The systemic amyloidoses. N Engl J Med. 1997;337:898–909. doi: 10.1056/NEJM199709253371306. [DOI] [PubMed] [Google Scholar]
- 4.Lachmann HJ, Goodman HJ, Gilbertson JA, et al. Natural history and outcome in systemic AA amyloidosis. N Engl J Med. 2007;356:2361–2371. doi: 10.1056/NEJMoa070265. [DOI] [PubMed] [Google Scholar]
- 5.Petre S, Shah IA, Gilani N. Review article: gastrointestinal amyloidosis: clinical features, diagnosis and therapy. Aliment Pharmacol Ther. 2008;27:1006–1016. doi: 10.1111/j.1365-2036.2008.03682.x. [DOI] [PubMed] [Google Scholar]
- 6.Sipe JD, Benson MD, Buxbaum JN, et al. Amyloid fibril protein nomenclature: 2012 recommendations from the Nomenclature Committee of the International Society of Amyloidosis. Amyloid. 2012;19:167–170. doi: 10.3109/13506129.2012.734345. [DOI] [PubMed] [Google Scholar]
- 7.Cowan AJ, Skinner M, Seldin DC, et al. Amyloidosis of the gastrointestinal tract: a 13-year, single-center, referral experience. Haematologica. 2013;98:141–146. doi: 10.3324/haematol.2012.068155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Madsen LG, Gimsing P, Schiodt FV. Primary (AL) amyloidosis with gastrointestinal involvement. Scand J Gastroenterol. 2009;44:708–711. doi: 10.1080/00365520902783717. [DOI] [PubMed] [Google Scholar]
- 9.Gertz MA, Comenzo R, Falk RH, et al. Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): a consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis, Tours, France, 18-22 April 2004. Am J Hematol. 2005;79:319–328. doi: 10.1002/ajh.20381. [DOI] [PubMed] [Google Scholar]
- 10.Tada S, Iida M, Iwashita A, et al. Endoscopic and biopsy findings of the upper digestive tract in patients with amyloidosis. Gastrointest Endosc. 1990;36:10–14. doi: 10.1016/s0016-5107(90)70913-3. [DOI] [PubMed] [Google Scholar]
- 11.Tada S. Diagnosis of gastrointestinal amyloidosis with special reference to the relationship with amyloid fibril protein. Fukuoka Igaku Zasshi. 1991;82:624–647. [PubMed] [Google Scholar]
- 12.Hayman SR, Lacy MQ, Kyle RA, Gertz MA. Primary systemic amyloidosis: a cause of malabsorption syndrome. Am J Med. 2001;111:535–540. doi: 10.1016/s0002-9343(01)00919-6. [DOI] [PubMed] [Google Scholar]
- 13.Obici L, Perfetti V, Palladini G, Moratti R, Merlini G. Clinical aspects of systemic amyloid diseases. Biochim Biophys Acta. 2005;1753:11–22. doi: 10.1016/j.bbapap.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 14.Menke DM, Kyle RA, Fleming CR, Wolfe JT, 3rd, Kurtin PJ, Oldenburg WA. Symptomatic gastric amyloidosis in patients with primary systemic amyloidosis. Mayo Clin Proc. 1993;68:763–767. doi: 10.1016/s0025-6196(12)60634-x. [DOI] [PubMed] [Google Scholar]
- 15.James DG, Zuckerman GR, Sayuk GS, Wang HL, Prakash C. Clinical recognition of Al type amyloidosis of the luminal gastrointestinal tract. Clin Gastroenterol Hepatol. 2007;5:582–588. doi: 10.1016/j.cgh.2007.02.038. [DOI] [PubMed] [Google Scholar]
- 16.Gould M, Zarrin-Khameh N, Sellin J. Small bowel amyloidosis. Curr Gastroenterol Rep. 2013;15:350. doi: 10.1007/s11894-013-0350-4. [DOI] [PubMed] [Google Scholar]
- 17.Kumar S, Dispenzieri A, Lacy MQ, et al. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J Clin Oncol. 2012;30:989–995. doi: 10.1200/JCO.2011.38.5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gertz MA. Immunoglobulin light chain amyloidosis: 2013 update on diagnosis, prognosis, and treatment. Am J Hematol. 2013;88:416–425. doi: 10.1002/ajh.23400. [DOI] [PubMed] [Google Scholar]
- 19.Kourelis TV, Kumar SK, Gertz MA, et al. Coexistent multiple myeloma or increased bone marrow plasma cells define equally high-risk populations in patients with immunoglobulin light chain amyloidosis. J Clin Oncol. 2013;31:4319–4324. doi: 10.1200/JCO.2013.50.8499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Desikan KR, Dhodapkar MV, Hough A, et al. Incidence and impact of light chain associated (AL) amyloidosis on the prognosis of patients with multiple myeloma treated with autologous transplantation. Leuk Lymphoma. 1997;27:315–319. doi: 10.3109/10428199709059685. [DOI] [PubMed] [Google Scholar]
- 21.Dember LM. Amyloidosis-associated kidney disease. J Am Soc Nephrol. 2006;17:3458–3471. doi: 10.1681/ASN.2006050460. [DOI] [PubMed] [Google Scholar]