

Abstract
Coenzyme B12-dependent enzymes such as ethanolamine ammonia lyase have remarkable catalytic power and some unique properties that enable detailed analysis of the reaction chemistry and associated dynamics. By selectively deuterating the substrate (ethanolamine) and/or the β-carbon of the 5′-deoxyadenosyl moiety of the intrinsic coenzyme B12, it was possible to experimentally probe both the forward and reverse hydrogen atom transfers between the 5′-deoxyadenosyl radical and substrate during single-turnover stopped-flow measurements. These data are interpreted within the context of a kinetic model where the 5′-deoxyadenosyl radical intermediate may be quasi-stable and rearrangement of the substrate radical is essentially irreversible. Global fitting of these data allows estimation of the intrinsic rate constants associated with CoC homolysis and initial H-abstraction steps. In contrast to previous stopped-flow studies, the apparent kinetic isotope effects are found to be relatively small.
Keywords: coenzyme B12, enzymes, ethanolamine ammonia lyase, kinetic isotope effects, reaction mechanisms
Introduction
Coenzyme B12-dependent enzymes1, 2 catalyse a range of reactions, including radical rearrangement3 and elimination4, 5 chemistry, and reductive dehalogenation.6 In those enzymes catalysing radical-based chemistry, the unusual Co=C bond to the upper axial ligand of coenzyme B12 (5′-deoxyadenosylcobalamin) breaks homolytically to form a 5′-deoxyadenosyl radical, which then abstracts a hydrogen atom from the substrate.7 In the case of many lyase and mutase enzymes such as ethanolamine ammonia lyase (EAL), the resulting substrate radical undergoes an irreversible rearrangement, after which a hydrogen atom is returned from the 5′-deoxyadenosine to the product radical (Scheme 1).8
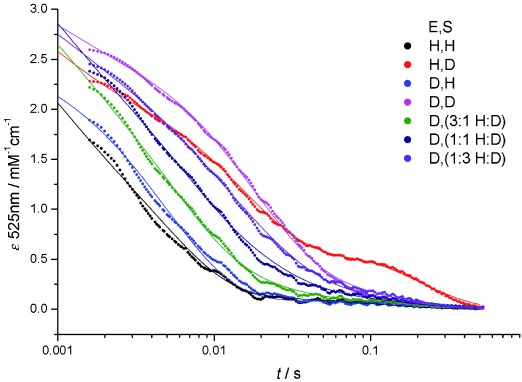
Figure 1.
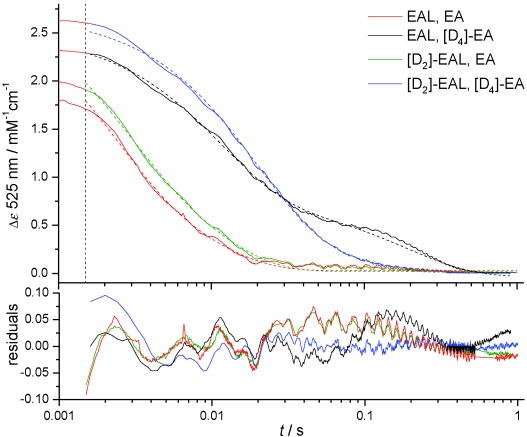
Averaged stopped-flow transients showing the change in EAL or [D2]EAL absorbance at 525 nm upon mixing with saturating concentrations of EA or [D4]EA (0.5 mM post-mixing) at 277 K. The vertical dotted line indicates the stopped-flow dead-time of about 1.5 ms and only data collected at ≥1.5 ms were used for fitting to a double exponential function (dotted lines). Fitting parameters are given in Table 1.
Scheme 1.
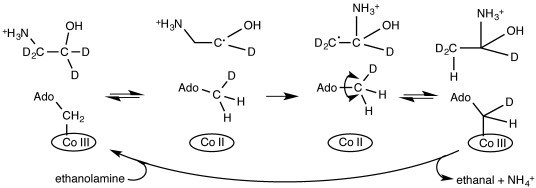
Simplified reaction mechanism of EAL showing how the 5′-deoxyadenosyl Cβ of the intrinsic coenzyme B12 can become deuterated upon (multiple) turnover of the enzyme with deuterated ethanolamine.20 Note that for simplicity, Co=C homolysis and H abstraction from the substrate to 5′-deoxyadenosyl radical are drawn here as a concerted step.
Kinetic isotope effects (KIEs) can uniquely probe the nature of an H-transfer reaction’s transition state, and have been very effectively employed in the study of enzyme mechanism.9 Inflated KIEs also remain the definitive experimental test for the occurrence of quantum mechanical H-tunnelling (QMT)10, 11 and have been used to probe the coupling of protein dynamics to chemistry (for examples see references 12–14). A number of studies have identified apparently inflated primary (1°) KIEs on the H-transfer step(s) that follows Co=C bond homolysis in a number of B12-dependent enzymes.15–19 However, the interpretation and comparison of these experiments is difficult due to the kinetic complexity associated with these multistep, reversible reactions.
Initially, QMT was evoked in B12-dependent enzymatic H-transfer due to the apparent magnitude of primary (1°) deuterium KIEs (ca. 10–40) estimated from simple exponential fitting of stopped-flow data.15–19 The oxidation state change from cob(III)alamin to cob(II)alamin, which occurs during homolysis, is likely to be the only change that can be resolved in the UV–visible region on the stopped-flow timescale.21 Consequently, the observation of a KIE on this signal suggests that there is a significant degree of kinetic coupling between Co=C bond homolysis and subsequent H-abstraction step(s). However, the stopped-flow transients are further complicated by the fact that when enzymes such as glutamate mutase16 and EAL19 are mixed with deuterated substrates, an unusual reaction transient is observed, with what appear to be a burst or lag phase “hump” at longer timescales. If H-abstraction is not the fully rate-limiting of the two coupled steps, there is scope for the intrinsic KIEs to be even larger than those reported. However, studies with glutamate mutase using rapid-quench HPLC and mass spectrometry analysis revealed modest intrinsic 1° H/2H KIEs of 2.4 for l-glutamate22 and 4.1 for methyl aspartate.23 The authors also suggested that the biphasic nature of stopped-flow transients may include contributions from multiple turnovers in the second, “lag” phase, and emphasized the need for caution when analysing such complex data.22 Non-inflated intrinsic 1° KIEs (H/2H KIE<ca. 7) do not preclude a significant QMT contribution to the H transfer,24 and related rapid-quench and mass spectrometry studies suggest a significant QMT contribution in the case of glutamate mutase.25, 26 Isotopic labelling of the 5′-deoxyadenosyl 5′-Cβ with 3H gives a large, inverse secondary (2°) KIE on the coupled homolysis/H-abstraction reaction with protiated substrate (KIE=0.72).27 This 2° KIE approaches unity with deuterated substrate (kHD/kTD=1.05; that is, on a deuterium background), consistent with motion of the secondary hydrogen atoms on the 5′-deoxyadenosyl being coupled to that of the transferring hydrogen in a transition state that involves a significant QMT contribution.25
Clearly there is some uncertainly in the precise kinetic mechanism of these B12-dependent enzymes. Here, we show that by using various combinations of isotopically labelled ethanolamine substrate and EAL-bound B12, it is possible to investigate the coupling and control of Co=C cleavage and H-abstraction steps during the EAL-catalysed pre-steady state reaction using relatively simple stopped-flow spectroscopy.
Results and Discussion
The 5′-deoxyadenosyl group of holoEAL (EAL-B12) was specifically deuterated by treating holo-EAL (apo-EAL freshly reconstituted with B12) with a large excess of deuterated ethanolamine substrate ([D4]EA; Scheme 1) prior to a size-exclusion purification of the B12-deuterated EAL ([D2]EAL). The deuteration state of the 5′-deoxyadenosine is fairly stable8 and the 2H does not appear to wash out if the sample is kept in the dark in the absence of substrate. Nevertheless, subsequent experiments were performed immediately after preparation of [D2]EAL.
Stopped-flow experiments were performed by mixing an excess of EA or [D4]EA with EAL or [D2]EAL (Figure 1). At 298 K the reaction occurs rapidly relative to the stopped-flow mixing time (ca. 1.5 ms),19 so a significant portion of the reaction transient can be lost in the instrument dead-time. This was minimized here by performing all measurements at 277 K. The transients observed upon mixing EAL with [D4]EA are visually similar to previous reports of pre-steady state stopped-flow experiments with B12-dependent enzymes and deuterated substrates,16, 19 displaying a prominent “hump” at longer (ca. 30–500 ms) timescales. This feature is largely abolished in the [D2]EA transients, suggesting that this approach may be a useful method for simplifying the analysis of stopped-flow transients used to measure substrate KIEs on B12-dependent enzymes.1
Initially, transients in Figure 1 were fitted to a double-exponential function, which gives acceptable results except in the case of the long time-scale “hump” observed upon mixing EAL with [D4]EA. Apparent rate constants and observed KIEs are given in Table 1 and Table 2, respectively. There are significant, but modest KIEs observed on the faster rate constant (kobs1), but not the slower kobs2 (data not shown). The magnitude of the KIE observed on kobs1 is much smaller than previous reports,18 but is similar to the observed KIE on the reaction with the slower substrate S-2-aminopropanol.181, 2
Table 1.
| E, S[b] | 2H species | kobs1 [s−1)] | kobs2 [s−1] | fA1[c] |
|---|---|---|---|---|
| H,H | – | 236.1±11.9 | 4.0±1.1 | 0.94 |
| H,D | [D4]EA | 87.2±3.6 | 5.1±0.2 | 0.70 |
| D,H | [D2]EAL | 191.9±5.2 | 5.7±1.4 | 0.93 |
| D,D | [D2]EAL, [D4]EA | 48.2±4.4 | 6.1±3.8 | 0.88 |
Averaged transients are presented in Figure 1. Individual transients (typically ca. 10) were each fitted to a double exponential function with the averages ±1 standard deviation of rate constants presented here.
The deuteration state of the 5′-deoxyadenosyl Cβ (E) and the substrate (S).
The relative fraction of the amplitude of kobs1, that is, A1/(A1+A2).
Table 2.
Observed KIEs on kobs1 values from Table 1.
| E,S/E,Sa | KIEobs1 |
|---|---|
| H,H/H,D | 2.71±0.18 |
| H,H/D,H | 1.23±0.08 |
| H,H/D,D | 4.90±0.51 |
| D,H/D,D | 3.98±0.40 |
| H,D/D,D | 1.81±0.18 |
| (H,D/D,D)/(H,H/D,H) | 1.47±0.24 |
The same nomenclature as Table 1 is used, for example, H,H/H,D is the substrate 1° KIE.
Proton inventory-style stopped-flow experiments were also performed by mixing [D2]EAL with mixtures of EA and [D4]EA (Figure 2). A linear correlation between the observed KIE on kobs1 and [D4]EA concentration was obtained, which may be indicative of a single proton involved in the rate-limiting step.28 However, both kobs1 and kobs2 are apparent rate constants, which are likely to be different convolutions of some/all chemical steps up to, and including, the first irreversible step (on the timescale of the experiment) after Co=C cleavage. In order to better analyse these data and to estimate intrinsic KIE(s), a more realistic kinetic model is required.2
Figure 2.
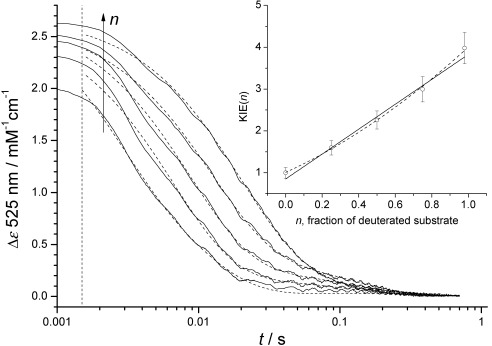
Proton inventory-style stopped-flow experiment performed by mixing [D2]EAL with various mixtures of EA and [D4]EA (0.5 mM total post-mixing concentration) at 277 K. The transients are fitted to a double-exponential function (dotted lines) and the arrow shows the direction of increasing n, the mole fraction of [D4]EA. The observed KIE on kobs1 (inset) versus fraction of deuterated EA (n) fitted to a linear (solid line) and quadratic (dashed line) equation. Rate constants and KIEs are given in Table S1 in the Supporting Information.
A proposed reaction mechanism for the steps observed during the stopped-flow experiments is shown in Scheme 2. As the transients are not affected by decreasing the substrate concentration to low μM concentrations (data not shown), substrate binding is likely to be faster and substrate release much slower than other chemical steps. Consequently, for simplicity we will assume in the following analysis that the EAL⋅EA complex is formed in the experimental dead-time and substrate dissociation and rebinding does not occur on the experimental timescale (ca. 1 s). While only the cob(III)alamin to cob(II)alamin transition is directly observed in UV/Vis stopped-flow experiments (e.g., A to B or A′ to B′ in Scheme 2),21 the resulting transients will be a convolution of the reversible Co=C homolysis (k1) and H atom abstraction (k2) steps, as well as the following irreversible8 substrate radical rearrangement (k3). Thus, in principle, some information can be inferred on the formation and decay of the 5′-deoxyadenosyl radical (e.g., B and B′) and substrate radical (C) intermediates species despite our inability to directly observe these species in our stopped-flow experiment.
Scheme 2.
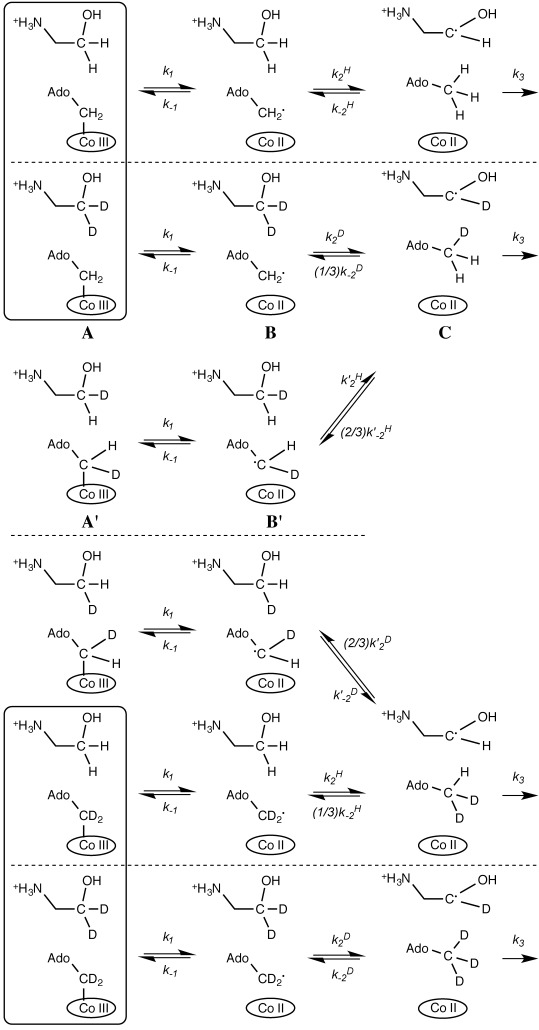
Proposed 3-step reaction mechanism for the pre-steady state reaction of EAL or [D2]EAL with EA and/or [D4]EA, showing Co=C cleavage (k1), H. abstraction (k2) and subsequent irreversible substrate radical rearrangement (k3). In a 2-step mechanism, k1 and k2 are concerted (called k2 in Table 3). The horizontal dotted lines delineate groups of species that cannot interconvert in our kinetic model and those species in boxes are starting species: E,S=H,H, H,D, D,H and D,D, respectively from top to bottom.
A major complication to the reaction mechanism in Scheme 2 is “scrambling” of the deuteration state of the 5′-deoxyadenosine 5′-Cβ in species C and the equivalent species in the EA versus [D2]EAL experiments. As the rotation of the 5′-methyl group is likely to be much faster than the pre-steady state chemistry, both deuterium and protium radicals can be transferred back from the 5′-deoxyadenosine to the substrate radical (via k−2 or k′−2) when [D4]EA and EAL or EA and [D2]EAL react (Scheme 2). This reaction branching will generate two different isotopologues of species A and B in each case. If substrate dissociation and rebinding are not kinetically relevant, further branching is restricted by the stereospecificity of the H. abstraction, as only the pro-S hydrogen of EA is expected to transfer to/from the 5′-deoxyadenosyl species.29, 30 A KIE will be observed on the branching as only one of the k−2 steps involves a deuterium transfer. Further, the apparent rate will be reduced by the fraction of reacting species: if methyl rotation is much faster than H transfer, then during a single turnover, 1/3 of the 5′-deoxyadenosine Cβ hydrogen atoms will be deuterium for the reaction of [D4]EA versus EAL or 2/3 for EAL versus [D2]EAL.
To investigate whether intrinsic rate constants can be estimated from the stopped-flow experiments, the combined data in Figures 1 and 2 were reanalysed using a complete, 3-step kinetic model describing Scheme 2 (model details given in the Experimental Section). A 2-step model, where Co=C cleavage and H abstraction from the substrate by 5′-deoxyadenosyl are concerted, was also considered. The data were globally fit (Figure 3 and Figure S1 in the Supporting Information) with shared rate constants.
Figure 3.
Global fitting of the resampled data taken from Figures 1 and 2 to the 3-step mechanism described in Scheme 2. Fitting parameters are given in Table 3 and Table S2 in the Supporting Information and more detail is given in the Experimental Section.
The 2-step and 3-step models give fits that are essentially indistinguishable over the experimental timescale (Figure S1 in the Supporting Information), and tabulated fitting parameters are given in Table 3 and Table S2 in the Supporting Information. While it is not necessary to describe the stopped-flow transients using a (3-step) model where the 5′-deoxyadenosyl radical is a bone fide intermediate, our data do not rule this out and suggest that in either case there is tight kinetic coupling of Co=C homolysis (k1) and initial H abstraction (k2) reactions. An experimental approach that can directly detect the 5′-deoxyadenosyl and/or substrate radical (e.g., time-resolved EPR)31, 32 is likely required to definitely determine whether the adenosyl radical is a stable intermediate under these conditions.3
Table 3.
Global fitting parameters from Figure 3.[a]
| 2-step | 3-step | |
|---|---|---|
| k1 | – | 562±61 |
| k−1 | – | 582±83 |
| k2 | 208.8±1.4[b] | 497±68 |
| k′2 | 19.6±0.9 | 45.0±4.7 |
| k −2 | 16.9±1.7 | 24.3±2.6 |
| k′−2 | 11.2±0.3 | 11.0±0.3 |
| k3 | 12.2±0.4 | 10.3±0.4 |
| KIE2[c] | 3.9±0.1 | 4.6±0.2 |
| KIE−2 | 2.2±0.3 | 3.7±0.4 |
| KIE′−2[d] | 39.4±6.1 | 37.5±5.2 |
[a] Full fitting parameters for the 2-step and 3-step models are given in Table S2 in the Supporting Information. [b] All rates constants in s−1. [c] This KIE is shared between the k2 and k′2 rate constants. [d] This KIE is not likely to be intrinsic. See the text for more details.
During the global fitting of the data in Figure 3, it was possible to obtain good fits if the value of the KIE on k2 and k′2 is shared (KIE2 in Table 3). This approach ignores the effect of α- and β-secondary isotope effects arising from deuteration of the substrate and 5′-deoxyadenosine, respectively. However, these are expected to be much smaller than the 1° KIE and are not expected to be reliably resolved in our stopped-flow experiments. There is no significant difference between the magnitudes of the shared KIE2 (ca. 4) determined using the 2-step and 3-step models and the value of this KIE is in good agreement with that obtained by fitting the [D4]EAL versus [D2]EA transients to a double-exponential function (Table 2). The KIE on the reverse H transfer (KIE−2) is also of a similar magnitude (ca. 2–4) to KIE2 and it appears that the KIEs on H transfer between the 5′-deoxyadenosyl and substrate radicals in EAL are likely to be less than the semi-classical limit of about 7.10 Consequently, there is no direct evidence from these values for QMT during the H atom transfer from substrate to the 5′-deoxyadenosyl radical.
The global fitting in Figure 3 did not converge unless the KIEs on k−2 and k′−2 were allowed to differ. The observed KIE on k′−2 (KIE′−2) appears to be about 40, whereas KIE−2 is 2–4 (Table 3). It is quite likely that Scheme 2 oversimplifies the “scrambling” reaction by describing a convolution of the return hydrogen transfer with slower active-site rearrangement (and fast methyl rotation). Indeed, we have previously shown that the active site glutamate287 residue may reorient during turnover to control the path of the 5′-deoxyadenosyl radical before binding the substrate.33–35 If the reorientation of E287 occurs on a slower timescale than H transfer (during k−2 and/or k′−2) then these protein dynamics could effectively gate the H radical transfer steps. As protein dynamics are expected to be isotope-insensitive, gating should reduce the magnitude of the apparent KIE. Due to these complications, we cannot confidently determine the absolute magnitude of the KIE on the return H atom transfer from substrate radical to 5′-deoxyadenosine. However, the global fitting does give insight into the origin of the long timescale “hump” observed in the [D4]EA versus EAL transients.
In Figure 4 the formation and decay of the 5′-deoxyadenosyl radical species is modelled using the best-fit parameters for the 3-step model (Table 3 and Table S2 in the Supporting Information). The “hump” is seen to arise from the formation of the “scrambled” 5′-deoxyadenosyl radical (B′ and equivalent species in Scheme 2). In the 2-step model, the equivalent mechanism (H or D atom return from substrate radical to 5′-deoxyadeonine) also reproduces this “hump” feature. Consequently, the long timescale “hump” in transients observed upon mixing other B12-enzymes with deuterated substrates (or vice versa) is likely to arise from reversibility in the H-transfer from substrate to 5′-deoxyadenosyl radical, rather than from multiple turnovers of the enzyme. Deuteration of the B12 Cβ prior to stopped-flow KIE measurements minimises the effect of the “scrambling” as this can only occur upon mixing with EA, not [D4]EA, and the extent of the branching to A′ and B′ is minimised by the KIE on k′2.4
Figure 4.
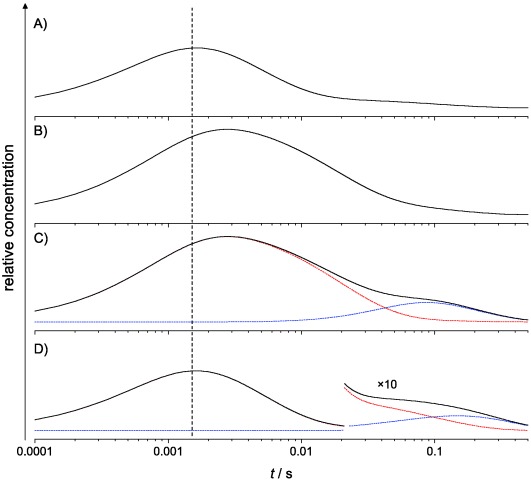
Apparent evolution of the 5′-deoxyadenosyl radical concentration modelled from the best-fit parameters for the 3-step model fitting in Figure 3. Transients are for: A) EA versus EAL, B) [D4]EA versus [D2]EAL, C) [D4]EA versus EAL, and D) EA versus [D2]EAL. In C and D the black transients are convolutions of the contributions from the initial H or D abstraction (red) and the “scrambled” partially deuterated 5′-deoxyadenosyl radical (blue). The vertical dotted line indicates the stopped-flow dead-time of about 1.5 ms.
Conclusion
We have selectively deuterated the Cβ of the 5′-deoxyadenosyl moiety of the intrinsic coenzyme B12 in EAL prior to stopped-flow KIE measurements with [D4]EA. This approach significantly simplifies the analysis of the resulting transients and appears to allow reliable 1° KIEs to be determined from simple (multi)exponential fitting of the data. Global fitting of stopped-flow transients to kinetic models allows the estimation of some intrinsic rate constants and KIEs on the reversible H-transfer steps preceding substrate radical rearrangement in EAL. No direct evidence for QMT during H-transfer was found (i.e., the KIEs are not inflated). The data can also be described by a 3-step model where Co=C bond homolysis and H-abstraction from the substrate occur sequentially, consistent with recent computational studies on a coenzyme B12-dependent mutase.36 This approach allows us to model the concentration of the 5′-deoxyadenosyl radical over the course of a single enzyme turnover, and the “hump” observed in some stopped-flow transients is shown to arise from “scrambling” of the deuteration state of the 5′-deoxyadenosyl moiety.
Experimental Section
All reagents were analytical grade and purchased from Sigma–Aldrich (Gillingham, UK) except for [D4]ethanolamine, which was purchased from Goss Scientific Instruments Limited (Crewe, UK). Recombinant EAL from Salmonella enterica was prepared as described previously.19, 34 Holo-EAL was prepared freshly on the day of experiments by reconstituting EAL with excess coenzyme B12 prior to removal of unbound B12 by size-exclusion chromatography using a 10 DG desalting column (BioRad, Hemel Hempstead, UK). [D2]EAL was prepared by treating 10–20 μM of holo-EAL with 2 mM [D4]EA for 10 min prior to separation of products and unreacted substrate with a 10 DG desalting column. [D2]EAL was then brought to the desired concentration for stopped-flow using 10 kDa molecular weight cut-off centrifugal filter devices. All preparations were performed on ice in the dark, while all kinetic measurements were performed in 20 mM HEPES pH 7.5 at 277 K. Stopped-flow experiments were performed essentially as described previously19 using an Applied Photophysics (Leatherhead, UK) SX.18 MV-R stopped-flow spectrophotometer with approximately 30 μM EAL and 500 μM ethanolamine post-mixing. The absorbance change accompanying interconversion between cob(III)alamin and cob(II)alamin was monitored at 525 nm.
The stopped-flow transients in Figures 1 and 2 were globally fit to the kinetic model shown in Scheme 2. To ensure even weighting, transients for each experiment were averaged (as shown in Figures 1 and 2) and then linearly interpolated before resampling. For each transient, 200 data points were taken at equal distance over a logarithmic timescale between 1.6 and 525 ms using Mathematica 9.0 (Wolfram Research, Inc., Champaign, IL). COPASI37 was used for model construction and data fitting.
The [D4]EA versus EAL reaction in Scheme 2 is described by:
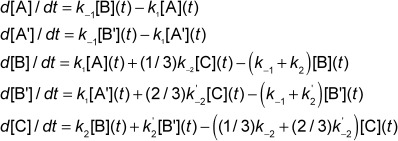 |
1 |
The other reactions were modelled similarly, and the EA versus EAL and [D4]EA versus [D2]EAL reactions are simplified by the lack of “scrambling” and thus do not contain A′ and B′ species. The global model contains four each A, B and C species and two each A′ and B′ species.
A 2-step mechanism where homolysis and H abstraction are concerted is similarly described by:
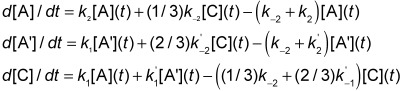 |
2 |
In this case, there are four each A and C species and two A′ species.
To globally fit the seven transients in Figures 1 and 2 to either model, all species were treated locally while the rate constants were shared globally. Three global KIEs were fitted for, as described in the main text. A single global extinction coefficient for the sum of all six A and A′ species in each experiment was fitted for, but the relative starting concentration of each species was allowed to vary between 0.7–1.3 across each experiment to allow for uncertainty in active enzyme concentration. The start time was also allowed to vary by ±2 ms to allow for variation in mixing times. Best-fit values are given in Table S2 in the Supporting Information.
Acknowledgments
S.H. is a Biotechnology and Biological Sciences Research Council (BBSRC) David Phillips Fellow (BB/H021523/1). N.S.S. is a Royal Society Wolfson Research Merit Award holder and an Engineering and Physical Sciences Research Council Established Career Fellow in Catalysis (EP/J020192/1). J.R. was funded by a BBSRC studentship.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/chem.201500958.
References
- [1].Reed GH. Curr. Opin. Chem. Biol. 2004;8:477–483. doi: 10.1016/j.cbpa.2004.08.008. [DOI] [PubMed] [Google Scholar]
- [2].Brown KL. Chem. Rev. 2005;105:2075–2149. doi: 10.1021/cr030720z. [DOI] [PubMed] [Google Scholar]
- [3].Banerjee R. Chem. Rev. 2003;103:2083–2094. doi: 10.1021/cr0204395. [DOI] [PubMed] [Google Scholar]
- [4].Toraya T. Chem. Rev. 2003;103:2095–2127. doi: 10.1021/cr020428b. [DOI] [PubMed] [Google Scholar]
- [5].Toraya T. Arch. Biochem. Biophys. 2014;544:40–57. doi: 10.1016/j.abb.2013.11.002. [DOI] [PubMed] [Google Scholar]
- [6].Payne KA, Quezada CP, Fisher K, Dunstan MS, Collins FA, Sjuts H, Levy C, Hay S, Rigby SE, Leys D. Nature. 2015;517:513–516. doi: 10.1038/nature13901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Marsh ENG, Patterson DP, Li L. ChemBioChem. 2010;11:604–621. doi: 10.1002/cbic.200900777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Weisblat DA, Babior BM. J. Biol. Chem. 1971;246:6064–6071. [PubMed] [Google Scholar]
- [9].Kohen A, Limbach HH. Isotope Effects In Chemistry and Biology. CRC; 2006. [Google Scholar]
- [10].Bell R. The Tunnel Effect in Chemistry. Chapman and Hall; 1980. [Google Scholar]
- [11].Allemann RK, Scrutton NS. Quantum Tunnelling in Enzyme-Catalysed Reactions. Cambridge: RSC; 2009. [Google Scholar]
- [12].Hay S, Scrutton NS. Nat. Chem. 2012;4:161–168. doi: 10.1038/nchem.1223. [DOI] [PubMed] [Google Scholar]
- [13].Klinman JP. Acc. Chem. Res. 2015;48:449–456. doi: 10.1021/ar5003347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kohen A. Acc. Chem. Res. 2015;48:466–473. doi: 10.1021/ar500322s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Padmakumar R, Padmakuma R, Banerjee R. Biochemistry. 1997;36:3713–3718. doi: 10.1021/bi962503g. [DOI] [PubMed] [Google Scholar]
- [16].Marsh ENG, Ballou DP. Biochemistry. 1998;37:11864–11872. doi: 10.1021/bi980512e. [DOI] [PubMed] [Google Scholar]
- [17].Chowdhury S, Banerjee R. J. Am. Chem. Soc. 2000;122:5417–5418. [Google Scholar]
- [18].Bandarian V, Reed GH. Biochemistry. 2000;39:12069–12075. doi: 10.1021/bi001014k. [DOI] [PubMed] [Google Scholar]
- [19].Jones AR, Hay S, Woodward JR, Scrutton NS. J. Am. Chem. Soc. 2007;129:15718–15727. doi: 10.1021/ja077124x. [DOI] [PubMed] [Google Scholar]
- [20].Sato K, Orr JC, Babior BM, Abeles RH. J. Biol. Chem. 1976;251:3734–3737. [PubMed] [Google Scholar]
- [21].Hollaway MR, White HA, Joblin KN, Johnson AW, Lappert MF, Wallis OC. Eur. J. Biochem. 1978;82:143–154. doi: 10.1111/j.1432-1033.1978.tb12005.x. [DOI] [PubMed] [Google Scholar]
- [22].Cheng M-C, Marsh ENG. Biochemistry. 2005;44:2686–2691. doi: 10.1021/bi047662b. [DOI] [PubMed] [Google Scholar]
- [23].Yoon M, Kalli A, Lee H, Håkansson K, Marsh ENG. Angew. Chem. Int. Ed. 46:8455–8459. doi: 10.1002/anie.200702448. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007;119 [Google Scholar]
- [24].Pang J, Hay S, Scrutton NS, Sutcliffe MJ. J. Am. Chem. Soc. 2008;130:7092–7097. doi: 10.1021/ja800471f. [DOI] [PubMed] [Google Scholar]
- [25].Cheng M-C, Marsh ENG. Biochemistry. 2007;46:883–889. doi: 10.1021/bi0616908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Yoon M, Song H, Hakansson K, Marsh ENG. Biochemistry. 2010;49:3168–3173. doi: 10.1021/bi1001695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Cheng M-C, Marsh ENG. Biochemistry. 2004;43:2155–2158. doi: 10.1021/bi036122w. [DOI] [PubMed] [Google Scholar]
- [28].Venkatasubban KS, Schowen RL. CRC Critical Reviews in Biochemistry. 1984;17:1–44. doi: 10.3109/10409238409110268. [DOI] [PubMed] [Google Scholar]
- [29].Diziol P, Haas H, Retey J, Graves SW, Babior BM. Eur. J. Biochem. 2005;106:211–224. doi: 10.1111/j.1432-1033.1980.tb06012.x. [DOI] [PubMed] [Google Scholar]
- [30].Shibata N. J. Biol. Chem. 2010;285:26484–26493. doi: 10.1074/jbc.M110.125112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Zhu C, Warncke K. J. Am. Chem. Soc. 2010;132:9610–9615. doi: 10.1021/ja907769g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wang M, Warncke K. J. Am. Chem. Soc. 2013;135:15077–15084. doi: 10.1021/ja404467d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Jones AR, Hardman SJO, Hay S, Scrutton NS. Angew. Chem. Int. Ed. 50:10843–10846. doi: 10.1002/anie.201105132. [DOI] [PubMed] [Google Scholar]
- [34].Russell HJ, Jones AR, Hay S, Greetham GM, Towrie M, Scrutton NS. Angew. Chem. Int. Ed. 51:9306–9310. doi: 10.1002/anie.201202502. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012;124 [Google Scholar]
- [35].Jones AR, Levy C, Hay S, Scrutton NS. FEBS J. 2013;280:2997–3008. doi: 10.1111/febs.12223. [DOI] [PubMed] [Google Scholar]
- [36].Bucher D, Sandala GM, Durbeej B, Radom L, Smith DM. J. Am. Chem. Soc. 2012;134:1591–1599. doi: 10.1021/ja207809b. [DOI] [PubMed] [Google Scholar]
- [37].Hoops S, Sahle S, Gauges R, Lee C, Pahle J, Simus N, Singhal M, Xu L, Mendes P, Kummer U. Bioinformatics. 2006;22:3067–3074. doi: 10.1093/bioinformatics/btl485. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.